

Abstract
LSD1 (KDM1A) is a histone demethylase that plays both oncogenic and tumor suppressor roles in breast cancer. However, the exact contexts under which it plays these opposite functions remain largely elusive. By characterizing its role in luminal breast epithelial cells, here we show that inhibition of LSD1 by both genetic and pharmacological approaches increases their invasion and migration, whereas its inhibition by genetic approach, but not by pharmacological approach, impairs their proliferation/survival. Induced loss of LSD1 in luminal cells in a mouse model of luminal breast cancer, MMTV-PyMT, leads to a profound increase in lung metastasis. Mechanistically, LSD1 interacts with GATA3, a key luminal-specific transcription factor (TF), and their common target genes are highly related to breast cancer. LSD1 positively regulates GATA3 expression. It also represses expression of TRIM37, a breast epithelial oncogene encoding a histone H2A ubiquitin ligase, and ELF5, a key TF gene for luminal progenitors and alveolar luminal cells. LSD1-loss also leads to reduced expression of several cell-cell adhesion genes (e.g., CDH1, VCL, CTNNA1), possibly via TRIM37-upregulation and subsequently TRIM37-mediated repression. Collectively, our data suggest LSD1 largely plays a tumor suppressor role in luminal breast cancer and the oncogenic program associated with LSD1-inhibition may be suppressed via TRIM37-inhibition.
Keywords: LSD1, KDM1A, epigenetic regulator, histone demethylase, luminal breast cancer, invasion, migration, metastasis, estrogen receptor, ER, GATA3, TRIM37, ELF5, cell adhesion, CDH1, E-cadherin, CTNNA1, VCL
Introduction
It has become increasingly recognized that both genetic and epigenetic events can contribute to cancer development [1]. Epigenetic de(regulation) in cancer can be in the forms of genetic lesions (e.g., overexpression, deletion, or mutation) of epigenetic regulators that control histone modifications or DNA methylation/demethylation, as well as of global alternations in the epigenetic landscape of cancer cells [2]. Since epigenetic changes are reversible and epigenetic regulators are often proteins with enzymatic activities, they are attractive targets for therapeutic intervention in cancer [3]. However, since epigenetic regulators can regulate expression of many target genes and depending on different cellular contexts, they can play distinct roles as either tumor suppressors or oncoproteins. In order to target them therapeutically for treating or preventing cancer, it is important to define their roles during tumorigenesis in a context-dependent manner.
LSD1 (also known as KDM1A) is a histone demethylase that removes methyl groups from mono- and di-methylated lysine 4 on histone H3 (H3K4me1/me2) [4]. It is often found in multi-protein complexes that function as transcription repressors (e.g., CoREST or NuRD repressor complexes) [5, 6]. It can also function as a transcription activator when in complex with androgen or estrogen receptor (AR or ER) via demethylation of mono- and di-methylated lysine 9 on histone H3 (H3K9me1/me2) [7-9]. Furthermore, LSD1 can demethylate non-histone proteins (e.g., p53 [10], DNMT1 [11]). High levels of LSD1 expression have been found correlated with poor prognosis in multiple human cancer types (e.g., prostate cancer, neuroblastoma, lung cancer, leukemia) [12]. In breast cancer, LSD1 was found highly expressed in ER-negative breast tumors and high LSD1 expression could predict poorer outcomes [13]; elevated LSD1 expression was also found during progression from ductal carcinoma in situ to invasive ductal breast cancer [14]. However, roles of LSD1 in breast cancer remain contradicting. In one study, it was found that as a subunit of the NuRD complex, LSD1 suppressed invasion and metastatic potential of MDA-MB-231 breast cancer cells in vitro and in vivo [6], thus arguing LSD1 as a tumor suppressor. In other studies, inhibition of LSD1 was shown to lead to reduced growth of MDA-MB-231 cells [15], as well as of MDA-MB-453, MCF7, and T47D breast cancer cell lines [13, 16]. Lastly, LSD1 has also been linked to breast cancer stem cells (CSCs), by positively regulating CSC-like properties of tumor cells from the MMTV-Wnt1 mouse model as well as those of human breast cancer cell lines BT549 and MCF7, and its knockdown significantly inhibited tumor growth from MCF7 cells in vivo [17]. All these studies suggested that LSD1 might also function as an oncoprotein in breast cancer.
As a histone-modifying enzyme, LSD1 does not bind DNA directly and DNA-binding transcription factors (TFs) recruit LSD1 to its target genes (e.g., TAL1 [18]). Thus, depending on specific cellular contexts, LSD1 is expected to control expression of distinct target genes when recruited by different lineage-specific TFs. In order to understand differential roles of LSD1 in breast cancer, it is essential to identify such lineage-specific TFs that interact with LSD1. As target cells of breast tumorigenesis, the mammary epithelium is composed of luminal and basal mammary epithelial cells (MECs). Correlating with these two MEC types, breast cancers can be categorized into multiple intrinsic subtypes based on expression profiling [19, 20]. Among them, luminal and basal-like breast cancers are the two major subtypes, which differ in their MEC differentiation patterns, expression of lineage-specific TFs, and patient outcomes. In basal-like breast cancer cells, LSD1 has been shown to interact with SNAIL1 and SNAIL2 (SLUG), two basal TFs involved in epithelial-mesenchymal transition (EMT) [21, 22]. In this study, we focused on luminal breast cancer cells and defined roles of LSD1 in maintaining expression of cell-cell adhesion genes in them and in suppression of luminal breast cancer cell invasion, migration and metastasis, possibly via the luminal lineage-specific TF, GATA3, and their common targets.
Results
LSD1 and GATA3 participate in the same complexes
In ER+ luminal cells, LSD1 was shown previously as a key epigenetic regulator to preclude unliganded ERα from effective binding and from functioning as ligand-independent, constitutive activator [9]. In luminal MECs, in addition to ERα, GATA3 is a DNA-binding master regulatory TF that plays an essential role in specifying and maintaining the luminal fate [23, 24]. We hypothesized that LSD1 might work together with key luminal-specific TFs, such as GATA3, to sustain the differentiation state of luminal MECs. To test this hypothesis, particularly to determine whether LSD1 may be recruited to DNA via GATA3, we first examined its potential interaction with GATA3 in luminal breast cancer cells. By reciprocal co-immunoprecipitation (co-IP) experiments in nuclear extracts from a luminal breast cancer cell line, MCF7, we found that LSD1 interacted with GATA3, and vice versa (Fig. 1a-b). We also confirmed their interaction by co-IP in another luminal breast cancer cell line, T47D (Supplementary Fig. S1a). By co-immunofluorescence (co-IF) staining, we further confirmed that LSD1 and GATA3 co-localized in the nuclei of both MCF7 and T47D cells (Supplementary Fig. S1b). By a structure-function study, we found that the SWIRM domain of LSD1 is required for the interaction between LSD1 and GATA3 (Fig. 1c). In human breast cancers, point mutations of LSD1 (i.e., KDM1A) are found in its SWIRM domain or amino oxidase (AO) enzymatic domain (based on cbioportal, Supplementary Fig. S1c). To determine whether point mutations in the SWIRM domain affect the interaction of LSD1 with GATA3, we introduced a mutation, R251Q, to its SWIRM domain; by the GST pull-down assay, we found that R251Q almost entirely abolished the interaction of LSD1 SWIRM domain with GATA3 (Fig. 1d). This data suggests that while mutation in the AO domain of LSD1 is expected to affect its demethylase activity, mutation in its SWIRM domain impairs its interaction with a key luminal TF, GATA3, and both of which may lead to loss-of-function of LSD1. Lastly, by co-IP, we found that GATA3 interacted not only with LSD1 but also with other components of the CoREST complex (Fig. 1e). By gel filtration chromatography, we further confirmed that both GATA3 and LSD1 co-migrated with these CoREST components in MCF7 cells (Fig. 1f). Collectively, these data suggest that in luminal breast cancer cells, LSD1 participates in the multi-protein complexes containing both GATA3 and CoREST components.
Figure 1. LSD1 interacts with GATA3 in luminal breast cancer cells.
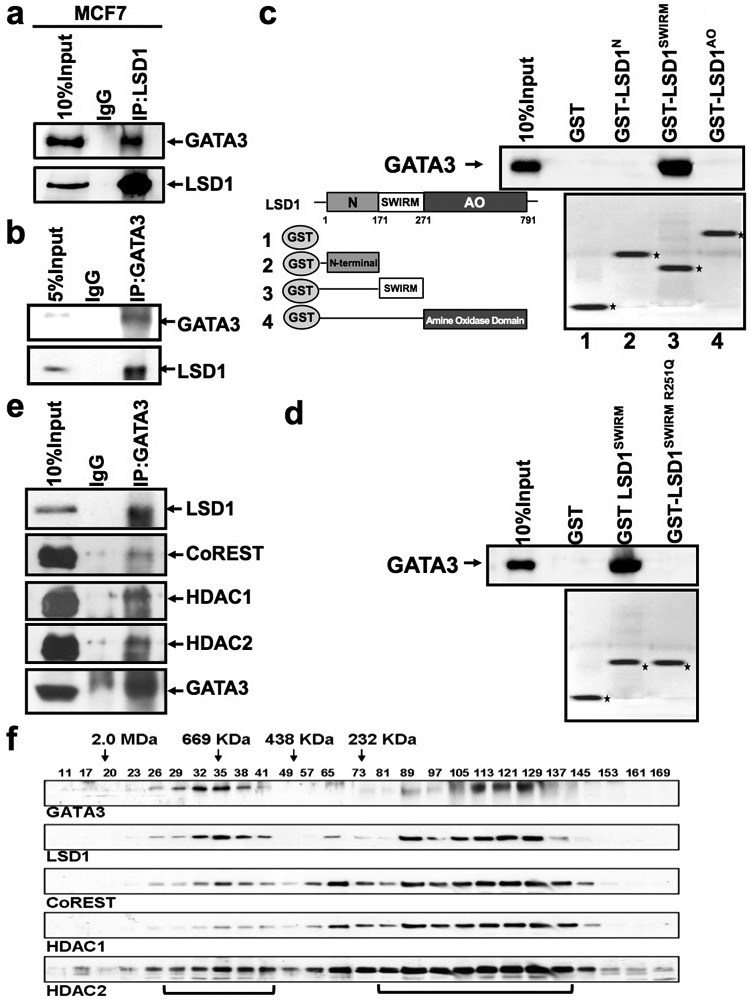
(a-b) MCF7 cell nuclear extracts were immunoprecipitated by an anti-LSD1 antibody (a) or an anti-GATA3 antibody (b), then subjected to Western blotting analysis with anti-LSD1 and anti-GATA3 antibodies.
(c-d) C-Left: schematic representations of the structure of LSD1 and the GST-LSD1 fusion proteins used in GST pull-down assays; c-Right: GST, GST-LSD1N, GST-LSD1SWIRM and GST-LSD1AO, and d: GST, GST-LSD1SWIRM, GST-LSD1SWIRM R251Q fusion proteins, were pre-adsorbed to glutathione-Sepharose beads, and incubated with MCF7 nuclear extracts; proteins that were bound to LSD1 were eluted and resolved with an SDS-PAGE gel.
(e) MCF7 cell nuclear extracts were immunoprecipitated by an anti-GATA3 antibody and subjected to Western blotting analysis with anti-LSD1, anti-CoREST, anti-HDAC1, anti-HDAC2, and anti-GATA3 antibodies.
(f) Nuclear extracts from MCF7 cells were fractionated through the SephacrylS-300 HR column. The fractions were collected and analyzed by Western blot using the indicated antibodies.
LSD1 and GATA3 control a common set of programs in luminal MECs
To study common genes regulated directly or indirectly by LSD1 and GATA3 in luminal breast cells, we used siRNAs to knock down their expression in MCF7 cells and then performed microarray expression profiling (Fig. 2a). The effective knockdown was confirmed by Western blot analysis (Supplementary Fig. 2a). Compared to gene expression in the control cells, we found expression levels of ~1,796 or 1,524 genes were significantly affected in MCF7 cells with LSD1 or GATA3 knockdown, respectively, with ~443 genes affected by both (Fig. 2b). To determine gene programs and pathways affected by LSD1 and GATA3 knockdown in MCF7 cells, we performed gene set enrichment analysis (GSEA) [25]. By GSEA, we found common gene sets downregulated upon either LSD1 or GATA3 knockdown included those related to cell-cell adhesion, E-cadherin stabilization, cell cycle, and steroid hormone biosynthesis; common upregulated gene sets included those related to Integrin pathway, Met pathway, and Notch signaling (Fig. 2c-d and Supplementary Fig. S2b-d). We also observed a previously generated gene set for conserved genes in human and mouse mammary stem cells (MaSC) (i.e., basal cells [26]) was commonly upregulated in MCF7 cells with either LSD1 or GATA3 knockdown (Supplementary Fig. S2b), including MaSC genes such as ITGB1, TCF4, CAV1, TMEM121, SPRED1 and POU3F1 (Supplementary Fig. S2c). To validate downregulation of cell adhesion-related pathways, we checked expression levels of several genes in the E-cadherin stabilization pathway, such as CDH1 (encoding E-cadherin), VCL (encoding vinculin), and CTNNA1 [encoding catenin alpha 1 (α-catenin)], upon either LSD1 or GATA3 knockdown. By Western blot, we confirmed that all these three genes were expressed at lower levels (protein) in knockdown cells (Fig. 2e).
Figure 2. LSD1 and GATA3 co-regulate breast cancer-related target genes.
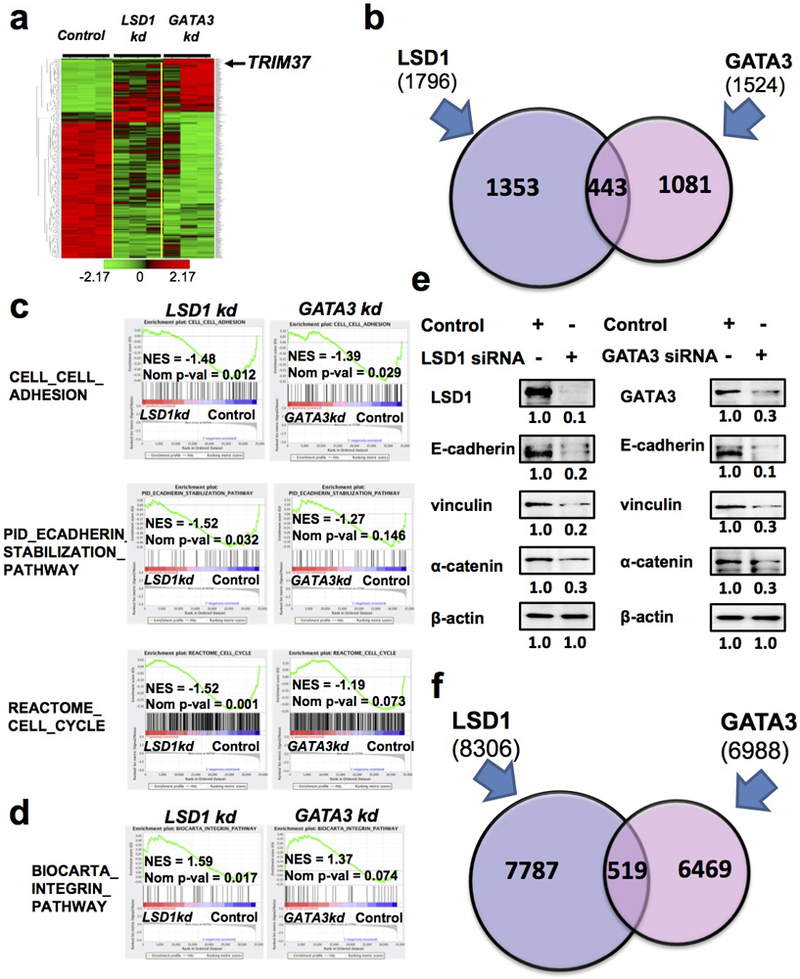
(a) Hierarchical clustering of genes altered by LSD1 knockdown or GATA3 knockdown in MCF7 cells.
(b) Numbers of genes altered by either LSD1 knockdown or GATA3 knockdown in MCF7 cells. A set of 443 genes are significantly affected by both LSD1 and GATA3 knockdown.
(c-d) Common gene sets downregulated (c) or upregulated (d) in MCF7 cells upon either LSD1 or GATA3 knockdown.
(e) Western blotting analysis showing downregulation of E-cadherin, vinculin and α-catenin in MCF7 cells upon either LSD1 or GATA3 knockdown.
(f) Integrated analysis of ChIP-seq data for GATA3 and LSD1 in MCF7 cells showing 519 peaks in common.
To identify common target genes of LSD1 and GATA3, we performed integrated analysis of two published genome-wide chromatin immunoprecipitation (ChIP) dataset, including the ENCODE ChIP-sequencing (ChIP-seq) data for GATA3 in MCF7 cells (www.encodeproject.org) and a ChIP-seq data for LSD1 in MCF7 cells [27]. After re-processing, we identified 8,306 LSD1 peaks and 6,988 GATA3 peaks. Although we only identified a small subset of peaks in common (519) (Fig. 2f and Supplementary Data 1), gene enrichment analysis of genes close to genomic regions of these common peaks revealed that they were highly enriched for pathways related to breast cancer (Supplementary Data 2), suggesting LSD1 and GATA3 control expression of common genes highly related to the biology of breast epithelial cells and breast cancer. From these potential common targets of LSD1 and GATA3, we selected several genes known to be involved in regulating breast epithelial cells and/or in breast cancer and found that the overlapped LSD1 and GATA3 binding peaks are located at either their promoter or potential enhancer regions (Supplementary Fig. S2e). We validated their expression levels in MCF7 cells upon either LSD1 or GATA3 knockdown and observed their upregulation in both cases (Supplementary Fig. S2f), suggesting LSD1 and GATA3 may work together to repress their expression.
LSD1 suppresses invasion and migration of luminal breast cancer cells
So far we found that both LSD1 and GATA3 positively regulate expression of cell adhesion genes (e.g., CDH1, CTNNA1) in luminal cells (Fig. 2e). As loss of E-cadherin (encoded by CDH1) can lead to EMT, leading to increased migration and invasiveness of breast epithelial cells [28], we measured invasion of MCF7 cells upon LSD1 or GATA3 knockdown and found in both cases, their invasiveness was significantly increased (Fig. 3a-b). Pharmacological inhibition of LSD1 further confirmed that loss of the LSD1 activity in MCF7 cells led to their increased invasion, similar to that of LSD1 knockdown (Fig. 3c-d). We validated the specificity of the two LSD1 inhibitors (GSK-LSD1, Pargyline) we used here and confirmed that they led to increased levels of H3K4me2 (i.e., a substrate of LSD1 demethylase) in MCF7 cells (Supplementary Fig. S3a). Furthermore, we performed the wound-healing assay in the cultured MCF7 cells with either LSD1 knockdown or inhibitor treatment; in both cases, we observed increased cell migration (Fig. 3e-h). In addition to their invasion and migration, we also measured their growth rates. Consistent with downregulation of the cell cycle-related gene sets (Fig. 2c), we found that proliferation/survival of MCF7, T47D and MCF10A cell lines were all reduced upon LSD1 knockdown (Supplementary Fig. S3b-c). However, pharmacological inhibition of LSD1 in MCF7 cells did not consistently change the proliferation/survival of MCF7 cells (Supplementary Fig. S3d). These data suggest that LSD1 suppresses invasion and migration of luminal breast cancer cells, possibly in a demethylase activity-dependent manner, whereas its potential role in supporting cell proliferation/survival may be independent of the demethylase activity.
Figure 3. LSD1 suppresses invasion/migration of luminal breast cancer cells.
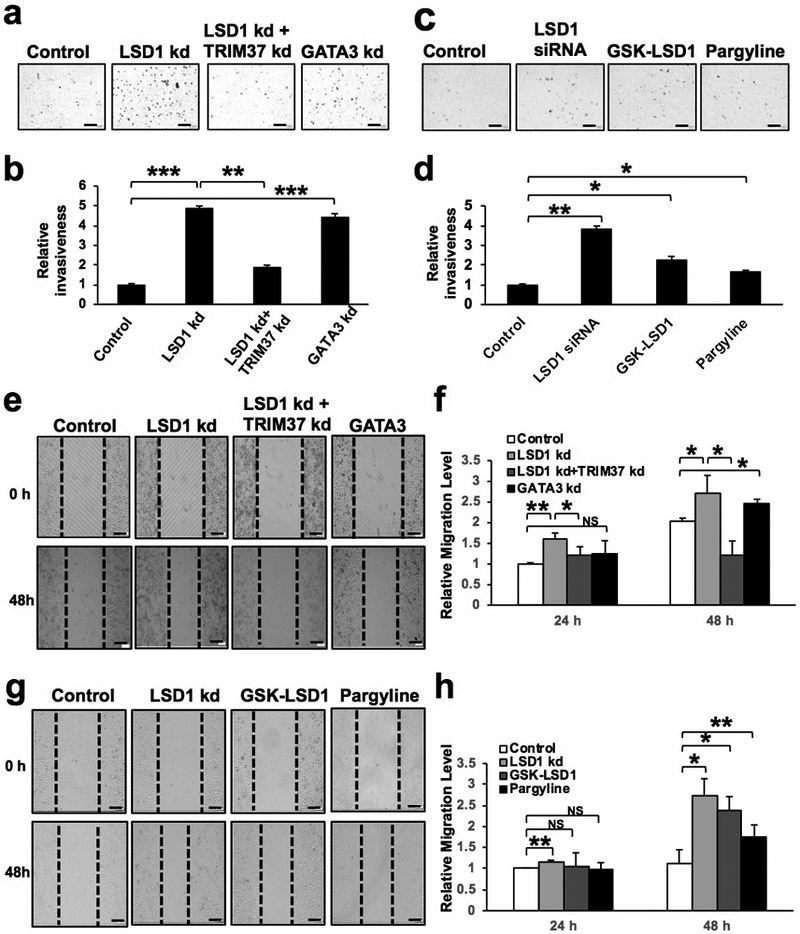
(a) Representative pictures of the Transwell invasion assay for MCF7 cells upon LSD1, GATA3 or TRIM37 knockdown. Of note, knockdown of both LSD1 and TRIM37 reverted the invasion phenotype induced by LSD1 knockdown alone. Scale bars = 200μm.
(b) Quantification for (a). Data represent mean ± SEM. P-values: Student t-test (2-sided), n=4, ** indicates P<0.01, *** indicates P<0.001.
(c) Representative pictures of the Transwell invasion assay for MCF7 cells upon LSD1 knockdown (as control) or inhibitor treatment. Scale bars = 200μm.
(d) Quantification for (c). Data represent mean ± SEM. P-values: Student t-test (2-sided), n=3, * indicates P<0.05, ** indicates P<0.01.
(e) Representative picture of the Scratch Wound Healing assay for MCF-7 cells upon LSD1, GATA3 or TRIM37 knockdown. Knockdown of both LSD1 and TRIM37 rescued the increased migration phenotype induced by LSD1 knockdown alone. Scale bars = 200μm.
(f) Quantification for (e). Data represent mean ± SEM. P-values: Student t-test (2-sided), n=4, * indicates P<0.05, ** indicates P<0.01.
(g) Representative picture of the Scratch Wound Healing assay for MCF-7 cells upon LSD1 knockdown (as control) or inhibitor treatment. Scale bars = 200μm.
(h) Quantification for (g). Data represent mean ± SEM. P-values: Student t-test (2-sided), n=4, * indicates P<0.05, ** indicates P<0.01, NS: not significant.
GATA3 is a target of LSD1 positively regulated by it
While studying LSD1 knockdown in MCF7 cells, we noticed that the expression level of GATA3 was reduced (Supplementary Fig. S2a). We further confirmed downregulation of GATA3 at both the transcript and protein levels by using several additional siRNAs for LSD1 (Fig. 4a: mRNA/qRT-PCR; 4b-c: protein/Western and quantification; Supplementary Fig. S4a: protein/co-IF staining). In addition to GATA3, we found that ELF5, which encodes for a master regulatory TF for mammary luminal progenitors and alveolar luminal cells [29, 30], was upregulated in MCF7 cells upon LSD1 knockdown (at both mRNA and protein levels, Fig. 4a-c). We further confirmed downregulation of GATA3 and upregulation of ELF5 by generating stable LSD1 knockdown MCF7 cells using two different shRNAs (Supplementary Fig. S4b). To determine if restored LSD1 expression could rescue downregulation of GATA3 upon LSD1 knockdown, we used a codon-changed LSD1 expression construct that is resistant to the LSD1 siRNA; upon its expression in MCF7 cells transfected with LSD1 siRNA, we found that GATA3 expression was restored (Fig. 4d), thus demonstrating that the suppressive effect of LSD1 siRNA on GATA3 expression is not through off-target effects of LSD1 siRNA. In addition, overexpression of LSD1 in MCF7 cells caused further upregulation of GATA3 expression (Fig. 4e); by Western blot, we confirmed upregulation of GATA3 and downregulation of ELF5 (protein levels, Fig. 4f-g) upon LSD1 overexpression. To test whether GATA3 is a direct target gene of LSD1, we investigated if LSD1 (as well as GATA3 itself) could bind the control region of the GATA3 gene. By ChIP analysis, we found that both LSD1 and GATA3 could bind the same promoter region of GATA3 (two sites, Fig. 4h). Additionally, knockdown of LSD1 caused increased H3K9 dimethylation (H3K9me2, another substrate of LSD1 demethylase) at the same two sites of the GATA3 promoter region (Supplementary Fig. S4c). Lastly, to determine whether ELF5 is also a target of LSD1, we performed ChIP analysis in MCF7 cells and identified two LSD1 binding sites in the promoter region of ELF5 (Supplementary Fig. S4d). Collectively, these data suggest that GATA3 and ELF5 are direct targets positively or negatively regulated by LSD1 in luminal breast cells, respectively.
Figure 4. LSD1 positively regulates GATA3 expression.
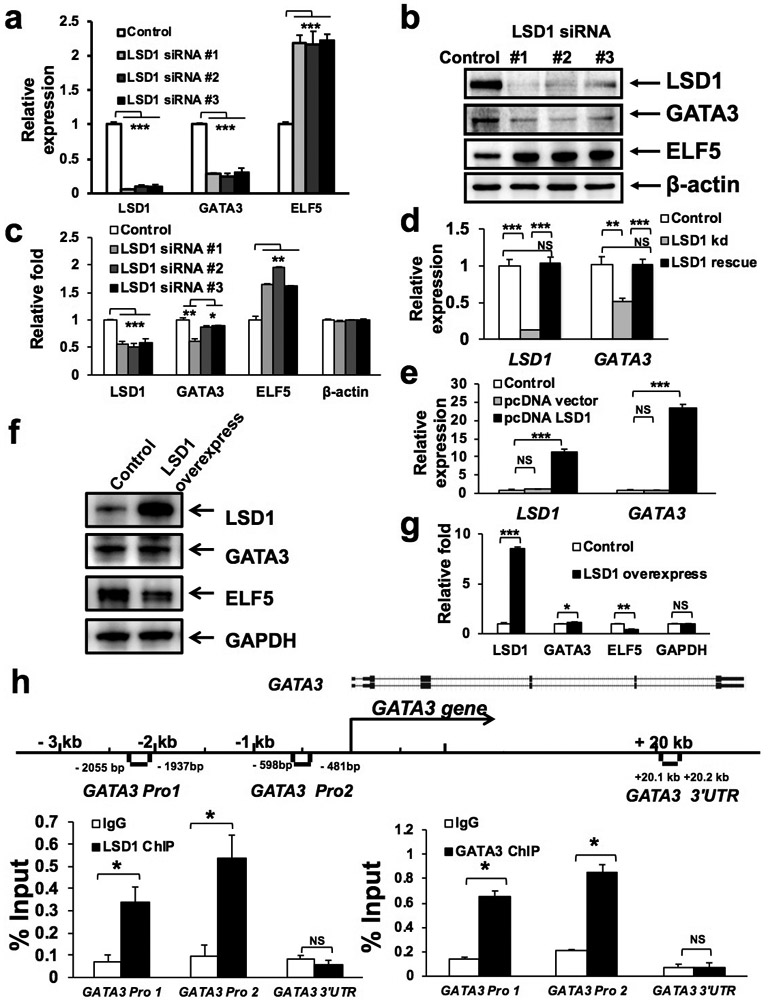
(a-b) qRT-PCR (a) and Western blotting (b) analyses showing downregulation of GATA3 and upregulation of ELF5 in MCF7 cells upon knockdown of LSD1 using three different siRNAs. Data represent mean ± SEM. P-values: Student t-test (2-sided), n=3, *** indicates P<0.001.
(c) Quantification for (b). Data represent mean ± SEM. P-values: Student t-test (2-sided), n=3, * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001.
(d) qRT-PCR analysis showing restoration of GATA3 expression in MCF7 cells with LSD1 knockdown, upon co-transfection with a LSD1 rescue construct resistant to the LSD1 siRNA. Data represent mean ± SEM. P-values: Student t-test (2-sided), n=3, * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, NS: not significant.
(e-f) qRT-PCR (e) and Western blotting (f) analyses showing increased expression of GATA3 in MCF7 cells upon overexpression of LSD1. Data represent mean ± SEM. P-values: Student t-test (2-sided), n=3, *** indicates P<0.001, NS: not significant.
(g) Quantification for (f). Data represent mean ± SEM. P-values: Student t-test (2-sided), n=3, * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, NS: not significant.
(h) ChIP with anti-LSD1 (left) or anti-GATA3 (right) antibody in MCF7 cells, followed by PCR with primers specific to either the GATA3 promoter (Pro1, 2) region or its 3’ untranslated region (UTR), as indicated in the schematic diagram (for the GATA3 gene). Data represent mean ± SEM. P-values: Student t-test (2-sided), n=3, * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, NS: not significant.
LSD1 represses its target gene TRIM37
LSD1 is best known as a transcription repressor by demethylating H3K4me2 and H3K4me1, and by working together with another histone demethylase, JARID1B, which demethylates H3K4me3 and H3K4me2 (H3K4me3 is a histone mark for active transcription) [31]. To identify key target genes repressed by both LSD1 and GATA3, we focused on a recently identified oncogene in breast cancer, TRIM37, which encodes a histone H2A ubiquitin ligase that is associated with polycomb repressive complex 2 (PRC2) and is thus involved in gene repression [32]. From our microarray data, we found that expression of TRIM37 was notably increased in MCF7 cells upon either LSD1 or GATA3 knockdown (Fig. 2a). Our integrated analysis of LSD1 and GATA3 ChIP data in MCF7 cells also revealed TRIM37 as a common target of both (Supplementary Data 1 and Fig. S2e). To confirm the negative correlation between LSD1/GATA3 and TRIM37 expression, we tested several breast cell lines, including MCF7, which contains 17q23 genomic amplification (a region harboring TRIM37) [32], as well as MCF10A and T47D, which do not have 17q23 amplification. By Western blot, we confirmed that TRIM37 is expressed at the highest level in MCF7 cells and lowest level in MCF10A cells (Supplementary Fig. S5a). We knocked down either LSD1 or GATA3 in these three cell lines. By qRT-PCR, we found that LSD1 knockdown led to profound upregulation of TRIM37 expression in all three cell lines; GATA3 knockdown also caused notable upregulation of TRIM37 expression in both MCF10A and T47D cells (Fig. 5a). By Western blot analysis, we found that knockdown of LSD1 led to a profound increase in the level of TRIM37 protein in MCF10A and T47D cells (and to a lesser degree, in MCF7 cells) (Fig. 5b). This increase was accompanied by an increase in the level of H2A ubiquitination (due to the histone H2A ubiquitin ligase activity of TRIM37) (Fig. 5b, Ub-H2A). Upregulation of TRIM37 expression upon LSD1 knockdown was further confirmed in MCF7 cells by co-IF staining (Supplementary Fig. S5b). Furthermore, we found that knockdown of GATA3 also led to upregulation of TRIM37 protein and increased level of H2A ubiquitination, most notably in MCF10A and T47D cells (Fig. 5c).
Figure 5. LSD1 and GATA3 represses TRIM37 expression.

(a) qRT-PCR analysis showing TRIM37 expression in MCF7, T47D and MCF10A breast cells upon either LSD1 or GATA3 knockdown. Data represent mean ± SEM. P-values: Student t-test (2-sided), n=3, *** indicates P<0.001.
(b-c) Western blotting analyses showing LSD1, TRIM37, GATA3, Ub-H2A, H2A, H3K4me2, H3, H4, and β-actin expression in MCF7, MCF10A, or T47D cells with either LSD1 knockdown (b) or GATA3 knockdown (c).
(d) ChIP with anti-LSD1 (upper) or anti-GATA3 (lower) antibody in MCF7 cells, followed by PCR with primers specific to either the TRIM37 promoter region (Pro1, 2, 3) or 3’ untranslated region (UTR) in TRIM37, as indicated in the schematic diagram (for the TRIM37 gene). Data represent mean ± SEM. P-values: Student t-test (2-sided), n=3, ** indicates P<0.01, NS: not significant.
(e) Western blotting analysis showing restoration of E-cadherin, vinculin and α-catenin expression in MCF7 cells with LSD1 knockdown, upon simultaneous TRIM37 knockdown.
To determine whether LSD1 and GATA3 bind the control region of TRIM37, we performed ChIP analysis in MCF7 cells and indeed observed binding of both LSD1 and GATA3 to the promoter region of TRIM37 [three sites, including the +451 site (Pro 3) revealed from the integrated analysis of LSD1 and GATA3 ChIP-seq data (Supplementary Data 1), Fig. 5d]. In addition, knockdown of either LSD1 or GATA3 caused increased H3K4 dimethylation (H3K4me2) at the promoter region of TRIM37 (Supplementary Fig. S5c), suggesting GATA3 may suppresses TRIM37 expression through LSD1-mediated demethylation of H3K4me2. Furthermore, to verify whether TRIM37 is a direct target of both LSD1 and GATA3, we performed double ChIP assay and confirmed that LSD1 and GATA3 bind to the same TRIM37 promoter region together (Supplementary Fig. S5d). Moreover, to test whether LSD1 was indeed recruited by GATA3, we performed ChIP assay for LSD1 in GATA3 knockdown cells and found that the binding of LSD1 to the TRIM37 promoter region was abolished upon GATA3 knockdown (Supplementary Fig S5e). Of note, as the level of TRIM37 de-repression upon LSD1 knockdown was notably higher than that upon GATA3 knockdown (Fig. 5a-c), this suggests that LSD1 may also repress TRIM37 expression using a GATA3-independent mechanism. Collectively, these data suggest that TRIM37 is a direct target of LSD1 and is repressed by it in GATA3-dependent and/or independent manner.
As knockdown of LSD1 in MCF7 cells led to downregulation of cell adhesion molecules such as E-cadherin, vinculin and α-catenin, which was accompanied by increased invasion and migration, we tested whether this phenotype is mediated by TRIM37. Upon knockdown of both LSD1 and TRIM37 in MCF7 cells, we found that expression levels of E-cadherin, vinculin and α-catenin were restored to almost the WT levels (Fig. 5e). Importantly, LSD1 and TRIM37 double knockdown significantly reduced the invasion and migration of MCF7 when compared to that of LSD1 knockdown alone (Fig. 3a-b, e-f, to levels more comparable to those of controls). These data suggest that LSD1 restricts invasion and migration of luminal breast cancer cells in part via suppression of TRIM37; loss of LSD1 may lead to increased invasiveness and migration of luminal cells in part via TRIM37-mediated suppression of cell adhesion genes (e.g., CDH1, VCL, CTNNA1, as these genes have been reported as ChIP targets of TRIM37 [32]).
LSD1 suppresses metastasis of luminal breast cancer in vivo
Since increased invasion and migration of luminal breast cancer cells with GATA3-loss correlates with their increased metastasis potential [33, 34], we asked whether LSD1-loss could also increase metastasis of luminal breast cancer cells. To test this, we established a mouse model by breeding the Lsd1 conditional knockout model (Lsd1L) to an autochthonous luminal breast cancer mouse model, MMTV-PyMT, together with a conditional Cre-reporter, Rosa26-LSL-YFP (R26Y). Lsd1 was inactivated in PyMT tumor cells via intraductal injection of Cre-expressing adenovirus under the control of the Keratin 8 promoter (Ad-K8-Cre), an approach we established [35-37]. The conditional R26Y reporter was included to detect MECs undergo Cre-mediated recombination (i.e., YFP+ cells). Intriguingly, induced loss of LSD1 in MMTV-PyMT females significantly shortened their survival, particularly when Ad-K8-Cre was injected at younger ages (e.g., 6-weeks, Fig. 6a). The reduced survival was apparently due to a significant increase in metastatic burdens in their lungs (Fig. 6b-c). By co-IF staining, we confirmed that while LSD1 protein expression could still be detected in the lung metastasis from the Ad-K8-Cre-injected MMTV-PyMT;R26Y control females, its expression was lost in the lung metastasis from the Ad-K8-Cre-injected MMTV-PyMT;Lsd1L/L;R26Y females (Fig. 6c, right). qRT-PCR analysis of primary tumor cells (i.e., sorted YFP+ tumor cells) also confirmed loss of Lsd1 expression and revealed profound downregulation of Gata3 and upregulation of Trim37 and Elf5 expression, as well as downregulation of Cdh1, Ctnna1 and Vcl expression in PyMT tumor cells with induced loss of LSD1 (Fig. 6d). Of note, it was shown recently that overexpression of Elf5 in the same MMTV-PyMT mouse model also led to an increase in the size and number of lung metastasis [38]. Together, these data support that LSD1 is a suppressor of metastasis in luminal breast cancer.
Figure 6. LSD1 is a metastasis suppressor.
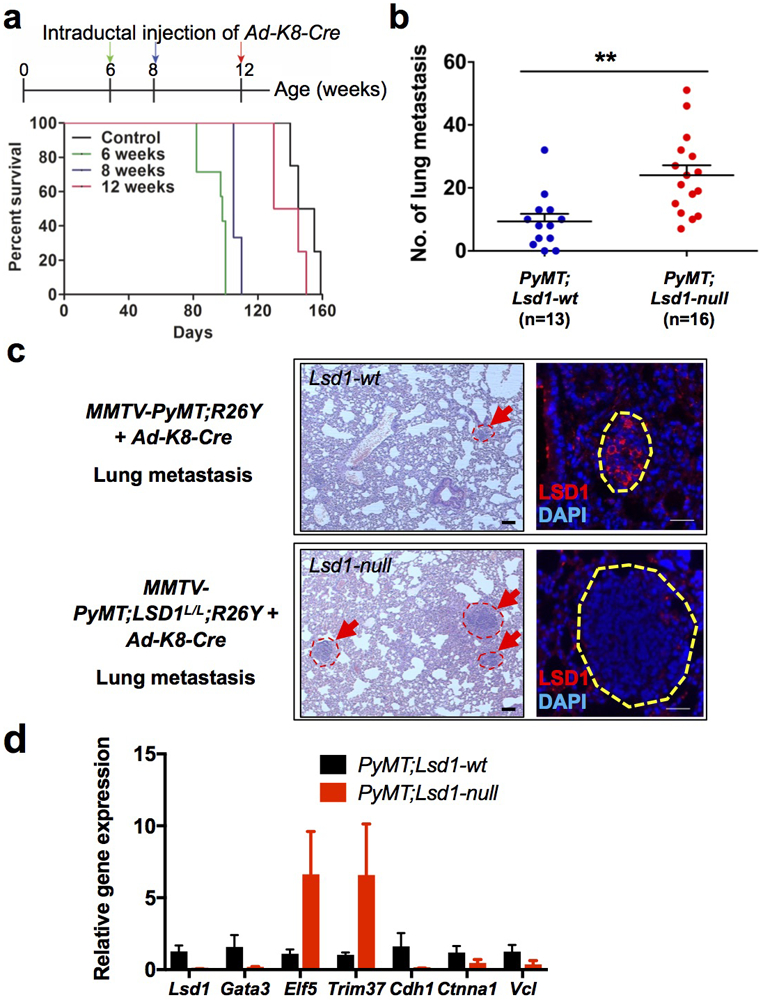
(a) Kaplan-Meier curves showing reduced survival of MMTV-PyMT;Lsd1L/L;R26Y female mice with intraductal injection of Ad-K8-Cre at 6, 8, 12-weeks of age (n=7, 3, 4, respectively); when mice were injected at older ages (e.g., 12-weeks), they have already developed mammary tumors and the efficiency of infection by the injected adenovirus was probably reduced due to larger sizes of tumors. Control represents MMTV-PyMT;R26Y female mice (n=4) without intraductal injection of Ad-K8-Cre.
(b) Numbers of metastatic lesions found in the lungs of MMTV-PyMT;Lsd1L/L;R26Y mice (PyMT;Lsd1-null) and MMTV-PyMT;R26Y control mice (PyMT;Lsd1-wt) with intraductal injection of Ad-K8-Cre at ~4 weeks of age. Animals were analyzed ~2-3 months after injection. Data represent mean ± SEM, **: P<0.01.
(c) Left: Hematoxylin and eosin (H&E) pictures showing representative metastatic lesions (arrows) detected in lungs of mice with the indicated genotypes (as in b), scale bars = 50μm. Right: IF staining for LSD1 on lung metastasis from mice with the indicated genotypes, scale bars = 25μm.
(d) qRT-PCR analysis (normalized to Gapdh) of sorted YFP+ cells from PyMT primary tumors with or without LSD1-loss. Data represent mean ± SEM, n=3 or 4 independently sorted samples.
LSD1 is a suppressor of human luminal breast cancer
Our data suggest that LSD1 works together with GATA3 to control luminal breast epithelial genes, including GATA3 itself, as well as those related to cell-cell adhesion (e.g., CDH1), and to repress expression of TRIM37, which is an oncogene associated with luminal breast cancer [32] (Fig. 7a). Thus, similar to GATA3, LSD1 may be a tumor suppressor under the setting of luminal breast cancer. To seek clinical support for LSD1 as a luminal breast tumor suppressor, we examined the TCGA breast cancer dataset [39] and found that among 962 breast cancer cases in this dataset, seven contain either homozygous deletion or missense mutations of LSD1 (of note, all these missense mutations affect either its SWIRM or AO domain, Supplementary Fig. S1c). Among these cases, at least five are positive for ER (Fig. 7b). Importantly, these seven cases exhibited significantly worse outcomes compared to the rest of cases (patient death in four out of seven cases, Fig. 7b; Supplementary Fig. S6a). When expanded our analysis to all cases with LSD1 deletions (i.e., both homozygous and heterozygous deletions) and mutations, we found that they still exhibited worse outcomes compared to those without LSD1 alteration (Supplementary Fig. S6b). Furthermore, by using the bc-GenExMiner online tool, we found that lower LSD1 expression is associated with reduced survival of patients with ER+ luminal A breast cancers (Fig. 7c-e); conversely, higher TRIM37 expression is associated with reduced survival of patients with luminal A cancers in the same cohorts (Fig. 7f-h). To determine if expression levels of LSD1 and TRIM37 are inversely correlated in patient samples, we checked the correlation of their expression levels in the METABRIC cohort [40, 41]. We focused on the ER+ cases in this cohort (1459 samples) and observed a negative correlation between their expression levels (Supplementary Fig. S6c). Lastly, we examined LSD1 in metastatic breast cancers and found that among those cases with LSD1 abnormalities, the majority of them (i.e., ~1.5% of all these metastatic cases) carried either homozygous deletion or mutations of LSD1 (Supplementary Fig. S6d, both mutations are missense or truncation mutations affecting the LSD1 AO domain). Collectively, these analyses provide further support that LSD1 is a tumor suppressor under the setting of luminal breast cancer.
Figure 7. LSD1 is a suppressor of human luminal breast cancer and metastasis.
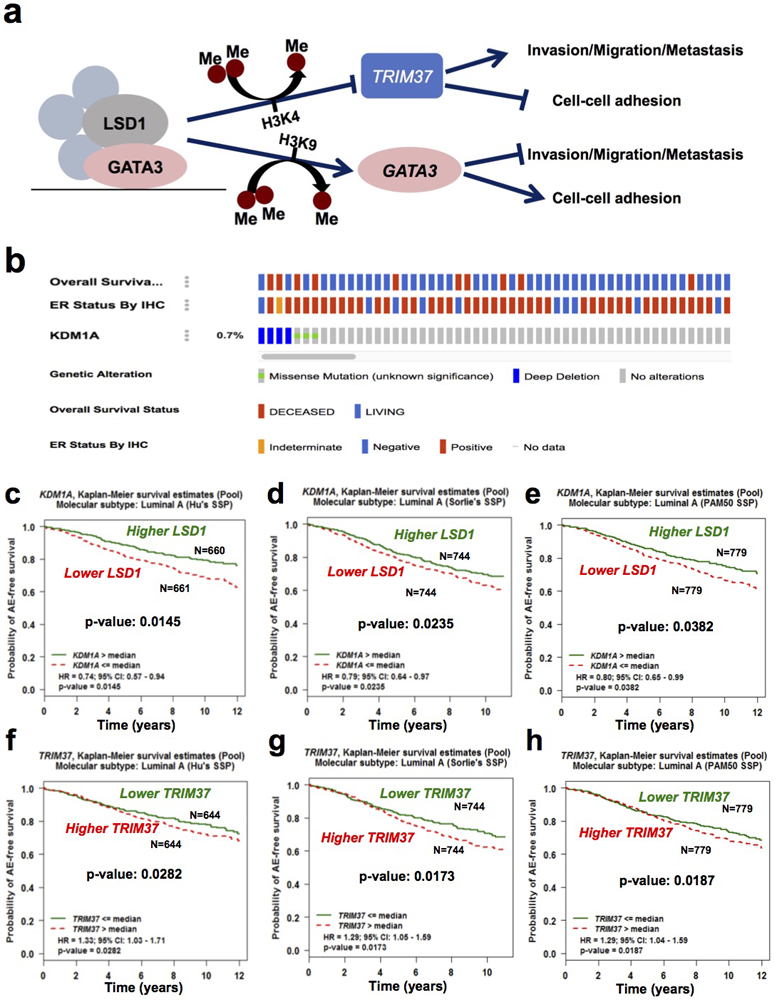
(a) A proposed model of LSD1 in maintaining luminal MEC differentiation (e.g., luminal epithelial cell adhesion gene expression) and in inhibiting their invasion, migration and possibly also metastasis.
(b) OncoPrint showing cases with LSD1 (KDM1A) deep deletion or mutation from the TCGA breast cancer cohort, their overall survival status and ER status, by using the cbioportal online tool.
(c-h) Kaplan-Meier curves showing lower levels of LSD1 (c-e) and higher levels of TRIM37 (f-h) are associated with worse any event (AE)-free survival in patients with luminal A breast cancers in three different cohorts [c, f: Hu’s SSP (Single Sample Predictor); d, g: Sorlie’s SSP; e, h: PAM50 SSP], based on bc-GenExMiner online tool.
Discussion
As a histone demethylase, LSD1 can be recruited by different TFs to repress or activate its targets genes, and by doing so, it can either function as an oncoprotein or a tumor suppressor in breast cancer development in a context-dependent manner. Here we reveal that in luminal breast cells, LSD1 largely plays a tumor suppressor role by maintaining expression of cell-cell adhesion genes and by suppressing invasion and migration. This role is mediated in part via the luminal TF GATA3, which is a master regulator for mature luminal cells and a well-known tumor suppressor in breast cancer [23, 24, 42, 43]. We show that LSD1 and GATA3 are in the same multi-protein complexes and directly regulate GATA3 expression, and that LSD1 represses expression of a key breast cancer oncogene, TRIM37, in part via GATA3. In addition, knockdown of LSD1 or GATA3 leads to reduced expression of key cell-cell adhesion genes, such as CDH1. LSD1 and GATA3 can regulate expression of these adhesion molecules either directly (e.g., CDH1 was reported as a target of GATA3 [34]) or indirectly; the latter can be achieved via repression of TRIM37, as multiple genes related to cell-cell adhesion, such as CDH1, VCL, CTNNA1, and CLDN3, were reported previously as potential target genes of TRIM37 (repressed by TRIM37, possibly via PRC2/EZH2) [32].
Although the majority of the published work so far has recognized LSD1 as an oncoprotein in breast cancer, it is important to note that the reported oncogenic role of LSD1 in breast cancer may be largely related to proliferation/survival of breast cancer cells, including the breast CSC subpopulation [13, 15-17]. LSD1 has also been linked to EMT in breast cancer cells [44, 45], but the breast cancer cells under investigation in these studies were ER− cells (e.g., basal-like breast cancer cells). In this study, we also found that LSD1 was required for growth of luminal breast cells, possibly in a demethylase activity-independent manner; genetic loss of LSD1 (rather than inhibition of its enzymatic activity) led to their reduced proliferation/survival (Supplementary Fig. S3b-c). In luminal breast cells, similar to GATA3 and ERα, LSD1 may work with them and together, these factors may serve as lineage-specific survival factors for luminal MECs; this is supported by the fact that induced loss of GATA3 in luminal cancer cells at the early stage of cancer development also leads to cell death [43], and that approaches targeting the ER program can lead to reduced proliferation and/or survival of ER+ luminal breast cancer cells (i.e., the basis for hormone therapy). It should be pointed out that our data showing that LSD1 can function as a tumor suppressor in luminal breast cancer is not inconsistent with its role as a breast cancer oncoprotein, as these may largely reflect different roles of this epigenetic regulator in regulating different biological processes (even within the same tumor), possibly by using different functional domains of the LSD1 protein.
As currently inhibitors for LSD1 are under clinical trial (e.g., in leukemia [46, 47]), to potentially use them for breast cancer treatment, it is important to define the correct context under which LSD1 plays an oncogenic role (versus a tumor suppressive role), so that therapeutic approaches targeting this demethylase can be applied precisely. Here we show that in addition to an oncogenic role, LSD1 can also play a tumor suppressor role in breast cancer, in particular in ER+ luminal breast cancer; relating to this, LSD1-inhibition can be oncogenic by increasing the invasiveness and migration of luminal breast cancer cells. As cancer cells within a tumor can be highly heterogeneous and exhibit plasticity, even under the setting that LSD1 mainly plays an oncogenic role (e.g., in basal-like breast cancer containing largely ER− cells), LSD1-inhibition may lead to unwanted oncogenic programs (e.g., increased invasion/migration) in some cancer cells. Thus, in addition to targeting the oncogenic programs driven by LSD1, it is also important to define novel therapeutic targets associated with the oncogenic pathways caused by LSD1-inhibition. We show that TRIM37 is such a target and its inhibition can suppress the increased invasiveness and migration of luminal breast cancer cells triggered by LSD1-inhibition. Overall, our study unveils a context-dependent tumor suppressor role of LSD1 in breast cancer and provides insights into how to target it therapeutically.
Materials and Methods
Cells culture
MCF7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Big Cabin, OK) supplemented with 10% fetal bovine serum (FBS), 100 IU ml−1 penicillin/streptomycin. T47D cells were cultured in Roswell Park Memorial Institute-1640 medium (RPMI-1640) (Gibco) with 10% FBS, 3.2 μg ml−1 insulin (Sigma) and 100 IU ml−1 penicillin/streptomycin. MCF10A cells were cultured in Ham’s F12 medium and Dulbecco’s modified Eagle’s medium with 2.5 mM l-glutamine (DMEM:F12, Gibco), 5% horse serum (Gibco), 20 ng ml−1 EGF (Peprotech), 100 ng ml−1 cholera toxin (Sigma), 10 μg ml−1 insulin (Sigma), 0.5 mg ml−1 hydrocortisone (Sigma) and 100 IU ml−1 penicillin/streptomycin. All cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained at 37°C and 5% CO2 in a humidified incubator.
Mouse models
The Lsd1L/L (B6.129-Kdm1atm1.1Sho/J) conditional knockout mouse line was obtained from Dr. Stuart Orkin (Harvard Medical School). Rosa26-LSL-YFP (R26Y) [B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J, Stock No: 006148] reporter mice and MMTV-PyMT [FVB/N-Tg(MMTV-PyVT)634Mul/J, Stock No: 002374] transgenic mice were obtained from The Jackson Laboratory (Bar Harbor, ME). To target luminal MECs in MMTV-PyMT;Lsd1L/L;R26Y or MMTV-PyMT;R26Y female mice, mice were anesthetized and Ad-K8-Cre adenovirus [diluted in injection medium (DMEM supplemented with 0.1% Bromophenol blue and 0.01M CaCl2)] were introduced into mammary ducts of their #4 inguinal glands via intraductal injection [36]. All experimental and control mice were female young adult or adult mice. All animal experiments were approved by Brigham and Women’s Hospital Institutional Animal Care and Use Committee (IACUC).
Western blot analysis
Cells were collected in ice-cold PBS, and lysed in ice-cold whole cell extraction buffer (WCEB) containing 25 mM β-glycerophosphate (pH 7.3), 5 mM EDTA, 2 mM EGTA, 5 mM β-mercaptoethanol, 1% Triton X-100, 0.1 M NaCl, and a protease inhibitor mixture (Roche Applied Science, Indianapolis, IN). The protein concentrations of the cell lysates were determined by use of Bradford methods and boiled in SDS sample buffer [50 mM Tris (pH 6.8), 100 mM DTT, 2% SDS, 0.1% bromophenol blue, 10% glycerol]. The proteins were separated on 8~10% SDS polyacrylamide gel and electro-transferred to polyvinylidene fluoride (PVDF) membrane. After blocking with 3% BSA-TBST, primary antibodies [ELF5: Abcam (Cambridge, MA) ab136119; 1:1000, GATA3: Abcam ab199428 or Millipore (Burlington, MA) 9076; 1:1000, LSD1: Abcam ab17721; 1:1000, TRIM37: Abcam ab95997; 1:1000, ERα: Santa Cruz (Dallas, TX) SC542; 1:1000, Ub-H2A: Cell Signaling (Danvers, MA) 8240S; 1:1000, H2A: Cell Signaling 2578S; 1:1000, H3K4me2: PTM Biolabs (Hangzhou, China) #PTM-612; 1:1000, H3: Cell Signaling 9715S; 1:1000, H4: Cell Signaling 2592S; 1:1000, GAPDH: Bioworld (Nanjing, China) AP0063; 1:5000] were detected using HRP-conjugated anti-rabbit antibodies and visualized on Tanon-5200 Chemiluminescent Imaging System (Tanon Science & Technology, Shanghai, China). β-actin (ZSGB-BIO TA-09; 1:1000) was also tested to confirm equal loading.
Quantitative RT-PCR
For breast cell lines, total RNA from cells was extracted with TRIzol reagent (Invitrogen, Waltham, MA) following the manufacturer’s instruction, and complementary DNA was synthesized using moloney murine leukemia virus (M-MLV) reverse transcriptase with random primers. cDNA was generated with BioTeke super RT kit (Bioteke, Beijing, China) according to the manufacture’s protocol. qRT-PCR was performed using SYBR Premix Ex TaqTM [TaKaRa (Beijing, China), RR820A]. The RT-PCR primer sequences are listed in Supplementary Data 3.
For mouse primary MECs, total RNAs from sorted YFP+ cells were purified by the Allprep DNA/RNA Micro kit (Qiagen, Germantown, MD), followed by amplification of RNA and generation of cDNA with QuantiTect Whole Transcriptome Kit (Cat. NO. 207043) according to manufacture’s protocol. qRT-PCR was performed using FastStart SYBR Green Master (Roche). The RT-PCR primer sequences are listed in Supplementary Data 3.
MTT assay
Cells were seeded in a 96-well plate to a final concentration of 5000 cells/well and transfected with siRNA as described above for 24 hours (h), 48 h and 72 h. For MTT assay, culture medium was removed and fresh medium was added to each well along with 10 μl of MTT solution [BestBio (Shanghai, China), BB-4201-500T]. After incubation at 37°C for 4 h, the medium was poured off and replaced by 150 ml of MTT solvent. The plates were read at wavelength of 490 nm using a microplate reader (BioTek, Winooski, VT). Six reduplicate wells were used for each treatment, and experiments were repeated three times.
Small interfering RNA
Small interfering RNA (siRNA) (Human GATA3 SMARTpool, Dharmacon, #E-003781-00, California, USA; Human LSD1 SMARTpool, Dharmacon, #E-009223-00, California, USA) and nonspecific control siRNA were obtained from Invitrogen and transfected into MCF7, T47D and MCF10A cells using Lipofectamine RNAiMAX (Invitrogen).
Nuclear extraction
3×107 Cells were collected and resuspended in PBS. Cells were centrifuged at 4 °C at 3500 rpm for 15 min, and subsequently 2000 rpm for 5 min. cells were resuspended in Buffer A (20 mM Hepes pH 7.9,10% Glycerol, 1.5 mM MgCl2, 0.2 mM EDTA pH 8.0, 0.5% NP-40, 1 mM DTT) to destroy cell membrane. After incubated 10 min on ice, cells were collected by centrifugation at 4°C at 2000 rpm for 5 min. The nuclear membranes were digested with Buffer B (20 mM Hepes pH 7.9, 10% Glycerol, 500 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA pH 8.0, 0.5% NP-40, 1 mM DTT), incubated 60 min on ice, and disturbed by vortexes every 5 min. After centrifugation at 12000 rpm for 30 min at 4 °C, the supernatant was transferred into dialysis bag and put into Buffer C (20 mM Hepes pH 7.9, 10% Glycerol, 200 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA pH 8.0, 0.5% NP-40, 1 mM DTT) overnight at 4 °C. Then the supernatant was collected by centrifugation at 12000 rpm for 30 min at 4 °C. The final supernatant was stored in −80 °C.
Co-immunoprecipitation (co-IP) Assay
Cell nuclear extract was pre-cleared with control IgG [Bioss (Boston, MA), bs-0295G-HRP] and protein A/G beads (Invitrogen, 10004D). Pre-cleared extract was then incubated with specific antibody (10 μg; LSD1, Abcam ab17721; GATA3, MILLIPORE 9076) or negative control IgG (Bioss, bs-0295G-HRP) overnight at 4 °C with rotation. Dynabeads (Invitrogen, 10004D) were added to the mixture and rotated for 3 h at 4 °C. After a brief centrifugation, the pellet was washed in RIPA buffer (1% DOC Sodium deoxgoholate, 0.1% SDS) 4-5 times at 4 °C for 10 min, and the immunoprecipitated protein complexes were analyzed by Western blot analysis with specific antibodies.
Site-directed Mutagenesis
LSD1 R251Q mutant was generated by site-directed mutagenesis kit [E0552S, NEB (Ipswich, MA)] and cloned into the pGEX vector. The primers used in the study were (sense) 5’- AGTCCACCAAGTTCACAGTT - 3’; (antisense) 5’- TGAACTGTGAACTTGGTGGAC - 3’.
Gel Filtration, GST Pull-Down Assays
MCF7 cell nuclear extract was prepared and fractionated as previously described [18, 48]. The eluted fractions were analyzed by Western Blot (GATA3: Millipore 9076; 1:1000, LSD1: Abcam ab17721; 1:1000, CoREST: Millipore 07455; 1:1000, HDAC1: Abcam ab7028; 1:1000, HDAC2: Abcam ab7029; 1:1000). GST fusion proteins were cloned into pGEX and transformed in the BL21 strain of E. coli. The successfully transformed monoclone was grown in LB liquid AmpR culture medium at 37°C with moderate agitation (OD600 0.6-0.8). Expression of GST fusion proteins was induced by 0.4 mmol/L IPTG with shaking at 37 °C for 5 h. The bacteria were collected by centrifugation, resuspended in STE (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1.5% Sodium lauroyl sarcosine) containing Protease Inhibitor (BestBio, BB-3301) and sonicated (sonication for 7 sec, followed by pausing for 40 sec for each cycle, totally 30 min). The supernatant was incubated with glutathione-sepharose-transferase (GST)-beads (Invitrogen, G2879) at 4 °C overnight. After a brief centrifugation, the beads were washed in PBS/0.1% Triton for 3-4 times (10 min each time) at 4 °C. The proteins that bound to the beads were resuspended in PBS/ 0.1% Triton/ 0.01% NaN3. Expression of GST-tagged LSD1 was confirmed by SDS-PAGE and Coomassie blue staining [Solarbio (Beijing, Chia), P1300-500]. Nuclear extracts (500 μg) were incubated with equal amount of GST-tagged LSD1 proteins at 4 °C overnight. The proteins that bound to the beads were eluted with SDS sample buffer after washing with RIPA buffer (1% DOC Sodium deoxgoholate, 0.1% SDS) for 3 times, then analyzed by SDS-PAGE and Western blot.
Flow cytometry
Mammary tumors were dissected and minced, and then incubated in digestion medium (DMEM/F12 with 2% Penicillin/Streptomycin, 0.1 mg ml−1 Gentamicin, 0.6% Nystatin, 2 mg ml−1 Collagenase A, 0.096 mg ml−1 Hyaluronidase) at 37°C with shaking for 1-1.5 hour. After digestion, the cells/tissues were treated sequentially with 0.25% trypsin/EDTA (37°C, 2 minutes), 5 mg ml−1 dispase with DNaseI (0.1mg ml−1, Sigma, St. Louis, MO) (37°C, 5 minutes), cold red blood cell (RBC) lysis buffer (on ice, 2-3 minutes). Between each treatment step, cells/tissues were washed with 1x PBS with 5% FCS. After treatment with the RBC lysis buffer, cells/tissues were filtered through 40 μm cell strainer and washed with 1x PBS/5% FCS, to obtain single cell suspension [49]. Flow cytometric (FACS) analysis was performed with an Accuri C6 analyzer (BD Biosciences, San Jose, CA) and analyzed with CFlow (BD Biosciences), or with a DXP11 flow cytometer and analyzed with FlowJo. FACS cell sorting was performed with a FACSAria sorter (BD Biosciences).
Immunofluorescence (IF) staining
MCF7 and T47D cells were seeded on slides at an appropriate density for 24h. After paraformaldehyde fixation, cells on slides were treated with 0.3% Triton X-100 for permeabilization of the plasma membrane and then immunostained with specific antibodies including anti-LSD1 (Abcam, ab90996 1:200 or ab17721; 1:200), anti-GATA3 (Abcam, ab199428; 1:200), anti-TRIM37 (Abcam, ab95997; 1:200), and DAPI (nuclear marker). The expression of target proteins (red or green) and DAPI (blue) were examined by fluorescence microscopy; areas of co-localization are shown in the merged images.
For the mouse model, IF staining was performed on lung tissue sections derived from the indicated mouse models injected with Ad-K8-Cre that were fixed in 10% formalin (Fisher Scientific, Hampton, NH) and embedded in paraffin. Antigen retrieval (Citrate buffer pH 6.0, 20 min boil in microwave oven, level 1) was performed prior to blocking. Primary antibody [anti-KDM1/LSD1 (ab17721, 1:500, Abcam, Cambridge, UK)] and the secondary antibody [goat anti-rabbit IgG conjugated with AF594 (A11037, 1:250)] were used. Slides were counterstained with DAPI (1 μg/ml).
Transwell and wound-healing assays
For transwell assays, the membranes of Transwell chambers (24-well insert; pore size, 8 μm; BD Biosciences) were coated with 20 μg/ml fibronectin (Sigma-aldrich) at 37 °C overnight. Cells (1×105) were resuspended with serum-free medium and plated in the top chamber. Inserts were placed in complete medium supplemented with serum. After 24 h, cells that invaded through the pores and migrated to the lower surface of the membrane were fixed and stained with 1% crystal violet (Solarbio). Photos were taken and the numbers of cells were counted.
For wound-healing assays, cells (5×105) were plated on 6-well plate and cultured to confluence. A wound was created by manually scraping the monolayer of cells with a 200 μl tip and washed with PBS. Cells were maintained in serum-free medium. The wounds were photographed at 0 and 48 h following scraping, and the sizes of the healing were measured with Image-Pro Plus and analyzed with GraphPad Prism. Nine photos were taken for each treatment. All the experiments were repeated three times.
ChIP and double ChIP
1×108 Cells were treated with 1% formaldehyde for 10 min for cross-link, and the cross-link was stopped by 0.125 M glycine for 5 min. Cells were washed with cold PBS, collected by centrifugation at 1500 rpm for 5 min at 4 °C and resuspended in ChIP lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8.0) containing protease inhibitor (BestBio, BB-3301). After incubated 60 min on ice, cells were sonicated to shear DNA to an average size of 200-1000 base pairs on ice (sonication for 5 sec, followed by pausing for 5s for, for a total of 3 min. The output of sonication is 40%W). The immunoprecipitating antibodies (10 μg; LSD1, Abcam ab17721, MILLIPORE 17-10531; GATA3, MILLIPORE 9076; H3K4me2, Abcam ab7766, MILLIPORE 17-677; H3K9me2: Abcam ab1220) were added to the supernatant and incubated overnight at 4 °C with rotation. Dynabeads (Invitrogen, 10004D) were added to the mixture and rotated for 6 h at 4 °C. The beads/antibody complexes were washed for 15 min on a rotating platform with the buffers as below: Low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 150 mM NaCl); High salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 500 mM NaCl); LiCl buffer (0.25 M LiCl, 1% NP-40, 1% SDC, 1 mM EDTA, 10 mM Tris-HCl pH8.0); TE buffer (20 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0). Elution buffer was added to treat protein beads, and the supernatant was taken into new tubes and incubated overnight at 65℃ for reversal of cross-linking. DNA was purified and the enrichment of specific genomic regions was determined by real-time quantitative PCR. Final results were represented as percentage of input chromatin. For double-ChIP, double-ChIP elution buffer (20 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 2% SDS, 15mM DTT) was added to treat the washed beads complexes, the supernatant was transferred into new tubes, diluted 20 times with ChIP dilution buffer (16.7 mM Tris-HCl pH 8.0, 1.2 mM EDTA pH 8.0, 0.01% SDS, 1.1% Triton X-100, 167 mM NaCl) supplemented with 50μg of BSA and protease inhibitor, and incubated with a second antibody overnight at 4℃. Dynabeads were added and the beads/antibody complexes was treated as described in the ChIP protocol. The ChIP primer sequences are listed in Supplementary Data 3.
LSD1 inhibitor treatment
LSD1 inhibitors GSK-LSD1 2HCL [Selleck (Houston, TX), S7574] and pargyline [Shanghai yuanye Bio-Technology Co., Ltd (Shanghai, China), C127543] were used to treat cells. Cells were seeded in 96-well plate at 500 cells/well, and treated with GSK LSD1 2HCL at 50 nm or paragyline at 50 μm for 24, 48 and 72 hours. Cell viabilities were analyzed with the MTT assay.
Microarray analysis of gene expression
Total RNA samples from three independent biological replicates (n = 3) were extracted using RNA Extraction kit (Invitrogen). The array study was performed using Human HT expression beadchip V4 array platform, which contains 47323 probes representing all functionally characterized genes in the human genome. The data were analyzed by Genome Studio (Illumina, San Diego, CA). Gene Set Enrichment Analysis (GSEA) was performed by using the standalone GSEA program (http://software.broadinstitute.org/gsea/downloads.jsp).
ChIP-seq data analysis
Raw reads were aligned to the human refence genome (assembly hg19) using Bowtie2 [50] with default parameters. Peaks were called with MACS2 [51] with default parameters. The co-localization of GATA3 and LSD1 peaks was performed using bedtools [52] intersect utility. The association rule was set as 5,000 bp upstream and 1,000 bp downstream of a gene body, with 1,000,000 bp max extension and curated regulatory domains included. Gene set enrichment analysis of overlapping regions for GATA3 and LSD1 was performed with the webapp GREAT [53] with default settings.
Statistics
Student’s t-test (two-tailed) was used to calculate the P values (unless otherwise indicated). Data were reported as mean ± SEM. No statistical method was used to pre-determine the sample size for mice. No samples or animals were excluded from the analysis. For analysis of lung metastasis, sample identity was blinded from histopathological assessment; otherwise no randomization or blinding was used in the in vivo studies.
Data availability
The microarray expression profiling data has been deposited in the Genbank GEO database (http://www.ncbi.nlm.nih.gov/geo/) under accession code GSE95165.
Supplementary Material
Acknowledgments
We thank Dr. Stuart Orkin for the Lsd1L/L conditional knockout mouse line, and Drs. Alan Cantor, Hui Huang and Andrew Woo (Boston Children’s Hospital) for help with gel filtration chromatography. This research was supported by Breakthrough Award from U.S. Department of Defense (W81XWH-15-1-0100), Pathway-to-Independence K99/R00 grant (CA126980) and R01 grant (CA222560) from NCI/NIH, Seed Grant (SG-0062-10) and Cancer Program Pilot Grant (DP-0137-13-00) from Harvard Stem Cell Institute, and Start-up Fund from Brigham and Women’s Hospital to ZL. This work was also supported by National Natural Science Foundation of China (NSFC31471356) and Jilin Province Science and Technology Development Project (20180101240JC, 20190701005GH) to XH.
Footnotes
Competing Interests statement: The authors declare no potential conflicts of interest.
Conflict of Interest
The authors declare no potential conflicts of interest.
References
- 1.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell 2013; 153: 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012; 150: 12–27. [DOI] [PubMed] [Google Scholar]
- 3.Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest 2014; 124: 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119: 941–953. [DOI] [PubMed] [Google Scholar]
- 5.Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005; 437: 432–435. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009; 138: 660–672. [DOI] [PubMed] [Google Scholar]
- 7.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005; 437: 436–439. [DOI] [PubMed] [Google Scholar]
- 8.Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 2007; 9: 347–353. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 2007; 128: 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007; 449: 105–108. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 2009; 41: 125–129. [DOI] [PubMed] [Google Scholar]
- 12.Lynch JT, Harris WJ, Somervaille TC. LSD1 inhibition: a therapeutic strategy in cancer? Expert Opin Ther Targets 2012; 16: 1239–1249. [DOI] [PubMed] [Google Scholar]
- 13.Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R et al. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010; 31: 512–520. [DOI] [PubMed] [Google Scholar]
- 14.Serce N, Gnatzy A, Steiner S, Lorenzen H, Kirfel J, Buettner R. Elevated expression of LSD1 (Lysine-specific demethylase 1) during tumour progression from pre-invasive to invasive ductal carcinoma of the breast. BMC Clin Pathol 2012; 12: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Y, Vasilatos SN, Boric L, Shaw PG, Davidson NE. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res Treat 2012; 131: 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Lu F, Wang J, Yin F, Xu Z, Qi D et al. Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep 2013; 5: 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Y, Wang Y, Yang XH, Kang T, Zhao Y, Wang C et al. The deubiquitinase USP28 stabilizes LSD1 and confers stem-cell-like traits to breast cancer cells. Cell Rep 2013; 5: 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu X, Li X, Valverde K, Fu X, Noguchi C, Qiu Y et al. LSD1-mediated epigenetic modification is required for TAL1 function and hematopoiesis. Proc Natl Acad Sci U S A 2009; 106: 10141–10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA et al. Molecular portraits of human breast tumours. Nature 2000; 406: 747–752. [DOI] [PubMed] [Google Scholar]
- 20.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001; 98: 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI et al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. Embo J 2010; 29: 1803–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG, Weiss SJ. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci U S A 2012; 109: 16654–16659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asselin-Labat ML, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol 2007; 9: 201–209. [DOI] [PubMed] [Google Scholar]
- 24.Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 2006; 127: 1041–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim E, Wu D, Pal B, Bouras T, Asselin-Labat ML, Vaillant F et al. Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res 2010; 12: R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benner C, Konovalov S, Mackintosh C, Hutt KR, Stunnenberg R, Garcia-Bassets I. Decoding a signature-based model of transcription cofactor recruitment dictated by cardinal cis-regulatory elements in proximal promoter regions. PLoS Genet 2013; 9: e1003906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JY, Park MK, Park JH, Lee HJ, Shin DH, Kang Y et al. Loss of the polycomb protein Mel-18 enhances the epithelial-mesenchymal transition by ZEB1 and ZEB2 expression through the downregulation of miR-205 in breast cancer. Oncogene 2014; 33: 1325–1335. [DOI] [PubMed] [Google Scholar]
- 29.Choi YS, Chakrabarti R, Escamilla-Hernandez R, Sinha S. Elf5 conditional knockout mice reveal its role as a master regulator in mammary alveolar development: failure of Stat5 activation and functional differentiation in the absence of Elf5. Dev Biol 2009; 329: 227–241. [DOI] [PubMed] [Google Scholar]
- 30.Oakes SR, Naylor MJ, Asselin-Labat ML, Blazek KD, Gardiner-Garden M, Hilton HN et al. The Ets transcription factor Elf5 specifies mammary alveolar cell fate. Genes Dev 2008; 22: 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Q, Shi L, Gui B, Yu W, Wang J, Zhang D et al. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res 2011; 71: 6899–6908. [DOI] [PubMed] [Google Scholar]
- 32.Bhatnagar S, Gazin C, Chamberlain L, Ou J, Zhu X, Tushir JS et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 2014; 516: 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Si W, Huang W, Zheng Y, Yang Y, Liu X, Shan L et al. Dysfunction of the Reciprocal Feedback Loop between GATA3- and ZEB2-Nucleated Repression Programs Contributes to Breast Cancer Metastasis. Cancer Cell 2015; 27: 822–836. [DOI] [PubMed] [Google Scholar]
- 34.Yan W, Cao QJ, Arenas RB, Bentley B, Shao R. GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition. J Biol Chem 2010; 285: 14042–14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao L, van Bragt MP, Li Z. A Long-Lived Luminal Subpopulation Enriched with Alveolar Progenitors Serves as Cellular Origin of Heterogeneous Mammary Tumors. Stem Cell Reports 2015; 5: 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tao L, van Bragt MPA, Laudadio E, Li Z. Lineage Tracing of Mammary Epithelial Cells Using Cell-Type-Specific Cre-Expressing Adenoviruses. Stem Cell Reports 2014; 2: 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tao L, Xiang D, Xie Y, Bronson RT, Li Z. Induced p53 loss in mouse luminal cells causes clonal expansion and development of mammary tumours. Nature communications 2017; 8: 14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gallego-Ortega D, Ledger A, Roden DL, Law AM, Magenau A, Kikhtyak Z et al. ELF5 Drives Lung Metastasis in Luminal Breast Cancer through Recruitment of Gr1+ CD11b+ Myeloid-Derived Suppressor Cells. PLoS Biol 2015; 13: e1002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Network TCGA. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486: 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature communications 2016; 7: 11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asselin-Labat ML, Sutherland KD, Vaillant F, Gyorki DE, Wu D, Holroyd S et al. Gata-3 negatively regulates the tumor-initiating capacity of mammary luminal progenitor cells and targets the putative tumor suppressor caspase-14. Mol Cell Biol 2011; 31: 4609–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kouros-Mehr H, Bechis SK, Slorach EM, Littlepage LE, Egeblad M, Ewald AJ et al. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell 2008; 13: 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi HJ, Park JH, Park M, Won HY, Joo HS, Lee CH et al. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO reports 2015; 16: 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng J, Xu G, Liu J, Zhang N, Li L, Ji J et al. Phosphorylation of LSD1 at Ser112 is crucial for its function in induction of EMT and metastasis in breast cancer. Breast Cancer Res Treat 2016; 159: 443–456. [DOI] [PubMed] [Google Scholar]
- 46.Mould DP, McGonagle AE, Wiseman DH, Williams EL, Jordan AM. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to date. Medicinal research reviews 2015; 35: 586–618. [DOI] [PubMed] [Google Scholar]
- 47.Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018; 33: 495–511 e412. [DOI] [PubMed] [Google Scholar]
- 48.Woo AJ, Moran TB, Schindler YL, Choe SK, Langer NB, Sullivan MR et al. Identification of ZBP-89 as a novel GATA-1-associated transcription factor involved in megakaryocytic and erythroid development. Mol Cell Biol 2008; 28: 2675–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML et al. Generation of a functional mammary gland from a single stem cell. Nature 2006; 439: 84–88. [DOI] [PubMed] [Google Scholar]
- 50.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods 2012; 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010; 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray expression profiling data has been deposited in the Genbank GEO database (http://www.ncbi.nlm.nih.gov/geo/) under accession code GSE95165.