

Abstract
Background
In many parts of the world, hepatitis A infection represents a significant cause of morbidity and socio‐economic loss. Whilst hepatitis A vaccines have the potential to prevent disease, the degree of protection afforded against clinical outcomes and within different populations remains uncertain. There are two types of hepatitis A virus (HAV) vaccine, inactivated and live attenuated. It is important to determine the efficacy and safety for both vaccine types.
Objectives
To determine the clinical protective efficacy, sero‐protective efficacy, and safety and harms of hepatitis A vaccination in persons not previously exposed to hepatitis A.
Search methods
We searched The Cochrane Hepato‐Biliary Group Controlled Trials Register, The Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library, MEDLINE, EMBASE, Science Citation Index Expanded, and China National Knowledge Infrastructure (CNKI) up to November 2011.
Selection criteria
Randomised clinical trials comparing HAV vaccine with placebo, no intervention, or appropriate control vaccines in participants of all ages.
Data collection and analysis
Data extraction and risk of bias assessment were undertaken by two authors and verified by a third author. Where required, authors contacted investigators to obtain missing data. The primary outcome was the occurrence of clinically apparent hepatitis A (infectious hepatitis). The secondary outcomes were lack of sero‐protective anti‐HAV immunoglobulin G (IgG), and number and types of adverse events. Results were presented as relative risks (RR) with 95% confidence intervals (CI). Dichotomous outcomes were reported as risk ratio (RR) with 95% confidence interval (CI), using intention‐to‐treat analysis. We conducted assessment of risk of bias to evaluate the risk of systematic errors (bias) and trial sequential analyses to estimate the risk of random errors (the play of chance).
Main results
We included a total of 11 clinical studies, of which only three were considered to have low risk of bias; two were quasi‐randomised studies in which we only addressed harms. Nine randomised trials with 732,380 participants addressed the primary outcome of clinically confirmed hepatitis A. Of these, four trials assessed the inactivated hepatitis A vaccine (41,690 participants) and five trials assessed the live attenuated hepatitis A vaccine (690,690 participants). In the three randomised trials with low risk of bias (all assessing inactivated vaccine), clinically apparent hepatitis A occurred in 9/20,684 (0.04%) versus 92/20,746 (0.44%) participants in the HAV vaccine and control groups respectively (RR 0.09, 95% CI 0.03 to 0.30). In all nine randomised trials, clinically apparent hepatitis A occurred in 31/375,726 (0.01%) versus 505/356,654 (0.18%) participants in the HAV vaccine and control groups respectively (RR 0.09, 95% CI 0.05 to 0.17). These results were supported by trial sequential analyses. Subgroup analyses confirmed the clinical effectiveness of both inactivated hepatitis A vaccines (RR 0.09, 95% CI 0.03 to 0.30) and live attenuated hepatitis A vaccines (RR 0.07, 95% CI 0.03 to 0.17) on clinically confirmed hepatitis A. Inactivated hepatitis A vaccines had a significant effect on reducing the lack of sero‐protection (less than 20 mIU/L) (RR 0.01, 95% CI 0.00 to 0.03). No trial reported on a sero‐protective threshold less than 10 mIU/L. The risk of both non‐serious local and systemic adverse events was comparable to placebo for the inactivated HAV vaccines. There were insufficient data to draw conclusions on adverse events for the live attenuated HAV vaccine.
Authors' conclusions
Hepatitis A vaccines are effective for pre‐exposure prophylaxis of hepatitis A in susceptible individuals. This review demonstrated significant protection for at least two years with the inactivated HAV vaccine and at least five years with the live attenuated HAV vaccine. There was evidence to support the safety of the inactivated hepatitis A vaccine. More high quality evidence is required to determine the safety of live attenuated vaccines.
Keywords: Humans; Hepatitis A; Hepatitis A/immunology; Hepatitis A/prevention & control; Hepatitis A Antibodies; Hepatitis A Antibodies/immunology; Hepatitis A Vaccines; Hepatitis A Vaccines/administration & dosage; Hepatitis A Vaccines/immunology; Randomized Controlled Trials as Topic; Time Factors; Vaccination; Vaccination/methods; Vaccines, Attenuated; Vaccines, Attenuated/administration & dosage; Vaccines, Attenuated/immunology; Vaccines, Inactivated; Vaccines, Inactivated/administration & dosage; Vaccines, Inactivated/immunology
Hepatitis A immunisation in persons not previously exposed to hepatitis A
Hepatitis A is a common, contagious viral disease in many low‐income countries. It is estimated that world wide, around 1.5 million people are affected each year. The hepatitis A virus is limited to man and several species of non‐human primates. It is transmitted primarily by faecal‐oral spread from person to person, or through ingestion of contaminated food or water. Since 1995, hepatitis A vaccines have been used to prevent hepatitis A in people not yet exposed to the hepatitis A virus. Only three of the included trials were considered to be at low risk of bias; that is, free from overestimation of benefits and underestimation of harm due to systemic errors. In persons not previously exposed to hepatitis A infection, hepatitis A vaccination with inactivated or live attenuated hepatitis A vaccines had a clear effect on reducing the risk of developing clinically apparent hepatitis A. The review also found that hepatitis A vaccines significantly reduce the risk of lacking protective antibodies against hepatitis A. The inactivated vaccine appears to be relatively safe. There were insufficient data to draw any conclusions on production of protective antibodies and adverse events for live attenuated vaccines.
Summary of findings
Summary of findings for the main comparison.
Hepatitis A vaccines for preventing clinical hepatitis A in those not previously exposed
| Hepatitis A vaccines for preventing hepatitis A in those not previously exposed | ||||||
| Patient or population: patients with preventing hepatitis A. Settings: pre‐exposure. Intervention: hepatitis A vaccines. | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (trials) | Quality of the evidence (GRADE) | Comment | |
| Assumed risk | Corresponding risk | |||||
| Control | Hepatitis A vaccines | |||||
| Clinically apparent hepatitis A (low risk of bias trials) Dienstag 1999 Follow‐up: 12 to 17 months | 4 per 1000 | 4 per 1000 (0 to 1) | RR 0.09 (0.03 to 0.30) | 41430 (3 studies) | ⊕⊕⊕⊕ high | |
| Clinically apparent hepatitis A (all included trials) Dienstag 1999 Follow‐up: 1 to 60 months | 1 per 1000 | 0 per 1000 (0 to 0) | RR 0.09 (0.05 to 0.17) | 732380 (9 studies) | ⊕⊝⊝⊝ very low | |
| Clinically apparent hepatitis A (inactivated HAV vaccines) low risk of bias trials Dienstag 1999 Follow‐up: 12 to 17 months | 4 per 1000 | 0 per 1000 (0 to 1) | RR 0.09 (0.03 to 0.30) | 41430 (3 studies) | ⊕⊕⊕⊕ high | |
| Clinically apparent hepatitis A (inactivated HAV vaccines) all included trials Dienstag 1999 Follow‐up: 12 to 17 months | 5 per 1000 | 1 per 1000 (0 to 1) | RR 0.12 (0.05 to 0.31) | 41690 (4 studies) | ⊕⊕⊝⊝ low | |
| Clinically apparent hepatitis A (live attenuated HAV vaccines) low risk of bias Dienstag 1999 | See comments | See comments | No included inactivated HAV vaccine study was at low risk of bias. | |||
| Clinically apparent hepatitis A (live attenuated HAV vaccines) all included trials Dienstag 1999 Follow‐up: 1 to 60 months | 1 per 1000 | 0 per 1000 (0 to 0) | RR 0.07 (0.03 to 0.17) | 690690 (5 studies) | ⊕⊝⊝⊝ very low | |
| Clinically apparent hepatitis A (Dose of live attenuated HAV vaccine, titre >= TCID50 ) low risk of bias Dienstag 1999 | See comment | See comment | No included inactivated HAV vaccine study was at low risk of bias. | |||
| Clinically apparent hepatitis A (Dose of live attenuated HAV vaccine, titre >= TCID50 ) all included trials Dienstag 1999 Follow‐up: 24 to 60 months | 9 per 1000 | 0 per 1000 (0 to 1) | RR 0.03 (0.02 to 0.06) | 62270 (2 studies) | ⊕⊝⊝⊝ very low | |
| Clinically apparent hepatitis A (Dose of live attenuated HAV vaccine, titre < TCID50 ) low risk of bias Dienstag 1999 | See comment | See comment | No included inactivated HAV vaccine study was at low risk of bias. | |||
| Clinically apparent hepatitis A (Dose of live attenuated HAV vaccine, titre < TCID50 ) all included trials Dienstag 1999 Follow‐up: 1 to 36 months | 0 per 1000 | 0 per 1000 (0 to 0) | RR 0.11 (0.05 to 0.26) | 628420 (3 studies) | ⊕⊝⊝⊝ very low | |
| Clinically apparent hepatitis A (low endemicity) low risk of bias Dienstag 1999 Follow‐up: 12 months | 71 per 1000 | 14 per 1000 (6 to 31) | RR 0.19 (0.08 to 0.42) | 1037 (1 study) | ⊕⊕⊕⊕ high | |
| Clinically apparent hepatitis A (low endemicity) all included trials Dienstag 1999 Follow‐up: 12 months | 71 per 1000 | 14 per 1000 (6 to 30) | RR 0.19 (0.08 to 0.42) | 1037 (1 study) | ⊕⊕⊕⊕ high | |
| Clinically apparent hepatitis A (high endemicity) low risk of bias Dienstag 1999 Follow‐up: 15 to 17 months | 3 per 1000 | 0 per 1000 (0 to 0) | RR 0.05 (0.01 to 0.17) | 40393 (2 studies) | ⊕⊕⊕⊕ high | |
| Clinically apparent hepatitis A (high endemicity) all included trials Dienstag 1999 Follow‐up: 1 to 60 months | 1 per 1000 | 1 per 1000 (0 to 0) | RR 0.08 (0.04 to 0.15) | 731343 (8 studies) | ⊕⊝⊝⊝ very low | |
| Clinically apparent hepatitis A (single dose regimen) low risk of bias Dienstag 1999 Follow‐up: 15 months | 124 per 1000 | 4 per 1000 (0 to 58) | RR 0.03 (0 to 0.47) | 274 (1 study) | ⊕⊕⊕⊕ high | |
|
Clinically apparent hepatitis A (single dose regimen) all included studies Dienstag 1999 Follow‐up: 1 to 60 months |
1 per 1000 | 0 per 1000 (0 to 0) | RR 0.07 (0.03 to 0.15) | 690707 (6 studies) | ⊕⊝⊝⊝ very low | |
| Clinically apparent hepatitis A (multiple doses) low risk of bias Dienstag 1999 Follow‐up: 12 to 17 months | 4 per 1000 | 0 per 1000 (0 to 1) | RR 0.11 (0.03 to 0.40) | 41156 (2 studies) | ⊕⊕⊕⊕ high | |
|
Clinically apparent hepatitis A (multiple doses) all included trials Dienstag 1999 Follow‐up: 12 to 17 months |
4 per 1000 | 1 per 1000 (0 to 1) | RR 0.14 (0.05 to 0.37) | 41416 (3 studies) | ⊕⊕⊕⊝ moderate | |
| Clinically apparent hepatitis A (follow‐up duration 1 to 12 months) low risk of bias Dienstag 1999 Follow‐up: 12 months | 71 per 1000 | 14 per 1000 (6 to 30) | RR 0.19 (0.08 to 0.42) | 1037 (1 study) | ⊕⊕⊕⊕ high | |
|
Clinically apparent hepatitis A (follow‐up duration 1 to 12 months) all included trials Dienstag 1999 Follow‐up: 1 to 12 months |
47 per 1000 | 9 per 1000 (5 to 17) | RR 0.20 (0.1 to 0.37) | 2377 (3 studies) | ⊕⊝⊝⊝ very low | |
|
Clinically apparent hepatitis A (follow‐up duration 13 to 24 months) low risk of bias Dienstag 1999 Follow‐up: 15 to 17 months |
3 per 1000 | 0 per 1000 (0 to 0) | RR 0.05 (0.01 to 0.17) | 40393 (2 studies) | ⊕⊕⊕⊕ high | |
|
Clinicially apparent hepatitis A (follow‐up duration 13 to 24 months) all included trials Dienstag 1999 Follow‐up: 15 to 24 months |
4 per 1000 | 0 per 1000 (0 to 1) | RR 0.06 (0.02 to 0.17) | 164776 (4 studies) | ⊕⊝⊝⊝ very low | |
| Clinically apparent hepatitis A (follow‐up duration 25 to 48 months) low risk of bias | See comments | See comments | Insufficients evidence | |||
|
Clinicially apparent hepatitis A (follow‐up duration 25 to 48 months) all included trials Dienstag 1999 Follow‐up: 36 months |
0 per 1000 (0 to 0) | RR 0.05 (0.02 to 0.13) | 564642 (1 study) | ⊕⊕⊝⊝ low | ||
| Clinically apparent hepatitis A (follow‐up duration 49 to 60 months) low risk of bias | See comments | See comments | Insufficient evidence | |||
|
Clinically apparent hepatitis A (follow‐up duration 49 to 60 months) all included trials Dienstag 1999 Follow‐up: 60 months |
67 per 1000 | 4 per 1000 (1 to 27) | RR 0.06 (0.01 to 0.41) | 585 (1 study) | ⊕⊕⊝⊝ low | |
|
All‐cause mortality low risk of bias Mortality Follow‐up: 17 months |
0 per 1000 (0 to 2) | RR 1.40 (0.62 to 3.16) | 585 (1 study) | ⊕⊕⊕⊕ high | ||
|
All‐cause mortality all included trials Mortality Follow‐up: 17 months |
0 per 1000 (0 to 2) | RR 1.40 (0.62 to 3.16) | 585 (1 study) | ⊕⊕⊕⊕ high | ||
| Hepatitis A‐related mortality | See comment | See comment | No trial directly addressed this as an outcome. | |||
| Lack of sero‐protection, (anti HAV IgG < 10mIU/L) (low risk of bias) Maiwald 1997 Follow‐up: 12 to 17 months | See comment | See comment | No study included this outcome. | |||
| Lack of sero‐protection, (anti HAV IgG < 10mIU/L) (all included trials) Maiwald 1997 Follow‐up: 12 to 17 months | See comment | See comment | No study included this outcome. | |||
| Lack of sero‐protection, (anti HAV IgG < 20mIU/L) (low risk of bias ) Maiwald 1997 Follow‐up: 17 months | 972 per 1000 | 10 per 1000 (0 to 29) | RR 0.01 (0.00 to 0.03) | 486 (1 study) | ⊕⊕⊕⊕ high | |
| Lack of sero‐protection, (anti HAV IgG < 20mIU/L) (all included trials) Maiwald 1997 Follow‐up: 12 to 17 months | 973 per 1000 | 10 per 1000 (0 to 29) | RR 0.01 (0 to 0.03) | 739 (2 studies) | ⊕⊕⊝⊝ low | |
| Non‐serious local adverse events (low risk of bias) inactivated HAV vaccine ICH‐GCP 1997 Follow‐up: 12 to 15 months | 85 per 1000 | 101 per 1000 (57 to 179) | RR 1.19 (0.67 to 2.11) | 1299 (2 studies) | ⊕⊕⊕⊕ high | |
| Non‐serious local adverse events (all included trials) ICH‐GCP 1997 Follow‐up: 12 to 15 months | 98 per 1000 | 118 per 1000 (84 to 166) | RR 1.21 (0.86 to 1.70) | 1559 (4 studies) | ⊕⊕⊝⊝ low | |
| Non‐serious systemic adverse events (low risk of bias) inactivated HAV vaccine ICH‐GCP 1997 Follow‐up: 12 to 15 months | 42 per 1000 | 45 per 1000 (28 to 74) | RR 1.09 (0.67 to 1.78) | 1299 (3 studies) | ⊕⊕⊕⊕ high | |
|
Non‐serious systemic adverse events (all included studies) inactivated HAV vaccine
ICH‐GCP 1997 Follow‐up: 12 to 15 months |
65 per 1000 | 64 per 1000 (45 to 92) | RR 0.98 (0.68 to 1.41) | 1599 (4 studies) | ⊕⊕⊝⊝ low | |
| Non‐serious local adverse events (low risk of bias) live attenuated HAV vaccine ICH‐GCP 1997 | See comment | See comment | Insufficient evidence | |||
| Non‐serious local adverse events (all included studies) live attenuated HAV vaccine ICH‐GCP 1997 | See comment | See comment | Insufficient evidence | |||
| Non‐serious systemic adverse events (low risk of bias) live attenuated HAV vaccine ICH‐GCP 1997 Follow‐up: 12 to 17 months | See comment | See comment | Insufficient evidence | |||
| Non‐serious systemic adverse events (all included studies) live attenuated HAV vaccine ICH‐GCP 1997 | See comment | See comment | Insufficient evidence | |||
| *The basis for the assumed risk (eg, the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Background
Hepatitis A virus (HAV) is a non‐enveloped positive stranded RNA picornavirus, which is primarily transmitted by the faecal‐oral route (Lemon 1997a). HAV is a significant cause of morbidity and social‐economic losses in many parts of the world (Hollinger 1996; Berge 2000). Poor sanitation and crowded living conditions often result in subclinical infection early in life, which confers lifelong immunity. With improved sanitation and hygiene, hepatitis A infections can be avoided in childhood. Consequently, the number of adults susceptible to the disease increases. This is important given that adults are more likely to have clinically apparent disease and have a higher mortality than children following infection (WHO 2000). Currently, an estimated 1.5 million people worldwide become infected with clinically‐evident hepatitis A each year (WHO 2000). Despite this, the overall mortality appears to be low (0.1% in children less than 14 years) (WHO 2000). The incidence of hepatitis A is closely related to economic development, socio‐economic status, and geographical conditions. Sero‐epidemiological studies show that the prevalence of anti‐HAV antibodies in the general population varies from 15% to close to 100% in different parts of the world (Bell 2002). Anti‐HAV prevalence estimates suggest that middle‐income regions in Asia, Latin America, Eastern Europe, and the Middle East currently have an intermediate or low level of endemicity (Jacobsen 2010).
In regions of low disease endemicity, vaccination against hepatitis A is often recommended for individuals with increased risk of contracting the infection. At particular risk are travellers to areas of intermediate or high endemicity, users of illicit injection drugs, people with haemophilia or other clotting factor disorders, laboratory workers exposed to hepatitis A, workers exposed to non‐human primates, and susceptible individuals with chronic liver diseases (Bell 2005).
Description of the condition
Hepatitis A virus is excreted in the bile and shed in the stools of infected persons (Lemon 1997a). Peak excretion occurs during the two weeks before the onset of jaundice; the concentration of virus in the stool drops after jaundice appears. The average incubation period of hepatitis A is around 28 days (range 15 to 50 days) (Routenberg 1979). Cytotoxic T lymphocytes have been reported to be involved in liver injury caused by acute HAV infection (Vallbracht 1989). This suggests that immune injury is the major pathogenic mechanism of acute HAV infection (Ida 2002).
The course of hepatitis A infection is extremely variable. In children under five years of age 80% to 95% of infections are asymptomatic, while in adults 70% to 95% of infections result in clinical illness (Purcell 2005). Fulminant hepatitis occurs rarely (less than 1% overall), but rates are higher with increasing age and in those with underlying chronic liver disease, including those with chronic hepatitis B and C infections (Yao 1988).
Description of the intervention
Inactivated hepatitis A vaccine
Binn et al were the first to produce an inactivated HAV vaccine from a HAV propagated in cell culture, subsequently purified and inactivated by exposure to formalin (Binn 1986). The vaccine was first licensed in 1992 and became commercially available in 1995. Since then, several inactivated vaccines have been developed and evaluated in non‐human primate models and in human clinical trials of HAV infection. Currently, four inactivated monovalent HAV vaccines are commercially available (Havrix®, Vaqta®, Avaxim®, and Epaxal®) and include antigen prepared from different strains of the HAV. The antigen content is not standardised, and the units by which the antigen is expressed are different for each vaccine (Bell 2005). Because of the different assays used and the absence of an international antigen reference agent, it is not currently possible to compare the antigen content among vaccines. The current inactivated vaccines appear to provide protection against all the many genotypes of hepatitis A and can be used interchangeably (Bell 2005).
Live attenuated hepatitis A vaccine
In order to produce the live attenuated vaccine, the disease producing 'wild type' virus is first modified in the laboratory. This 'attenuated' virus still retains the ability to replicate and stimulate a host immune response but should not cause clinical disease on vaccination. The H2 strain live attenuated vaccine first became available in China in 1992 (Mao 1989). Hu et al then went on to successfully developed a LA‐1 strain live attenuated vaccine (Hu 1988). These two live attenuated vaccines are now divided into subgroups according to an antigen titre of less than or more than or equal to 106.5 TCID50 (Wang 2008). The TCID50 dilution assay quantifies the amount of virus required to produce a cytopathic effect in 50% of inoculated tissue culture cells. Due to distinct differences in assay methods, principles and other infectivity, assay results are not equivalent.
How the intervention might work
Antibodies that persist following vaccination have long been considered as the principle marker of protection against hepatitis A infection. In extensive studies of children and adults, the inactivated hepatitis A vaccine has been found to be highly immunogenic. More recent evidence suggests that protection against HAV is not dependent on antibodies alone (Schmidtke 2005). A more complete understanding of immunity suggests that long‐term protection also occurs through immune cell memory. Others have demonstrated that the HAV vaccine induces a cell mediated response in addition to antibody production (Wang 2000; Wang 2001). Some have suggested that protection conferred by cellular immunity may persist long after anti‐HAV immunoglobulin G (IgG) is no longer detectable in the plasma of people; and following re‐exposure to HAV, vaccine recipients may undergo an anamnestic response that may prevent overt clinical disease (Van Damme 2003). Virosomal vaccines such as Epaxal® are thought to target both macrophages and influenza primed antigen presenting cells, in turn stimulating T and B cell proliferation (Gluck 2002). It is important to note that immunoglobulin M (IgM) class antibodies to HAV can occasionally be detected by standard assays after vaccination (Lemon 1997b). This means that even in the absence of hepatitis A infection, it may be possible to detect IgM post‐vaccination, thus leading to the false conclusion that the individual is infected with hepatitis A.
Why it is important to do this review
The World Health Organization (WHO) makes recommendations for prevention of hepatitis A by vaccination in given populations (WHO 2000). A systematic review of inactivated hepatitis A vaccines has previously been published. This included eight trials studying the effectiveness and safety of hepatitis A vaccines in adults and children (four containing efficacy outcomes, three containing only safety outcomes, and a single study containing efficacy and adverse events outcomes) (Demicheli 2003). The review did not proceed to meta‐analysis and concluded that despite poor design and reporting of trials there was convincing evidence of the effectiveness and safety of inactivated HAV vaccines. No conclusion was made for the effects and safety of the live attenuated HAV vaccine because of the inclusion of a single, poorly reported trial. A meta‐analysis of live attenuated vaccine that identified 13 trials has also been published (Wang 2008). The authors concluded that live attenuated hepatitis A vaccine has good protective efficacy but they did not review vaccine safety. A Cochrane systematic review of hepatitis A vaccination does not exist. So we conducted a systematic review on the effectiveness and safety of vaccination for preventing hepatitis A as set out in our pre‐published Cochrane protocol.
Objectives
To assess the beneficial and harmful effects of pre‐exposure hepatitis A vaccines (inactivated and live attenuated) in adults and children.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised clinical trials irrespective of blinding, publication status, language, or unit of randomisation (individuals or population‐based, that is, cluster randomised clinical trials). Quasi‐randomised studies were excluded for all outcomes other than adverse events. We excluded historical controlled studies.
Types of participants
People of any age or ethnic origin who were at the stage of pre‐exposure to hepatitis A.
Types of interventions
Experimental: any type of inactivated hepatitis or live attenuated A vaccine, irrespective of route of administration, dosage, or schedule.
Control: placebo, no intervention, or any vaccine other than HAV vaccine.
Trials assessing different HAV vaccines or different schedules of the same HAV vaccine were also considered.
Types of outcome measures
Primary outcomes
The outcome measures of this review were sought at maximal follow‐up.
All‐cause mortality.
Mortality from hepatitis A.
Occurrence of hepatitis A. The diagnosis must be based on the serological detection of IgM class antibodies to HAV, elevated serum aminotransferase levels, and clinical symptoms such as fatigue, malaise, headache, myalgias, nausea, vomiting, loss of appetite, jaundice (Dienstag 1999).
Secondary outcomes
Lack of sero‐protection:
number of vaccine recipients with anti‐HAV antibody titre less than 10 mIU/L in accordance with the WHO standard reference serum at maximum follow‐up (Wiedermann 1992);
number of vaccine recipients with anti‐HAV antibody titre less than 20 mIU/L in accordance with the WHO standard reference serum at maximum follow‐up (Maiwald 1997).
Number and types of adverse events. Two types of adverse events were analysed: serious adverse events and non‐serious (local and systemic) adverse events. Serious adverse events were any untoward medical occurrence that resulted in death, were life‐threatening, required hospitalisation or prolongation of hospitalisation, resulted in persistent or significant disability, caused a congenital anomaly or birth defect; or was an event that might have jeopardised the patient or required intervention to prevent one of these former adverse events (ICH‐GCP 1997).
Search methods for identification of studies
Electronic searches
We searched The Cochrane Hepato‐Biliary Group Controlled Trials Register (Gluud 2012), The Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library, MEDLINE, EMBASE, Science Citation Index Expanded, and China National Knowledge Infrastructure (CNKI) (Royle 2003) up to November 2011. Search strategies with the time span of the searches are given in Appendix 1.
Searching other resources
We searched for further potentially relevant trials by cross‐checking the reference lists of published randomised clinical trials and systematic reviews. We used the results of journals searched by hand, for example, the Vaccine journal, the results of which are included in CENTRAL. A full list of journals handsearched by the Cochrane Hepato‐Biliary Group is available in the Group's Module, published in The Cochrane Library (Gluud 2012).
Data collection and analysis
Two authors (GI and JH) independently inspected the abstract of each reference identified by the search and determined the potential relevance of each publication. For potentially relevant publications, or in cases of disagreement, we obtained the full paper and independently inspected it, and applied the inclusion criteria. Duplicate publications on trials were not excluded but listed with the main publication in Included studies. Where uncertainties remained about the duplication of published trials, efforts were made to contact the corresponding author.
Selection of studies
Two authors (GI and JH) independently selected trials to be included in the review according to the pre‐specified selection criteria. Any disagreement was solved by discussion. Where we were unable to resolve disagreements through discussion, we added the publication to those 'awaiting assessment' and contacted the authors of the study for clarification. In the event of no reply from the authors, a third review author (DP) checked the publication to solve disagreements. We documented our justification for excluding studies from the review.
Data extraction and management
Two authors (GI and JH) independently extracted data from the included trials. In the event of any disagreement between the two review authors, a third review author (DP) also extracted the data. We documented our decisions and, where necessary, contacted the trial authors for clarification.
Data on all participants irrespective of compliance or follow‐up were sought to allow intention‐to‐treat analyses. In case a randomised clinical trial had a cross‐over design, we considered data only from the first period.
We identified trials by the name of the first author and year in which the trial was first published. We extracted, checked, and recorded the following data.
Characteristics of the trial
Date
Location and setting of the trial
Publication status
Generation of the allocation sequence
Allocation concealment method
Blinding methods
Characteristics of the participants
Number of participants in each group
Age, sex, nationality, ethnic group, and any risk category
Previous immunisation status (if known)
Presence of immunodeficiency
Baseline comparability
Characteristics of the interventions
Type of vaccine
Type of control
Dose
Immunisation schedule
Route of administration
Characteristics of outcome measures
Primary and secondary outcome measures (as above)
Any adverse events
Length of follow‐up
Loss to follow‐up (drop‐outs) before end of trial
Assessment of risk of bias in included studies
The authors followed the instructions given in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and the Cochrane Hepato‐Biliary Group Module (Gluud 2012) to assess the risk of bias of the included trials.
Due to the risk of biased overestimation of beneficial intervention effects in randomised clinical trials with inadequate methodological quality (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008), we examined the influence of the validity of the included studies on the results by evaluating bias risk domains (Higgins 2011). Where information was not available in the published trial, we made attempts to contact the authors in order to assess the trials correctly.
Sequence generation
Low risk of bias: sequence generation has been achieved using computer random number generation or a random number table.
Unclear: the trial is described as randomised but the method of sequence generation is not specified.
High risk of bias: the sequence generation method was not, or may not be, random. Quasi‐randomised studies, those using dates, names, or admittance numbers in order to allocate patients, are inadequate and were excluded for the assessment of benefits but not for harms.
Allocation concealment
Low risk of bias: allocation was controlled by a central and independent randomisation unit, sequentially numbered, opaque and sealed envelopes or similar, so that intervention allocations could not be foreseen in advance of, or during, enrolment.
Unclear: the trial is described as randomised but the method used to conceal the allocation is not described, so that intervention allocations might have been foreseen in advance of, or during, enrolment.
High risk of bias: the allocation sequence was known to the investigators who assigned participants. Quasi‐randomised studies were excluded for the assessment of benefits but not for harms.
Blinding
Low risk of bias: the trial is described as blinded, the parties that were blinded and the method of blinding were described, so that knowledge of allocation was adequately prevented during the trial.
Unclear: the trial is described as double blind but the method of blinding was not described, so that knowledge of allocation was possible during the trial.
High risk of bias: the trial was not blinded, so that the allocation was known during the trial.
Incomplete outcome data reporting
Low risk of bias: the numbers and reasons for drop‐outs and withdrawals in all intervention groups were described, or it was specified that there were no drop‐outs or withdrawals.
Unclear: the report gave the impression that there had been no drop‐outs or withdrawals, but this was not specifically stated.
High risk of bias: the number or reasons for drop‐outs and withdrawals were not described.
Selective outcome reporting
Low risk of bias: pre‐defined, or clinically relevant and reasonably expected outcomes, were reported on.
Unclear: not all pre‐defined, or clinically relevant and reasonably expected outcomes, were reported on or were not reported fully, or it is unclear whether data on these outcomes were recorded or not.
High risk of bias: one or more clinically relevant and reasonably expected outcomes was not reported on; data on these outcomes were likely to have been recorded.
Other sources of bias
Low risk of bias: if the trial appeared to be free of other sources of bias.
Unclear risk of bias: if there was insufficient information to assess whether other sources of bias were present.
High risk of bias: if it is likely that potential sources of bias related to specific design used, early termination due to some data‐dependent process, lack of sample size or power calculation, or other risks were present.
Trials in which sequence generation, allocation concealment, blinding, outcome data reporting, and selective outcome reporting were reported adequately, with no other source of bias (as defined above), were considered as low risk of bias (Kjaergard 2001; Wood 2008). Trials with one or more domains judged as unclear or at high risk of bias were considered as trials with high risk of bias (Kjaergard 2001; Wood 2008). We also reported on whether the investigators had performed a sample‐size calculation and used intention‐to‐treat analysis.
Measures of treatment effect
The statistical package Review Manager (RevMan 2011) provided by The Cochrane Collaboration was used for the statistical analyses. Dichotomous data were presented as relative risk (RR) and continuous outcomes as mean difference (MD), both with 95% confidence intervals (CI).
Unit of analysis issues
We assessed the outcome of cluster randomised clinical trials at the level of the group, thereby keeping the unit of analysis the same as the unit of randomisation.
Dealing with missing data
In the event that data were missing, we added these trials to those 'awaiting assessment' and contacted the authors of the trial for clarification. In the event of no reply from the authors within six months, we imputed a replacement value for the missing data (assuming all were poor outcomes). We then performed a sensitivity analysis to assess how sensitive results were to reasonable changes in the assumptions that had been made. The impact of any missing data on the finding is fully discussed.
Assessment of heterogeneity
We initially assessed heterogeneity of the results of the trials by inspecting the graphical presentations and by calculating a test of heterogeneity (Chi2 test). However, we were aware of the fact that the Chi2 test has a poor ability to detect statistically significant heterogeneity among trials. Therefore, we also quantified the impact of heterogeneity in the meta‐analysis using a measure of the degree of inconsistency in the studies' results (Higgins 2011). This measure, the I2 statistic, describes the percentage of total variation across trials that is due to heterogeneity rather than the play of chance (Higgins 2011). The values of I2 lie between 0% and 100%, and a simplified categorisation of heterogeneity is: not important (0% to 40%), moderate (30% to 60%), substantial (50% to 90%) and considerable (75% to 100%); as defined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Assessment of reporting biases
Assessment of bias
We used funnel plots to provide a visual assessment of whether treatment estimates were associated with trial size. We explored bias according to Egger's methods (Egger 1997).
Data synthesis
Stratified analyses were undertaken to provide three estimates of intervention effect: trials at low risk of bias, trials at high risk of bias, and all included trials (Higgins 2011). We analysed dichotomous data by calculating the risk ratio (RR) for each trial with the uncertainty in each result being expressed using 95% confidence intervals (CI). The random‐effects and the fixed‐effect models were used. In case of discrepancy in the results of the two models, we reported the results with both models. If the results were not statistically different, we reported the results with the random‐effects model.
Using the method of the Newcombe‐Wilson hybrid score (not continuity corrected (Newcombe 1998)) and the corresponding 95% CI, we estimated the number of people required to receive hepatitis A vaccine to avoid one person with clinical hepatitis A (number needed to prevent one patient with hepatitis A (NNP)). This number was calculated for all primary outcomes.
Trial sequential analysis
Trial sequential analysis is a statistical method which assesses the risk of random error caused by sparse data and formal or informal repetitive testing of accumulating data. Meta‐analyses do not address the risk of introducing random errors. When few, small trials are combined a in meta‐analysis, the risk of introducing random errors increases due to sparse data and due to multiplicity when conducting cumulative meta‐analyses. We employed trial sequential analysis to address the risk of random errors for the primary outcomes including all‐cause mortality, mortality from hepatitis A, and occurrence of hepatitis A (Brok 2008; Wetterslev 2008; Brok 2009; Wetterslev 2009; Thorlund 2009; Thorlund 2010; CTU 2011; Thorlund 2011).
Subgroup analysis and investigation of heterogeneity
Analysis of the randomised clinical trials was carried out in Review Manager 5.1 (RevMan 2011), with estimates of relative risks (RR) and 95% confidence intervals (CI).
Subgroup analyses were conducted according to pre‐specified characteristics of trials that were considered clinically relevant. We used the test for interaction to estimate the difference between two subgroups (Altman 2003).
Subgroup analyses were conducted for the following.
Doses (high compared to low dose).
Number of doses of vaccine (single dose compared to two or three doses).
Endemicity (high, intermediate, low as per WHO criteria (WHO 2000)).
Vaccine type (inactivated, live attenuated).
Length of follow‐up (1 to 12 months, 13 to 24 months, 25 to 36 months, 37 to 48 months, 49 to 60 months).
Trials with low risk of bias compared to trials with high risk of bias.
Small trials (sample size less than 100 participants) compared to larger trials (sample size at or more than 1000 participants).
Sensitivity analysis
We performed a sensitivity analysis for the clinically defined hepatitis A outcome measure in order to assess the robustness of the findings to different aspects of the trials' methodology: trials with low risk of bias compared to trials with high risk of bias; exclusions after randomisation (reported compared to not reported); and sample size (less than 1000 compared to equal to or more than 1000).
Results
Description of studies
Results of the search
Using the pre‐defined strategies described in Appendix 1, we identified 13,255 references (without removing duplicates): 123 from the Cochrane Hepato‐Biliary Group Controlled Trials Register, 137 from CENTRAL in The Cochrane Library, 3769 from MEDLINE, 6783 from EMBASE, 1366 from Science Citation Index Expanded, and 1077 from CNKI. We identified another 105 references following handsearches of the Vaccine journal or through screening the references from the retrieved studies. After the removal of duplicate references, 8121 references remained. After screening the abstracts for each of these references, 7989 publications were excluded and 132 full articles were retrieved. We excluded 119 references with justifications provided in the Characteristics of excluded studies table. A PRISMA flow diagram is provided in Figure 1.
Figure 1.
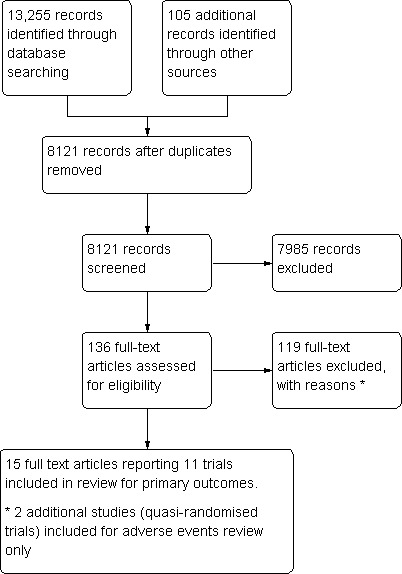
PRISM Study flow diagram.
Included studies
Eleven studies described in 17 publications met the predefined inclusion criteria (Riedemann 1992; Werzberger 1992; Innis 1994; Jiang 1995; Yuan 1995; Wu 1996; Li 2000; Meng 2000; Jiang 2001; Mayorga Pérez 2003; Luo 2004). Four studies were published in English and seven in Chinese. The four trials published in English all used inactivated HAV vaccine as the experimental intervention, while five of the seven studies published in Chinese used live attenuated HAV vaccine. Participants totaled 814,945 across all 11 included studies. The sample size ranged from 260 (Riedemann 1992) to 564,642 participants (Li 2000). Inactivated and live attenuated vaccines were compared with either placebo, no intervention, or inactive against hepatitis A comparator (hepatitis B virus (HBV) or typhoid vaccines).
Four trials used cluster randomised trials with school classes as the unit of randomisation (Jiang 1995; Yuan 1995; Li 2000; Meng 2000). None of the cluster trials specified the total number of units of randomisation. Both commercially available and experimental HAV vaccines were compared with placebo and inactive controls. The dosage of HAV vaccine varied between trials. As stated in the introduction, the assays through which the antigen content of inactivated HAV vaccines were determined varied between manufacturers. This, together with the absence of an international reference standard, meant that a direct comparison was not possible. The duration of follow‐up ranged from one month (Jiang 2001) to 60 months (Meng 2000; Luo 2004) among the included trials.
Two of the included trials were quasi‐randomised studies and were excluded for all outcomes other than that of adverse events (Wu 1996; Luo 2004).
Excluded studies
After screening the abstracts of each of the 8121 references (duplicates removed), 7985 publications were excluded. A further 119 publications were excluded after reviewing the full text of the publication. The most common reasons for exclusion was inappropriate comparator intervention. Six trials were quasi‐randomised clinical studies but did not include adverse events as an outcome, nor did they mention any adverse events in their publications (Zhang 1994; Lin 1997; Xu 1998; Gong 2000; Zhang 2001; Xu 2002). The characteristics of excluded trials are provided (Characteristics of excluded studies).
Risk of bias in included studies
A summary of judgements made by the review authors on the risk of bias for each trial is provided in Figure 2. Only three of the nine included randomised trials were judged to be of low risk of bias (Werzberger 1992; Innis 1994; Mayorga Pérez 2003) when assessed against the domains allocation sequence generation, allocation concealment, blinding, incomplete outcome data reporting, selective outcome reporting, and other risks of bias. The trials with low risk of bias assessed inactivated hepatitis A vaccine. All the trials assessing live attenuated hepatitis A vaccine had high risk of bias. See Figure 2 and Figure 3 for a summary of the results.
Figure 2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study. Please note that Luo 2004 and Wu 1996 were quasi‐randomised studies and were included in the adverse events review only.
Figure 3.
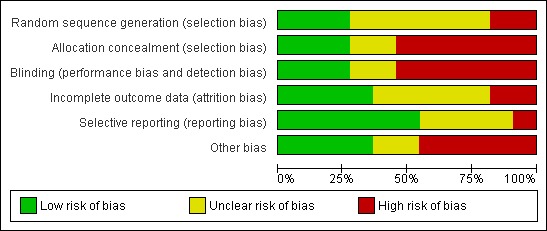
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
All trials stated that they had been randomised. Three trials used adequate methods for generating the allocation sequence (Werzberger 1992; Innis 1994; Mayorga Pérez 2003). The methodology by which the allocation sequence was produced was unclear in six trials. Two studies used methods that were considered to be at high risk of bias, each of which used date of birth to allocate participants (Wu 1996; Luo 2004). These two studies were included in the adverse events review only.
Three trials reported adequate allocation concealment methods (Werzberger 1992; Innis 1994; Mayorga Pérez 2003). However, allocation concealment methodology was considered unclear in two trials (Meng 2000; Luo 2004) and was judged inadequate in the remaining six trials.
Blinding
Five trials stated they were double blinded (Riedemann 1992; Werzberger 1992; Innis 1994; Mayorga Pérez 2003; Luo 2004). Of these only two provided adequate evidence to demonstrate that participants and key investigators were fully blinded, by providing a full discussion of blinding methodology (Werzberger 1992; Mayorga Pérez 2003). Blinding was judged unclear in three trials and at high risk of bias in six trials.
Incomplete outcome data
The inclusion of all randomised participants in the subsequent analysis was determined based on the primary outcome of the trial in question. A number of the inactivated HAV vaccine trials gave the impression that there had been no drop‐outs or withdrawals, although this was not explicitly stated (Jiang 1995; Yuan 1995; Wu 1996; Meng 2000; Jiang 2001; Luo 2004). One of these trials also gave the impression that only those participants who developed clinical hepatitis A were actively followed up (Jiang 1995). Outcome data reporting was considered adequate in three trials (Riedemann 1992; Werzberger 1992; Mayorga Pérez 2003), unclear in five trials, and was considered inadequate in three trials.
Selective reporting
Seven trials were judged to be at low risk of bias, reporting on relevant and reasonably expected outcomes (Riedemann 1992; Werzberger 1992; Innis 1994; Yuan 1995; Wu 1996; Mayorga Pérez 2003). Three trials were judged to be unclear (Li 2000; Meng 2000; Luo 2004) and one at high risk of bias (Jiang 2001).
Other potential sources of bias
Four trials were judged to be at low risk for other potential sources of bias (Riedemann 1992; Werzberger 1992; Innis 1994; Mayorga Pérez 2003). Two trials were judged to be at unclear risk (Li 2000; Jiang 2001) and five at high risk of bias (Jiang 1995; Yuan 1995; Wu 1996; Meng 2000; Luo 2004).
Three trials were cluster randomised clinical trials, and for each the unit of randomisation was either a kindergarten or a school class (Jiang 1995; Yuan 1995; Meng 2000). Given that cluster randomised clinical trials methodologically vary from those trials where the unit of randomisation is an individual, we followed the CONSORT statement to assess these trials for risk of bias (www.consort‐statement.org) (Campbell 2004). Assessment domains included: rationale for cluster randomisation; how cluster randomisation was incorporated in the sample size calculation; how clustering was incorporated into analyses; and records of how both individuals and clusters flowed through the trial (Campbell 2004). All three cluster randomised clinical trials failed to provide any rationale for adopting this trial design. Details for the flow of both participants and clusters were not provided in any of the three trials, neither did they account for clustering in sample size calculations or subsequent analyses.
In one trial, a number of participants were analysed in a group different to that originally allocated due to participants in the control group incorrectly receiving the intervention (Jiang 1995).
Effects of interventions
See: Table 1
Nine randomised clinical trials including 728,850 participants addressed the primary outcome of clinically confirmed development of hepatitis A. The trials compared HAV vaccine versus: 1) no intervention, 2) isotonic saline solution, 3) HBV vaccine, or 4) typhoid vaccines. The follow‐up duration ranged from one month to five years.
Primary outcomes
All‐cause mortality
HAV vaccines versus no intervention, inactive control, or placebo
Only one trial at low risk of bias reported on the outcome of all‐cause mortality (Innis 1994). This trial demonstrated that all‐cause death occurred in 14/20,028 (0.07%) and 10/20,091 (0.05%) participants in the HAV vaccine and control groups respectively (RR 0.14, 95% CI 0.62 to 3.16) (Analysis 5.1). No trials at high risk of bias reported on the outcome of all‐cause mortality.
Analysis 5.1.
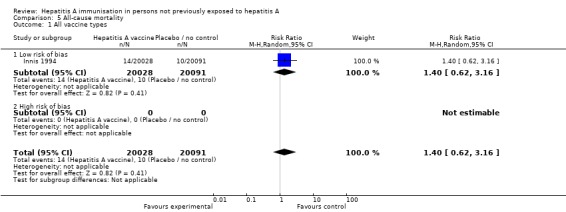
Comparison 5 All‐cause mortality, Outcome 1 All vaccine types.
Hepatitis A related mortality
HAV vaccines versus no intervention, inactive control, or placebo
No trials reported on the outcome of hepatitis A related mortality.
Clinically apparent hepatitis A
HAV vaccines versus no intervention, inactive control, or placebo (trials with low risk of bias)
Three randomised clinical trials were considered to be at low risk of bias (Werzberger 1992; Innis 1994; Mayorga Pérez 2003). Clinically confirmed hepatitis A occurred in 9/20,684 (0.04%) and 92/20,746 (0.44%) participants in the HAV vaccine and control groups respectively (RR 0.09, 95% CI 0.03 to 0.30). The relative risk reduction (RRR) was 0.90 (95% CI 0.81 to 0.95). The number needed to treat (NNT) was 250 participants (95% CI 323 to 199) (Analysis 1.1). Trial sequential analysis was conducted for the three trials (Figure 4). The required information size was 55,961 participants based on the a priori assumption of a relative risk reduction of 90%; an event proportion of 0.1% in the control arm; an alpha of 5%; a beta of 80%; and a heterogeneity correction for the calculation of the required information size. The cumulated Z‐curve crossed the traditional boundary of 5% significance and the trial sequential alpha spending monitoring boundary.
Analysis 1.1.
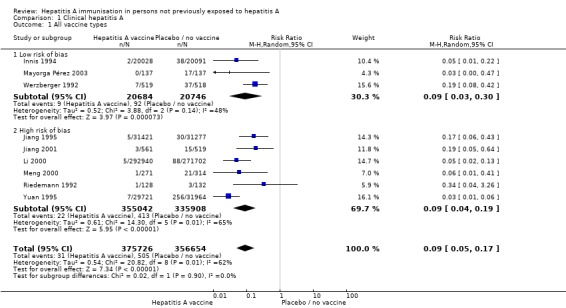
Comparison 1 Clinical hepatitis A, Outcome 1 All vaccine types.
Figure 4.
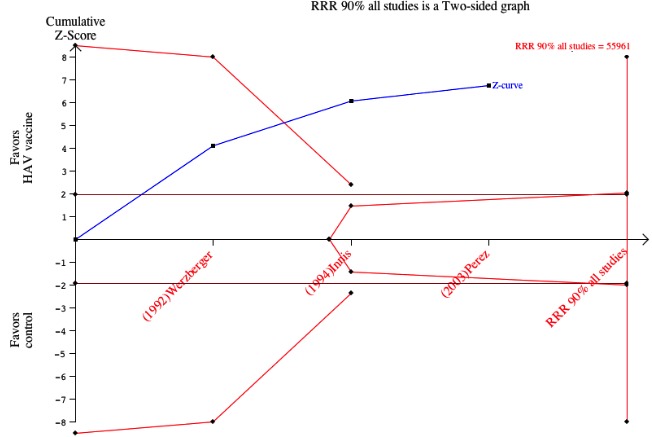
Trial sequential analysis for trials with low risk of bias. The required information size was 55,961 participants based on the a priori assumption of a relative risk reduction of 90%; an event proportion of 0.1% in the control arm; an alpha of 5%; a beta of 80%; and a heterogeneity correction for the calculation of the required information size. The cumulated Z‐curve crosses the traditional boundary of 5% significance and the trial sequential alpha spending monitoring boundary.
HAV vaccines versus no intervention, inactive control or placebo (all included trials)
Clinical hepatitis A occurred in 31/375,726 (0.01%) and 505/356,654 (0.14%) participants in the HAV vaccine and control groups respectively (RR 0.09, 95% CI 0.05 to 0.17). The RRR was 0.94 (95% CI 0.92 to 0.96). The NNT was 749 participants (95% CI 826 to 683). We conducted trial sequential analysis of the nine included randomised clinical trials (Figure 5). The required information size was 60,421 participants based on the a priori assumption of a relative risk reduction of 90%; an event proportion of 0.1% in the control arm; an alpha of 5%; a beta of 80%; and a heterogeneity correction for the calculation of the required information size. The cumulative Z‐curve crossed both the traditional boundary of 5% significance and the trial sequential alpha spending monitoring boundary, and eventually exceeded the required information size. This implies that there is firm evidence for an effect of 90% risk ratio reduction when adjusted for multiple testing on accumulating data.
Figure 5.
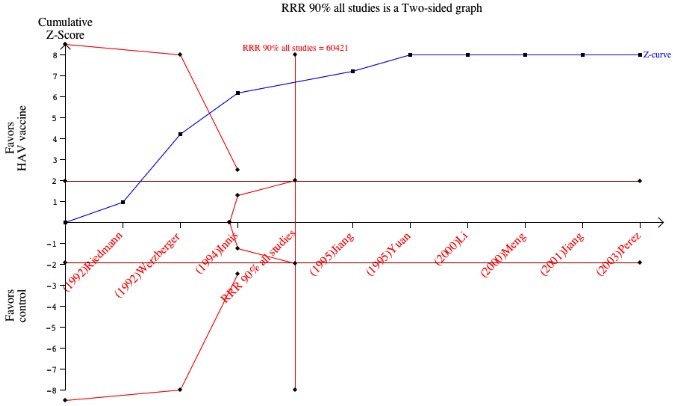
Trial sequential analysis for clinical and contemporarily laboratory confirmed hepatitis A (all included studies). The required information size was 60421 participants based on the a priori assumption of a relative risk reduction of 90%; an event proportion of 0.1% in the control arm; an alpha of 5%; a beta of 80%; and a heterogeneity correction for the calculation of the required information size. The cumulated Z‐curve crosses the traditional boundary of 5% significance and the trial sequential alpha spending monitoring boundary.
As specified in the protocol, subgroup analyses based on vaccine type were carried out: vaccine type, dose, regimen, endemicity, and follow‐up duration.
Inactivated HAV vaccines (three trials with low risk of bias)
Three randomised clinical trials were considered to be at low risk of bias (Werzberger 1992; Innis 1994; Mayorga Pérez 2003). Clinically confirmed hepatitis A occurred in 9/20,684 (0.04%) and 92/20,746 (0.44%) participants in the HAV vaccine and control groups respectively (RR 0.09, 95% CI 0.03 to 0.30) (Analysis 1.2). The relative risk reduction (RRR) was 0.90 (95% CI 0.81 to 0.95). The NNT was 250 participants (95% CI 323 to 199).
Analysis 1.2.
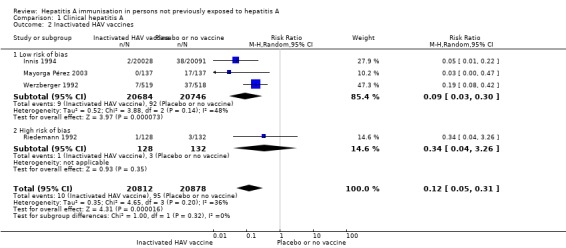
Comparison 1 Clinical hepatitis A, Outcome 2 Inactivated HAV vaccines.
Inactivated HAV vaccines (three trials with low risk of bias and one with high risk of bias)
A meta‐analysis of four trials demonstrated that clinical hepatitis A occurred in 10/20,812 (0.05%) and 95/20,978 (0.45%) participants in the inactivated HAV vaccine and control groups respectively (RR 0.12, 95% CI 0.05 to 0.31) (Analysis 1.2). The RRR was 0.89 (95% CI 0.80 to 0.95). The NNT was 247 participants (95% CI 319 to 197).
Live attenuated HAV vaccines (trials with low risk of bias)
No included randomised clinical trial using live attenuated HAV vaccine was at low risk of bias (Analysis 1.3).
Analysis 1.3.
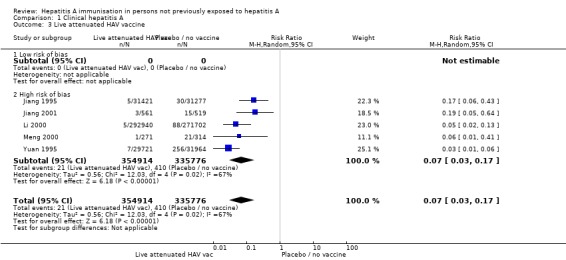
Comparison 1 Clinical hepatitis A, Outcome 3 Live attenuated HAV vaccine.
Live attenuated HAV vaccines (five trials with high risk of bias)
A meta‐analysis of all five trials demonstrated that clinical hepatitis A occurred in 21/354,914 (0.01%) and 410/335,776 (0.12%) participants in the live attenuated HAV vaccine and control groups respectively (RR 0.07, 95% CI 0.03 to 0.17) (Analysis 1.3). The RRR was 0.95 (95% CI 0.93 to 0.97). The NNT was 860 participants (95% CI 957 to 777).
Dose of HAV vaccine (low risk of bias)
No included randomised clinical trials of live attenuated HAV vaccine were at low risk of bias (Analysis 1.4; Analysis 1.5).
Analysis 1.4.
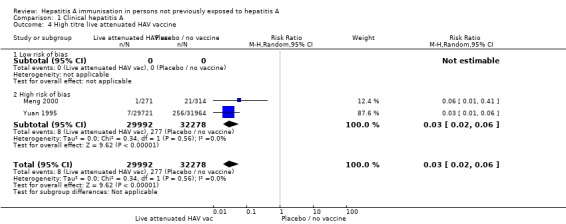
Comparison 1 Clinical hepatitis A, Outcome 4 High titre live attenuated HAV vaccine.
Analysis 1.5.
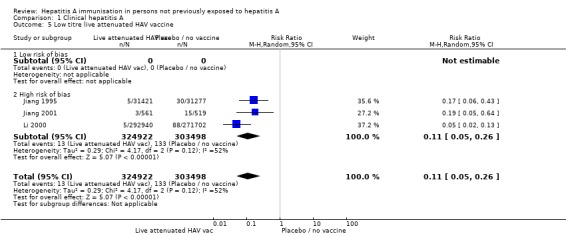
Comparison 1 Clinical hepatitis A, Outcome 5 Low titre live attenuated HAV vaccine.
Dose of HAV vaccine (all included trials)
For inactivated HAV vaccine, assays to determine antigen content are not standardised between manufacturers. Together with the absence of an international reference, the dose of inactivated HAV vaccine cannot be compared at present. A meta‐analysis of two trials demonstrated that clinical hepatitis A occurred in 8/29,992 (0.01%) and 277/32,278 (0.86%) participants in the live attenuated HAV vaccines (titre ≥ 106.5 TCID50) and control groups (RR 0.03, 95% CI 0.02 to 0.06) (Analysis 1.4; Analysis 1.5). The RRR was 0.97 (95% CI 0.94 to 0.99). The NNT was 120 participants (95% CI 136 to 107). A meta‐analysis of three trials demonstrated that clinical hepatitis A occurred in 13/324,922 (0.004%) and 133/303,498 (0.04%) participants in the live attenuated hepatitis A vaccines (titres < 106.5TCID50) and control group respectively (RR 0.11, 95% CI 0.05 to 0.26, random‐effects model). The RRR was 0.91 (95% CI 0.84 to 0.95). The NNT was 2511 participants (95% CI 3085 to 2079).
HAV vaccine in regions of high, intermediate, and low endemicity (trials with low risk of bias)
Data from three randomised clinical trials (Werzberger 1992; Innis 1994; Mayorga Pérez 2003) were available for subgroup analysis comparing HAV vaccine with no intervention, placebo, or inactive control in areas of high and low endemicity as defined by the WHO. A meta‐analysis of two trials (Innis 1994; Mayorga Pérez 2003) at low risk of bias conducted in a region of high endemicity demonstrated that clinical hepatitis A occurred in 2/20,165 (0.01%) and 55/20,228 (0.27%) participants in the inactivated HAV vaccine and control groups respectively (RR 0.05, 95% CI 0.01 to 0.17) (Analysis 1.6). The RRR was 0.96 (95% CI 0.85 to 0.99). The NNT was 381 participants (95% CI 516 to 291). No trial was conducted in a region of intermediate endemicity. Werzberger 1992 was the only trial at low risk of bias conducted in a region of low endemicity. Werzberger 1992 reported that clinical hepatitis A occurred in 7/519 and 37/518 participants in the inactivate HAV vaccine and control groups respectively (RR 0.19, 95% CI 0.08 to 0.42) (Analysis 1.7). The RRR was 0.8 (95% CI 0.58 to 0.92). The NNT was 17 participants (95% CI 29 to 12).
Analysis 1.6.
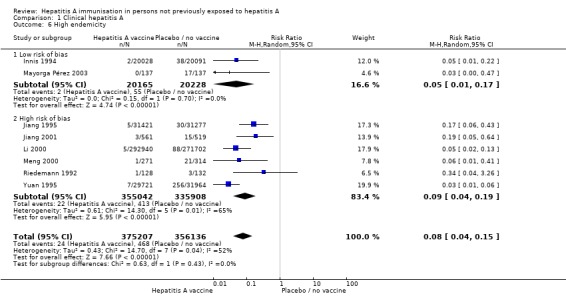
Comparison 1 Clinical hepatitis A, Outcome 6 High endemicity.
Analysis 1.7.
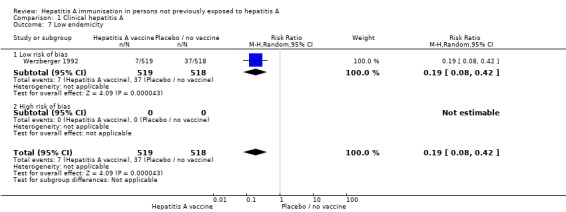
Comparison 1 Clinical hepatitis A, Outcome 7 Low endemicity.
HAV vaccine in regions of high, intermediate, and low endemicity (all included trials)
Data from all nine trials were available for subgroup analysis comparing HAV vaccine with no intervention, placebo or inactive control in areas of high, intermediate, and low endemicity as defined by the WHO. Eight trials compared HAV vaccine versus no intervention, placebo or inactive control in areas of high endemicity. A meta‐analysis of eight trials demonstrated that clinically confirmed hepatitis A occurred in 24/375,207 (0.01%) and 468/356,136 (0.13%) participants in the HAV vaccine and control groups respectively (RR 0.08, 95% CI 0.04 to 0.15). The RRR was 0.95 (95% CI 0.93 to 0.97). The NNT was 799 participants (95% CI 883 to 727). One trial compared HAV vaccine versus placebo in areas of low endemicity (Werzberger 1992). Werzberger 1992 reported that clinical hepatitis A occurred in 7/519 (0.01%) and 37/518 (0.07%) participants in the HAV vaccine and control groups respectively (RR 0.19, 95% CI 0.08 to 0.42) (Analysis 1.7). The RRR was 0.81 (95% CI 0.58 to 0.92). The NNT was 17 participants (95% CI 29 to 12).
Number of HAV vaccine doses (trials with low risk of bias)
Data from two randomised clinical trials (Innis 1994; Mayorga Pérez 2003) were available for subgroup analysis comparing HAV vaccine with no intervention, placebo, or inactive control according to dosing regimen (single dose or two or more doses). Mayorga Pérez 2003 was the only trial at low risk of bias conducted using a single dose regimen. Mayorga Pérez 2003 reported that clinical hepatitis A occurred in 0/137 (0%) and 17/137 (12.41%) participants in the inactivated HAV vaccine and control groups respectively (RR 0.03, 95% CI 0.00 to 0.47) (Analysis 1.8). The RRR was 0.99 (95% CI ‐1.93 to 1). The NNT was 8 participants (95% CI 14 to 5). Two trials at low risk of bias were conducted using multiple HAV vaccine doses (Werzberger 1992; Innis 1994). A meta‐analysis of these two trials demonstrated that clinical hepatitis A occurred in 9/20,547 and 75/20,609 participants in the inactivate HAV vaccine and control groups respectively (RR 0.11, 95% CI 0.03 to 0.40) (Analysis 1.9). The RRR was 0.88 (95% CI 0.76 to 0.94). The NNT was 312 participants (95% CI 422 to 241).
Analysis 1.8.
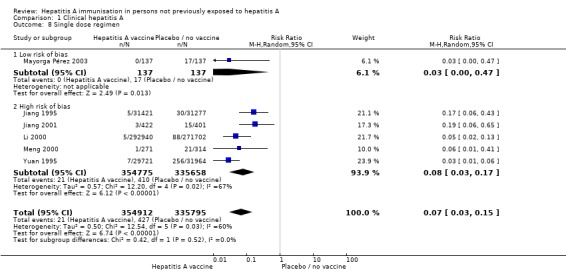
Comparison 1 Clinical hepatitis A, Outcome 8 Single dose regimen.
Analysis 1.9.
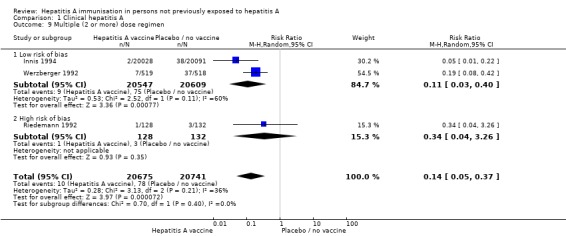
Comparison 1 Clinical hepatitis A, Outcome 9 Multiple (2 or more) dose regimen.
Number of HAV vaccine doses (all included trials)
Data from all nine trials were available for subgroup analysis comparing HAV vaccine with no intervention, placebo, or inactive control according to dosing regimen (single dose, and two or more doses). A meta‐analysis of six trials demonstrated that participants with clinical hepatitis A occurred in 21/354,912 (0.01%) and 427/335,795 (0.13%) participants in the single HAV vaccine and control groups respectively (RR 0.07, 95% CI 0.03 to 0.15). The RRR was 0.95 (95% CI 0.93 to 0.97). The NNT was 824 participants (95% CI 915 to 746) (Analysis 1.8). A meta‐analysis of three trials demonstrated that clinical hepatitis A occurred in 10/20,675 (0.05%) and 78/20,741 (0.38%) participants in the multiple HAV vaccine dose and control groups respectively (RR 0.14, 95% CI 0.05 to 0.37). The RRR was 0.87 (95% CI 0.75 to 0.93). The NNT was 305 participants (95% CI 412 to 236) (Analysis 1.9). All trials included in the multiple dosing regimen used three doses (no trial used two doses).
Follow‐up duration (trials with low risk of bias)
Data from three trials were available for subgroup analysis comparing HAV vaccine (inactivated or live attenuated) with no intervention, placebo or inactive control according to follow‐up duration (1 to 12 months, 13 to 24 months, 25 to 36 months, 37 to 48 months, 49 to 60 months). Werzberger 1992 was the only randomised clinical trial at low risk of bias conducted in the 1 to 12 month interval. Werzberger 1992 reported that clinical hepatitis A occurred in 7/519 and 37/518 participants in the inactivated HAV vaccine and control groups respectively (RR 0.19, 95% CI 0.08 to 0.42). The RRR was 0.81 (95% CI 0.58 to 0.92). The NNT was 17 participants (95% CI 29 to 12) (Analysis 1.10).
Analysis 1.10.
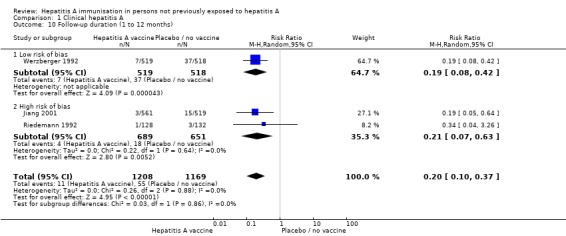
Comparison 1 Clinical hepatitis A, Outcome 10 Follow‐up duration (1 to 12 months).
Two trials at low risk of bias conducted an analysis in the 13 to 24 month interval (Innis 1994; Mayorga Pérez 2003). A meta‐analysis of these two trials demonstrated that clinical hepatitis A occurred in 2/20,165 (0.01%) and 55/20,228 (0.27%) participants in the inactivated HAV vaccine and control groups respectively (RR 0.05, 95% CI 0.01 to 0.17). The RRR was 0.96 (95% CI 0.85 to 0.99). The NNT was 381 participants (95% CI 516 to 291) (Analysis 1.11). No trial at low risk of bias was conducted in the 25 to 36 month, 37 to 48 month, and 49 to 60 month intervals.
Analysis 1.11.
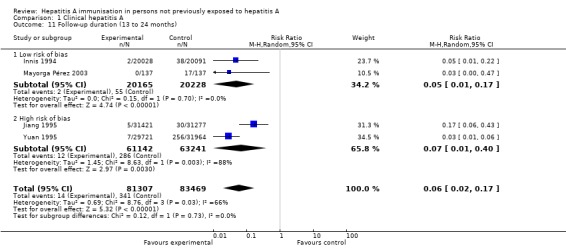
Comparison 1 Clinical hepatitis A, Outcome 11 Follow‐up duration (13 to 24 months).
Follow‐up duration (all included trials)
Data from all nine trials were available for subgroup analysis comparing HAV vaccine (inactivated and live attenuated with no intervention, placebo or inactive control according to follow‐up duration (1 to 12 months, 13 to 24 months, 25 to 36 months, 37 to 48 months, and 49 to 60 months).
A meta‐analysis of three trials in the 1 to 12 month interval (Riedemann 1992; Werzberger 1992; Jiang 1995) demonstrated that clinically confirmed hepatitis A occurred in 11/1208 (0.91%) and 55/1169 (4.70%) participants in the HAV vaccine and control groups respectively (RR 0.20, 95% CI 0.10 to 0.38). The RRR was 0.81 (95% 0.63 to 0.90). The NNT was 26 participants (95% CI 40 to 19). A meta‐analysis of four trials in the 13 to 24 month interval (Innis 1994; Jiang 1995; Yuan 1995; Mayorga Pérez 2003) demonstrated that clinically confirmed hepatitis A occurred in 14/81,307 (0.02%) and 341/83,469 (0.41%) in the HAV vaccine and control groups respectively (RR 0.06, 95% CI 0.02 to 0.21). The RRR was 0.96 (95% 0.93 to 0.98). The NNT was 255 participants (95% CI 287 to 229). One trial (Li 2000) in the 25 to 36 month interval demonstrated that clinically confirmed hepatitis A occurred in 5/292,940 (0.002%) and 88/271,702 (0.03%) participants in the HAV vaccine and control groups respectively (RR 0.05, 95% CI 0.02 to 0.13). The RRR was 0.95 (95% CI 0.87 to 0.98). The NNT was 3259 participants (95% CI 4137 to 2614) (Analysis 1.12). No trial was conducted in the 37 to 48 month interval. One trial (Meng 2000) in the 49 to 60 month interval demonstrated that clinically confirmed hepatitis A occurred in 1/271 (0.36%) and 21/314 (6.69%) participants in the HAV vaccine and control groups respectively (RR 0.06, 95% CI 0.01 to 0.41). The RRR was 0.95 (95% CI 0.60 to 0.99). The NNT was 15 participants (95% CI 29 to 10) (Analysis 1.13).
Analysis 1.12.
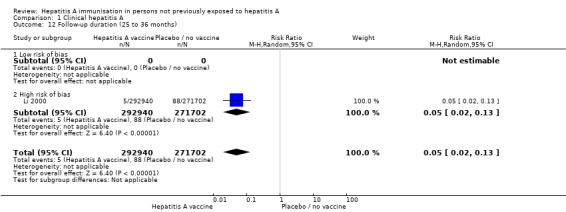
Comparison 1 Clinical hepatitis A, Outcome 12 Follow‐up duration (25 to 36 months).
Analysis 1.13.
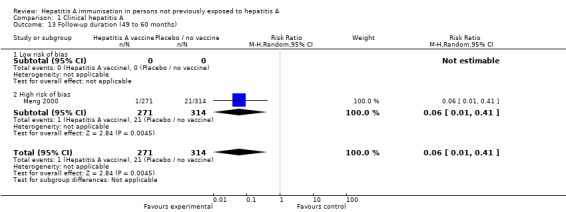
Comparison 1 Clinical hepatitis A, Outcome 13 Follow‐up duration (49 to 60 months).
Sensitivity analyses
Trial size Only one trial at low risk of bias had less than 1000 participants (Mayorga Pérez 2003). Here, clinical hepatitis A occurred in 0/137 (0%) and 17/137 (12.4%) participants in the HAV vaccine and control groups respectively (RR 0.03, 95% CI 0.00 to 0.47). A meta‐analysis of all trials with 1000 or more participants demonstrated that clinical hepatitis A occurred in 30/375,461 (0.01%) and 485/356,288 (0.14%) participants in the HAV vaccine and control groups respectively (RR 0.08, 95% CI 0.04, to 0.16) (Analysis 4.1). For all trials with less than 1000 participants, a meta‐analysis of two trials at low risk of bias demonstrated that clinical hepatitis A occurred in 1/265 (0.38%) and 20/269 (7.43%) participants in the HAV vaccine and control groups respectively. Significance was reached for the fixed‐effect model (RR 0.07, 95% CI 0.01 to 0.38) but not the random‐effects model (RR 0.11, 95% CI 0.01 to 1.62) (Analysis 4.2).
Analysis 4.1.
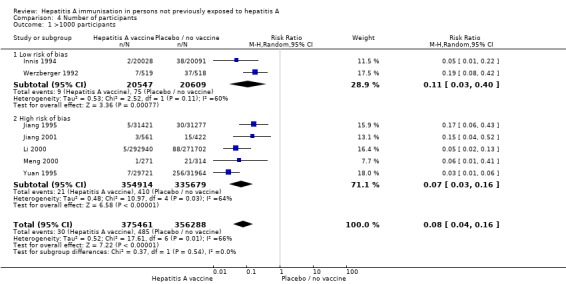
Comparison 4 Number of participants, Outcome 1 >1000 participants.
Analysis 4.2.
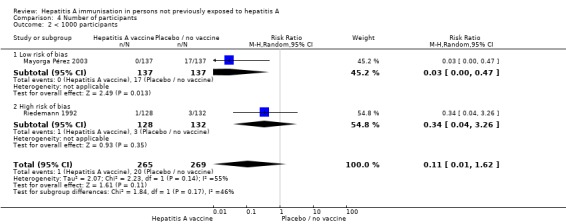
Comparison 4 Number of participants, Outcome 2 < 1000 participants.
Risk of bias Two trials with low risk of bias had 1000 or more participants (Werzberger 1992; Innis 1994). A meta‐analysis of these two trials demonstrated that clinically confirmed hepatitis A occurred in 9/20,547 (0.04%) versus 75/20,609 (0.36%) participants in the HAV vaccine and control groups respectively (RR 0.11, 95% CI 0.03 to 0.40).
The effect of adjustment and non‐adjustment for cluster randomisation indicated that the results remained significant, with significant heterogeneity.
Funnel plot
In this preliminary analysis the plot for clinical protective efficacy was investigated. Given the relatively small number of trials (nine) no statistical test for funnel plot asymmetry was used (Egger 1997; Egger 1998). Visual inspection of the funnel plot suggested a degree of asymmetry about the pooled RR line with a slight paucity of small negative trials (Figure 6). It is also important to remember that in addition to publication bias the asymmetry could represent true heterogeneity, poor methodological quality, artefact, or chance (Sackett 1979).
Figure 6.
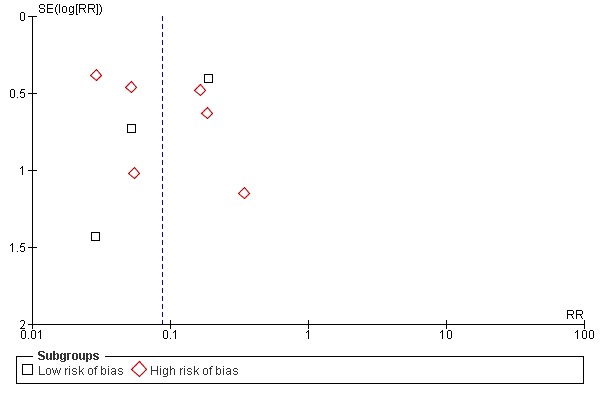
Funnel plot of comparison: 1 Clinical hepatitis A, outcome: 1.1 All vaccine types.
Secondary outcomes
Lack of anti‐HAV IgG sero‐protection
Seven of the nine randomised clinical trials included anti‐HAV IgG as an outcome measure. Four trials used the inactivated HAV vaccine as the intervention (Riedemann 1992;Werzberger 1992;Innis 1994; Mayorga Pérez 2003). Three trials used the live attenuated HAV vaccine (Yuan 1995; Li 2000;Meng 2000). Of these, three did not measure anti‐HAV IgG in the control group of the trial (Werzberger 1992;Yuan 1995; Li 2000). Two trials reported this outcome as the geometric mean titre (Riedemann 1992; Mayorga Pérez 2003). No trial defined anti‐HAV IgG greater than 10 mIU/L as an outcome. Two randomised clinical trials examining the inactivated hepatitis A vaccine defined anti‐HAV IgG greater than 20 mIU as an outcome (Riedemann 1992; Innis 1994).
Inactivated HAV vaccine (trials with low risk of bias)
Only one trial that met the inclusion criteria for sero‐protection was at low risk of bias (Innis 1994). This trial examined the inactivated hepatitis A vaccine and defined anti‐HAV IgG greater than 20mIU as an outcome. Lack of sero‐protection was demonstrated in 2/238 (0.84%) and 241/248 (97.18%) participants in the HAV vaccine and control groups respectively (RR 0.01, 95% CI 0.00, 0.03) (Analysis 2.1).
Analysis 2.1.
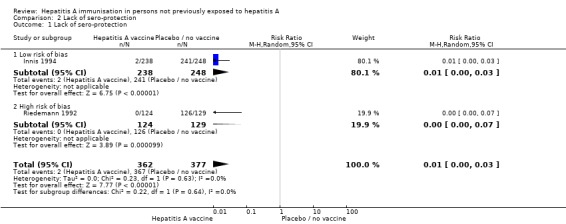
Comparison 2 Lack of sero‐protection, Outcome 1 Lack of sero‐protection.
Inactivated HAV vaccine (all included trials)
Two randomised clinical trials examining the inactivated hepatitis A vaccine defined anti‐HAV IgG greater than 20 mIU as an outcome (Riedemann 1992; Innis 1994). At this threshold a meta‐analysis of these two trials showed that lack of sero‐protection was demonstrated in 2/362 (0.55%) and 367/377 (97.35%) participants in the HAV vaccine and control groups respectively (RR 0.01, 95% CI 0.00, 0.03) (Analysis 2.1). The follow‐up duration in these two trials was 17 and 7 months. A sensitivity analysis excluding the trial with the shorter follow‐up duration found that the result remained significant. No trial investigating live attenuated vaccine met the inclusion criteria.
Inactivated HAV vaccine (trials with low risk of bias, all included studies)
No randomised clinical trial using the live attenuated vaccine met the inclusion criteria for sero‐protection as pre‐defined in our review protocol.
Adverse events
Serious adverse events (inactivated HAV vaccine)
No serious adverse events or deaths were reported in any participants receiving live attenuated HAV vaccines.
Non‐serious local adverse events (inactivated HAV vaccine)
Four trials reported the outcome of local adverse events (pain, tenderness, warmth or redness) (Riedemann 1992;Werzberger 1992;Innis 1994;Mayorga Pérez 2003). The total number of non‐serious local adverse was not available for one study (Innis 1994). A meta‐analysis of those trials at low risk of bias (Werzberger 1992; Mayorga Pérez 2003) demonstrated that non‐serious local adverse events occurred in 61/652 (9.36%) versus 65/647 (10.04%) participants in the HAV vaccine group and the control group respectively (RR 1.19, 95% CI 0.67 to 2.1) (Analysis 3.1). All trials using the inactivated HAV vaccine (Riedemann 1992; Werzberger 1992; Innis 1994; Mayorga Pérez 2003) demonstrated that non‐serious local adverse events occurred in 89/780 (11.41%) versus 76/779 (9.76%) participants in the HAV vaccine group and the control group respectively (RR 1.21, 95% CI 0.86 to 1.70) (Analysis 3.1).
Analysis 3.1.
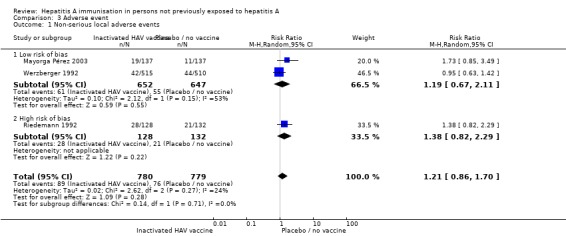
Comparison 3 Adverse event, Outcome 1 Non‐serious local adverse events.
Non‐serious systemic adverse events (inactivated HAV vaccine)
Systemic adverse events were reported in four trials (Riedemann 1992; Werzberger 1992; Innis 1994; Wu 1996; Mayorga Pérez 2003). The total number of non‐serious local adverse was not available for two studies (Innis 1994; Wu 1996). A meta‐analysis of the trials at low risk of bias (Werzberger 1992; Mayorga Pérez 2003) demonstrated that systemic adverse events occurred in 29/652 (4.45%) versus 27/647 (4.17%) participants in the HAV vaccine group and the control group respectively (RR 1.09, 95% CI 0.67 to 1.78). A meta‐analysis of all trials (Riedemann 1992; Werzberger 1992; Innis 1994; Mayorga Pérez 2003) demonstrated that local adverse events occurred in 49/780 (6.28%) versus 51/799 (6.38%) participants in the HAV vaccine group and the control group respectively (RR 0.98, 95% CI 0.68 to 1.41) (Analysis 3.2).
Analysis 3.2.
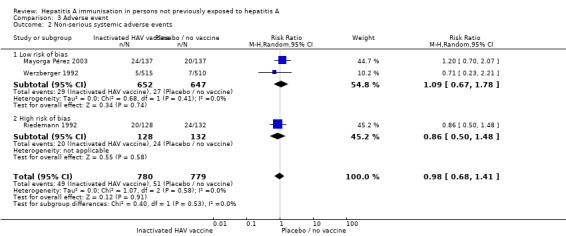
Comparison 3 Adverse event, Outcome 2 Non‐serious systemic adverse events.
Serious adverse events (live attenuated HAV vaccine)
Only one trial reported a narrative description of a serious adverse event. Yuan 1995 reported that one participant developed fever and was admitted to hospital for both treatment and observation. The participant had incorrectly received three doses of the live attenuated HAV vaccine. No further data on serious adverse events were available.
Non‐serious local adverse events (live attenuated HAV vaccine)
One quasi‐randomised study (Luo 2004) reported on the local adverse events but data on the numbers affected were not available.
Non‐serious systemic adverse events (live attenuated HAV vaccine)
One trial described "high grade fever" in one participant and "low grade fever" in 22 participants receiving the live attenuated HAV vaccine (Jiang 1995). No further comparative data were available for this trial. One quasi‐randomised controlled study described one participant as developing a "skin eruption" that required treatment with anti‐histamines and another participant developing fever after vaccination with live attenuated vaccine. No further data on non‐serious adverse events were available (Wu 1996).
Discussion
This review has attempted to address the fundamental question whether HAV vaccines protect people from hepatitis A. The findings of this review demonstrate the paucity of trials that actually go on to evaluate clinical protective efficacy. In contrast, there are a large number of trials that use surrogate measures to imply protection. Clearly, these are important issues for both clinicians and policy makers to be aware of.
Summary of main results
Compared with placebo, inactive control or no intervention, inactivated HAV vaccine demonstrated a significant protective effect against clinical hepatitis A. The same seems to be true for live attenuated (HAV) vaccine but, regarding this vaccine, all trials had a high risk of bias. The size of the trials and the consistency of results across a variety of different populations provided convincing evidence that HAV vaccine is effective for preventing clinically apparent hepatitis A.
There are inevitable potential limitations of the outcome criteria used in this review. Firstly, anti‐HAV IgM can be detected two to three weeks post‐vaccination. Despite continued improvement in the design of assays used to detect anti‐HAV IgM, false positives may occur as a result of IgM anti‐IgG rheumatoid factor (Roque‐Afonso 2010). Secondly, the identification of clinically apparent hepatitis A is dependent on the precise assessment of signs and symptoms of each participant; this is a potential source of error. Finally, there is an ongoing debate as to what level of anti‐HAV IgG confers protection. Although the WHO currently indicates that the level should be set between 10 to 20 mIU/L, they also acknowledge that it remains uncertain whether levels less than 10 mIU/ml could also be considered protective (WHO 2010a).
Subgroup analysis determined that HAV vaccine is effective in regions of both high and low endemicity. No trial was conducted in regions of intermediate endemicity. This may be an important omission given that the existing policy supporting widespread vaccination is focused on regions of intermediate endemicity. It should be highlighted that the outcomes of this review are based on participants susceptible to HAV rather than whole populations. Whilst we have reported NNT for susceptible individuals, we have not reported numbers needed to vaccinate (NNV). This is because NNV incorporates the issues of herd immunity and annual event rates in unvaccinated individuals, which will vary from region to region.
This review found that vaccination with the inactivated HAV vaccine had a significant effect on conferring sero‐protective anti‐HAV IgG. Together with the finding from the clinical efficacy review it could be argued that these findings support the theory that protection depends on the production of protective antibodies. However, we emphasise that this review was not designed to explore how antibody levels changed over time (or otherwise) and how this relates to long‐term clinical protection. It follows that we suggest caution when interpreting these results. It has been reported that protection based on cellular immunity could persist after anti‐HAV antibodies become undetectable (Wang 2007). Once again, this highlights the need for trials to measure both clinical and laboratory outcomes and not just unvalidated surrogate outcomes alone (Gluud 2007).
There is a lack of trials with low risk of bias to conclude whether or not live attenuated HAV vaccine has a significant risk of any adverse events in comparison to placebo, adequate control, or no intervention. A number of studies investigating adverse events in live attenuated HAV vaccine used non‐comparative study designs with non‐standardised definitions of what constituted an adverse event. This paucity of high quality data for the live attenuated HAV vaccine is of particular concern given the theoretical possibility of virulent atavism where the attenuated virus reverts back to its 'wild type'. The WHO highlight the need to conduct rigorous high‐quality post‐marketing surveillance in selected communities to measure and monitor safety and adverse reactions (WHO 2010b). For the inactivated HAV vaccine, no significant difference was noted for either local or serious adverse events when compared to placebo, appropriate control, or no vaccine. Although only one trial looked at this outcome, the trial itself had low risk of bias and was appropriately powered.
Overall completeness and applicability of evidence
Although a systemic approach was adopted toward identifying trials for this review, it is possible that some trials may have been missed. However, the findings from this review are broadly in keeping with the systematic reviews conducted by Demicheli et al (Demicheli 2003) and the meta‐analysis conducted by Wang et al (Wang 2008). Randomised clinical trials are not the only way that the effectiveness of a vaccine can be determined and results from randomised clinical trials should be interpreted together with other relevant data gathered over time. The epidemiology of the disease provides the essential context, including how epidemics occur and patterns of susceptibility change. Further observations of morbidity and mortality figures after the vaccine was introduced, and changes seen in disease epidemiology when a vaccination program is stopped, are also of vital importance. It is important to recognise that a number of factors can influence transmission patterns in a given population such as hygiene, herd immunity, and prevalence of the virus.
Quality of the evidence
The nine included trials were of variable methodological quality when assessed for adequacy of allocation sequence generation, allocation concealment, blinding and inclusion of all randomised participants. Some trials presented the results of subgroups without presenting the unadjusted results for the whole intervention and control groups. The revised CONSORT statement recommends that unadjusted data should be reported if adjusted analyses are presented (Campbell 2004). It is, therefore, possible that non‐significant results from trials are under‐represented in the meta‐analyses.
Potential biases in the review process
Three cluster randomised clinical trials were identified, none of which adequately adjusted for the effects of cluster randomisation (Jiang 1995;Yuan 1995; Meng 2000). Cluster randomisation presents a number of challenges when it comes to systematic reviews containing such trials. Much has been written regarding the methodological quality of cluster randomised clinical trials and their use in meta‐analysis (Ukoumunne 1999;Adams 2004;Campbell 2004). Campbell 2004 reminds us that these trials are complex to design and calls for careful attention to analysis and statistical power. Because this meta‐analysis includes cluster randomised clinical trials with variable cluster types, it is essential to appreciate how this may effect those interventions being evaluated. For example, by conferring herd immunity the vaccine may be more effective when given to all individuals in a group than when only given to a third of the people. It is also important to be aware that those participants within a cluster have a tendency to respond in the same manner and their data cannot be assumed to be independent of each other. None of the included cluster randomised clinical trials made the necessary adjustment for clustering in their analysis. Instead, they incorrectly analysed results as though the individual was the allocation unit. If clusters are simply ignored, then this 'unit‐of‐analysis error' results in artificially small P values (Whiting‐O'Keefe 1984). This may result in false positive conclusions about the efficacy of the intervention. To address this, an estimated intra‐cluster correlation coefficient was applied as suggested by the CHBG (Gluud 2012). However, the sensitivity analyses demonstrated that both before and after adjustment the pooled result reached significance for all analyses involving the clustered trials.
Agreements and disagreements with other studies or reviews
This review is the first systematic review conducted using the Cochrane Collaboration methodology to investigate the clinical protective efficacy of vaccines for use against hepatitis A ,and strengthens previous conclusions about vaccine effectiveness. A previous systematic review (without meta‐analysis) of inactivated HAV vaccine has been published (Demicheli 2003). This included eight trials and examined the effectiveness and safety of HAV vaccines in children and adults. The review concluded that although the included trials were of poor design, there was evidence to support the clinical efficacy and safety of the inactivated HAV vaccine. No conclusion could be made for the efficacy and safety of the live attenuated HAV vaccine as only one trial of poor methodological quality, that is, high risk of bias, was identified. A meta‐analysis of live attenuated vaccine has also been published (Wang 2008). The meta‐analysis identified 13 studies and concluded that live hepatitis A vaccine has good protective efficacy. The review did not address vaccine safety.
Authors' conclusions
Inactivated hepatitis A vaccines can markedly decrease the risk of contracting hepatitis A in susceptible individuals. Live attenuated hepatitis A vaccines seems to offer similar effects, but the results are at risk of bias. Immunisation should be a priority for persons at increased risk of acquiring hepatitis A. This meta‐analysis demonstrated significant protection for at least two years with the inactivated HAV vaccine and at least five years with the live attenuated HAV vaccine. This review provided evidence to support the safety of the inactivated HAV vaccine. However, there was insufficient evidence to comment on the safety of live attenuated vaccines.
Based on the evidence available in this review, we suggest the following are important considerations for future research.
There remains a need for the development of a standard assay to determine and compare antigen content of the inactivated HAV vaccine between manufacturers.
Standardised definitions of adverse events as defined by the ICH‐GCP should be followed when evaluating vaccines in future trials (ICH‐GCP 1997).
There remains a need for well‐designed randomised clinical trials with a comparative trial design together with high‐quality post‐marketing surveillance looking at benefits and adverse events following vaccination with live attenuated HAV vaccines.
Clarification over the absolute protective level of anti‐HAV should be sought through systematic observation. In particular, clarification is required on whether anti‐HAV levels less than 10 mIU/ml could be protective.
There remains a need for large, well‐constructed randomised clinical trials in regions of intermediate endemicity.
The duration of protection following a single dose of vaccine should be investigated further.
Future trials should consider reporting patients with hepatitis by time since vaccination. The utility of this approach was demonstrated by Werzberger 1992.
If cluster randomisation is used then the total number of randomisation units should be stated in the text.
Future trials ought to be reported according to the CONSORT guidelines (www.consort‐statement.org).
Acknowledgements
We would like to thank Dimitrinka Nikolova and Sarah Klingenberg from the Cochrane Hepato‐Biliary Group for guidance and support throughout the review process. We would also like to thank China/Asia on Demand for additional translation support.
We would also like to thank our contact editor, JianPing Liu, and peer reviewers Steven Todd Wiesmar, and Yutong Fei for their helpful and constructive comments.
Protocol: Peer reviewers: Steven Todd Wiesmar, Switzerland; Yutong Fei, China. Contact editor: JianPing Liu, China.
Review: Peer reviewers: Steven Todd Wiesmar, Switzerland. Contact editors: JianPing Liu, China; Christian Gluud, Denmark.
Appendices
Appendix 1. Search strategy
| Database | Time Span | Search Strategy |
| The Cochrane Hepato‐Biliary Group Controlled Trials Register | November 2011 | ('hepatitis A' OR 'hep A') AND ((vaccine* AND (attenuated OR inactivated)) OR vaccin* OR immuni* OR inoculat*) |
| Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library | Issue 4, 2011 | #1 MeSH descriptor Vaccination explode all trees #2 MeSH descriptor Immunization explode all trees #3 ((vaccine* AND (attenuated OR inactivated)) OR vaccin* OR immuni* OR inoculat*) #4 (#1 OR #2 OR #3) #5 MeSH descriptor Hepatitis A explode all trees #6 hepatitis A OR hep A #7 (#5 OR #6) #8 (#4 AND #7) |
| MEDLINE (Ovid SP) | 1950 to November 2011 | 1 (HEPATITIS‐A VACCINES OR HEPATITIS‐A OR hepatitis A) 2 (VACCINES‐ATTENUATED OR VACCINES‐INACTIVATED) 3 1 AND 2 4 MeSH descriptor VACCINATION explode all trees 5 1 AND 4 6 MeSH descriptor IMMUNIZATION explode all trees 7 1 AND 6 8 (vaccin* OR immuni* OR inoculate) 9 1 AND 8 |
| EMBASE (Ovid SP) | 1980 to November 2011 | 1 (HEPATITIS‐A VACCINES OR HEPATITIS‐A OR hepatitis A).mp [mp= title, abstract or Keywords in all products] 2 (VACCINES‐ATTENUATED OR VACCINES‐INACTIVATED OR VACCINES).mp [mp= title, abstract or Keywords in all products] 3 1 and 2 4 Exp VACCINATION 5 1 and 4 6 Exp IMMUNIZATION 7 1 and 6 8 (vaccin* OR immuni*OR inoculate*) 9 1 and 8 |
| Science Citation Index Expanded | 1900 to November 2011 | # 5 #4 AND #3 # 4 TS=(random* OR blind* OR placebo* OR meta‐analysis) # 3 #2 AND #1 # 2 TS=((vaccine* AND (attenuated OR inactivated)) OR vaccin* OR immuni* OR inoculat*) # 1 TS=('hepatitis A' OR 'hep A') |
Data and analyses
Comparison 1.
Clinical hepatitis A
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All vaccine types | 9 | 732380 | Risk Ratio (M‐H, Random, 95% CI) | 0.09 [0.05, 0.17] |
| 1.1 Low risk of bias | 3 | 41430 | Risk Ratio (M‐H, Random, 95% CI) | 0.09 [0.03, 0.30] |
| 1.2 High risk of bias | 6 | 690950 | Risk Ratio (M‐H, Random, 95% CI) | 0.09 [0.04, 0.19] |
| 2 Inactivated HAV vaccines | 4 | 41690 | Risk Ratio (M‐H, Random, 95% CI) | 0.12 [0.05, 0.31] |
| 2.1 Low risk of bias | 3 | 41430 | Risk Ratio (M‐H, Random, 95% CI) | 0.09 [0.03, 0.30] |
| 2.2 High risk of bias | 1 | 260 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.04, 3.26] |
| 3 Live attenuated HAV vaccine | 5 | 690690 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.03, 0.17] |
| 3.1 Low risk of bias | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 High risk of bias | 5 | 690690 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.03, 0.17] |
| 4 High titre live attenuated HAV vaccine | 2 | 62270 | Risk Ratio (M‐H, Random, 95% CI) | 0.03 [0.02, 0.06] |
| 4.1 Low risk of bias | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 High risk of bias | 2 | 62270 | Risk Ratio (M‐H, Random, 95% CI) | 0.03 [0.02, 0.06] |
| 5 Low titre live attenuated HAV vaccine | 3 | 628420 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.05, 0.26] |
| 5.1 Low risk of bias | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 High risk of bias | 3 | 628420 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.05, 0.26] |
| 6 High endemicity | 8 | 731343 | Risk Ratio (M‐H, Random, 95% CI) | 0.08 [0.04, 0.15] |
| 6.1 Low risk of bias | 2 | 40393 | Risk Ratio (M‐H, Random, 95% CI) | 0.05 [0.01, 0.17] |
| 6.2 High risk of bias | 6 | 690950 | Risk Ratio (M‐H, Random, 95% CI) | 0.09 [0.04, 0.19] |
| 7 Low endemicity | 1 | 1037 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.08, 0.42] |
| 7.1 Low risk of bias | 1 | 1037 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.08, 0.42] |
| 7.2 High risk of bias | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 8 Single dose regimen | 6 | 690707 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.03, 0.15] |
| 8.1 Low risk of bias | 1 | 274 | Risk Ratio (M‐H, Random, 95% CI) | 0.03 [0.00, 0.47] |
| 8.2 High risk of bias | 5 | 690433 | Risk Ratio (M‐H, Random, 95% CI) | 0.08 [0.03, 0.17] |
| 9 Multiple (2 or more) dose regimen | 3 | 41416 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.05, 0.37] |
| 9.1 Low risk of bias | 2 | 41156 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.03, 0.40] |
| 9.2 High risk of bias | 1 | 260 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.04, 3.26] |
| 10 Follow‐up duration (1 to 12 months) | 3 | 2377 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.10, 0.37] |
| 10.1 Low risk of bias | 1 | 1037 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.08, 0.42] |
| 10.2 High risk of bias | 2 | 1340 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.07, 0.63] |
| 11 Follow‐up duration (13 to 24 months) | 4 | 164776 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.02, 0.17] |
| 11.1 Low risk of bias | 2 | 40393 | Risk Ratio (M‐H, Random, 95% CI) | 0.05 [0.01, 0.17] |
| 11.2 High risk of bias | 2 | 124383 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.01, 0.40] |
| 12 Follow‐up duration (25 to 36 months) | 1 | 564642 | Risk Ratio (M‐H, Random, 95% CI) | 0.05 [0.02, 0.13] |
| 12.1 Low risk of bias | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 12.2 High risk of bias | 1 | 564642 | Risk Ratio (M‐H, Random, 95% CI) | 0.05 [0.02, 0.13] |
| 13 Follow‐up duration (49 to 60 months) | 1 | 585 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.01, 0.41] |
| 13.1 Low risk of bias | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13.2 High risk of bias | 1 | 585 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.01, 0.41] |
Comparison 2.
Lack of sero‐protection
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lack of sero‐protection | 2 | 739 | Risk Ratio (M‐H, Random, 95% CI) | 0.01 [0.00, 0.03] |
| 1.1 Low risk of bias | 1 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 0.01 [0.00, 0.03] |
| 1.2 High risk of bias | 1 | 253 | Risk Ratio (M‐H, Random, 95% CI) | 0.00 [0.00, 0.07] |
Comparison 3.
Adverse event
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Non‐serious local adverse events | 3 | 1559 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.86, 1.70] |
| 1.1 Low risk of bias | 2 | 1299 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.67, 2.11] |
| 1.2 High risk of bias | 1 | 260 | Risk Ratio (M‐H, Random, 95% CI) | 1.38 [0.82, 2.29] |
| 2 Non‐serious systemic adverse events | 3 | 1559 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.68, 1.41] |
| 2.1 Low risk of bias | 2 | 1299 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.67, 1.78] |
| 2.2 High risk of bias | 1 | 260 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.50, 1.48] |
Comparison 4.
Number of participants
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 >1000 participants | 7 | 731749 | Risk Ratio (M‐H, Random, 95% CI) | 0.08 [0.04, 0.16] |
| 1.1 Low risk of bias | 2 | 41156 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.03, 0.40] |
| 1.2 High risk of bias | 5 | 690593 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.03, 0.16] |
| 2 < 1000 participants | 2 | 534 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.01, 1.62] |
| 2.1 Low risk of bias | 1 | 274 | Risk Ratio (M‐H, Random, 95% CI) | 0.03 [0.00, 0.47] |
| 2.2 High risk of bias | 1 | 260 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.04, 3.26] |
Comparison 5.
All‐cause mortality
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All vaccine types | 1 | 40119 | Risk Ratio (M‐H, Random, 95% CI) | 1.40 [0.62, 3.16] |
| 1.1 Low risk of bias | 1 | 40119 | Risk Ratio (M‐H, Random, 95% CI) | 1.40 [0.62, 3.16] |
| 1.2 High risk of bias | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Differences between protocol and review
Trial sequential analysis was performed following recommendations from the CHBG. This was in keeping with CHBG methodological recommendations at the time of undertaking this review.
Historical controlled trials were excluded.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Randomised clinical trial. | |
| Participants | Children age 1 to 16 attending primary school. 40,119 participants were recruited. Of these 38,157 participants entered surveillance and became the analysed cohort. | |
| Interventions | 1. Inactivated HAV vaccine (Havrix, SmithKine Beecham) (0,1,12 months). 20,028 participants were randomised to this group. 19,037 participants received all doses as per protocol and entered surveillance. 2. Recombinant HBV vaccine (Engerix‐B, SmithKine Beecham (0,1,12 months). 20,091 participants were randomised to this group. 19,120 participants received all doses as per protocol and entered surveillance. |
|
| Outcomes | 1. Clinical hepatitis A (signs/symptoms consistent with hepatitis A, alanine aminotransferase levels of 45 U/L or higher, and IgM to HAV). Participants with the disease were identified by evaluating school absences of 2 or more days. 2. Adverse events (anaphylaxis, pain, headache, disturbed sleep, fever, local tenderness, redness/swelling, rash). (Measured in subgroup of 1181 participants). 3. Immunogenicity (anti‐HAV IgG). |
|
| Notes | Location: Thailand. Endemicity: high. Follow‐up: 532 days, multiple treatment. Drop out : 6533 participants (33,586/40,119 (84%) at 532 days). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated block randomisation (block size 10). |
| Allocation concealment (selection bias) | Low risk | A colour code was used to label hepatitis A and control vaccines. Neither parents of participants nor investigators knew the code. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Described as double blind with adequate detail provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The numbers and reasons for dropouts and withdrawals in all intervention groups were described. |
| Selective reporting (reporting bias) | Low risk | Expected outcomes reported on. |
| Other bias | Low risk | Trial appears to be free of other sources of bias. |
| Methods | Cluster randomised clinical trial. | |
| Participants | School children age 0 to 18 years. 62,698 participants recruited. | |
| Interventions | 1. Attenuated live HAV vaccine (0 months) (H2 TCID50 106.0 and LA‐1 TCID50 105.5. 31,421 participants were randomised to this group. 2. Unvaccinated. 31, 277 participants were randomised to this group. Additional note: 760 participants were vaccinated by mistake and 2081 participants who should have received the vaccine did not receive it. |
|
| Outcomes | 1. Clinical hepatitis A: Clinical symptoms and physical signs of hepatitis A, increase of ALT, and IgM‐HAV positive. 2. Adverse events. |
|
| Notes | Location: Liuzhou, China. Endemicity: high. Follow‐up: 2 years. Drop out: unclear. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial is described as randomised but the method of sequence generation is not specified. School class used as unit of randomisation. |
| Allocation concealment (selection bias) | High risk | Allocation sequence is known to the investigators who assigned participants. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The final analysis includes all randomised participants. The report gave the impression that there had been no dropouts or withdrawals, but this was not specifically stated. However, only 32,148 of those in the intervention group were actively followed‐up (51%). For the remaining participants clinical hepatitis A was identified through infectious disease reporting systems and hospital records. Furthermore, 760 participants were vaccinated by mistake and 2081 participants who should have been vaccinated were not. The discussion of how the above impacts on outcome data was considered inadequate. |
| Selective reporting (reporting bias) | Unclear risk | Immunogenicity (anti‐HAV IgG) not reported on. |
| Other bias | High risk | Did not adjust for cluster randomisation. |
| Methods | Randomised clinical trial. | |
| Participants | Healthy school children, age 7 to 12 years. 1080 participants were recruited. | |
| Interventions | 1. Attenuated live HAV vaccine (0 months) (LA‐1 strain TCID50 high titre). 561 participants were randomised to this intervention. Only 422 participants were vaccinated. 2. HBV vaccine 519 participants were randomised to this intervention. Only 401 participants received the vaccine. |
|
| Outcomes | 1. Clinical Hepatitis A: as per Law of the People's Republic of China on the prevention and treatment of infectious disease (August, 1990), clinical symptoms and physical signs of hepatitis A, increase of ALT, and IgM‐HAV positive. 2. Adverse events. |
|
| Notes | Location: Shanghai, China. Endemicity: high. Follow‐up: 1 month. Drop out: unclear. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States individuals were randomly divided into two groups, but the method of sequence generation is not specified. |
| Allocation concealment (selection bias) | High risk | The trial is unblinded, so that the allocation was known during the trial. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Although the number of participant dropouts is provided the reason for this is not fully provided. A total of 823/1080 (76%) completed the trial. No intention‐to‐treat analysis was conducted. |
| Selective reporting (reporting bias) | High risk | Immunogenicity (anti‐HAV IgG) and adverse events not reported on. |
| Other bias | Unclear risk | Insufficient information to assess whether other sources of bias are present. |
| Methods | Cluster randomised clinical trial. | |
| Participants | Healthy primary and secondary school students seronegative for HAV. 564,442 participants were recruited. | |
| Interventions | 1. Attenuated live HAV vaccine (0 months) (H2 TCID50 106.0 and H2 strain TCID50 106.7). 292,740 participants received the vaccine. 2. Unvaccinated. 271,702 participants were unvaccinated controls. | |
| Outcomes | 1. Clinical hepatitis A: as per Law of the People's Republic of China on the prevention and treatment of infectious. disease (August, 1990), clinical symptoms and physical signs of hepatitis A, increase of ALT, and IgM‐HAV positive. 2. Immunogenicity (anti‐HAV IgG). |
|
| Notes | Location: Shanghai, China. Endemicity: high. Follow‐up: 3 years. Drop out: unclear. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States clusters were randomly divided into two groups but the method of sequence generation is not specified. |
| Allocation concealment (selection bias) | High risk | The trial is not blinded, so that the allocation was known during the trial. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The final analysis includes all randomised participants. The report gave the impression that there had been no dropouts or withdrawals, but this was not specifically stated. |
| Selective reporting (reporting bias) | Unclear risk | Adverse events not reported on. |
| Other bias | Unclear risk | Insufficient information to assess whether other sources of bias are present. |
| Methods | A quasi‐randomised study described as a randomised controlled trial by its authors. We have included the study for the report of adverse events only. | |
| Participants | Children age 1 to 12, sero‐negative for anti‐HAV: 30,040 participants recruited. | |
| Interventions | 1. Attenuated live HAV vaccine (0 months) (LA‐1 strain TCID50 106.5). 15,779 participants received this vaccine. 2. Typhoid vaccine. 14,261 participants received this vaccine. |
|
| Outcomes | 1. Clinical hepatitis A : signs and symptoms, IgM‐HAV positive. 2. Adverse events. |
|
| Notes | Location: Guangxi, China. Endemicity: high. Follow‐up: 5 years. Drop out: unclear. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | The sequence is generated according to odd or even data of birth month. Participants born in even month who refused placebo were put into a third group. Also a number of participants declined vaccination as a fee was charged for the vaccine whilst others were wrongly vaccinated. |
| Allocation concealment (selection bias) | Unclear risk | The trial is described as randomised. However, the method used to conceal the allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | States parents of children and medical staff did not know grouping detail. However, the method of blinding was not fully discussed. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The final analysis includes all randomised participants. The report gave the impression that there had been no dropouts or withdrawals, but this was not specifically stated. |
| Selective reporting (reporting bias) | Unclear risk | Immunogenicity (anti‐HAV IgG) not reported on. |
| Other bias | High risk | Those participants with hepatitis A were identified through epidemic reports and medical records rather than active follow‐up of all participants. |
| Methods | Randomised clinical trial. | |
| Participants | Children 1.5 to 6 years who were negative for antibodies to HAV. 274 participants were recruited. | |
| Interventions | 1. Inactivated HAV vaccine (Virosome) (0 months). 137 participants received this vaccine. 2. Placebo (virosome suspension). 137 participants received the placebo. |
|
| Outcomes | 1. IgM‐HAV positive. 2. Clinical hepatitis: IgM‐HAV; glutamic‐pyruvic transaminase level of at least twice the normal upper limit, and 1 or more clinical sign consistent with hepatitis: icterus, clay‐coloured; stools, or dark urine. 3. Adverse events: pain, redness, swelling, induration, malaise, fatigue. 4. Immunogenicity (anti‐HAV IgG). |
|
| Notes | Location: Nicaragua. Endemicity: high. Follow‐up: 15 months. Drop out: 2 participants. Addtional notes: denominator for intention‐to‐treat analysis reported as 136 participants for both inactivated and placebo groups. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation code was centrally prepared with an equal probability of assignment to the groups, by use of random permuted blocks of size 4. |
| Allocation concealment (selection bias) | Low risk | Numbered syringes containing vaccine or placebo with identical appearance were used. Investigators enrolling children in Nicaragua allocated the next available number at entry into the trial. Neither the participants nor research team were aware of group assignments. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Numbered syringes containing vaccine or placebo with identical appearance were used. Investigators enrolling children in Nicaragua allocated the next available number, at entry into the trial. Neither the participants or research team were aware of group assignments. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The numbers and reasons for dropouts and withdrawals in all intervention groups were described. An intention‐to‐treat analysis was conducted and described in adequate detail. |
| Selective reporting (reporting bias) | Low risk | Expected outcomes reported on. |
| Other bias | Low risk | Trial appears to be free of other sources of bias. |
| Methods | Cluster and individually randomised clinical trial. | |
| Participants | Children age 1 to 12. 12,036 participants were recruited. | |
| Interventions | 1. Attenuated live HAV vaccine (0 months) (H2 strain TCID50 106.5). 5551 participants were received the vaccine 2. Unvaccinated. 6485 participants went unvaccinated as controls. |
|
| Outcomes | 1. Clinical hepatitis A (signs / symptoms consistent with hepatitis A, rise in ALT and IgM to HAV). 2. Immunogenicity (anti‐HAV IgG). |
|
| Notes | Location: Hebei, China. Endemicity: high. Follow‐up: 5 years. Drop out: unclear. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial is described as randomised but the method of sequence generation is not specified. |
| Allocation concealment (selection bias) | Unclear risk | The trial is described as randomised. However, the method used to conceal the allocation is not described. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The final analysis includes all randomised participants. The report gave the impression that there had been no dropouts or withdrawals. However, this was not specifically stated. However, those participants with clinical hepatitis A were identified through epidemic reports and medical records rather than active follow‐up of all participants. |
| Selective reporting (reporting bias) | Unclear risk | Does not report on adverse events. |
| Other bias | High risk | Did not adjust for cluster randomisation. |
| Methods | Randomised controlled trial. | |
| Participants | Children aged 6 to 15 years in good health; normal ALT/AST. 260 participants were recruited. | |
| Interventions | 1. Inactivated HAV vaccine (Havrix, GSK) (0,1,6 months). 128 participants received the vaccine. 2. Recombinant HBV vaccine (Engerix‐B GSK (identical). 132 participants received the vaccine. |
|
| Outcomes | 1. Clinical hepatitis A (IgM to HAV). 2. Averse events (fever, headache, malaise, anorexia, nausea, vomiting, any symptoms). 3. Immunogenicity (anti‐HAV IgG). |
|
| Notes | Location: Chile Endemicity: high. Follow‐up: 12 months, multiple treatment. Trial conducted under increased hygiene measure as result of earlier cholera outbreak. Drop out: 4 participants. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial is described as randomised, but the method of sequence generation is not specified. |
| Allocation concealment (selection bias) | High risk | A colour code was used to label hepatitis A and control vaccines. From the description provided those involved in the trial could have determined the allocation sequence. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double blind. However, the method of blinding not fully described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The numbers and reasons for dropouts and withdrawals in all intervention groups were described. |
| Selective reporting (reporting bias) | Low risk | Expected outcomes reported on. |
| Other bias | Low risk | Trial appears to be free of other sources of bias. |
| Methods | Randomised clinical trial. | |
| Participants | Seronegative children aged 2 to 16 years. 1037 participants were recruited. | |
| Interventions | 1. Inactivated HAV vaccine (Merck) (0,1,6 months). 519 participants received the vaccine. 2. Placebo (aluminium hydroxide dilutant). 518 participants received the vaccine. |
|
| Outcomes | 1. Clinical hepatitis A (one or more signs/symptoms consistent with hepatitis A; ALT twice level of normal during episodic illness due to no other cause; IgM to HAV). 2. Adverse events (fever, headache, malaise, anorexia, nausea, vomiting, any symptoms). 3. Immunogenicity (anti‐HAV IgG). | |
| Notes | Location: New York (Hasidic Jewish community). Endemicity: low. Follow‐up: 12 months. Drop out: none. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Sequence generation achieved using random number generation. |
| Allocation concealment (selection bias) | Low risk | Randomisation code was not revealed to any study personnel, the manufacturer's research staff, participants or their parents/guardians until the termination of the study. When labelled with the numbers the vials of the vaccine and placebo were indistinguishable. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Randomisation code was not revealed to any study personnel, the manufacturer's research staff, participants or their parents/guardians until the termination of the study. When labelled with the numbers the vials of the vaccine and placebo were indistinguishable. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | It was specified that there were no dropouts or withdrawals. |
| Selective reporting (reporting bias) | Low risk | Expected outcomes reported on. |
| Other bias | Low risk | Trial appears to be free of other sources of bias. |
| Methods | A quasi‐randomised study described as a multi‐centre individually randomised controlled trial by its authors. We have included the study for the report of adverse events only. | |
| Participants | Class 1 to 3 elementary students and kindergarten class students, sero‐negative for anti‐HAV: 54,746 participants were recruited. | |
| Interventions | 1. Attenuated live HAV vaccine (0 months) (H2 TCID50 104.0). 17,733 participants received the vaccine 2. Attenuated live HAV vaccine (0 months) (LA‐1 strain TCID50 105.5). 18,397 participants received this vaccine. 3. Unvaccinated: 18,616 participants went unvaccinated as controls. |
|
| Outcomes | 1. Clinical hepatitis A: as per Law of the People's Republic of China on the prevention and treatment of infectious disease (August, 1990), clinical symptoms and physical signs of hepatitis A, increase of ALT, and IgM‐HAV positive. 2. Immunogenicity (anti‐HAV IgG). 3. Adverse events. |
|
| Notes | Location: Taicang, Zhang, Jiagang, Kushan, Cahngshu, Dongtai in China. Endemicity: high. Follow‐up: unclear ‐ at least 507 days. Drop out: unclear. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | For individuals the sequence was generated according to odd or even data of birth month. |
| Allocation concealment (selection bias) | High risk | Unblinded trial. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that there had been no dropouts or withdrawals. However, this was not specifically stated. |
| Selective reporting (reporting bias) | Low risk | Expected outcomes reported on. |
| Other bias | High risk | It was whether all included participants were screened for anti‐HAV IgG. Only 4269 participants were actively followed up for serological testing. Hospital reports, epidemic reports and school absence registers were used to identify the remaining participants with clinical hepatitis A. Furthermore 4026 participants who were grouped into vaccine group were not vaccinated and 761 persons in the control group were wrongly vaccinated. There is little discussion of how the above impacts on outcome data. |
| Methods | Cluster randomised clinical trial. | |
| Participants | Factory workers and primary students. Total initially recruited is unclear. | |
| Interventions | 1. Attenuated live HAV vaccine (0 months) LA‐1 strain TCID50 106.5. 29,721 participants received the vaccine. 2. Unvaccinated. 31,964 participants. |
|
| Outcomes | 1. Clinical hepatitis A (signs/symptoms consistent with hepatitis A, and IgM to HAV). 2. Immunogenicity (anti‐HAV IgG). 3. Adverse events. |
|
| Notes | Location: Kunming, Zhejiang, Changchun, China. Follow‐up: 2 years. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States students were randomly assigned numbers and odd number were allocated to intervention group. For the factory groups the sequence generation method is unclear. |
| Allocation concealment (selection bias) | High risk | Allocation sequence was known to the investigators who assigned participants. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Paper does adequately describe number of participants initially included and randomised. The report gave the impression that there had been no dropouts or withdrawals, but this was not specifically stated. |
| Selective reporting (reporting bias) | Low risk | Expected outcomes reported on. |
| Other bias | High risk | Did not adjust for cluster randomisation. |
ALT: Alanine aminotransferase. AST: Aspartate aminotransferase. HAV: Hepatitis A virus. IgG: Immunoglobulin G. IgM: Immunoglobulin M.
TCID50: Median tissue culture infective dose; that amount of a pathogenic agent that will produce a pathological change in 50% of cell cultures.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abarca 2001a | Non‐randomised clinical trial eliciting the serological effectiveness of a two dose inactivated HAV vaccine regimen (Havrix, GSK ) (inadequate comparator). |
| Abarca 2001b | Non‐randomised clinical trial eliciting the serological effectiveness of a two dose inactivated HAV vaccine regimen (Havrix, GSK) (inadequate comparator intervention). |
| Ambrosch 1994 | Non‐randomised clinical trial eliciting the serological effectiveness in three groups : combined HAV + HBV vaccine versus separate HAV / HBV vaccine versus individual HAV / HBV vaccine regimen (inadequate comparator intervention). |
| Andre 2001 | Editorial review looking in response to a randomized, cross‐over, controlled comparison of two inactivated hepatitis A vaccines (Epaxal, Crucell) and (Havrix, GSK). |
| Anonymous 2008 | Randomised control trial comparing inactivated HAV vaccine (VAQTA, Sanofi Pasteur) versus HAV Immunoglobulin (inadequate comparator intervention) |
| Armstrong 1993 | Editorial review looking at inactivated HAV vaccine (VATQA, Merck). |
| Arslan 2001 | Non‐randomised clinical trial eliciting the serological effectiveness of a two dose inactivated HAV vaccine (VAQTA, Sanofi Pasteur) regimen in liver transplant recipients (inadequate comparator intervention). |
| Ashur 1999 | Randomised controlled trial comparing varying doses of inactivated HAV vaccine (Havrix, GSK) and (VAQTA, Sanofi Pasteur) (inadequate comparator intervention). |
| Beran 2000 | Randomised trial comparing three lots of combined HAV + typhoid vaccine (Hepatyrix, GSK) (inadequate comparator intervention). |
| Beran 2003 | Randomised controlled trial comparing inactivates HAV (Avaxim, Sanofi Pasteur) + combined HAV / typhoid vaccine (Vivaxam, Sanofi Pasteur) versus inactivated HAV (Avaxim, Sanofi Pasteur) (inadequate comparator intervention). |
| Black 2004 | Passive observational study. All participants received inactivated HAV vaccine (VATQA, Sanofi Pasteur). |
| Bovier 1999 | Randomised cross over clinical trial comparing virosomal HAV vaccine (Epaxal, Crucell) + Yellow fever vaccine (Stamaril, Sanofi Pasteur) versus virosomal HAV vaccine (Epaxal, Crucell) (inadequate comparator intervention). |
| Bovier 2005 | Randomised cross over clinical trial comparing virosomal HAV vaccine (Epaxal, Crucell) versus inactivated HAV vaccine (Havrix, GSK) (inadequate comparator intervention) |
| Branconier 1999 | Randomised control trial comparing inactivated HAV vaccine produced by different manufacturers (Havrix, GSK) and (VAQTA, Sanofi Pasteur) (inadequate comparator intervention). |
| Brown 1999 | Randomised control trial comparing inactivated HAV vaccine versus HAV Immunoglobulin versus inactivated HAV vaccine + immunoglobulin (inappropriate comparator intervention). |
| Bruguera 1996 | Clinical trial comparing yellow fever + HAV vaccine versus HAV vaccine (inadequate comparator intervention). |
| Bryan 1998 | Randomised control trial comparing inactivated HAV vaccine versus inactivated HAV from different manufacturers (Havrix, GSK) and (VAQTA, Sanofi Pasteur) (inadequate comparator intervention). |
| Bryan 2000 | Randomised cross over trial. Four groups comparing combinations of inactivated HAV vaccine from two manufacturers (Havrix, GSK) and (VAQTA, Sanofi Pasteur) (inadequate comparator intervention). |
| Burgess 2001 | Randomised control trial comparing two dosing regimens for combined HAV + HBV vaccine (Twinrix, GSK) (inadequate comparator intervention). |
| Chen 1997 | Randomised control trial comparing two dosing regimens for inactivated HAV vaccine (VAQTA, Sanofi Pasteur) (inadequate comparator intervention). |
| Clarke 2001 | A randomised control trial comparing two inactivated HAV vaccines (Avaxim, Sanofi Pasteur) or (VAQTA, Sanofi Pasteur) when given as a booster to patients primed with inactivated HAV vaccine (Avaxim, Sanofi Pasteur) (inadequate comparator intervention). |
| Clarke 2006 | Randomised control trial comparing virosomal adjuvant HAV vaccine (Epaxal, Crucell) with inactivated vaccine (Havrix, GSK) (inadequate comparator intervention). |
| Connor 2001 | Randomised control trial comparing boosting effect of inactivated HAV vaccine from different manufacturers (Havrix, GSK) or (VAQTA, Sanofi Pasteur) in patients already primed with inactivated HAV vaccine (Havrix, GSK) (inadequate comparator intervention). |
| Connor 2007 | Randomised control trial eliciting the serological effectiveness in two groups : combined HAV + HBV vaccine versus separate HAV / HBV vaccine (inadequate comparator intervention). |
| Dagan 2007 | Randomised control trial comparing virosomal adjuvant HAV vaccine (Epaxal, Crucell) + childhood vaccinations versus inactivated HAV (Havrix, GSK) + childhood vaccinations versus adjuvant HAV vaccine alone (Havrix, GSK) (inadequate comparator intervention). |
| De Artaza 2009 | Non‐randomised clinical study looking at adverse event and serological efficacy of HAV vaccine and HBV vaccine in patients with chronic liver disease (inadequate comparator intervention). |
| DeFraites 1995 | Randomised control trial comparing three varying vaccination regimens with inactivated HAV vaccine (inadequate comparator intervention). |
| Diaz 1996 | Editorial review looking at inactivated HAV vaccine. |
| Dulat 1996 | Non randomised clinical trial looking at seroprevalence rates following vaccination with inactivated HAV. |
| El‐Karaksy 2006 | Non randomised clinical trial looking at adverse events and serological efficacy of inactivated HAV (Havrix, GSK) in children with chronic liver disease. |
| Fafi‐Kremer 2008 | Editorial discussion of trial comparing inactivated HAV vaccine versus HAV immunoglobulin in post exposure patients. |
| Ferreira 2003 | Non randomised clinical trial looking at adverse events and serological efficacy of inactivated HAV (Havrix, GSK) in children with chronic liver disease. |
| Ferreira 2004 | Non randomised clinical trial looking at adverse events and serological efficacy of inactivated HAV (Havrix, GSK) in children with Down syndrome. |
| Fisch 1996 | Non randomised clinical trial comparing three administration routes of inactivated HAV vaccine. |
| Fleischmann 2002 | Non randomised clinical trial comparing two administration routes of inactivated HAV vaccine (Havrix, GSK) in patients with end stage renal failure. |
| Gil 1996 | Randomised control trail comparing inactivated HAV vaccine (SmithKine Beecham) versus inactivated HAV vaccine (GSK) + yellow fever vaccine (Pasteur Merieux) (inadequate comparator intervention). |
| Goilav 1995 | Randomised control trial comparing two inactivated HAV vaccines from different manufacturers (Sanofi Pasteur) (GSK) (inadequate comparator intervention). |
| Gong 2000 | Quasi randomised trial comparing inactivated HAV with placebo control. Did not include adverse events as an outcome. |
| Gong 2003 | Quasi randomised trial comparing inactivated HAV with placebo control. Did not include adverse events as an outcome. |
| Goubau 1992 | Randomised control trial comparing inactivated HAV vaccine produced from two different strains (inadequate comparator intervention). |
| Guptan 2002 | Randomised controlled trial eliciting the serological effectiveness in two groups: combined HAV / HBV vaccine versus combined HAV / HBV vaccine (Twinrix, GSK) (inadequate comparator intervention). |
| Holzer 1996 | Randomised control trial comparing virosomal adjuvant HAV vaccine (Epaxal, Crucell) with inactivated vaccine (Harvix, GSK) (inadequate comparator intervention). |
| Hornick 2001 | Randomised control trial comparing three booster regimens of inactivated HAV vaccine (VAQTA, Sanofi Pasteur) (inadequate comparator intervention). |
| Jiang 2008 | Randomised control trial eliciting the serological effectiveness in four groups: three consecutive lots of Healive® and one HAV vaccine control (inadequate comparator intervention). |
| Joines 2001 | Randomised control trial eliciting the serological effectiveness in two groups : combined HAV + HBV vaccine (Twinrix, GSK) versus separate HAV / HBV vaccine (Havrix, GSK) and (Engerix, GSK) (inadequate comparator intervention). |
| Kallinowski 1992 | Randomised control trial comparing three consecutive lots of inactivated HAV vaccine (GSK) (inadequate comparator intervention). |
| Kallinowski 2000 | Randomised control trial eliciting the serological effectiveness in two groups: combined HAV + HBV vaccine (Twinrix, GSK) versus separate HAV / HBV vaccine (Havrix+Engerix, GSK) or (VAQTA+ H‐B‐Vax, Sanofi Pasteur) (inappropriate comparator intervention). |
| Kallinowski 2003 | Randomised control trial eliciting the serological effectiveness in two groups in patients with chronic Hepatitis C : combined HAV + HBV vaccine versus separate HAV / HBV vaccine (inappropriate comparator intervention). |
| Landry 2000 | Non randomised controlled trial eliciting the serological effectiveness inactivated HAV vaccine regimen (Havrix, GSK) in travellers. |
| Lee 1999 | Randomised cross over trial. Four groups comparing combinations of inactivated HAV vaccine from two manufacturers (inappropriate comparator intervention). |
| Lee 2000 | Non randomised phase III clinical trial of inactivated HAV vaccine (AVAXIM, Sanofi Pasteur). |
| Leroux‐Roels 1996 | Randomised control trial comparing three lots of combined HAV + HBV (inappropriate comparator intervention). |
| Lin 1997 | Quasi randomised trial comparing live attenuate HAV vaccine with unvaccinated control. Did not consider adverse events as an outcome. |
| Lo 1996 | Randomised control trial comparing three inactivated HAV vaccine (Merck) regimens (inappropriate comparator intervention). |
| Lolekha 2003 | Randomised control trial comparing three inactivated HAV vaccine regimens (Avaxim, Sanofi Pasteur) (inappropriate comparator intervention). |
| Lopez 2007 | Randomised control trial comparing two dosing regimens for inactivated HAV vaccine (Avaxim, Sanofi Pasteur) (inappropriate comparator intervention). |
| Lu 1999 | Randomised control trial comparing two dosing regimens for inactivated HAV vaccine (VAQTA, Sanofi Pasteur) (inappropriate comparator intervention). |
| Mallet 2000 | Randomised control trial comparing hexavalent combined vaccine (Hexavac, Sanofi Pasteur) with reference vaccine (Pentavax + H‐B‐Vax II, Sanofi Pasteur) (inappropriate comparator intervention). |
| Newcomer 1994 | Non randomised clinical trial comparing 3 groups of inactivated HAV vaccine (VAQTA, Sanofi Pasteur) with varying protein content (inappropriate comparator intervention) |
| Nothdruft 2002 | Randomised control trial eliciting the serological effectiveness in two groups: combined HAV + HBV vaccine (Twinrix, GSK) versus separate HAV / HBV vaccine (Havrix + Engrix, GSK) (inappropriate comparator intervention). |
| Orr 2006 | Randomised controlled trial comparing varying dosing regimens of inactivated HAV vaccine (VATQA, Sanofi Pasteur) or (Avaxim, Sanofi Pasteur) (inappropriate comparator intervention). |
| Overbosch 2005 | Randomised controlled trial comparing combined HAV + typhoid vaccine (Viatim, Sanofi Pasteur) versus combined HAV vaccine followed by HAV + typhoid vaccine (inappropriate comparator intervention). |
| Poovorawan 1995 | Uncontrolled clinical trail eliciting serological effectiveness and adverse events in participants receiving virosomal HAV vaccine. |
| Poovorawan 1996 | Randomised controlled trial comparing varying dosing regimens of inactivated HAV vaccine (Havrix, GSK) (inappropriate comparator intervention). |
| Poovorawan 1998 | Randomised controlled trial comparing varying dosing regimens of inactivated HAV vaccine (inappropriate comparator intervention). |
| Prado 2002 | Randomised clinical trial comparing 3 lots of combined HAV + HBV vaccine (Twinerix, GSK) (inappropriate comparator intervention). |
| Proell 2003 | Randomised clinical trial comparing varying combined HAV + HBV + typhoid vaccine (Twinrix, GSK) and (Typherix, GSK) versus separate HAV + HBV / typhoid vaccine (Twinrix, GSK) and (Typherix, GSK) (inappropriate comparator intervention). |
| Ragni 2000 | Randomised control trial comparing inactivated HAV vaccines (Havrix, GSK) given by different routes (inappropriate comparator intervention). |
| Rajan 1995 | Randomised trial comparing three lots of inactivated HAV (Havrix, GSK) (inappropriate comparator intervention). |
| Rajan 1996 | Randomised trial comparing three lots of inactivated HAV (Havrix, GSK) (inappropriate comparator intervention). |
| Ramonet 2002 | Randomised control trial eliciting the serological effectiveness in three groups: combined HAV + HBV vaccine versus individual HAV / HBV vaccine regimen (Harvix, Engerix, GSK) (inappropriate comparator intervention). |
| Ran 1993 | Non randomised clinical trial eliciting the serological effectiveness live attenuated HAV vaccine (H2 strain) administered by different routes (inappropriate comparator intervention). |
| Ren 2002 | Randomised control trial comparing two dosing regimens for inactivated HAV vaccine (Havrix, GSK) (inappropriate comparator intervention). |
| Rieger 2004 | Randomised control trial eliciting the serological effectiveness in two groups: combined HAV + HBV vaccine versus separate HAV / HBV vaccine(inappropriate comparator intervention). |
| Roberton 2005 | Randomised control trial eliciting the serological effectiveness in two groups: combined HAV + HBV vaccine (Twinrix, GSK) versus separate HAV / HBV vaccine (GSK) (inappropriate comparator intervention). |
| Sagliocca 1999 | Randomised control trial comparing HAV versus unvaccinated control. Adequate control but post exposure study. |
| Shen 2008 | Non randomised clinical trial eliciting the serological effectiveness of inactivated HAV vaccine regimen (Healive, Sinovac Biotech). |
| Shouval 1993 | Randomised control trial comparing HAV immunoglobulin versus HAV Immunoglobulin + vaccine (inappropriate comparator intervention). |
| Shouval 1997 | Randomised control trial comparing inactivated HAV vaccine produced by different manufacturers (VAQTA, Sanofi‐Pasteur) and (Havrix, GSK) (inappropriate comparator intervention). |
| Shouval 1999 | Editorial review looking at HAV vaccine for secondary prevention. |
| Soysal 2007 | Randomised control trial comparing interchangeability of inactivated HAV vaccines produced by different manufacturers (Havrix, GSK) (VAQTA, Sanofi Pasteur) (following an initial dose of (Avaxim, Sanofi Pasteur) (inappropriate comparator intervention). |
| Srivastava 2008 | Editorial on randomised control trial comparing virosomal HAV vaccine (VAQTA, Sanofi Pasteur ) versus HAV Immunoglobulin (inappropriate comparator intervention). |
| Stojanov 2007 | Randomised control trial comparing two inactivated HAV vaccine regimens (VAQTA, Sanofi Pasteur) when given with a hexavalent vaccine (Hexavac, Sanofi Pasteur) (inappropriate comparator intervention). |
| Su 2000 | Randomised control trial comparing injection site pain tools (inappropriate comparator intervention). |
| Theilmann 1992 | Randomised control trial comparing three lots of inactivated HAV vaccine. (inappropriate comparator intervention). |
| Thoelen 1999 | Editorial review looking at combined HAV + HBV vaccine (Twinrix, GSK). |
| Thompson 1998 | Randomised control trial comparing two lots of combined HAV + HBV (Twinrix, GSK) (inappropriate comparator intervention). |
| Tilzey 1992 | Randomised control trial comparing two vaccination schedules with inactivated HAV vaccine (inappropriate comparator intervention). |
| Tilzey 1996 | Randomised control trial eliciting the serological effectiveness and safety of inactivated HAV vaccine (Havrix, GSK) in two groups : HIV positive patients and negative patients with haemophilia. (inappropriate comparator intervention). |
| Tong 1993 | Randomised control trial comparing three vaccination schedules with inactivated HAV vaccine (Havrix, GSK) (inappropriate comparator intervention). |
| Tsai 2000 | Randomised control trial eliciting the serological effectiveness in three groups : combined HAV + HBV vaccine (GSK) versus separate HAV / HBV vaccine (Havrix, Engerix, GSK) (inappropriate comparator intervention). |
| Van Damme 1994a | Randomised control trial comparing three vaccination schedules with inactivated HAV vaccine (inappropriate comparator intervention). |
| Van Damme 1994b | Uncontrolled clinical trail eliciting serological effectiveness and adverse events in participants receiving inactivated HAV vaccine (Havrix, GSK). |
| Van Damme 1996 | Uncontrolled clinical trail eliciting serological effectiveness and adverse events in participants receiving inactivated HAV vaccine. |
| Van der Wielen 2006 | Randomised control trial eliciting the serological effectiveness combined HAV + HBV vaccine (Twinrix, GSK) versus separate HAV / HBV vaccine (Havrix, Engerix, GSK) and (VAQTA, H‐B VAX, Sanofi Pasteur) (inappropriate comparator intervention). |
| Van der Wielen 2007 | Randomised trial comparing inactivated HAV vaccines produced by different manufacturers (Havrix, GSK) and (Epaxal, Crucell) (inappropriate comparator intervention). |
| Van Hervk 1999 | Randomised control trial comparing combined HAV / HBV vaccine (Twinrix, GSK) versus HAV Immunoglobulin (inappropriate comparator intervention). |
| Vidor 1996 | Randomised control trial comparing results from radio‐immunoassay and enzyme linked immunosorbent assay following vaccination with inactivated HAV vaccine (Havrix, GSK) (inappropriate comparator intervention). |
| Vidor 1998 | Randomised control trial comparing results from radio‐immunoassay and enzyme linked immunosorbent assay following vaccination with inactivated HAV vaccine (Avaxim, Sanofi Pasteur) (inappropriate comparator intervention). |
| Vodopija 1997 | Randomised control trial comparing HAV vaccine (Havrix, GSK) + typhoid vaccine (Typhim Vi, Sanofi Pasteur) versus separate HAV / Typhoid vaccines (inappropriate comparator intervention). |
| Wagner 1993 | Randomised control trial comparing HAV vaccine versus HAV Immunoglobulin + HAV vaccine (inappropriate comparator intervention). |
| Wallace 2001 | Randomised control trial comparing interchangeability of inactivated HAV vaccine produced by different manufacturers (Havrix, GSK) (VAQTA, Sanofi Pasteur) following an initial dose of inactivated HAV vaccine (Havrix, GSK) (inappropriate comparator intervention) . |
| Wang 2004 | Randomised control trial comparing inactivated HAV vaccine (Havrix, GSK) versus live attenuated HAV vaccine (Zhepu, Pukang Biotechnological Co) (inappropriate comparator intervention). |
| Weinberg 2006 | Non randomised clinical trial eliciting the serological effectiveness of a three dose regimen of inactivated HAV vaccine (Havrix, GSK) . |
| Westblom 1994 | Randomised controlled trial comparing varying doses and schedules of inactivated HAV vaccine (inappropriate comparator intervention). |
| Wiedermann 1994 | Randomised control trial comparing inactivated HAV vaccine (Havrix, GSK) stored at different temperatures (inappropriate comparator intervention). |
| Wiedermann 1997 | Randomised controlled trial comparing varying doses of inactivated HAV vaccine (inappropriate comparator intervention). |
| Williams 2000 | Randomised control trial comparing inactivated HAV vaccine (Havrix, GSK) administered via two different routes (inappropriate comparator intervention). |
| Wolters 2009 | Non randomised clinical trial eliciting the serological effectiveness of combined HAV + HBV vaccine (Twinrix, GSK). Does not consider primary outcome. |
| Xu 1998 | Quasi randomised trial comparing inactivated HAV with Typhoid fever Vi vaccine and unvaccinated controls. Did not include adverse events as an outcome. |
| Xu 2002 | Quasi randomised trial comparing inactivated HAV with placebo control. Did not include adverse events as an outcome. |
| Zanetti 1997 | Randomised control trial comparing inactivated HAV vaccine (Sanofi Pasteur) versus HAV Immunoglobulin versus inactivated HAV vaccine (inappropriate comparator intervention). |
| Zhang 1994 | Quasi randomised trial comparing inactivated HAV with unvaccinated control. Did not include adverse events as an outcome. |
| Zhang 2001 | Quasi randomised trial comparing inactivated HAV with unvaccinated control. Did not include adverse events as an outcome. |
| Zhao 2000 (a) | Abstract. Non randomised clinical trial eliciting the clinical effectiveness of live attenuated HAV vaccine regimen. |
| Zhao 2000 (b) | Non randomised clinical trial eliciting the clinical effectiveness of live attenuated HAV vaccine (H2 strain) regimen. |
| Zuckerman 1996 | Randomised control trial comparing two mono‐dose HAV vaccines (inappropriate comparator intervention). |
| Zuckerman 1997 | Randomised control trial comparing inactivated HAV vaccine produced by different manufacturers (Avaxim, Sanofi Pasteur and Havrix, GSK) (inappropriate comparator intervention). |
| Zuckerman 1998 | Randomised control trial comparing inactivated HAV vaccine produced by different manufacturers (Havrix, SmithKine Beecham) (Avaxim, Sanofi Pasteur) following initial vaccination with inactivated HAV vaccine (Havrix, GSK) (inappropriate comparator intervention). |
Contributions of authors
The protocol was written by Greg Irving (GI) and John Holden (JH). Both authors approved the final version of the protocol. Daniel Pope (DP) is the guarantor of the protocol. The search, assessment for risk of bias and data extraction and analysis were undertaken by GI and JH. RY double‐checked searches and data extraction for studies published in Chinese. The review was written by GI and JH. Both authors have approved the final version of the review. DP is a guarantor of the review.
Sources of support
Internal sources
New Source of support, Not specified.
External sources
Univeristy of Liverpool: Department of Public Health, UK.
Declarations of interest
None known
New
References
References to studies included in this review
- Innis B, Snitbhan R, Kunasol R, Laorakpongse P, Poopatanakool T, Kozik C, et al. Field efficacy trial of inactivated hepatitis A vaccine among children in Thailand (an extended abstract). Vaccine 1992;10:S159. [Google Scholar]; Innis B, Snitbhan R, Kunasol R, Laorakpongse P, Poopatanakool T, Kozik C, et al. Protection against hepatitis A by an inactivated vaccine. Journal of the American Medical Association 1994;271(17):1328‐34. [PubMed] [Google Scholar]
- Jiang S, Huang J, Chen J. The epidemiological efficacy assessment of attenuated live hepatitis A vaccine in Masses in Liuzhou. Chinese Journal of Epidemiology 1995;16(3):140‐2. 7648636 [Google Scholar]
- Jiang W, Niu X. Observation on the efficacy of attenuated live hepatitis A vaccine's vaccination contingency. Modern Preventative Medicine 2001;28(1):59‐61. [Google Scholar]
- Li Y, Wu H, Xu T, Yuan G, Zhang A, Zhou T. Observation of immunogenicity and epidemiological efficacy assessment of attenuated live hepatitis A vaccine. Chinese Journal of Public Health 2000;16(8):737‐8. [Google Scholar]
- Luo D, Li R, Gong J. Epidemiological efficacy of standardized live attenuated hepatitis A vaccine (LA‐1 strain). Chinese Journal of Vaccination and Immunization 2004;10(2):210‐2. [Google Scholar]
- Mayorga Pérez O, Herzog C, Zellmeyer M, Loáisiga A, Frösner G, Egger M. Efficacy of virosome hepatitis A vaccine in young children in Nicaragua: randomized placebo‐controlled trial. The Journal of Infectious Diseases 2003;188(5):671‐7. [DOI] [PubMed] [Google Scholar]
- Meng Z, Yao J, Zhao Y. Observation on the immunization effects of attenuated live hepatitis A vaccine. National Medical Journal of China 2000;80(1):9‐11. [PubMed] [Google Scholar]
- Riedemann S, Reinhardt G, Frosner G, Ibarra H, Moraleda L, Hering V, et al. Placebo‐controlled efficacy study of hepatitis A vaccine in Valdivia, Chile. Vaccine 1992;10 Suppl:152‐5. [DOI] [PubMed] [Google Scholar]
- Werzberger A, Kuter B, Mensch B, Lewis J, Miller W, Brown L, et al. Persistence of protection after a protective efficacy field trial of an inactivated hepatitis A vaccine [AASLD abstract]. Hepatology 1992;16(2 Pt 2):64A. [Google Scholar]; Werzberger A, Kuter B, Nalin D. Six years follow up after hepatitis A vaccination. New England Journal of Medicine 1998;338:1160. [DOI] [PubMed] [Google Scholar]; Werzberger A, Kuter B, Shouval D, Mensch B, Brown L, Wiens B, et al. Anatomy of a trial: a historical view of the Monroe inactivated hepatitis A protective efficacy trial. Journal of Hepatology 1993;18(2):S46‐S50. [DOI] [PubMed] [Google Scholar]; Werzberger A, Mensch B, Kuter B, Brown L, Lewis J, Sitrin R, et al. A controlled trial of a formalin‐inactivated hepatitis A vaccine in healthy children. New England Journal of Medicine 1992;327(7):453‐7. [DOI] [PubMed] [Google Scholar]
- Wu W, Xu Z, Xia J, Ouyang P, Min X, Lin X, et al. Assessment of attenuated live hepatitis A vaccines protective efficacy on spot. Chinese Journal of Public Health 1996;12(12):535‐6. [Google Scholar]
- Yuan Q, Luo S, Wu X, Chen D, Ding S, Wang Y, et al. Observations on the immunization effects of attenuated live hepatitis A vaccine. National Medical Journal of China 1995;80(1):9‐11. [Google Scholar]
References to studies excluded from this review
- Abarca K, Ibanez I, Flores J, Vial PA, Safary A, Potin M, et al. Efficacy of hepatitis A vaccination in children aged 12 to 24 months. Archives of Medical Research 2001;32(35):468‐72. [DOI] [PubMed] [Google Scholar]
- Abarca K, Ibanez I, Flores J, Vial PA, Safary A, Potin M, et al. Vaccination against hepatitis A in children aged 12 to 24 months [corrected]. Archives of Medical Research 2001;32(3):468‐72. [DOI] [PubMed] [Google Scholar]
- Ambrosch F, Wiedermann G, André F, Delem A, Gregor H, Hofmann H. Clinical and immunological investigation of a new combined hepatitis A and hepatitis B vaccine. Journal of Medical Virolology 1994;44(4):452. [DOI] [PubMed] [Google Scholar]
- Andre F. Randomized, cross‐over, controlled comparison of two inactivated hepatitis A vaccines. Vaccine 2001;20(23‐24):292‐3. [DOI] [PubMed] [Google Scholar]
- Anonymous. Is the hepatitis A vaccine an alternative for post‐exposure prophylaxis?. Journal of Family Practice 2008;57(2):82. [PubMed] [Google Scholar]
- Armstrong M, Giesa P, Davide J, Redner F, Waterbury J, Rhoad A, et al. Development of the formalin‐inactivated hepatitis A vaccine, VAQTA from the live attenuated virus strain CR326F. Journal of Hepatology 1993;18 Suppl(2):20‐6. [DOI] [PubMed] [Google Scholar]
- Arslan M, Wiesner R, Poterucha J, Zein N. Safety and efficacy of hepatitis Avaccination in liver transplantation recipients. Transplantation 2001;72(2):272‐6. [DOI] [PubMed] [Google Scholar]
- Ashur Y, Adler R, Rowe M, Shouval D. Comparison of immunogenicity of two hepatitisA vaccines ‐ VAQTA and HAVRIX ‐ in young adults. Vaccine 1999;17(18):2290‐6. [DOI] [PubMed] [Google Scholar]
- Beran J, Beutels M, Levie K, Damme P, Dieussaert I, Gillet M, et al. A single dose, combined vaccine against typhoid fever and hepatitis A: consistency, immunogenicity and reactogenicity. Journal of Travel Medicine 2000;7(5):246‐52. [DOI] [PubMed] [Google Scholar]
- Beran J, Chlibek R, Weber F. A combined dual‐chamber typhoid/hepatitis A vaccine as a booster dose in hepatitis A primed adults. Vaccine 2003;21(32):4650‐4. [DOI] [PubMed] [Google Scholar]
- Black S, Shinefield H, Hansen J, Lewis E, Su L, Coplan P. A post‐licensure evaluation of the safety of inactivated hepatitis A vaccine (VAQTA, Merck) in children and adults. Vaccine 2004;22(5‐6):766‐72. [DOI] [PubMed] [Google Scholar]
- Bovier P, Althaus B, Glueck R, Chippaux A, Loutan L. Tolerance and immunogenicity of the simultaneous administration of virosome hepatitis A and yellow fever vaccines. Journal of Travel Medicine 1999;6(4):228‐33. [DOI] [PubMed] [Google Scholar]
- Bovier PA, Farinelli T, Loutan L. Interchangeability and tolerability of a virosomal and an aluminium‐absorbed hepatitis A vaccine. Vaccine 2005;23(19):2424‐9. [DOI] [PubMed] [Google Scholar]
- Braconier J, Wennerholm S, Norrby S. Comparative immunogenicity and tolerance of Vaqta and Havrix. Vaccine 1999;17(17):2181‐4. [DOI] [PubMed] [Google Scholar]
- Brown L, Gress J, Harris K, Wiens B, Nalin D. Concurrent administration of inactivated hepatitis A vaccine with immune globulin in healthy adults. Vaccine 1999;17(11‐12):1468‐73. [DOI] [PubMed] [Google Scholar]
- Bruguera M, Bayas J, Vilella A, Tural C, González A, Vidal J, et al. Immunogenicity and reactogenicity of a combined hepatitis A and B vaccine in young adults. Vaccine 1996;14(15):1407‐11. [DOI] [PubMed] [Google Scholar]
- Bryan J, Henry C, Hoffman A, South‐Paul J, Smith J, Cruess D, et al. Randomized comparison of two licensed hepatitis A vaccines. American Journal of Tropical Medicine and Hygiene 1998;59:215. [Google Scholar]
- Bryan J, Henry C, Hoffman A, South‐Paul J, Smith J, Cruess D, et al. Randomized, cross‐over, controlled comparison of two inactivated hepatitis A vaccines. Vaccine 2000;19(7‐8):743‐50. [DOI] [PubMed] [Google Scholar]
- Burgess M, Rodger A, Waite S, Collard F. Comparative immunogenicity and safety of two dosing schedules of a combined hepatitis A and B vaccine in healthy adolescent volunteers: an open, randomised study. Vaccine 2001;19(32):4835‐41. [DOI] [PubMed] [Google Scholar]
- Chen X, Bülbül Q, Gast G, Loon A, Nalin D, Hattum J. Immunogenicity of two versus three injections of inactivated hepatitis A vaccine in adults. Journal of Hepatology 1997;26(2):260‐4. [DOI] [PubMed] [Google Scholar]
- Clarke P, Kitchin N, Souverbie F. A randomised comparison of two inactivated hepatitis A vaccines, Avaxim and Vaqta, given as a booster to subjects primed with Avaxim. Vaccine 2001;19(31):4429‐33. [DOI] [PubMed] [Google Scholar]
- Clarke P, Adams P, Ibáñez R, Herzog C. Rate, intensity, and duration of local reactions to a virosome‐adjuvant vs. an aluminium‐absorbed hepatitis A vaccine in UK travellers. Travel Medicine and Infectious Disease 2006;4(6):313‐8. [DOI] [PubMed] [Google Scholar]
- Connor B, Phair J, Sack D, McEniry D, Hornick R, Banerjee D, et al. Randomized, double‐blind study in healthy adults to assess the boosting effect of Vaqta or Havrix after a single dose of Havrix. Clinical Infectious Diseases 2001;32(3):396‐401. [DOI] [PubMed] [Google Scholar]
- Connor BA, Blatter MM, Beran J, Zou B, Trofa AF. Rapid and sustained immune response against hepatitis A and B achieved with combined vaccine using an accelerated administration schedule. Journal of Travel Medicine 2007;14(1):9‐15. [DOI] [PubMed] [Google Scholar]
- Dagan R, Amir J, Livni G, Greenberg D, Abu‐Abed J, Guy L, et al. Concomitant administration of a virosome‐ adjuvanted hepatitis a vaccine with routine childhood vaccines at age twelve to fifteen months: a randomized controlled trial. The Pediatric Infectious Disease Journal 2007;26(9):787‐93. [DOI] [PubMed] [Google Scholar]
- Artaza Varasa T, Sánchez Ruano JJ, García Vela A, Gómez Rodríguez R, Romero Gutiérrez M, Cruz Pérez G, et al. Efficacy and safety of vaccination against hepatitis A and B in patients with chronic liver disease. Gastroenterology and Hepatology 2009;32(7):483‐8. [DOI] [PubMed] [Google Scholar]
- DeFraites RF, Feighner BH, Binn LN, Kanjarpane DD, Delem AD, MacArthy PO, et al. Immunization of US soldiers with a two‐dose primary series of inactivated hepatitis A vaccine: early immune response, persistence of antibody, and response to a third dose at 1 year. Journal of Infectious Diseases 1995;171 Suppl(1):61‐9. [DOI] [PubMed] [Google Scholar]
- Diaz L, Bender B. New vaccines: hepatitis A and varicella. Internal Medicine 1996;17(17):25‐35. [Google Scholar]
- Dulat C, Defrance JP, Ille H. Hepatitis A virus immune status of Antilles military personnel: incidence after prophylaxis. Médecine Tropicale 1996;56(3):255‐8. [PubMed] [Google Scholar]
- El‐Karaksy HM, El‐Hawary MI, El‐Koofy NM, El‐Sayed R, El‐Raziky MA, Mansour SA, et al. Safety and efficacy of hepatitis A vaccine in children with chronic liver disease. World Journal of Gastroenterology 2006;12(45):7337‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fafi‐Kremer S, Stoll‐Keller F, Baumert T. Efficient post‐exposure prophylaxis by hepatitis A vaccine. Hepatology 2008;47(44):1416‐8. [DOI] [PubMed] [Google Scholar]
- Ferreira CT, Silveira T, Vieira S, Taniguchi A, Pereira‐Lima J. Immunogenicity and safety of hepatitis A vaccine in children with chronic liver disease. Journal of Pediatric Gastroenterology and Nutrition 2003;37(3):258‐61. [DOI] [PubMed] [Google Scholar]
- Ferreira CT, Leite JC, Taniguchi A, Vieira SM, Pereira‐Lima J, Silveira TR. Immunogenicity and safety of an inactivated hepatitis A vaccine in children with Down syndrome. Journal of Pediatric Gastroenterology and Nutrition 2004;39(4):337‐40. [DOI] [PubMed] [Google Scholar]
- Fisch A, Cadilhac P, Vidor E, Prazuck T, Dublanchet A, Lafaix C, et al. Immunogenicity and safety of a new inactivated hepatitis A vaccine: a clinical trial with comparison of administration route. Vaccine 1996;14(12):1132‐6. [DOI] [PubMed] [Google Scholar]
- Fleischmann EH, Kruppenbacher J, Bock HL, Weber M. Active immunization against hepatitis A in dialysis patients. Nephrology, Dialysis and Transplantation 2002;17(10):1825‐8. [DOI] [PubMed] [Google Scholar]
- Gil A, González A, Dal‐Ré R, Calero J. Interference assessment of yellow fever vaccine with the immune response to a single‐dose inactivated hepatitis A vaccine (1440 EL.U.). A controlled study in adults. Vaccine 1996;14(11):1028‐30. [DOI] [PubMed] [Google Scholar]
- Goilav C, Zuckerman J, Lafrenz M, Vidor E, Lauwers S, Ratheau C. Immunogenicity and safety of a new inactivated hepatitis A vaccine in a comparative study. Journal of Medical Virology 1995;46(3):287‐92. [DOI] [PubMed] [Google Scholar]
- Gong J, Li R, Yang J. Protective efficacy of large scale immunization with a live attenuated hepatitis A vaccine (LA‐1 strain). Guangxi Journal of Preventative Medicine 2000;6(5):257‐9. [Google Scholar]
- Gong J, Li R, Yang J. Long term immunogenicity and protective efficacy of a live attenuated hepatitis A vaccine (LA‐1 strain). World Chinese Journal of Digestology 2003;11(6):693‐6. [Google Scholar]
- Goubau P, Gerven V, Safary A, Delem A, Knops J, D'Hondt E. Effect of virus strain and antigen dose on immunogenicity and reactogenicity of an inactivated hepatitis A vaccine. Vaccine 1992;10 Suppl(1):114‐8. [DOI] [PubMed] [Google Scholar]
- Guptan R, Thakur V, Safary A, Sarin S. Immunogenicity and reactogenicity of a combined high dose hepatitis A and hepatitis B vaccine, compared to that of Twinrix in healthy Indian children. Vaccine 2002;20(16):2101‐6. [DOI] [PubMed] [Google Scholar]
- Holzer BR, Hatz C, Schmidt‐Sissolak D, Glück R, Althaus B, Egger M. Immunogenicity and adverse effects of inactivated virosome versus alum‐adsorbed hepatitis A vaccine: a randomized controlled trial. Vaccine 1996;14(10):982‐6. [DOI] [PubMed] [Google Scholar]
- Hornick R, Tucker R, Kaplan K, Eves K, Banerjee D, Jensen E, et al. A randomized study of a flexible booster dosing regimen of VAQTA in adults: safety, tolerability, and immunogenicity. Vaccine 2001;19(32):4727‐31. [DOI] [PubMed] [Google Scholar]
- Jiang W, Chen J, Wang X, Wang Y, Liu Y, Chen W, et al. Immunogenicity and safety of three consecutive lots of a new preservative‐free inactivated hepatitis A vaccine (Healive®): A double blind, randomised and controlled trial. Vaccine 2008;26:2297‐301. [DOI] [PubMed] [Google Scholar]
- Joines RW, Blatter M, Abraham B, Xie F, Clercq N, Baine Y, et al. A prospective, randomized, comparative US trial of a combination hepatitis A and B vaccine (Twinrix) with corresponding monovalent vaccines (Havrix and Engerix‐B) in adults. Vaccine 2001;19(32):4710‐9. [DOI] [PubMed] [Google Scholar]
- Kallinowski B, Gmelin K, Kommerell B, Bock HL, Clemens R, Scheiermann N, et al. Immunogenicity, reactogenicity and consistency of a new, inactivated hepatitis A vaccine ‐ a randomized multi centre study with three consecutive vaccine lots. Vaccine 1992;10(8):500‐1. [DOI] [PubMed] [Google Scholar]
- Kallinowski B, Knöll A, Lindner E, Sänger R, Stremmel W, Vollmar J, et al. Can monovalent hepatitis A and B vaccines be replaced by a combined hepatitis A/B vaccine during the primary immunization course?. Vaccine 2000;19(1):16‐22. [DOI] [PubMed] [Google Scholar]
- Kallinowski B, Jilg W, Buchholz L, Stremmel W, Engler S. Immunogenicity of an accelerated vaccination regime with a combined hepatitis A/B vaccine in patients with chronic hepatitis C. Gastroenterology 2003;41(10):983‐90. [DOI] [PubMed] [Google Scholar]
- Landry P, Tremblay S, Darioli R, Genton B. Inactivated hepatitis A vaccine booster given >/=24 months after the primary dose. Vaccine 2000;19(4‐5):399‐402. [DOI] [PubMed] [Google Scholar]
- Lee S, Chan C, Yu M, Wang Y, Chang F, Lo K, et al. A two dose combined hepatitis A and B vaccine in Chinese youngsters. Journal of Medical Virology 1999;59(1):1‐4. [PubMed] [Google Scholar]
- Lee C, Huang L, Lee P, Chiu HH, Dumas R, Milcamps B, et al. Immunogenicity and safety of an inactivated hepatitis A vaccine in Taiwanese adults and children. Southeast Asian Journal of Tropical Medicine and Public Health 2000;31(1):29‐36. [PubMed] [Google Scholar]
- Leroux‐Roels G, Moreau W, Desombere I, Safary A. Safety and immunogenicity of a combined hepatitis A and hepatitis B vaccine in young healthy adults. Scandinavian Journal of Gastroenterology 1996;31(10):1027‐31. [DOI] [PubMed] [Google Scholar]
- Lin F, Gu X, Wang F. Assessment on the spot of attenuated live hepatitis A vaccine’s efficacy. Acta Academiae Medicinae Suzhou 1997;17(5):868‐9. [Google Scholar]
- Lo Y, Lee S, Wong K, Lim W. Inactivated hepatitis A vaccine in healthy Chinese adults. Journal of Internal Medicine 1996;239(2):173‐6. [DOI] [PubMed] [Google Scholar]
- Lolekha S, Pratuangtham S, Punpanich W, Bowonkiratikachorn P, Chimabutra K, Weber F. Immunogenicity and safety of two doses of a paediatric hepatitis A vaccine in Thai children: comparison of three vaccination schedules. Journal of Tropical Pediatrics 2003;49(6):333‐9. [DOI] [PubMed] [Google Scholar]
- López E, Contrini M, Xifró M, Cattaneo M, Zambrano B, Dumas R, et al. Hepatitis A vaccination of Argentinean infants: comparison of two vaccination schedules. Vaccine 2007;25(1):102‐8. [DOI] [PubMed] [Google Scholar]
- Lu MY, Chang MH, Tsai KS, Chen DS. Hepatitis A vaccine in healthy adults: a comparison of immunogenicity and reactogenicity between two‐ and three‐dose regimens. Vaccine 1999;17(1):26‐30. [DOI] [PubMed] [Google Scholar]
- Mallet E, Fabre P, Pines E, Salomon H, Staub T, Schödel F, et al. Immunogenicity and safety of a new liquid hexavalent combined vaccine compared with separate administration of reference licensed vaccines in infants. The Pediatric Infectious Disease Journal 2000;19(12):1119‐27. [DOI] [PubMed] [Google Scholar]
- Newcomer W, Rivin B, Reid R, Moulton LH, Wolff M, Croll J. Immunogenicity, safety and tolerability of varying doses and regimens of inactivated hepatitis A virus vaccine in Navajo children. The Pediatric Infectious Disease Journal 1994;13(7):640‐2. [DOI] [PubMed] [Google Scholar]
- Nothdurft H, Dietrich M, Zuckerman J, Knobloch J, Kern P, Vollmar J, et al. A new accelerated vaccination schedule for rapid protection against hepatitis A and B. Vaccine 2002;20(7‐8):1157‐62. [DOI] [PubMed] [Google Scholar]
- Orr N, Klement E, Gillis D, Sela T, Kayouf R, Derazne E, et al. Long‐term immunity in young adults after a single dose of inactivated hepatitis A vaccines. Vaccine 2006;20:4328‐32. [DOI] [PubMed] [Google Scholar]
- Overbosch D, Peyron F, Picot N, Varichon JP, Dumas R, Chambonneau L, et al. Combined typhoid fever and hepatitis Avaccine: comparison of immunogenicity and safety to concomitant monovalent vaccine over 3 years. Journal of Travel Medicine 2005;12(6):319‐26. [DOI] [PubMed] [Google Scholar]
- Poovorawan Y, Theamboonlers A, Chumdermpadetsuk S, Glück R, Cryz SJ Jr. Safety, immunogenicity, and kinetics of the immune response to a single dose of virosome‐formulated hepatitis A vaccine in Thais. Vaccine 1995;13(10):891‐3. [DOI] [PubMed] [Google Scholar]
- Poovorawan Y, Theamboonlers A, Safary A. Single‐dose hepatitis A vaccination: comparison of different dose levels in adolescents. Vaccine 1996;14(12):1092‐4. [DOI] [PubMed] [Google Scholar]
- Poovorawan Y, Kosuwon P, Sutra S, Theamboonlers A, Vimolket T, Safary A. Comparison of the reactogenicity and immunogenicity of two different dose levels of hepatitis A vaccine in healthy children and adolescents. Asian Pacific Journal of Allergy and Immunology 1998;16(2):111‐7. [PubMed] [Google Scholar]
- Prado V, Riedemann S, Ibarra H, Potin M. Immunogenicity and reactogenicity of a combined hepatitis A and B vaccine in healthy Chilean subjects. International Journal of Infectious Diseases 2002;6(2):129‐33. [DOI] [PubMed] [Google Scholar]
- Proell S. Immunisation against hepatitis A, hepatitis B and typhoid fever via combined vaccination: rationale, immunogenicity and reactogenicity. BioDrugs 2003;17(1):10. [DOI] [PubMed] [Google Scholar]
- Ragni M, Lusher J, Koerper MA, Manco‐Johnson M, Krause D. Safety and immunogenicity of subcutaneous hepatitis A vaccine in children with haemophilia. Haemophilia 2000;6(2):98‐103. [DOI] [PubMed] [Google Scholar]
- Rajan E, al Bloushi S, O'Farrell B, Shattock A, Courtney M, Safary A, et al. Two year old hepatitis A vaccine (Havarix) retain its immunogenicity. Irish Journal of Medical Sciences 1995;164(2):184‐5. [Google Scholar]
- Rajan E, al Bloushi S, O'Farrell B, Shattock A, Courtney M, Safary A. Two year old hepatitis A vaccine is as good as new. Vaccine 1996;14(15):1439‐41. [DOI] [PubMed] [Google Scholar]
- Ramonet M, Silveira T, Lisker‐Melman M, Rüttimann R, Pernambuco E, Cervantes Y, et al. A two‐dose combined vaccine against hepatitis A and hepatitis B in healthy children and adolescents compared to the corresponding monovalent vaccines. Archives of Medical Research 2002;33(1):67‐73. [DOI] [PubMed] [Google Scholar]
- Ran L, Wang D, Duan Q, Yan T, Liu Q, Luo Y, et al. Safety and immunogenicity of live attenuated hepatitis A virus vaccine (H2 strain) in humans. Chininese Medical Journal 1993;106(8):604‐7. [PubMed] [Google Scholar]
- Ren A, Feng F, Ma J, Xu Y, Liu C. Immunogenicity and safety of a new inactivated hepatitis A vaccine in young adults: a comparative study. Chinese Medical Journal 2002;115(10):1483‐5. [PubMed] [Google Scholar]
- Rieger MA, Hofmann F, Michaelis M. Simultaneous vaccination against hepatitis A and B: results of an open, randomized study from the occupational health point of view. International Journal of Occupational Medicine Environmental Health 2004;17(3):379‐91. [PubMed] [Google Scholar]
- Roberton D, Marshall H, Nolan T, Sokal E, Díez‐Domingo J, Flodmark C, et al. Reactogenicity and immunogenicity profile of a two‐dose combined hepatitis A and B vaccine in 1‐11‐year‐old children. Vaccine 2005;23(43):5099‐105. [DOI] [PubMed] [Google Scholar]
- Sagliocca L, Amoroso P, Stroffolini T, Adamo B, Tosti ME, Lettieri G, et al. Efficacy of hepatitis A vaccine in prevention of secondary hepatitis A infection: a randomised trial. Lancet 1999;353(9159):1136‐9. [DOI] [PubMed] [Google Scholar]
- Shen Y, Gu X, Zhou J. Protective effect of inactivated hepatitis A vaccine against the outbreak of hepatitis A in an open rural community. World Journal of Gastroenterology 2008;14(7):2771‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval D, Ashur Y, Adler R, Lewis J, Armstrong M, Davide J, et al. Single and booster dose responses to an inactivated hepatitis A virus vaccine: comparison with immune serum globulin prophylaxis. Vaccine 1993;11(1):S9‐S14. [DOI] [PubMed] [Google Scholar]
- Shouval D, Adler R. Comparison of immunogenicity of two hepatitis A vaccines ‐ VAQTA and HAVRIX in young adults. Hepatology 1997;26(4):432a. [DOI] [PubMed] [Google Scholar]
- Shouval D, Ashur Y. Hepatitis A vaccine for secondary hepatitis A infection. Lancet 1999;354(9175):342‐3. [DOI] [PubMed] [Google Scholar]
- Soysal A, Gokçe I, Pehlivan T, Bakir M. Interchangeability of a hepatitis A vaccine second dose: Avaxim 80 following a first dose of Vaqta 25 or Havrix 720 in children in Turkey. European Journal of Pediatrics 2007;166(6):533‐9. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Aggarwal R. Hepatitis A vaccine versus immunoglobulin for post‐exposure prophylaxis. National Medical Journal of India 2007;20(6):302‐3. [PubMed] [Google Scholar]
- Stojanov S, Liese JG, Belohradsky BH, Vandermeulen C, Hoppenbrouwers K, Wielen M, et al. Administration of hepatitis A vaccine at 6 and 12 months of age concomitantly with hexavalent (DTaP‐IPV‐PRP approximately T‐HBs) combination vaccine. Vaccine 2007;25(43):7549‐58. [DOI] [PubMed] [Google Scholar]
- Su L, Tucker R, Frey SE, Gress J, Chan I, Kuter B, et al. Measuring injection‐site pain associated with vaccine administration in adults: a randomised, double‐blind, placebo‐ controlled clinical trial. Journal of Epidemiology and Biostatistics 2000;5(6):359‐65. [PubMed] [Google Scholar]
- Theilmann L, Kallinowski B, Gmelin K, Hofmann F, Scheiermann N, Wohland B, et al. Reactogenicity and immunogenicity of three different lots of a hepatitis A vaccine. Vaccine 1992;10 Suppl:132‐4. [DOI] [PubMed] [Google Scholar]
- Thoelen S, Damme P, Leentvaar‐Kuypers A, Leroux‐Roels G, Bruguera M, Frei P, et al. The first combined vaccine against hepatitis A and B: an overview. Vaccine 1999;17(13):1657‐62. [DOI] [PubMed] [Google Scholar]
- Thompson S, Norris M. Immunogenicity and reactogenicity of a combined hepatitis A‐hepatitis B vaccine in adolescents. International Journal of Infectious Diseases 1998;2(4):193‐6. [DOI] [PubMed] [Google Scholar]
- Tilzey A, Palmer S, Barrow S, Perry K, Tyrrell H, Safary A, et al. Clinical trial with inactivated hepatitis A vaccine and recommendations for its use. BMJ (Clinical Research Ed.) 1992;304(6837):1272‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilzey A, Palmer S, Harrington C, O'Doherty M. Hepatitis A vaccine responses in HIV‐positive persons with haemophilia. Vaccine 1996;14(11):1039‐41. [DOI] [PubMed] [Google Scholar]
- Tong M, Co R, Bellak C. Hepatitis A vaccination. Western Journal of Medicine 1993;158(6):602‐5. [PMC free article] [PubMed] [Google Scholar]
- Tsai I, Chang M, Chen H, Ni Y, Lee P, Chiu T, et al. Immunogenicity and reactogenicity of the combined hepatitis A and B vaccine in young adults. Vaccine 2000;19(4‐5):437‐41. [DOI] [PubMed] [Google Scholar]
- Damme P, Matheï C, Thoelen S, Meheus A, Safary A, André F. Single dose inactivated hepatitis A vaccine: rationale and clinical assessment of the safety and immunogenicity. Journal of Medical Virology 1994;44(4):435‐41. [DOI] [PubMed] [Google Scholar]
- Damme P, Thoelen S, Cramm M, Groote K, Safary A, Meheus A. Inactivated hepatitis A vaccine: reactogenicity, immunogenicity, and long‐term antibody persistence. Journal of Medical Virology 1994;44(4):446‐51. [DOI] [PubMed] [Google Scholar]
- Damme P, Thoelen S, Cramm M, Meheus A. Safety and immunogenicity of a high‐potency inactivated hepatitis A vaccine. Journal of Travel Medicine 1996;3(2):83‐90. [DOI] [PubMed] [Google Scholar]
- Wielen M, Damme P, Chlibek R, Smetana J, Sonnenburg F. Hepatitis A/B vaccination of adults over 40 years old: comparison of three vaccine regimens and effect of influencing factors. Vaccine 2006;24(26):5509‐15. [DOI] [PubMed] [Google Scholar]
- Wielen M, Vertruyen A, Froesner G, Ibáñez R, Hunt M, Herzog C, et al. Immunogenicity and safety of a pediatric dose of a virosome‐adjuvanted hepatitis A vaccine: a controlled trial in children aged 1‐16 years. The Pediatric Infectious Disease Journal 2007;26(8):705‐10. [DOI] [PubMed] [Google Scholar]
- Herck K, Damme P, Collard F, Thoelen S. Two‐dose combined vaccination against hepatitis A and B in healthy subjects aged 11‐18 years. Scandinavian Journal of Gastroenterology 1999;34(12):1236‐40. [DOI] [PubMed] [Google Scholar]
- Vidor E, Fritzell B, Plotkin S. Clinical development of a new inactivated hepatitis A vaccine. Infection 1996;24(6):447‐58. [DOI] [PubMed] [Google Scholar]
- Vidor E, Ratheau C, Briantais P, Vuillier D. Comparison of two immunization schedules with an inactivated hepatitis A vaccine (AvaximTM). Journal of Travel Medicine 1998;5(4):167‐72. [DOI] [PubMed] [Google Scholar]
- Vodopija I I, Baklaic Z, Vodopija R, Clemens R. Immune response to hepatitis A vaccine combined or given simultaneously with typhoid fever vaccine. Journal of Travel Medicine 1997;4(3):114‐17. [DOI] [PubMed] [Google Scholar]
- Wagner G, Lavanchy D, Darioli R, Pécoud A, Brulein V, Safary A, et al. Simultaneous active and passive immunization against hepatitis A studied in a population of travellers. Vaccine 1993;11(10):1027‐32. [DOI] [PubMed] [Google Scholar]
- Wallace M. Hepatitis A vaccine interchangeability. Currrent Gastroenterolgy Research and Practice 2001;3(4):283. [PubMed] [Google Scholar]
- Wang X, Xu Z, Yao X, Tian M, Zhou L, He L, et al. Immune responses of anti‐HAV in children vaccinated with live attenuated and inactivated hepatitis A vaccines. Vaccine 2004;22(15‐16):1941‐5. [DOI] [PubMed] [Google Scholar]
- Weinberg A, Gona P, Nachman S, Defechereux P, Yogev R, Hughes W, et al. Antibody responses to hepatitis A virus vaccine in HIV‐infected children with evidence of immunologic reconstitution while receiving highly active antiretroviral therapy. The Journal of Infectious Diseases 2006;193(2):302‐11. [DOI] [PubMed] [Google Scholar]
- Westblom T, Gudipati S, DeRousse C, Midkiff B, Belshe R. Safety and immunogenicity of an inactivated hepatitis A vaccine: effect of dose and vaccination schedule. The Journal of Infectious Diseases 1994;169(5):996‐1001. [DOI] [PubMed] [Google Scholar]
- Wiedermann G, Ambrosch F. Immunogenicity of an inactivated hepatitis A vaccine after exposure at 37 degrees C for 1 week. Vaccine 1994;12(5):401‐2. [DOI] [PubMed] [Google Scholar]
- Wiedermann G, Kundi M, Ambrosch F, Safary A, D'Hondt E, Delem A. Inactivated hepatitis A vaccine: long‐ term antibody persistence. Vaccine 1997;15(6‐7):612‐5. [DOI] [PubMed] [Google Scholar]
- Williams J, Fox‐Leyva L, Christensen C, Fisher D, Schlicting E, Snowball M, et al. Hepatitis A vaccine administration: comparison between jet‐injector and needle injection. Vaccine 2000;18(18):1939‐43. [DOI] [PubMed] [Google Scholar]
- Wolters B, Müller T, Ross RS, Clauberg R, Werfel U, Roggendorf H, et al. Comparative evaluation of the immunogenicity of combined hepatitis A and B vaccine by a prospective and retrospective trial. Human Vaccination 2009;5(4):248‐53. [DOI] [PubMed] [Google Scholar]
- Xu Z, Li R, Meng Z. Immunogenicity and efficacy trials of live attenuated hepatitis A vaccines. National Medical Journal of China 1998;78(4):254‐6. [PubMed] [Google Scholar]
- Xu Z, Li R, Meng Z, Zhang Y, Gong J. Immunogenicity and efficacy of two live attenuated hepatitis A vaccines (H(2) strains and LA‐1 strains). National Medical Journal of China 2002;82(10):678‐81. [PubMed] [Google Scholar]
- Zanetti A, Pregliasco F, Andreassi A, Pozzi A, Viganò P, Cargnel A, et al. Does immunoglobulin interfere with the immunogenicity to Pasteur Merieux inactivated hepatitis A vaccine?. Journal of Hepatology 1997;26(1):25‐30. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ma J, Han C. Primary research on efficacy of attenuated live hepatitis A vaccine. Chinese Journal of Epidemiology 1994;13(6):341‐3. [Google Scholar]
- Zhang Y, Liu X, Ma J. A field evaluation of the epidemiological efficacy of an attenuated live hepatitis A vaccine (H2 strain). Chinese Journal of Preventative Medicine 2001;35(6):363‐5. [PubMed] [Google Scholar]
- Zhao Y, Meng Z, et al. Protective efficacy of H2 strain live attenuated hepatitis A vaccines in an outbreak of hepatitis A. Zhonghua Yu Fang Yi Xue Za Zhi 2000;34(3):144‐6. [PubMed] [Google Scholar]
- Zhao Y, Meng Z, Xu Z, Guo J, Chai S, Duo C. H2 strain attenuated live hepatitis A vaccines: Protective efficacy in a hepatitis A outbreak. World Journal of Gastroenterology 2000;6(6):829‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerman J, Peyron F. Comparison of two monodose inactivated hepatitis A vaccines [EASL abstract]. Journal of Hepatology 1996;Suppl 1:103. [Google Scholar]
- Zuckerman J, Peyron F, Wallon M, Gay F, Delolme H, Briantais P, et al. Comparison of the safety and immunogenicity of two inactivated hepatitis A vaccines. Advances in Therapy 1997;14(3):116‐24. [Google Scholar]
- Zuckerman J, Kirkpatrick C, Huang M. Immunogenicity and reactogenicity of Avaxim (160 AU) as compared with Havrix (1440 EL.U) as a booster following primary immunization with Havrix (1440 EL.U) against hepatitis A. Journal of Travel Medicine 1998;5(1):18‐22. [DOI] [PubMed] [Google Scholar]
Additional references
- Adams G, Gulliford M, Ukoumunne O, Eldridge S, Chinn S, Campbell M. Patterns of intra‐cluster correlation from primary care research to inform study design and analysis. Journal of Clinical Epidemiology 2004;57(8):785‐94. [DOI] [PubMed] [Google Scholar]
- Altman D, Bland M. Interaction revisited: the difference between two estimates. BMJl (Clinical Research Ed.) 2003;326:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell B. Global epidemiology of hepatitis A: implications for control strategies. In: Margolis H, Alter M, Liang J editor(s). Viral Hepatitis and Liver disease. London: International Medical Press, 2002:359‐65. [Google Scholar]
- Bell B. [Hepatitis A] Prevention. In: Thomas H, Lemon S, Zuckerman A editor(s). Viral Hepatitis. 3rd Edition. Oxford: Backwell Publishing, 2005:126‐45. [Google Scholar]
- Berge J, Drennan D, Jacobs R, Jakins A, Meyerhoff A, Stubblefield W, et al. The cost of hepatitis A infections in American adolescents and adults in 1997. Hepatology 2000;31(2):469‐73. [DOI] [PubMed] [Google Scholar]
- Binn L, Bancroft W, Lemon S, Marchwicki R, LeDuc J, Trahan C, et al. Preparation of a prototype inactivated Hepatitis A virus vaccine from infected cell cultures. Journal of Infectious Diseases 1986;153:743‐56. [DOI] [PubMed] [Google Scholar]
- Brok J, Thorlund K, Gluud C, Wetterslev J. Trial sequential analysis reveals insufficient information size and potentially false positive results in many meta‐analyses. Journal of Clinical Epidemiology 2008;61(8):763‐9. [DOI] [PubMed] [Google Scholar]
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive‐‐Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [DOI] [PubMed] [Google Scholar]
- Campbell M, Elbourne D, Altman D. The CONSORT statement: extension to cluster randomised trials. BMJ (Clinical Research Ed.) 2004;328:702‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhagen Trial Unit. TSA ‐ Trial Sequential Analysis. http://ctu.dk/tsa/ (accessed 8 June 2012).
- Demicheli V, Tiberti D. The effectiveness and safety of hepatitis A vaccine: A systematic review. Vaccine 2003;21:2242‐5. [DOI] [PubMed] [Google Scholar]
- Dienstag J. Hepatitis A. In: Bircher J, Benhamou J, McIntyre N, Rizzetto M, Rodes J editor(s). Oxford Textbook of Clinical Hepatology. 1. Oxford: Oxford University Press, 1999. [Google Scholar]
- Egger M, Smith D, Schneider M, Minder C. Bias in meta‐analysis detected by a simple graphical test. BMJ (Clinical Research Ed) 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger M, Smith G. Meta‐analysis bias in location and selection of studies. BMJ (Clinical Research Ed.) 1998;316(7124):61‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück R, Metcalfe I. New technology platforms in the development of vaccines for the future. Vaccine 2002;20(5):B10‐6. [DOI] [PubMed] [Google Scholar]
- Gluud C, Brok J, Gong Y, Koretz R. Hepatology may have problems with putative surrogate outcome measures. Journal of Hepatology 2007;46(4):734‐42. [DOI] [PubMed] [Google Scholar]
- Gluud C, Nikolova D, Klingenberg SL, Alexakis N, Als‐Nielsen B, Colli A, et al. Cochrane Hepato‐Biliary Group. About The Cochrane Collaboration (Cochrane Review Groups (CRGs)). 2012, Issue 5. Art. No.: LIVER.
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Colloboration, 2011. Available from www.cochrane‐handbook.org.
- Hollinger F, Ticehurst J. Hepatitis A virus. In: Fields B, Knipe D, Howley P editor(s). Fields Virology. 3rd Edition. Philadelphia: Lippincott ‐ Raven, 1996:735‐82. [Google Scholar]
- Hu M, Sun B, Zhou Y. Research on attenuated live hepatitis A vaccine. Shanghai Medical Journal 1988;11(11):653‐6. [Google Scholar]
- International Conference on Harmonisation Expert Working Group. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice1997 CFR & ICH Guidelines. Vol. 1, PA 19063‐2043, USA: Barnett International, 1997. [Google Scholar]
- Ida S, Tachikawa N, Nakajima A, Daikoku M, Yano M, Kikuchi Y, et al. Influence of immunodeficiency virus type 1 infection on acute hepatitis A virus infection. Clinical Infectious Diseases 2002;34:379‐85. [DOI] [PubMed] [Google Scholar]
- Jacobsen K, Wiersma S. Hepatitis A virus seroprevalence by age and world region, 1990 and 2005. Vaccine 2010;28(41):6653‐7. [DOI] [PubMed] [Google Scholar]
- Kjaergard L, Villumsen J, Gluud C. Reported methodological quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [DOI] [PubMed] [Google Scholar]
- Lemon S. Hepatitis A virus: epidemiology, diagnosis, and prevention. Clinical Chemistry 1997;43(8(B)):1494‐9. [PubMed] [Google Scholar]
- Lemon S, Murphy P, Provost P, Chalikonda I, Davide J, Schofield T, et al. Immunoprecipitation and virus neutralization assays demonstrate qualitative differences between protective antibody responses to inactivated hepatitis A vaccine and passive immunization with immune globulin. Journal of Infectious Diseases 1997;176:9‐19. [DOI] [PubMed] [Google Scholar]
- Maiwald H, Jilg W, Bock H, Löscher T, Sonnenburg F. Long‐term persistence of anti‐HAV antibodies following active immunization with hepatitis A vaccine. Vaccine 1997;15(4):346‐8. [DOI] [PubMed] [Google Scholar]
- Mao J, Dong D, Zhang H, Chen N, Zhang X, Huang H, et al. Primary study of attenuated live hepatitis A vaccine (H2 strain) in humans. Journal of Infectious Disease 1989;159(4):621‐4. [DOI] [PubMed] [Google Scholar]
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352(9128):609‐13. [DOI] [PubMed] [Google Scholar]
- Newcombe R. Two‐sided confidence intervals for the single proportion: comparison of seven methods. Statistics in Medicine 1998;17:857‐72. [DOI] [PubMed] [Google Scholar]
- Purcell R, Emerson S. Natural history and experimentation models. In: Thomas H, Lemon S, Zuckerman A editor(s). Viral Hepatitis. 3rd Edition. Oxford: Blackwell publishing, 2005:109‐25. [Google Scholar]
- The Nordic Cochrane Centre. The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Cochrane Collaboration, 2011.
- Roque‐Afonso A. Hepatitis A virus: serology and molecular diagnosis. Future Virology 2010;5(2):233‐42. [Google Scholar]
- Routenberg J, Dienstag J, Harrison W, Kilpatrick M, Hooper R, Chisari F, et al. Foodborne epidemics of hepatitis A: clinical and laboratory features of acute and protracted illness. American Journal of the Medical Sciences 1979;278:123‐37. [PubMed] [Google Scholar]
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591‐603. [DOI] [PubMed] [Google Scholar]
- Sackett D. Bias in analytic research. Journal of Chronic Disease 1979;32(1):51‐63. [DOI] [PubMed] [Google Scholar]
- Schmidtke P, Habermehl P, Knuf M, Meyer CU, Sänger R, Zepp F. Cell mediated and antibody immune response to inactivated hepatitis A vaccine. Vaccine 2005;23(44):5127‐32. [DOI] [PubMed] [Google Scholar]
- Schulz K, Chalmers I, Hayes R, Altman D. Empirical evidence of bias. JAMA 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta‐analyses. International Journal of Epidemiology 2009;38(1):276‐86. [DOI] [PubMed] [Google Scholar]
- Thorlund K, Anema A, Mills E. Interpreting meta‐analysis according to the adequacy of sample size. An example using isoniazid chemoprophylaxis for tuberculosis in purified protein derivative negative HIV‐infected individuals. Clinical Epidemiology 2010;2:57‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. User manual forTrial Sequential Analysis (TSA). http://ctu.dk/tsa/files/tsa_manual.pdf 2011 (accessed 30 April 2012).
- Ukoumunne O, Gulliford M, Chinn S, Sterne J, Burney P, Donner A. Methods in health service research, Evaluation of heath interventions at area and organisation level. BMJ (Clinical Research Ed.) 1999;319(7206):376‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallbracht A, Maier K, Stierhof Y, Wiedmann K, Flehmig B, Fleischer B. Liver‐derived cytotoxic T cells in hepatitis A virus infection. Journal of Infectious Diseases 1989;160(2):209‐17. [DOI] [PubMed] [Google Scholar]
- Damme P, Banatvala J, Fay O, Iwarson S, McMahon B, Herck K, et al. Hepatitis A booster after vaccination: is there a need ?. Lancet 2003;362:1065‐71. [DOI] [PubMed] [Google Scholar]
- Wang X, Ma J, Zhang Y, Zhang Y, Han C, Xing Z, et al. Primary study on immunologic effect of live attenuated hepatitis A vaccine (H2 strain) after booster dose. Zhong Hua Liu Xing Bing Xue Za Zhi 2000;2:124‐7. [PubMed] [Google Scholar]
- Wang X, Ma J, Zhang Y. Immunogenicity and long‐term persistence of anti‐HAV in groups with different attenuated and inactivated hepatitis A vaccine dosage. Zhong Hua Liu Xing Bing Xue Za Zhi 2001;22(2):111‐3. [PubMed] [Google Scholar]
- Wang X, Xu Z, Ma J, Seidlein L, Zhang Y, Hao ZY, et al. Long‐term immunogenicity after single and booster dose of a live attenuated hepatitis A vaccine: Results from 8‐year follow‐up. Vaccine 2007;25(3):446‐9. [DOI] [PubMed] [Google Scholar]
- Wang H, Sui H. The meta analysis of protective efficacy of attenuated live hepatitis A vaccine. Chinese Journal of Vaccines and Immunization 2008;14(1):1‐6. [Google Scholar]
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61:64‐75. [DOI] [PubMed] [Google Scholar]
- Wetterslev J, Thorlund K, Brok J, Gluud C. Estimating required information size by quantifying diversity in a random‐effects meta‐analysis. BMC Medical Research Methodology2009 [Epub]; Vol. 9, issue 86. [DOI] [PMC free article] [PubMed]
- Whiting‐O'Keefe Q, Henke C, Simborg D. Choosing the correct unit of analysis in Medical Care experiments. Medical Care 1984;22(12):1101‐14. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Department of Communicable Disease Surveillance and Response. Hepatits A. http://www.who.int/csr/disease/hepatitis/HepatitisA_whocdscsredc2000_7.pdf 2000 (accessed 8 June 2012).
- World Health Organization. SAGE hepatitis A working group report. Buenos Aires: World Health Organiszation, 2010. [Google Scholar]
- World Health Organization. Global Advisory Committee on Vaccine Safety: Live attenuated hepatitis A vaccine. WHO Weekly Epidemiological Record 2010;85:289. [Google Scholar]
- Wiedermann G, Ambrosch F, André F, D'Hondt E, Delem A, Safary A. Persistence of vaccine‐induced antibody to hepatitis A virus. Vaccine 1992;10 Suppl 1:129. [DOI] [PubMed] [Google Scholar]
- Wood L, Egger M, Gluud L, Schulz K, Jüni P, Altman G, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ (Clinical Research Ed.) 2008;336:601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G. Clinical history and natural history of viral Hepatitis A in Sahanghai epidemic. Viral Heptitis and Liver disease. Hollinger F, Lemon S. Baltimore: Williams and Wilkins, 1991:76‐8. [Google Scholar]