

Abstract
Background
Dementia with Lewy bodies (DLB) is the second most common subtype of neurodegenerative dementia in humans following Alzheimer’s disease (AD). Present clinical diagnosis of DLB has high specificity and low sensitivity and finding potential biomarkers of prodromal DLB is still challenging. MicroRNAs (miRNAs) have recently received a lot of attention as a source of novel biomarkers.
Methods
In this study, using serum miRNA expression of 478 Japanese individuals, we investigated potential miRNA biomarkers and constructed an optimal risk prediction model based on several machine learning methods: penalized regression, random forest, support vector machine, and gradient boosting decision tree.
Results
The final risk prediction model, constructed via a gradient boosting decision tree using 180 miRNAs and two clinical features, achieved an accuracy of 0.829 on an independent test set. We further predicted candidate target genes from the miRNAs. Gene set enrichment analysis of the miRNA target genes revealed 6 functional genes included in the DHA signaling pathway associated with DLB pathology. Two of them were further supported by gene-based association studies using a large number of single nucleotide polymorphism markers (BCL2L1: P = 0.012, PIK3R2: P = 0.021).
Conclusions
Our proposed prediction model provides an effective tool for DLB classification. Also, a gene-based association test of rare variants revealed that BCL2L1 and PIK3R2 were statistically significantly associated with DLB.
Keywords: Dementia with Lewy bodies, Risk prediction model, microRNAs, Single nucleotide polymorphism
Background
Dementia with Lewy bodies (DLB) is the second most common subtype of neurodegenerative dementia in humans following Alzheimer’s disease (AD) [1] and accounts for around 4.6% of all dementia cases [2]. The main pathological lesions in DLB are Lewy bodies and neurites, containing abnormal α-synuclein (α Syn) [3]. The characteristic features of DLB are different to those in AD, with less marked memory impairment and more severe impairments of visuospatial, attentional and frontal-executive functions [3].
Present clinical diagnosis of DLB has high specificity and low sensitivity [4], and finding DLB patients in the prodromal phase is still challenging. An accurate diagnosis of DLB at the prodromal stage would be an important advance in the pharmacological management, as cholinesterase inhibitors (ChEIs) have good responsiveness for patients with Lewy body dementia (LBD) including DLB and Parkinson’s disease dementia (PDD), although a careful monitoring of treatment compliance and side effects is required [5]. Therefore, as potential biomarkers for prodromal DLB are required in clinical implication, our findings might enable DLB to be one of the most treatable neurodegenerative disorders.
MicroRNAs (miRNAs) are small non-coding RNAs, which play key roles in many biological or pathological processes by regulating the expression of their target transcripts. Previous studies have reported that alterations in miRNA expression have be associated with several neurodegenerative diseases [6–8]. We also reported potential biomarkers for earlier diagnosis and therapeutic intervention through comprehensive miRNA expression analyses and constructed a risk prediction model using the biomarkers based on supervised principal component analysis (PCA) logistic regression, a machine learning (ML) method [9].
Current studies for disease prediction models implemented several ML methods. For example, Lebedev et al. reported a random forest model that predicted MCI-to-AD conversion with high accuracy using morphometric measures from 3D brain MRI images and clinical information [10]. Wei et al. developed a promising support vector machine method that detected persons with diabetes and pre-diabetes using comprehensive clinical information [11]. We have also reported several efficient risk prediction models for not only type II diabetes based on a penalized regression method (LASSO) incorporating clinical information and genetic data [12], but also postoperative overall survival and disease-free survival in patients with breast cancer based on a Cox proportional hazard model [13]. However, there is, as of yet, no clear consensus of which ML method is most appropriate for application to disease prediction models.
Here, we applied several ML methods to comprehensive miRNA expression data of serum samples, composed of DLB patients and individuals with cognitive normal function (referred to as normal controls: NC) and investigated an optimal risk prediction model from among these ML methods. We used 10-fold cross-validation on a training set consisting of half of the Japanese individuals, separated from a test set. We constructed risk prediction models using four ML methods, penalized regression [14–16], random forest (RF) [17], support vector machine (SVM) [18], and gradient boosted decision tree (GBDT) [19], and evaluated the predictive performance of the ML models on an independent test set. This final model based on GBDT showed better accuracy than the model based on supervised PCA logistic regression constructed in our previous studies [9].
Methods
Clinical samples
All of 457 serum subjects and their clinical data were obtained from the National Center for Geriatrics and Gerontology (NCGG) Biobank. The total set of subjects was composed of 169 DLB patients and 288 normal control (NC) subjects with normal cognitive function [9]. The DLB subjects were diagnosed on basis of the criteria of the fourth report of the DLB Consortium [20]. The NC subjects were confirmed with a Mini-Mental State Examination (MMSE) score ≥ 27. The APOE ε4 genotype and MMSE score of all subjects were available. These miRNA expression data are publicly available through the Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information (GSE120584, http://www.ncbi.nlm.nih.gov/projects/geo/).
Sixty-nine DLB cases and 2008 NCs used in the genetic association studies were also selected from the subjects enrolled in the NCGG Biobank. All subjects were ≥ 60 years in age and were genotyped using Japonica arrays [21]. We excluded all SNPs with a genotype call rate < 0.99, a Hardy-Weinberg equilibrium p-value < 1.0 × 10−3 in NCs or a minor allele frequency (MAF) < 0.01.
Target gene annotation using miRNAs
The target genes of miRNAs were determined using the microRNA Target Prediction and Functional Study Database (miRDB version 5.0, [22], where MirTarget V3 predicted the miRNA-target genes with a prediction score in the range between 0 and 100. In this study, target genes with a score of > 90 were used in further gene-based association studies.
Parameter selection in several machine learning methods
Top-ranked miRNAs were detected using a logistic regression method after adjustment for age, sex and APOE ε4 genotype on the training set. All ML model optimizations were performed against each pre-selected top-ranked p miRNA using 10-fold cross-validation on the training set. The hyper-parameter optimizations were implemented in the scikit-learn library (version 0.19.1) in Python. Precision, recall, F-measure, and accuracy were used to evaluate the four ML methods: penalized regression, RF, SVM, GBDT. The precision, recall, F-measure, and accuracy were calculated using the counts of true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN) as follows:
Random forest (RF)
Using the best prediction model based on random forest method, we investigated the optimal combination of the following five hyper-parameters: maxdepth, ntree, minsplit, minsample, and criterion. The maxdepth parameter is maximum number of levels in each decision tree (maxdepth = 2, 3, 4, 5). The ntree parameter is the number of decision trees (ntree = 100, 200, 300, 400, 500, 600, 700, 800, 900, and 1000). The minsplit parameter is minimum number of data points placed in a node before the node is split (minsplit = 2, 3, 5, 10, 15, 20). The minsample parameter is the minimum number of samples in a leaf (minsample = 1, 3, 5). The criterion parameters, ‘gini impurity’ and ‘information gain entropy’, are used for splitting the data.
Penalized regression
We implemented ridge regression [14], elastic net [15] and lasso methods [16], known as penalized regression methods. The phenotype of subject i = 1, ⋯, n was set as the dependent variables (case = 1, control = 0) and the expression Xi, j of each miRNA j = 1, ⋯, m for a subject i. Let Xi = (X1, ⋯, Xp) be the values of pre-selected top-ranked p miRNA for a subject i and let l(β; γi, Xi) be the logistic log-likelihood:
where , and α was set to 1 for lasso, 0 for ridge regression, and 0 to 0.9 at 0.1 intervals for elastic net, and λ are selected using 10-fold cross-validation. For the best prediction model based on penalized regression methods, we investigated optimal combinations of above two hyper-parameters: α and λ.
Support vector machine (SVM)
To construct prediction models, we applied a support vector machine method with the radial basis function (RBF) kernel defined as:
We investigated the optimal combinations of the above two hyper-parameters: gamma (γ) and cost (C). The parameters C and γ affect model complexity and model smoothness, respectively. Increasing C and γ cause over-fitting, and range of those parameters we implemented were C = {2−15, 2−14, …, 214, 215} and γ = {2−15, 2−14, …, 214, 215}. The terms β and were then coefficients of classifier resulting from a separating hyperplane and the amount of misclassified data.
Gradient boosting decision tree (GBDT)
Significantly different from the similar ensemble method, random forest, the tree-based models of GBDT were trained sequentially, and each base model was updated to correct the error produced by its previous tree models, called a learning rate. To optimize parameters in this method, we examined combinations of the following five hyper-parameters: maxdepth, ntree, minsplit, minsample, and learning rate. The first four parameters were the same as those used in random forest. The learning rate was optimized across {0.001, 0.01, 0.05, 0.1, 0.2}.
Results
Data collection of Japanese individuals
We split the 457 Japanese individuals (169 DLB cases, 288 controls) into a training set of 229 individuals (85 DLB cases, 144 controls) and a test set of 228 individuals (84 DLB cases, 144 controls). This separation was performed to result in a similar distribution in the age between the training and test sets (Table 1).
Table 1.
Average age, sex and APOE ε4 genotype information in the training and test data
| Training data set | Test data set | |||||||
|---|---|---|---|---|---|---|---|---|
| Phenotype | #Sample | Age | Sex (Male) | APOE ε4a | #Sample | Age | Sex (Male) | APOE ε4a |
| DLB | 85 | 79.5 | 0.45 | 0.34 | 84 | 79.5 | 0.36 | 0.30 |
| NC | 144 | 71.7 | 0.49 | 0.22 | 144 | 71.8 | 0.56 | 0.15 |
aAPOE ε4 shows the average of the number of APOE ε4 genotype
Comparison of classifier performance
All approaches were performed using a data set of the p most significant miRNAs in a stepwise manner (p ≤ 500). The most significant miRNAs (top-ranked miRNAs) were determined in nine-tenths of entire training set using a logistic regression method. Note that top-ranked miRNAs were determined for each cross-validation step. The adjusted model was constructed using the nine-tenths of the training set and was evaluated using the remaining one-tenth. Four ML methods, penalized regression (ridge regression, elastic net and least absolute shrinkage and selection operator: LASSO), RF, SVM, and GBDT, were used for model construction. Using 10-fold cross validation estimation, we determined the optimal number of miRNAs for the final model construction for each ML method (Fig. 1). Final models were constructed using the complete training set. The number of top-ranked miRNAs and the tuning hyper-parameters are shown in Table 2. The adjusted models constructed with the entire training set were then evaluated on a completely independent test set (Fig. 2). Among the four MLs, a final risk prediction model based on the GBDT method achieved the highest accuracy of 0.829 when pre-selecting the top-ranked 216 miRNAs and three clinical features. The other methods were 0.825 for penalized regression with 434 miRNAs, 0.820 for SVM with 27 miRNAs, and 0.789 for RF with 60 miRNAs (Table 2 and Fig. 2). The hyper-parameters used in the final risk prediction model with GBDT were then optimized: (maxdepth, ntree, minsplit, minsample, learning rate) = (4, 200, 20, 5, 0.1) (see the Methods).
Fig. 1.

Outline of the risk prediction model construction and the validation
Table 2.
Hyperparameter values in each final model
| Method | #top-ranked miRNA | Hyperparameter | Value |
|---|---|---|---|
| Penalized regression | 434 | α | 0.1 |
| λ | 0.10882 | ||
| RF | 60 | max depth | 4 |
| n tree | 100 | ||
| min split | 10 | ||
| min sample | 3 | ||
| SVM | 27 | C | 2.14355 |
| γ | 0.001122 | ||
| GBDT | 216 | max depth | 4 |
| n tree | 200 | ||
| min split | 20 | ||
| min sample | 5 | ||
| learning rate | 0.1 |
Fig. 2.

Precision, Recall, F-measure, and Accuracy values listed four ML methods. Performance of four ML methods on training (a) and test (b) data
We also constructed a GBDT risk prediction model using another feature selection algorithm, μHEM [23], publicly available at http://www.isical.ac.in/~bibl/results/mihem/mihem.html, and investigated whether this feature selection methodology can further improve the predictive ability of our model. The GBDT risk prediction model was performed using a data set of the top-ranked p miRNAs in a stepwise manner (p ≤ 500). This final risk prediction model using μHEM algorithm achieved an accuracy of 0.803 on an independent test set when pre-selecting the top-ranked 330 miRNAs and three clinical features. Although the final risk prediction model showed a lower accuracy than that using a logistic regression method (Additional file 1: Table S1), implementation of feature selection algorithms might contribute to further improvement of the GBDT risk prediction model.
We also compared the run time of the four ML methods on a 7-core Intel Xeon 2.40GHz CPU with 256 GB of memory. While runtimes for SVM and RF were independent of the number of top-ranked miRNAs used, the runtimes for GBDT and penalized regression increased with the number of top-ranked miRNAs. SVM was the fastest ML method, which spent 1.5 min to construct the risk prediction model when using top-ranked 500 miRNAs. RF took 30 min, GBDT took 2.2 h, and penalized regression took 2.9 h (Fig. 3). This result shows that difficulties may arise when implementing GBDT and penalized regression with larger numbers of top-ranked miRNAs in the risk prediction model construction compared with SVM and RF.
Fig. 3.
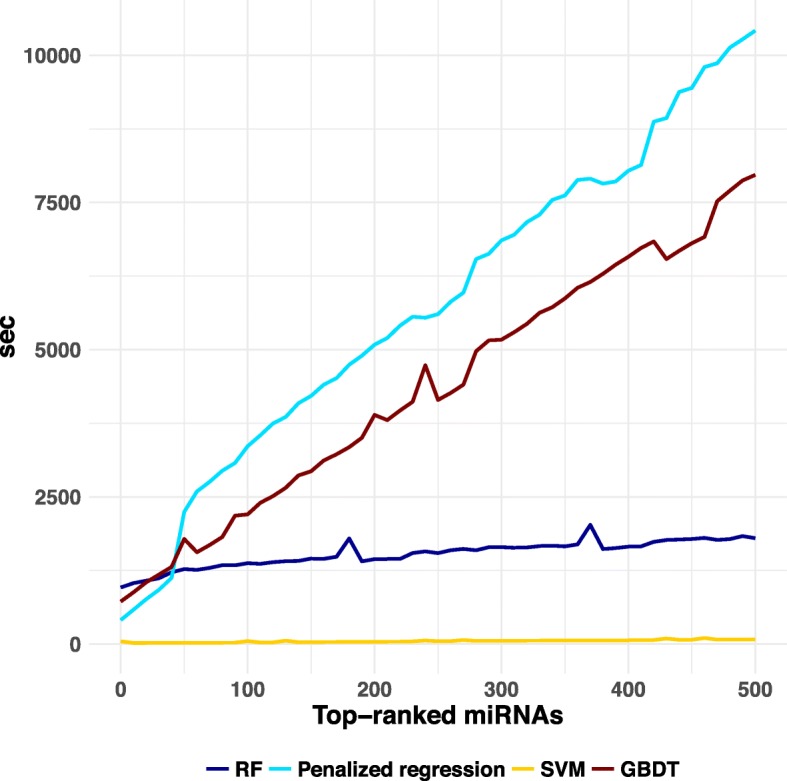
Runtimes of four ML methods
Effective features used in risk prediction model
The final GBDT risk prediction model was constructed by pre-selecting the 216 top-ranked miRNAs and three clinical features (‘age’, ‘APOE ε4 genotype’, and ‘sex’). Of the 219 features, 182 were used in the final risk prediction model construction as effective features with a feature importance > 0 (180 miRNAs and 2 clinical features: ‘age’ and ‘APOE ε4 genotype’) (Additional file 2: Table S2).
To examine the biological significance of our findings (180 miRNAs), we further predicted the miRNA functional target genes using miRDB [22]. The miRNAs were predicted to target 4119 genes (see the Methods), of which 423 genes were predicted by the 7 miRNAs (MIMAT0014984, MIMAT0027624, MIMAT0016852, MIMAT0023713, MIMAT0019849, MIMAT0022491, and MIMAT0007882) with a feature importance > 0.015 in the final GBDT risk prediction model (Additional file 2: Table S2). The rank of miRNAs’ feature importance in the final GBDT risk prediction model was also correlated with that of miRNAs chosen by the logistic regression method (Spearman’s ρ =0.21 and p-value = 0.006).
Functional pathways using gene set enrichment analysis (GSEA)
In order to elucidate any enrichment of functional units or categories, we applied GSEA to the 423 target genes of the 7 miRNAs described above. GSEA was performed using Ingenuity Pathways analysis software (IPA; Ingenuity Systems). We identified six statistically significant canonical pathways: protein kinase A signaling (21 genes), ERK/MAPK signaling (14 genes), molecular mechanisms of cancer (20 genes), p38 MAPK signaling (10 genes), glucocorticoid receptor signaling (18 genes), and docosahexaenoic acid (DHA) signaling (6 genes), with a q-value < 0.05 (Table 3 and Additional file 3: Table S3). One of them, the DHA signaling pathway (Fig. 4), has been reported to be associated with DLB pathology; high levels of alpha-synuclein oligomers were induced by high levels of DHA in vitro and in vivo [24]. This result suggests that six genes (PNPLA2, PIK3C2B, PIK3R2, GSK3A, GSK3B, and BCL2L1) included in this DHA signaling pathway, could be associated with DLB pathology (Fig. 4).
Table 3.
Canonical pathways associated with DLB pathology
| Canonical pathway | #genes related | q-value |
|---|---|---|
| protein kinase a signaling | 21 | 7.08E-3 |
| ERK/MAPK signaling | 14 | 7.08E-3 |
| molecular mechanisms of cancer | 20 | 7.6E-3 |
| p38 MAPK signaling | 10 | 7.6E-3 |
| glucocorticoid receptor signaling | 18 | 8.92E-3 |
| docosahexaenoic acid (DHA) signaling | 6 | 3.19E-2 |
Fig. 4.

Docosahexaenoic acid (DHA) signaling pathway detected by GSEA. The DHA signalling pathway was generated through the use of IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis)
Gene-based association studies using large numbers of SNP markers
To check the genetic associations of the six genes in the DHA signaling pathway, we examined genetic differences using single nucleotide polymorphism (SNP) markers from 69 DLB cases and 2008 controls. A gene-based association test of rare variants, SNP-set Kernel Association Test (SKAT) [25], was applied to the gene coding sequence six genes including 1 Mb of sequence up and downstream, since expression quantitative trait loci (eQTL) SNPs [26] have a major effect on gene expression regulation. Several thousand SNPs were used for the association tests, which showed statistically significant association with BCL2L1 (p-value = 0.012) and PIK3R2 (p-value = 0.021) (Table 4). Furthermore, the expression of these two genes was observed in several brain tissues in the Genotype-Tissue Expression (GTEx) database [27].
Table 4.
Gene-based association studies for the six genes in the DHA signaling pathway
| Gene on pathway | Gene symbol | #SNPs | p-value |
|---|---|---|---|
| PNPLA | PNPLA2 | 376 | 0.059 |
| PI3K | PIK3C2B | 560 | 0.915 |
| PIK3R2 | 364 | 0.021* | |
| GSK3 | GSK3A | 174 | 0.371 |
| GSK3B | 429 | 0.451 | |
| BCL-XL | BCL2L1 | 135 | 0.012* |
*statistically significant association
Discussion
Early diagnosis and therapeutic intervention could prevent severe disease manifestations in patients suffering from several diseases including DLB, and miRNAs have attracted a lot of attention as novel biomarkers [28–30]. In fact, risk prediction models using miRNA biomarkers have been developed for early diagnosis prediction in several types of dementia [9], including sporadic AD [31], as well as cancers [32, 33]. However, more accurate prediction models are required for practical clinical use.
To construct a more accurate risk prediction model for DLB, we, in this study, used comprehensive miRNA expression data of serum samples and applied several ML methods. We investigated which combination of ML method and miRNA sets resulted in the best predictive model. We found that the GBDT method achieved the highest accuracy among four ML methods examined, although the performance of all ML methods was similar (Fig. 2). Other studies have recently reported the powerful classification performance of GBDT [34], which produces a prediction model in the form of an ensemble of weak prediction models (decision trees), constructs the model in a stage-wise fashion, and generalizes them by allowing optimization of an arbitrary differentiable loss function. It has also been reported that for this ML model, the arbitrary customization of the loss function contributes to the recent success of prediction models [35].
Our final GBDT risk prediction model was constructed using 216 pre-selected top-ranked miRNAs, selected through a logistic regression method, and three clinical features. However, only 180 of the 216 miRNAs contributed to the risk prediction model construction as efficient features. Of the 180 miRNAs, 7 showed a high feature importance in the final GBDT model. GSEA using the target gens of the 7 miRNAs detected a significantly enriched biological pathway, the DHA signaling pathway, which has been previously reported to be associated with DLB pathology [24]. In particular, six target genes were involved in the biological pathway, two of which, BCL2L1 and PIK3R2, were further supported by gene-based association studies using a large number of SNP markers. BCL2L1 belongs to the family of BDL-2 proteins, which is involved in not only in the control of apoptosis, but also in mitochondrial damage protection [36], modulation of immune response [37], and DNA repair [38]. Borras et al. have reported that the over-expression of BCL2L1 in PBMCs was confirmed in centenarians, compared with septuagenarians and young people [39]. This evidence supports that BCL2L1 plays an important role in healthy aging. In other words, defects in BCL2L1 could exert an adverse influence on the healthy aging (e.g. cognitive impairment). On the other hand, PIK3R2 (phosphoinositide-3-kinase regulatory subunit 2) is a lipid kinase that functions in growth signaling pathways and a known as a tumor suppressor gene [40]. There are no reports of it being associated with DLB pathology. However, Shu et al. reported that the PI3K/ANK pathway containing PIK3R2 is involved in cognitive impairment [41]. These miRNAs were also used to perform DIANA-miRPath v3.0 [42], a web-based functional analysis tool incorporating biological pathways, and three statistically significant Kyoto Encyclopedia of Genes and Genomes (KEGG) biological pathways [43, 44] were detected with a q-value < 0.001: Metabolism of xenobiotics by cytochrome P450 (8 genes), Vasopressin-regulated water reabsorption (14 genes), and thyroid hormone signaling pathway (25 genes) (Additional file 4: Table S4). One of them, the thyroid hormone signaling pathway, has been reported to be associated with neurodegenerative diseases; the administration of thyroid hormone in AD model mice prevented cognitive deficit and improved the neurological function [45]. In future work, we will perform further refinement of our model, and investigations with larger sample size will further validate the effectiveness of this classifier.
The most efficient features have a weaker correlation between the ranking of feature importance of the final method and the top-ranked miRNAs than we expected (Spearman’s ρ =0.21). This implies that there is still room for improvement in our prediction model. One way may be to integrate functional units, such as metabolic pathways, into our final risk prediction model, as miRNAs with high feature importance in the GBDT were associated with several biological pathways. Another way may be to integrate interactions among miRNAs into our final risk prediction model. Interaction effects have been reported to increase the power of risk prediction models [46]. Finally, artificial intelligence (AI) technology, in particular deep learning, is a recent and fast-growing field of machine learning. AI technology could also contribute to an improvement of this risk prediction model. Some potential applications have been proposed for novel diagnostic and treatment options in medical imaging and genomics [47, 48]. However, this technology relies on large amounts of data to learn automatically, and it has certain advantages for dealing with big data [49, 50]. At least several thousand unique training data sets would be required for successful application [50]. Although at present, our data sets were too small to effectively apply these AI technologies, we expect that these technologies will contribute to improvement of our prediction model in the future.
Next generation sequencing technology (NGS) has enabled comprehensive detection of coding and non-coding RNAs as well as genetic variants. Integrative analysis of these genetic variations and gene expressions, such as expression quantitative trait loci (eQTL), has revealed potential target genes for associations of genetic susceptibility risk loci. We believe that these omics data would also play an important role for improvement of risk prediction models.
Conclusions
In this study, we investigated potential miRNA biomarkers using serum miRNA expression and constructed an optimal risk prediction model using several machine learning methods. The final risk prediction model based on a GBDT achieved an accuracy of 0.829 on an independent test set. GSEA of the miRNA candidate target genes revealed 6 functional genes in the DHA signaling pathway associated with DLB pathology. For two of them (BCL2L1 and PIK3R2), this was further supported by gene-based association studies using a large numbers of SNP markers. Our study provides an effective tool for DLB classification, and with further improvement, such as integrative analyses of genomic and/or transcriptomic data, it has the potential to contribute to practical clinical application in DLB.
Supplementary information
Additional file 1: Table S1. Hyperparameter values in the final GBDT model when using μHEM algorithm.
Additional file 2: Table S2. All features used in the final GBDT risk prediction model.
Additional file 3: Table S3. Genes and the annoatation related to the canonical pathways.
Additional file 4: Table S4. Genes including in the KEGG pathways.
Acknowledgements
We thank NCGG Biobank for providing the study materials, clinical information and technical support. All microarray data of miRNAs were provided by Toray Industries, Inc.
Abbreviations
- AD
Alzheimer’s disease
- AI
Artificial intelligence
- ChEIs
Cholinesterase inhibitors
- DHA
Docosahexaenoic acid
- DLB
Dementia with Lewy bodies
- eQTL
Expression quantitative trait loci
- FN
False negatives
- FP
False positives
- GBDT
Gradient boosted decision tree
- GEO
Gene Expression Omnibus
- GSEA
Gene set enrichment analysis
- GTEx
Genotype-Tissue Expression
- LBD
Lewy body dementia
- MAF
Minor allele frequency
- miRDB
microRNA Target Prediction and Functional Study Database
- miRNAs
MicroRNAs
- ML
Machine learning
- MMSE
Mini-Mental State Examination
- NC
Normal controls
- NGS
Next generation sequencing technology
- PCA
Principal component analysis
- PDD
Parkinson’s disease dementia
- RF
Random forest
- SKAT
SNP-set Kernel Association Test
- SNP
Single nucleotide polymorphism
- SVM
Support vector machine
- TN
True negatives
- TP
True positives
- α Syn
α-synuclein
Authors’ contributions
D.S. and S.A. developed the method and performed the analyses; Y.A., K.A.B., A.S. and T.T. provided the technical assistance; T.S. contributed to data acquisition and the analyses; D.S., T.O., K.O. and S.N. wrote the manuscript and organized this work. All authors contributed to and approved the final manuscript.
Funding
This study was supported by the “Development of Diagnostic Technology for Detection of miRNA in Body Fluids” grant from the Japan Agency for Medical Research and Development and New Energy and Industrial Technology Development Organization (to S.N., grant number JP17ae0101013). This work was also partially supported by Core Research for Evolutional Science and Technology grant from the Japan Science and Technology Agency (grant number JPMJCR1412), Japan Society for the Promotion of Science KAKENHI (grant numbers 17H06307 and 17H06299), the Japan Foundation for Aging and Health and the Takeda Science Foundation. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
All microarray data used in this study are publicly available through the GEO database accession number GSE120584 (https://www.ncbi.nlm.nih.gov/geo/). Other datasets generated in this study are provided from the corresponding author on reasonable request.
Ethics approval and consent to participate
The present study was approved by the ethics committee of the NCGG. All participants were voluntary and the written informed consent was obtained before registering to NCGG Biobank. We also confirmed that all experiments were performed in accordance with relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12920-019-0607-3.
References
- 1.Mueller C, Ballard C, Corbett A, Aarsland D. The prognosis of dementia with Lewy bodies. Lancet Neurol. 2017;16(5):390–398. doi: 10.1016/S1474-4422(17)30074-1. [DOI] [PubMed] [Google Scholar]
- 2.Kane JPM, Surendranathan A, Bentley A, Barker SAH, Taylor JP, Thomas AJ, Allan LM, McNally RJ, James PW, McKeith IG, et al. Clinical prevalence of Lewy body dementia. Alzheimers Res Ther. 2018;10(1):19. doi: 10.1186/s13195-018-0350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKeith I, Mintzer J, Aarsland D, Burn D, Chiu H, Cohen-Mansfield J, Dickson D, Dubois B, Duda JE, Feldman H, et al. Dementia with Lewy bodies. Lancet Neurol. 2004;3(1):19–28. doi: 10.1016/S1474-4422(03)00619-7. [DOI] [PubMed] [Google Scholar]
- 4.Nelson PT, Jicha GA, Kryscio RJ, Abner EL, Schmitt FA, Cooper G, Xu LO, Smith CD, Markesbery WR. Low sensitivity in clinical diagnoses of dementia with Lewy bodies. J Neurol. 2010;257(3):359–366. doi: 10.1007/s00415-009-5324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsunaga S, Kishi T, Yasue I, Iwata N. Cholinesterase Inhibitors for Lewy Body Disorders: A Meta-Analysis. Int J Neuropsychopharmacol. 2015;19(2). [DOI] [PMC free article] [PubMed]
- 6.Satoh J. MicroRNAs and their therapeutic potential for human diseases: aberrant microRNA expression in Alzheimer's disease brains. J Pharmacol Sci. 2010;114(3):269–275. doi: 10.1254/jphs.10R11FM. [DOI] [PubMed] [Google Scholar]
- 7.Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, Kelnar K, Kemppainen J, Brown D, Chen C, et al. Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008;14(1):27–41. doi: 10.3233/JAD-2008-14103. [DOI] [PubMed] [Google Scholar]
- 8.Tacutu R, Budovsky A, Yanai H, Fraifeld VE. Molecular links between cellular senescence, longevity and age-related diseases - a systems biology perspective. Aging (Albany NY) 2011;3(12):1178–1191. doi: 10.18632/aging.100413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shigemizu D, Akiyama S, Asanomi Y, Boroevich KA, Sharma A, Tsunoda T, Matsukuma K, Ichikawa M, Sudo H, Takizawa S, et al. Risk prediction models for dementia constructed by supervised principal component analysis using miRNA expression data. Commun Biol. 2019;2:77. doi: 10.1038/s42003-019-0324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lebedev AV, Westman E, Van Westen GJ, Kramberger MG, Lundervold A, Aarsland D, Soininen H, Kloszewska I, Mecocci P, Tsolaki M, et al. Random Forest ensembles for detection and prediction of Alzheimer's disease with a good between-cohort robustness. Neuroimage Clin. 2014;6:115–125. doi: 10.1016/j.nicl.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu W, Liu T, Valdez R, Gwinn M, Khoury MJ. Application of support vector machine modeling for prediction of common diseases: the case of diabetes and pre-diabetes. BMC Med Inform Decis Mak. 2010;10:16. doi: 10.1186/1472-6947-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shigemizu D, Abe T, Morizono T, Johnson TA, Boroevich KA, Hirakawa Y, Ninomiya T, Kiyohara Y, Kubo M, Nakamura Y, et al. The construction of risk prediction models using GWAS data and its application to a type 2 diabetes prospective cohort. PLoS One. 2014;9(3):e92549. doi: 10.1371/journal.pone.0092549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shigemizu D, Iwase T, Yoshimoto M, Suzuki Y, Miya F, Boroevich KA, Katagiri T, Zembutsu H, Tsunoda T. The prediction models for postoperative overall survival and disease-free survival in patients with breast cancer. Cancer Med. 2017;6(7):1627–1638. doi: 10.1002/cam4.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cule E, Vineis P, De Iorio M. Significance testing in ridge regression for genetic data. BMC Bioinformatics. 2011;12:372. doi: 10.1186/1471-2105-12-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Stat Soc B. 2005;67(2):301–320. doi: 10.1111/j.1467-9868.2005.00503.x. [DOI] [Google Scholar]
- 16.Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc B. 1996;58(1):267–288. [Google Scholar]
- 17.Breimen L. Random forests. Mach Learn. 2001;45(1):5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 18.Muller KR, Mika S, Ratsch G, Tsuda K, Scholkopf B. An introduction to kernel-based learning algorithms. IEEE Trans Neural Netw. 2001;12(2):181–201. doi: 10.1109/72.914517. [DOI] [PubMed] [Google Scholar]
- 19.Friedman JH. Stochastic gradient boosting. Comput Stat Dataa Anal. 2002;38(4):367–378. doi: 10.1016/S0167-9473(01)00065-2. [DOI] [Google Scholar]
- 20.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88–100. doi: 10.1212/WNL.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai Y, Mimori T, Kojima K, Nariai N, Danjoh I, Saito R, Yasuda J, Yamamoto M, Nagasaki M. Japonica array: improved genotype imputation by designing a population-specific SNP array with 1070 Japanese individuals. J Hum Genet. 2015;60(10):581–587. doi: 10.1038/jhg.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Database issue):D146–D152. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paul S, Maji P. muHEM for identification of differentially expressed miRNAs using hypercuboid equivalence partition matrix. BMC Bioinformatics. 2013;14:266. doi: 10.1186/1471-2105-14-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yakunin E, Loeb V, Kisos H, Biala Y, Yehuda S, Yaari Y, Selkoe DJ, Sharon R. Alpha-synuclein neuropathology is controlled by nuclear hormone receptors and enhanced by docosahexaenoic acid in a mouse model for Parkinson's disease. Brain Pathol. 2012;22(3):280–294. doi: 10.1111/j.1750-3639.2011.00530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89(1):82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong J, Mei S, Liu C, Xiang Y, Ye Y, Zhang Z, Feng J, Liu R, Diao L, Guo AY, et al. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res. 2018;46(D1):D971–D976. doi: 10.1093/nar/gkx861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang C, Zhu DX, Dong HJ, Zhou ZJ, Wang YH, Liu L, Fan L, Miao KR, Liu P, Xu W, et al. Serum microRNAs are promising novel biomarkers for diffuse large B cell lymphoma. Ann Hematol. 2012;91(4):553–559. doi: 10.1007/s00277-011-1350-9. [DOI] [PubMed] [Google Scholar]
- 29.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant KC, Allen A, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizuno H, Nakamura A, Aoki Y, Ito N, Kishi S, Yamamoto K, Sekiguchi M, Takeda S, Hashido K. Identification of muscle-specific microRNAs in serum of muscular dystrophy animal models: promising novel blood-based markers for muscular dystrophy. PLoS One. 2011;6(3):e18388. doi: 10.1371/journal.pone.0018388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maes OC, Chertkow HM, Wang E, Schipper HM. MicroRNA: implications for Alzheimer disease and other human CNS disorders. Curr Genomics. 2009;10(3):154–168. doi: 10.2174/138920209788185252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu W, Qin W, Atasoy U, Sauter ER. Circulating microRNAs in breast cancer and healthy subjects. BMC Res Notes. 2009;2:89. doi: 10.1186/1756-0500-2-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimomura A, Shiino S, Kawauchi J, Takizawa S, Sakamoto H, Matsuzaki J, Ono M, Takeshita F, Niida S, Shimizu C, et al. Novel combination of serum microRNA for detecting breast cancer in the early stage. Cancer Sci. 2016;107(3):326–334. doi: 10.1111/cas.12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou C, Yu H, Ding Y, Guo F, Gong XJ. Multi-scale encoding of amino acid sequences for predicting protein interactions using gradient boosting decision tree. PLoS One. 2017;12(8):e0181426. doi: 10.1371/journal.pone.0181426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson R, Tong Z. Learning nonlinear functions using regularized greedy Forest. IEEE Trans Pattern Anal Mach Intell. 2014;36(5):942–954. doi: 10.1109/TPAMI.2013.159. [DOI] [PubMed] [Google Scholar]
- 36.Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem. 2000;275(1):322–327. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- 37.Opferman JT, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nat Immunol. 2003;4(5):410–415. doi: 10.1038/ni0503-410. [DOI] [PubMed] [Google Scholar]
- 38.Fan S, Ma YX, Wang JA, Yuan RQ, Meng Q, Cao Y, Laterra JJ, Goldberg ID, Rosen EM. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3′ kinase. Oncogene. 2000;19(18):2212–2223. doi: 10.1038/sj.onc.1203566. [DOI] [PubMed] [Google Scholar]
- 39.Borras C, Abdelaziz KM, Gambini J, Serna E, Ingles M, de la Fuente M, Garcia I, Matheu A, Sanchis P, Belenguer A, et al. Human exceptional longevity: transcriptome from centenarians is distinct from septuagenarians and reveals a role of Bcl-xL in successful aging. Aging (Albany NY) 2016;8(12):3185–3208. doi: 10.18632/aging.101078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vallejo-Diaz J, Chagoyen M, Olazabal-Moran M, Gonzalez-Garcia A, Carrera AC. The opposing roles of PIK3R1/p85alpha and PIK3R2/p85beta in Cancer. Trends Cancer. 2019;5(4):233–244. doi: 10.1016/j.trecan.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 41.Shu Y, Zhang H, Kang T, Zhang JJ, Yang Y, Liu H, Zhang L. PI3K/Akt signal pathway involved in the cognitive impairment caused by chronic cerebral hypoperfusion in rats. PLoS One. 2013;8(12):e81901. doi: 10.1371/journal.pone.0081901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T, Hatzigeorgiou AG. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015;43(W1):W460–W466. doi: 10.1093/nar/gkv403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47(D1):D590–D595. doi: 10.1093/nar/gky962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu AL, Zhou CY, Chen X. Thyroid hormone prevents cognitive deficit in a mouse model of Alzheimer's disease. Neuropharmacology. 2010;58(4–5):722–729. doi: 10.1016/j.neuropharm.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 46.Cordell HJ. Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. 2009;10(6):392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gulshan V, Peng L, Coram M, Stumpe MC, Wu D, Narayanaswamy A, Venugopalan S, Widner K, Madams T, Cuadros J, et al. Development and validation of a deep learning algorithm for detection of diabetic retinopathy in retinal fundus photographs. JAMA. 2016;316(22):2402–2410. doi: 10.1001/jama.2016.17216. [DOI] [PubMed] [Google Scholar]
- 48.Ratner M. IBM's Watson Group signs up genomics partners. Nat Biotechnol. 2015;33(1):10–11. doi: 10.1038/nbt0115-10. [DOI] [PubMed] [Google Scholar]
- 49.Wu M, Yang Y, Wang H, Xu Y. A deep learning method to more accurately recall known lysine acetylation sites. BMC Bioinformatics. 2019;20(1):49. doi: 10.1186/s12859-019-2632-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Hyperparameter values in the final GBDT model when using μHEM algorithm.
Additional file 2: Table S2. All features used in the final GBDT risk prediction model.
Additional file 3: Table S3. Genes and the annoatation related to the canonical pathways.
Additional file 4: Table S4. Genes including in the KEGG pathways.
Data Availability Statement
All microarray data used in this study are publicly available through the GEO database accession number GSE120584 (https://www.ncbi.nlm.nih.gov/geo/). Other datasets generated in this study are provided from the corresponding author on reasonable request.