

Abstract
Background
Most methods that integrate network and mutation data to study cancer focus on the effects of genes/proteins, quantifying the effect of mutations or differential expression of a gene and its neighbors, or identifying groups of genes that are significantly up- or down-regulated. However, several mutations are known to disrupt specific protein-protein interactions, and network dynamics are often ignored by such methods. Here we introduce a method that allows for predicting the disruption of specific interactions in cancer patients using somatic mutation data and protein interaction networks.
Methods
We extend standard network smoothing techniques to assign scores to the edges in a protein interaction network in addition to nodes. We use somatic mutations as input to our modified network smoothing method, producing scores that quantify the proximity of each edge to somatic mutations in individual samples.
Results
Using breast cancer mutation data, we show that predicted edges are significantly associated with patient survival and known ligand binding site mutations. In-silico analysis of protein binding further supports the ability of the method to infer novel disrupted interactions and provides a mechanistic explanation for the impact of mutations on key pathways.
Conclusions
Our results show the utility of our method both in identifying disruptions of protein interactions from known ligand binding site mutations, and in selecting novel clinically significant interactions.Supporting website with software and data: https://www.cs.cmu.edu/~mruffalo/mut-edge-disrupt/.
Electronic supplementary material
The online version of this article (10.1186/s12885-019-5532-5) contains supplementary material, which is available to authorized users.
Keywords: TCGA, Breast cancer, Feature construction, Protein interaction
Background
The impact of DNA mutations on the severity and progress of cancer has been a long standing focus for systems biology. On the one hand, several mutations to key genes were shown to play a critical role in cancer development and progression [1–7]. However, most mutations observed in cancer patients are unique, seen only in the individual in which they were observed, making it hard to determine their impact and to differentiate between causal and driver mutations [8, 9]. To address this issue, several network analysis methods have been used to aggregate the impact of mutations within and across patients [10, 11]. These methods operate under the assumptions that genes in a specific neighborhood of an interaction graph likely share a function or a pathway and so mutations in these genes, even if unique, may inform us about the importance of that pathway to the specific type of cancer being studied. An example of such network based methods is network smoothing, which fuses network structure with prior knowledge, and produces a measure for each node that respects both the input data and the structure of the network [12]. Such smoothing methods are widely used, with applications ranging from identification of cancer genes [13, 14], identification of gained/lost cellular functions [15] and more [12].
Network smoothing methods are commonly used to quantify the proximity of each node in the network to a set of nodes of interest, e.g. genes that are mutated or differentially expressed in a sample. While successful in identifying cancer genes and pathways, these methods are limited to using a static network that is shared between samples, and are not designed to handle dynamic effects (such as changes in interactions between samples). Mutations may disrupt interactions between proteins through a variety of mechanisms: alteration of protein structure impacting its function [16–18], affecting the ability of a protein to bind DNA [19–22], impacting the regulation of a gene, affecting its translation or degradation efficiency [23–25] and more. Most work utilizing protein-protein interaction networks in cancer do not adjust the networks based on such individual mutation information [26–28]. Thus, there is a need for methods that can perform comprehensive genome-wide prediction of protein interaction disruption and can determine the impact of such disruption on the resulting pathways and networks.
To enable the identification of mutations that significantly alter edges in the network we extended network smoothing algorithms to smooth not just node values but also edge (interaction) values. We do this by adding a set of nodes that represent the edges, assigning an initial value to each of these nodes and then performing network smoothing on the (much larger) network. This network adjustment has some conceptual similarities with other graph operations such as graph powers, in which transitive edges are added to an existing network; double graphs, in which a graph is duplicated and “cross” edges are added for each original edge; and line graphs, which represent edges of the original graph as nodes. We discuss the algorithmic and run time implications of the combined node and edge smoothing method. We next applied our method to study over a thousand mutation profiles from TCGA breast cancer patients. As we show, the network smoothing method was able to prioritize a subset of the edges, based on the mutation information alone, that were both better at predicting survival across patients and correctly associated with known ligand binding mutations. We discuss some of the top interactions identified by the method and show that these indeed include mainly known cancer related genes. Finally, for the subset of the predicted edges for which we could find structural information we tested the impact of the mutation on the specific interaction predicted and show that the R2 correlation between the predicted and actual impact is high.
Methods
Pre-processing the omics data
We obtained somatic mutation and clinical data from breast cancer (BRCA) samples in TCGA [29], which we used to construct features for prediction of interaction disruption.
We constructed a binary mutation matrix M, with samples as rows and genes as columns. We use C(A) to denote the set of column labels of matrix A, so that e.g. C(M) is the set of genes that appear in the TCGA somatic mutation data. Similarly, we define R(A) as the set of row labels of matrix A, corresponding to the distinct samples (individuals) present in each data set.
The mutation matrices M are defined as
| 1 |
The TCGA BRCA data includes somatic mutations in 22,232 genes across 1081 samples, including missense mutations, nonsense mutations, frame shifts, and in-frame deletions and insertions. In addition to the condition specific omics data we also use general interaction datasets. Our primary results use the HIPPIE protein-protein interaction network [30] (version 2.0, released 2016-06-24), which contains confidence scores for 318,757 interactions between 17,204 proteins. We also evaluate our method using the STRING network (v10.5), using all edges included in the downloadable version of that network: 4,724,503 edges between 17,179 nodes. Edges in the STRING network must have a weight of at least 0.15 to be included in the downloadable version of the network; we use all available edges in this version of STRING. Note that the network smoothing procedure allows using these edges in a way that respects the degree of confidence in those protein interaction – low-weight edges contribute less to the result of the network smoothing operation (Additional file 1: Supporting Methods). Results using the STRING network are shown in Additional file 1.
Network construction and initial edge scores
Given an original PPI network G=(V, E,w), with V as the set of proteins, E as the set of edges, and edge weights w(u, v) on every edge {u, v}∈E, we create an adjusted network G′=(V′,E′,w′). With AdjG[v] as the adjacency list of v in the network G, we define V′ and E′:
| 2 |
That is, we add a dummy node uv in the middle of each edge {u, v}, as shown in Fig. 1. These dummy nodes in G′ represent edges in G, and allow assigning scores to each edge by extending current network smoothing procedures.
Fig. 1.

Simulation of the edge smoothing procedure. From left to right: the original protein-protein interaction network, the adjusted network with dummy nodes (squares) for each protein-protein edge, somatic mutations shown as black nodes, and the result of the network smoothing procedure applied to the adjusted network with dummy nodes. White and black nodes in the third panel show assignment of values 0 and 1 (respectively) to nodes, and the fourth panel shows continuous node values in [0,1], denoting the smoothed score for each protein and protein-protein interaction
We define initial weights for our new edges in G′ as:
| 3 |
Protein interaction networks often use edge weights w(u, v)∈[0,1] to denote the confidence in some edge (u, v), and one can naturally define the reliability of a path pst between nodes s and t as the product of edge weights along this path [31].
| 4 |
Our choice of edge weights preserves the reliability of any path between two nodes s and t representing proteins in the network G, giving the same reliability in G′ (Additional file 1: Supporting Methods). We also evaluate our method using an alternative assignment of edge weights, with w′(u, uv)=w′(uv, v)=w(u, v)/2 (Additional file 1: Supporting Results).
Once we assign an initial score to edges, we use our adjusted network G′ to perform a standard network smoothing procedure, as described in the following section.
Gene set network smoothing
Here we extend the network propagation/smoothing method described in Vanunu et al. [32] that was initially only focused on nodes to smooth edge scores as well. Given a network G=(V, E,w) with V as the set of proteins and new nodes for original edges, E as the set of edges linking proteins with new edge nodes, edge weights defined in Eq. 3, and a prior knowledge vector Y:V→[0,1] constructed from somatic mutation status, we compute a function F(v) that is both smooth over the network and accounts for the prior knowledge about each node. Note that we do not perform this network smoothing procedure directly on the protein-protein interaction network; we compute smoothed node scores for our modified network that contains dummy nodes corresponding to edges in the original network and thus allows for scoring edges as well as nodes (Additional file 1: Supporting Methods).
Ligand binding site mutations
The mutLBSgeneDB database [33] contains annotations for genes with ligand binding site (LBS) mutations, and we combine these annotations with TCGA somatic mutation data. Of the 1081 TCGA samples with somatic mutation data, 389 have at least one somatic mutation which is contained in the LBS database, and 102 of these samples contain more than one LBS mutation, giving a total of 550 LBS mutations across all samples, in 340 distinct genes. We use these selected ligand binding mutations to evaluate our ranking of interaction edges, in “Ligand binding site edge scoring” section.
Protein structure alteration prediction
We use protein structures deposited in the RCSB (Research Collaboratory for Structural Bioinformatics) PDB database [34], and perform automated queries to PDB for all ligand binding site mutations in our dataset. We select edges which have a ligand binding site mutation in at least one interacting protein, and for which both interacting proteins have structures in PDB. This produces 143 selected edges, across 24 distinct patients and 98 distinct proteins. For these edges, it is possible, in principle, to use structural alteration prediction to predict binding disruption – though the results of our PDB queries require manual filtering to be usable for this task.
The mutLBSgeneDB database [33] includes specific amino acid substitutions for ligand binding site mutations in TCGA samples. We use the PyMOL tool [35] (version 2.0.7) mutagenesis functionality to simulate the effect of these amino acid substitutions on the relevant protein structures. We then upload structures for these interacting pairs to the ClusPro 2.0 [36] web service to predict protein docking, running two docking prediction jobs for each interacting pair: wild type of both proteins, and the PyMOL-simulated mutated protein structure with wild type of its interacting partner.
Results
We evaluate our edge scoring method in multiple ways. First, we examine whether high-scoring edges (those that we predict to be more disrupted based on mutational scores) are more predictive of patient survival than random sets of other edges. We then test whether our edge scores show significant agreement with known ligand binding site mutations. Finally we perform simulations of protein docking with and without ligand binding site mutations, and compare our edge scores to a measure of the disruption of specific protein interactions.
Identification of top scoring edges
To identify mutations impacting network edges we extended network smoothing so that it can produce smoothed scores for edges as well. We applied our method to somatic mutation data from TCGA breast invasive carcinoma (BRCA) samples [29]. The dataset contains mutation and survival information for 1081 patients. We use version 2.0 of the HIPPIE protein interaction network [30] to construct an expanded interaction network. The HIPPIE 2.0 network H=(VH,EH) has |EH|=314727 edges between |VH|=17204 nodes (genes), and our adjusted network H′=(VH′,EH′) has |VH′|=|VH|+|EH|=331931 nodes connected by |EH′|=2|E|=629454 edges. The STRING v10.5 network S=(VS,ES) likewise contains |ES|=4724503 edges between |VS|=17179 nodes, and our adjusted network S′=(VS′,ES′) contains |VS′|=4741682 nodes and |ES′|=9449006 edges.
For each sample in the TCGA BRCA data, we compute a smoothed mutational score for all nodes in H′ or S′, using somatic mutations to assign initial labels to nodes. This produces a continuous score m[v]∈[0,1] for each v∈VH′ or , which represents the proximity of that protein or interaction to somatic mutations in that patient. For each patient, we compute the median and maximum score across all edges, and plot histograms of the median and maximum for the HIPPIE network (Fig. 2) and STRING network (Additional file 1: Figure S12).
Fig. 2.

Histograms of propagated edge scores. For each patient, scores are collapsed across all edges by computing the median or maximum edge score in that patient. a shows the distribution of the median edge score in each patient, and b shows the distribution of the maximum edge score in each patient
Evaluation of edge scoring procedure
To evaluate the scores assigned to edges, and to determine if they indeed highlight key mutations that impact disease progression, we used several complementary information sources. We first examined the association between our propagated edge scores and patient survival. For this, we fit a univariate Cox regression model for each edge in the network, relating patient survival to each edge’s propagated mutation scores across patients. Cox models are commonly used in survival analysis, as these allow for dealing with censored survival data, in which exact survival times are known for some samples, but only lower bounds are known for others (e.g. if the patient was alive at their last follow-up, but no further information is known) [37, 38]. We compute the R2 goodness-of-fit value for the Cox model fit to each edge, and evaluate the difference in survival fits between high-scoring edges and random selections of the remaining edges.
We collapse propagated edge values across patients by considering the 80th decile of propagated mutation scores for that edge, i.e. the ⌊1081/5⌋=216th-highest score for that edge across any patient. These 80th-decile scores produce a measure of network proximity of each edge to somatic mutations in at least 20% of patients, and we use these scores to produce a global ranking of edges across all patients. We test whether the top 1000 edges have significantly higher R2 values than a random sample of 1000 edges. For each of the random sets we perform a Mann-Whitney U test to determine whether our top edges have higher R2 values than randomly chosen edges (Fig. 3). As can be seen, when compared to most random selections top scoring edges obtain a significantly higher R2 value with survival indicating that mutations related to these edges indeed impact disease progression. We repeated this analysis with alternative edge scores w′=w/2 and using the STRING network (Additional file 1: S10 and S16). In both additional of this survival analysis, we again see that high-scoring edges show a significantly higher R2 value when compared to random selections.
Fig. 3.
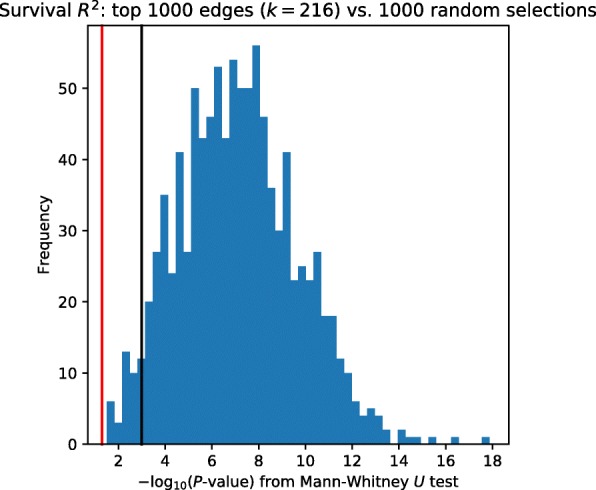
Histogram of Mann-Whitney U test P-values, comparing survival R2 values for top-scoring edges and 1000 sets of randomly-selected edges. The red vertical line shows P=0.05, the black vertical line shows P=0.001. P-values from the 1000 M–W tests are transformed to − log10-scale
Ligand binding site edge scoring
While survival analysis provides some evidence for the relevance of the high scoring edges, it does not provide any mechanistic explanation or support for these scores. To determine the relevance of the high scoring edge mutations to the interactions of the edge proteins (the two proteins on either side of the edge) we looked at a database of ligand binding site (LBS) mutations [33]. This database contains annotations for known ligand binding site mutations across the human genome, including additional cross-database references such as GO process terms, conservation information, and more. Every (gene,amino acid substitution) pair in this database is known to affect a ligand binding site in the protein product of that gene; we extract these pairs and use them to identify all somatic mutations in the TCGA BRCA cohort that are also listed in the mutLBSgeneDB database, allowing us to identify edges which are incident to these ligand binding site mutations.
Figure 4a shows our assignment of labels to edges: edges are assigned label 1 (shown in blue added node in the middle of the edge) if that edge is adjacent to a ligand binding site mutation (red), and 0 otherwise. This labeling of edges is imperfect; ideally we would label edges as 1 only if that specific interaction is disrupted by a ligand binding site mutation, but the mutLBSgeneDB database [33] does not contain data with this level of granularity.
Fig. 4.

a Edge labels for ligand binding site scoring. b Histograms of ROC AUC for selection of ligand binding site (LBS) mutation related edges. Scores from real LBS mutations are shown in blue, scores across the 100 shuffled LBS mutation assignments are shown in orange. Frequency values are normalized so that the total area under each histogram sums to 1
The total number of patient-model edges in our analysis is 314,727. Of these, only a small fraction are LBS edges, with counts per patient shown in Additional file 1: Figure S3. We consider each of the 389 patients with LBS mutations separately (details of mutation and gene counts in “Methods, and Ligand binding site mutations” sections), rank patients’ edges by propagated mutation scores, and evaluate this ranking through three separate measures: ROC AUC, normalized discounted cumulative gain (nDCG) [39, 40], and Spearman correlation P-values. For each of these measures, we compute the real ranking for each patient’s edges, with LBS mutations from the mutLBSgeneDB database, with histograms of ranking measures shown in blue in Fig. 4b and Additional file 1: Figures S4 and S5. We then generate 100 random sets by shuffling LBS assignments and computing the rankings of these random permutations. Note that as with other scale-free networks, shuffling a patient’s LBS mutations can have a large effect on the number of edges labeled 1 (shown in blue in Fig. 4a, since this depends on the degree of the nodes in the network. The performance across all 100 random permutations is shown in orange in Fig. 4b and Additional file 1: Figures S4 and S5. As can be seen, for all evaluation metrics we used the top ranked edges based on network propagated scores are significantly more associated with LBS mutations when compared to a random set of edges. We additionally used the Mann-Whitney U test to measure the difference in distributions between our top propagated edges and those obtained via shuffled mutations, for all three measures of the quality of this ranking. The difference between real and shuffled nDCG measures has M–W P=3.28×10−222, and likewise the ROC AUC and Spearman correlation P-value measures produce M–W P-values of 7.19×10−283 and 6.90×10−176, respectively.
Table 1 shows the unique interactions among the top 50 highest-scoring edges across all patients. The rank of each interaction is computed as the highest rank of that edge across all patients. The top-scoring edge here involves HDAC8, a class I histone deacetylase which is implicated as a therapeutic target in various diseases, including cancer [41, 42], and tumor suppressors TP53 [43, 44] and TP63 [45, 46] both score highly. Cytochrome P450 enzymes such as CYP2A7 and CYP2A13 score highly as well, and these genes are implicated in bladder cancer but not normally expressed in breast tissue [47, 48].
Table 1.
Unique interactions from the top 50 scoring edges based on the smoothed mutational score, pooled across all patients
| Gene 1 | Gene 2 | Prop. score | Top rank | References |
|---|---|---|---|---|
| STEAP1B | STEAP1 | 0.300938 | 1 | [49–51, 57] |
| TAS1R2 | TAS1R3 | 0.277285 | 2 | |
| SCGB3A2 | MARCO | 0.244833 | 3 | [52, 53] |
| CYP2A7 | CYP2A13 | 0.244117 | 4 | [47, 48] |
| CNGB1 | ABCA4 | 0.242088 | 6 | [58, 59] |
| PLXND1 | SEMA3E | 0.229689 | 12 | [60] |
| GSTA5 | GSTA2 | 0.211860 | 14 | [61] |
| UGT2B15 | UGT2A3 | 0.210076 | 15 | [62, 63] |
| CX3CR1 | CX3CL1 | 0.206455 | 16 | [64–66] |
| GFRA3 | ARTN | 0.204744 | 21 | [67–70] |
| GRID2 | GRID2IP | 0.202544 | 33 | [71] |
| PLXNC1 | SEMA7A | 0.199880 | 34 | [60, 72] |
| HAS2 | HAS3 | 0.199088 | 35 | [73, 74] |
| OBP2B | OBP2A | 0.197566 | 37 | [75, 76] |
| CD180 | LY86 | 0.195079 | 39 | [77, 78] |
| ZNF221 | ZNF225 | 0.193870 | 42 | [79, 80] |
| PSPN | GFRA4 | 0.192038 | 45 | [81, 82] |
| LIPE | FABP9 | 0.190075 | 47 | [83, 84] |
| KCNC1 | KCNC2 | 0.189430 | 48 | [85, 86] |
References refer to prior information about the involvement of these proteins in breast or other types of cancers. See Additional file 1: Table S2 for complete details and more information
Results for alternative edge weights w′=w/2 are shown in Additional file 1: Figures S7–S9, again with highly significant differences between real and shuffled edge selections (M–W P=1.59×10−225 for ROC AUC, P=5.02×10−213 for nDCG, and P=4.12×10−181 for Spearman correlation P-values). We likewise see highly significant differences between real and shuffled edge selections with the STRING network, shown in Additional file 1: Figures S13–S15. These figures show significantly higher ROC AUC and nDCG measures for selection of real LBS edges vs. shuffled LBS assignments (M–W P=1.12×10−230 and P=3.04×10−228, respectively), though selection of real LBS edges shows significantly lower Spearman correlation P-values than shuffled edge assignments (M–W P=1.12×10−230).
Protein structure alteration prediction
The above analysis focused on proteins with known ligand binding mutations. However, as mentioned the LBS database does not identify the interacting partner(s) that may are disrupted by the mutation. To test if we indeed can determine significant pairwise events that affect cancer prognosis we next examined the agreement between our patient specific edge disruption scores, the patient mutation profile and changes in predicted binding affinity between pairs of proteins, using the ClusPro 2.0 [36] tool. ClusPro 2.0 simulates protein docking using sampling of billions of conformations, followed by clustering of the lowest energy structures (Additional file 1: Supporting Methods). We started with 143 interactions which could potentially be simulated based on the availability of structure data for both proteins (“Methods” section). However, only a few of these pairs were actually usable for this analysis. While 98 distinct proteins had at least one structure available in PDB [34], few of these proteins had a comprehensive structure available for the entire protein, without including other molecules in complex. Such structure is required for an accurate docking of a pair. We eventually were able to test 14 pairs.
We used our propagated mutational scores to rank the pairs of proteins for which we could conceivably perform binding predictions, and hypothesized that higher propagated mutation scores would correlate with higher disruption of protein binding. To illustrate this analysis consider that the lowest-scoring (indicating little impact) interaction was the pair (YWHAG,SKP1), with YWHAG harboring a ligand binding site mutation causing amino acid substitution S46C; and the highest-scoring pair, (PTGIS,PTGS2), with a ligand binding site mutation in PTGIS that causes amino acid substitution F293L.
Additional file 1: Figure S6 shows the protein product of the YWHAG gene, both wild-type (left) and after using PyMOL [35] to simulate the amino acid change S46C (right). Some small differences in structure are visible, especially in the bottom-left of each structure, but this amino acid substitution shows little effect on the overall structure of the protein. Conversely, Fig. 5a shows the protein produced from the PTGIS gene, with left and right showing (respectively) wild-type and the predicted structure after amino acid substitution F293L. As can be seen, in agreement with our assigned higher score, Fig. 5a shows a much more significant alteration of protein structure, consistent with our increased prediction of edge disruption.
Fig. 5.
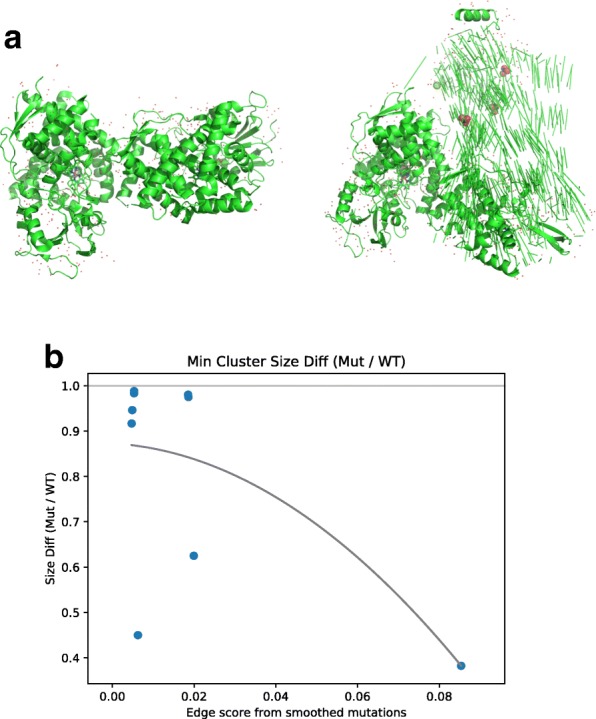
a Structure of prostaglandin I2 synthase, product of the PTGIS gene. Left: wild type, from PDB structure 2IAG, right: simulation of the impact of the high scoring edge mutation identified for this gene (amino acid substitution F293L). b Binding analysis of high and low scoring edges. For each edge we searched for protein structures for the two proteins connected by the edge in PDB. For pairs we found we simulated the impact of the mutation identified for that edge and used the ClusPro 2.0 docking tool to compare WT and mutated binding. Binding scores (y axis) represent ratio of maximum protein binding cluster with mutation vs. wild-type proteins. The lower the ratio the bigger the impact of the mutation. Curve is the best fit for a polynomial of degree 2. The curve indicates that as the edge score increases (x axis) the impact on binding increases as well
We used ClusPro 2.0 to predict binding affinity for all 14 usable pairs of proteins (Fig. 5b). We compute the binding affinity for each of the 14 pairs that we can test, by simulating docking for 1) the two wild-type protein structures, and 2) the simulated effect of the ligand binding site mutation in one protein with the wild-type structure of the other. For each pair of structures (wild-type and wild-type, or wild-type and simulated amino acid substitution), we run ClusPro twice, using each structure for both “receptor” and “ligand” in the ClusPro algorithm. For each {WT⇔WT,mut⇔WT} set of binding possibilities, we compute the ratio of the maximum binding cluster sizes between the mutated pair and the wild-type pair, and consider the minimum of the two ratios for the two assignments of receptor vs. ligand.
Results are shown in Fig. 5b where lower values indicate larger disruption in interaction. We see that the highest-scoring pair, (PTGIS,PTGS2), has the largest disruption in binding affinity, and that most low-scoring pairs have relatively small disruption in binding affinity. An order-2 polynomial fit for the points is shown in the figure.
Discussion
In this work, we introduce a method that allows for predicting the disruption of specific interactions in cancer patients using somatic mutation data and condition independent protein interaction networks as input. To do this, we extend traditional network smoothing techniques, which have been previously used to study cancer networks [12, 13, 32], and have also shown promise in the context of network dynamics [15]. Prior network smoothing techniques assigned scores to the nodes in a network based on the measured biological data.(for example mutation status or differential expression). We extended these techniques to assign scores to edges in addition to nodes.
We apply this method to somatic mutation data from the TCGA breast cancer [29] cohort, producing sample-specific scores for each protein-protein edge. We focus on breast cancer in this work due to the large number of samples, but note that our method is general and can be applied to any other cancer types as well. By using somatic mutation data as the prior knowledge vector in network smoothing methods (Supplementary Methods), we quantify the proximity of each protein-protein edge to somatic mutations in individual samples. We show that edges which score highly in at least 20% of samples show significantly higher association with patient survival when compared with random selections of lower-scoring edges. We evaluate the ability of our edge ranking to select interactions involving known ligand binding site mutations [33], and show that we consistently rank LBS mutation incident edges significantly higher than others when compared with random permutations of LBS mutations in each sample. Docking simulations based on the WT and mutants indicate that high scoring edges are indeed more likely to correspond to mutations that can significantly impact protein interactions.
The top 50 pairs ranked by their smoothed mutation scores is presented Table 1 and Additional file 1: Table S1. A number of the pairs and several proteins appear multiple times in different patients. We examined all 38 unique genes in the top 50 interacting pairs for known associations with cancer-related biological processes. As we show in Additional file 1: Table S2, 34 of these 38 genes are indeed known to be associated with at least one type of cancer, most of them with breast cancer and some others with ovarian, prostate or colon cancer. For example, STEAP1 is overexpressed in many cancers, including breast [49–51]. SCGB3A2 has been identified as a marker for pulmonary carcinoma in mice and humans [52], and MARCO has recently been identified as a possible candidate for targeted antibody therapy in non-small cell lung cancer [53].
Conclusions
While much of the analysis of coding region mutations focused on their impact on protein structure [17, 54–56], as we show many mutations are actually impacting interactions with key partners. Network smoothing performed across a cohort of patients can provide useful information about such alternation and a mechanistic explanation for the impact of these mutations on cell states. The fact that top scoring edges were significantly correlated with the ability to predict survival is a further indication for the impact that such changes in the interaction networks can cause. With better understanding of underlying causes that lead to cancer, our ability to address some of these issues with appropriate therapeutics would hopefully improve as well.
Additional file
Supplementary text. Results of alternative analyses, including using the STRING protein interaction network and using alternative edge weights. (PDF 670 kb)
Acknowledgments
We would like to acknowledge the TCGA consortium for access to breast cancer somatic mutation data, and clinical data for all cancer samples.
Funding
This work was supported in part by the National Science Foundation (grant number DBI-1356505 to ZBJ), by the U.S. National Institutes of Health (grants 1U54HL127624 to ZBJ and 1F32CA216937 to MMR) and by the Pennsylvania Department of Health (Health Research Nonformula Grant (CURE) Awards to ZBJ). No funding bodies played any role in the design of the study, the collection, analysis, and interpretation of data, or in writing the manuscript.
Availability of data and materials
Somatic mutation and survival data for TCGA BRCA samples are available at the NCI Genomic Data Commons (GDC) at https://gdc.cancer.gov/. Analysis scripts and generated data are available at https://www.cs.cmu.edu/~mruffalo/mut-edge-disrupt/.
Abbreviations
- AUC
Area under curve
- BRCA
Breast invasive carcinoma
- LBS
Ligand binding site
- M–W
Mann-Whitney (U test)
- nDCG
Normalized discounted cumulative gain
- PPI
Protein-protein interaction (network)
- RCSB
Research Collaboratory for Structural Bioinformatics
- ROC
Receiver operator characteristic
- TCGA
The cancer genome atlas WT: Wild-type
Authors’ contributions
MMR and ZBJ conceived the project, designed the experiments, interpreted the results, and prepared the manuscript. MMR implemented the computational/statistical methods. Both authors have read and approved the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Both authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Matthew Ruffalo, Email: mruffalo@cs.cmu.edu.
Ziv Bar-Joseph, Email: zivbj@cs.cmu.edu.
References
- 1.Karin M, Greten FR. NF- κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 2.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 3.Derynck R, Akhurst RJ, Balmain A. TGF- β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 4.Tirkkonen M, Johannsson O, Agnarsson BA, Olsson H, Ingvarsson S, Karhu R, et al. Distinct somatic genetic changes associated with tumor progression in carriers of BRCA1 and BRCA2 germ-line mutations. Cancer Res. 1997;57(7):1222–7. [PubMed] [Google Scholar]
- 5.Zheng L, Wang L, Ajani J, Xie K. Molecular basis of gastric cancer development and progression. Gastric Cancer. 2004;7(2):61–77. doi: 10.1007/s10120-004-0277-4. [DOI] [PubMed] [Google Scholar]
- 6.Iacobuzio-Donahue CA, Velculescu VE, Wolfgang CL, Hruban RH. AACR. Philadelphia: American Association for Cancer Research; 2012. Genetic basis of pancreas cancer development and progression: insights from whole-exome and whole-genome sequencing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T, et al. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78(4):703–11. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- 8.Vandin F, Raphael BJ, Upfal E. On the sample complexity of cancer pathways identification. J Comput Biol. 2016;23(1):30–41. doi: 10.1089/cmb.2015.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Kebir M, Oesper L, Acheson-Field H, Raphael BJ. Reconstruction of clonal trees and tumor composition from multi-sample sequencing data. Bioinformatics. 2015;31(12):i62–70. doi: 10.1093/bioinformatics/btv261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofree M, Shen JP, Carter H, Gross A, Ideker T. Network-based stratification of tumor mutations. Nat Methods. 2013;10(11):1108–15. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leiserson MD, Vandin F, Wu HT, Dobson JR, Raphael BR. Pan-cancer identification of mutated pathways and protein complexes. In: AACR: 2014.
- 12.Cowen L, Ideker T, Raphael BJ, Sharan R. Network propagation: a universal amplifier of genetic associations. Nat Rev Genet. 2017;18(9):551. doi: 10.1038/nrg.2017.38. [DOI] [PubMed] [Google Scholar]
- 13.Ruffalo M, Koyutürk M, Sharan R. Network-Based Integration of Disparate Omic Data To Identify “Silent Players” in Cancer. PLOS Comput Biol. 2015;11(12):e1004595. doi: 10.1371/journal.pcbi.1004595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Z, Zhang J, Yuan X, Liu Z, Liu B, Tuo S, et al. Network based stratification of major cancers by integrating somatic mutation and gene expression data. PloS ONE. 2017;12(5):e0177662. doi: 10.1371/journal.pone.0177662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patkar S, Magen A, Sharan R, Hannenhalli S. A network diffusion approach to inferring sample-specific function reveals functional changes associated with breast cancer. PLoS Comput Biol. 2017;13(11):e1005793. doi: 10.1371/journal.pcbi.1005793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas PJ, Qu BH, Pedersen PL. Defective protein folding as a basis of human disease. Trends Biochem Sci. 1995;20(11):456–9. doi: 10.1016/S0968-0004(00)89100-8. [DOI] [PubMed] [Google Scholar]
- 17.Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic acids research. 2005;33(suppl_2):W306–10. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Moult J. SNPs, protein structure, and disease. Human Mutat. 2001;17(4):263–70. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- 19.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 20.Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, et al. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16(3):307. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- 21.Pavletich NP, Chambers KA, Pabo CO. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993;7(12b):2556–64. doi: 10.1101/gad.7.12b.2556. [DOI] [PubMed] [Google Scholar]
- 22.Oitzl MS, Reichardt HM, Joëls M, de Kloet ER. Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc Natl Acad Sci. 2001;98(22):12790–5. doi: 10.1073/pnas.231313998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–59. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57(5):808–11. [PubMed] [Google Scholar]
- 25.Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, et al. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol. 2014;32(25):2718. doi: 10.1200/JCO.2014.55.5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarvagalla S, Cheung CHA, Tsai JY, Hsieh HP, Coumar MS. Disruption of protein–protein interactions: hot spot detection, structure-based virtual screening and in vitro testing for the anti-cancer drug target–survivin. Rsc Adv. 2016;6(38):31947–59. doi: 10.1039/C5RA22927H. [DOI] [Google Scholar]
- 27.Petta I, Lievens S, Libert C, Tavernier J, De Bosscher K. Modulation of protein–protein interactions for the development of novel therapeutics. Mol Ther. 2016;24(4):707–18. doi: 10.1038/mt.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Ivanov AA, Su R, Gonzalez-Pecchi V, Qi Q, Liu S, et al. The OncoPPi network of cancer-focused protein–protein interactions to inform biological insights and therapeutic strategies. Nat Commun. 2017;8:14356. doi: 10.1038/ncomms14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The Cancer Genome Atlas Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaefer MH, Fontaine JF, Vinayagam A, Porras P, Wanker EE, Andrade-Navarro MA. HIPPIE: Integrating Protein Interaction Networks with Experiment Based Quality Scores. PLoS ONE. 2012;7(2):e31826. doi: 10.1371/journal.pone.0031826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gitter A, Carmi M, Barkai N, Bar-Joseph Z. Linking the signaling cascades and dynamic regulatory networks controlling stress responses. Genome Res. 2013;23(2):365–76. doi: 10.1101/gr.138628.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol. 2010;6(1):e100641. doi: 10.1371/journal.pcbi.1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim P, Zhao J, Lu P, Zhao Z. mutLBSgeneDB: mutated ligand binding site gene DataBase. Nucleic Acids Res. 2016;45(D1):D256–63. doi: 10.1093/nar/gkw905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.8. 2015. https://pymol.org/2/. Accessed 21 Feb 2018.
- 36.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. The ClusPro web server for protein–protein docking. Nat Protocol. 2017;12(2):255. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365(9460):671–9. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 38.Lunn Mary, McNeil Don. Applying Cox Regression to Competing Risks. Biometrics. 1995;51(2):524. doi: 10.2307/2532940. [DOI] [PubMed] [Google Scholar]
- 39.Järvelin K, Kekäläinen J. Cumulated gain-based evaluation of IR techniques. ACM Trans Inf Syst (TOIS) 2002;20(4):422–46. doi: 10.1145/582415.582418. [DOI] [Google Scholar]
- 40.Burges C, Shaked T, Renshaw E, Lazier A, Deeds M, Hamilton N, et al. Proceedings of the 22nd international conference on Machine learning. International Conference on Machine Learning. La Jolla: ACM; 2005. Learning to rank using gradient descent. [Google Scholar]
- 41.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clinic Investig. 2014;124(1):30–9. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chakrabarti A, Oehme I, Witt O, Oliveira G, Sippl W, Romier C, et al. HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol Sci. 2015;36(7):481–92. doi: 10.1016/j.tips.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 43.Petitjean A, Achatz M, Borresen-Dale A, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26(15):2157. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 44.Olivier M, Langer A, Carrieri P, Bergh J, Klaar S, Eyfjord J, et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clinic Cancer Res. 2006;12(4):1157–67. doi: 10.1158/1078-0432.CCR-05-1029. [DOI] [PubMed] [Google Scholar]
- 45.Bougeard G, Limacher JM, Martin C, Charbonnier F, Killian A, Delattre O, et al. Detection of 11 germline inactivating TP53 mutations and absence of TP63 and HCHK2 mutations in 17 French families with Li-Fraumeni or Li-Fraumeni-like syndrome. J Med Genet. 2001;38(4):253–7. doi: 10.1136/jmg.38.4.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papageorgis P, Ozturk S, Lambert AW, Neophytou CM, Tzatsos A, Wong CK, et al. Targeting IL13Ralpha2 activates STAT6-TP63 pathway to suppress breast cancer lung metastasis. Breast Cancer Res. 2015;17(1):98. doi: 10.1186/s13058-015-0607-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iscan M, Klaavuniemi T, Ċoban T, Kapucuoġlu N, Pelkonen O, Raunio H. The expression of cytochrome P450 enzymes in human breast tumours and normal breast tissue. Breast Cancer Res Treat. 2001;70(1):47–54. doi: 10.1023/A:1012526406741. [DOI] [PubMed] [Google Scholar]
- 48.Nakajima M, Itoh M, Sakai H, Fukami T, Katoh M, Yamazaki H, et al. CYP2A13 expressed in human bladder metabolically activates 4-aminobiphenyl. Int J Cancer. 2006;119(11):2520–6. doi: 10.1002/ijc.22136. [DOI] [PubMed] [Google Scholar]
- 49.Maia CJ, Socorro S, Schmitt F, Santos CR. STEAP1 is over-expressed in breast cancer and down-regulated by 17 β-estradiol in MCF-7 cells and in the rat mammary gland. Endocrine. 2008;34(1-3):108–16. doi: 10.1007/s12020-008-9113-7. [DOI] [PubMed] [Google Scholar]
- 50.Moreaux J, Kassambara A, Hose D, Klein B. STEAP1 is overexpressed in cancers: a promising therapeutic target. Biochem Biophys Res Commun. 2012;429(3):148–55. doi: 10.1016/j.bbrc.2012.10.123. [DOI] [PubMed] [Google Scholar]
- 51.Gomes IM, Arinto P, Lopes C, Santos CR, Maia CJ. Urologic Oncology: Seminars and Original Investigations. Amsterdam: Elsevier; 2014. STEAP1 is overexpressed in prostate cancer and prostatic intraepithelial neoplasia lesions, and it is positively associated with Gleason score. [DOI] [PubMed] [Google Scholar]
- 52.Kurotani R, Kumaki N, Naizhen X, Ward JM, Linnoila RI, Kimura S. Secretoglobin 3A2/uteroglobin-related protein 1 is a novel marker for pulmonary carcinoma in mice and humans. Lung Cancer. 2011;71(1):42–8. doi: 10.1016/j.lungcan.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.La Fleur L, Boura VF, Alexeyenko A, Berglund A, Pontén V, Mattsson JS, et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int J Cancer. 2018;143(7):1741–52. doi: 10.1002/ijc.31545. [DOI] [PubMed] [Google Scholar]
- 54.Espinosa O, Mitsopoulos K, Hakas J, Pearl F, Zvelebil M. Deriving a mutation index of carcinogenicity using protein structure and protein interfaces. PloS one. 2014;9(1):e84598. doi: 10.1371/journal.pone.0084598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gauthier NP, Reznik E, Gao J, Sumer SO, Schultz N, Sander C, et al. MutationAligner: a resource of recurrent mutation hotspots in protein domains in cancer. Nucleic Acids Res. 2015;44(D1):D986–91. doi: 10.1093/nar/gkv1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siderius M, Jagodzinski F. Mutation Sensitivity Maps: Identifying Residue Substitutions That Impact Protein Structure Via a Rigidity Analysis In Silico Mutation Approach. J Comput Biol. 2018;25(1):89–102. doi: 10.1089/cmb.2017.0165. [DOI] [PubMed] [Google Scholar]
- 57.Gomes IM, Santos CR, Maia CJ. Expression of STEAP1 and STEAP1B in prostate cell lines, and the putative regulation of STEAP1 by post-transcriptional and post-translational mechanisms. Genes Cancer. 2014;5(3-4):142. doi: 10.18632/genesandcancer.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Havrysh K, Kiyamova R. 14New potential biomarkers for breast cancer prognosis. Ann Oncol. 2017;28(suppl7):mdx508.011. [Google Scholar]
- 59.Kazerounian S, Pitari GM, Shah FJ, Frick GS, Madesh M, Ruiz-Stewart I, et al. Proliferative signaling by store-operated calcium channels opposes colon cancer cell cytostasis induced by bacterial enterotoxins. J Pharmacol Exp Ther. 2005;314(3):1013–22. doi: 10.1124/jpet.105.089052. [DOI] [PubMed] [Google Scholar]
- 60.Luchino J, Hocine M, Amoureux MC, Gibert B, Bernet A, Royet A, et al. Semaphorin 3E suppresses tumor cell death triggered by the plexin D1 dependence receptor in metastatic breast cancers. Cancer Cell. 2013;24(5):673–85. doi: 10.1016/j.ccr.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 61.Kim SJ, Kim JS, Park ES, Lee JS, Lin Q, Langley RR, et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia. 2011;13(3):286–98. doi: 10.1593/neo.11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liang D, Meyer L, Chang DW, Lin J, Pu X, Ye Y, et al. Genetic variants in MicroRNA biosynthesis pathways and binding sites modify ovarian cancer risk, survival, and treatment response. Cancer Res. 2010;70(23):9765–76. doi: 10.1158/0008-5472.CAN-10-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wegman P, Elingarami S, Carstensen J, Stål O, Nordenskjöld B, Wingren S. Genetic variants of CYP3A5, CYP2D6, SULT1A1, UGT2B15 and tamoxifen response in postmenopausal patients with breast cancer. Breast Cancer Res. 2007;9(1):R7. doi: 10.1186/bcr1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shulby SA, Dolloff NG, Stearns ME, Meucci O, Fatatis A. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 2004;64(14):4693–8. doi: 10.1158/0008-5472.CAN-03-3437. [DOI] [PubMed] [Google Scholar]
- 65.Andre F, Cabioglu N, Assi H, Sabourin J, Delaloge S, Sahin A, et al. Expression of chemokine receptors predicts the site of metastatic relapse in patients with axillary node positive primary breast cancer. Ann Oncol. 2006;17(6):945–51. doi: 10.1093/annonc/mdl053. [DOI] [PubMed] [Google Scholar]
- 66.Tardáguila M, Mira E, García-Cabezas MA, Feijoo AM, Quintela-Fandino M, Azcoitia I, et al. CX3CL1 promotes breast cancer via transactivation of the EGF pathway. Cancer Res. 2013;73(14):4461–73. doi: 10.1158/0008-5472.CAN-12-3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cui J, Li F, Wang G, Fang X, Puett JD, Xu Y. Gene-expression signatures can distinguish gastric cancer grades and stages. PloS one. 2011;6(3):e17819. doi: 10.1371/journal.pone.0017819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao C, Cheng X, Li X, Tong B, Wu K, Liu Y. Prognostic significance of artemin and GFR α1 expression in laryngeal squamous cell carcinoma. Exp Ther Med. 2014;8(3):818–22. doi: 10.3892/etm.2014.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ito Y, Okada Y, Sato M, Sawai H, Funahashi H, Murase T, et al. Expression of glial cell line–derived neurotrophic factor family members and their receptors in pancreatic cancers. Surgery. 2005;138(4):788–94. doi: 10.1016/j.surg.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 70.Pandey V, Qian PX, Kang J, Perry JK, Mitchell MD, Yin Z, et al. Artemin stimulates oncogenicity and invasiveness of human endometrial carcinoma cells. Endocrinology. 2010;151(3):909–20. doi: 10.1210/en.2009-0979. [DOI] [PubMed] [Google Scholar]
- 71.Ngollo M, Lebert A, Daures M, Judes G, Rifai K, Dubois L, et al. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC cancer. 2017;17(1):261. doi: 10.1186/s12885-017-3256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saito T, Kasamatsu A, Ogawara K, Miyamoto I, Saito K, Iyoda M, et al. Semaphorin7A promotion of tumoral growth and metastasis in human oral cancer by regulation of G1 cell cycle and matrix metalloproteases: Possible contribution to tumoral angiogenesis. PloS one. 2015;10(9):e0137923. doi: 10.1371/journal.pone.0137923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Udabage L, Brownlee GR, Nilsson SK, Brown TJ. The over-expression of HAS2, Hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Exp Cell Res. 2005;310(1):205–17. doi: 10.1016/j.yexcr.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 74.Liu N, Gao F, Han Z, Xu X, Underhill CB, Zhang L. Hyaluronan synthase 3 overexpression promotes the growth of TSU prostate cancer cells. Cancer Res. 2001;61(13):5207–14. [PubMed] [Google Scholar]
- 75.Alves IT, Hartjes T, McClellan E, Hiltemann S, Böttcher R, Dits N, et al. Next-generation sequencing reveals novel rare fusion events with functional implication in prostate cancer. Oncogene. 2015;34(5):568. doi: 10.1038/onc.2013.591. [DOI] [PubMed] [Google Scholar]
- 76.Guo JC, Li CQ, Wang QY, Zhao JM, Ding JY, Li EM, et al. Protein-coding genes combined with long non-coding RNAs predict prognosis in esophageal squamous cell carcinoma patients as a novel clinical multi-dimensional signature. Mol BioSyst. 2016;12(11):3467–77. doi: 10.1039/C6MB00585C. [DOI] [PubMed] [Google Scholar]
- 77.Liu JC, Voisin V, Bader GD, Deng T, Pusztai L, Symmans WF, et al. Seventeen-gene signature from enriched Her2/Neu mammary tumor-initiating cells predicts clinical outcome for human HER2+:ER α- breast cancer. Proc Natl Acad Sci. 2012;109(15):5832–7. doi: 10.1073/pnas.1201105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrózek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clinic Oncol. 2010;28(14):2348. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jutras S, Bachvarova M, Keita M, Bascands JL, Mes-Masson AM, Stewart JM, et al. Strong cytotoxic effect of the bradykinin antagonist BKM-570 in ovarian cancer cells–analysis of the molecular mechanisms of its antiproliferative action. FEBS J. 2010;277(24):5146–60. doi: 10.1111/j.1742-4658.2010.07928.x. [DOI] [PubMed] [Google Scholar]
- 80.L’Espérance S, Bachvarova M, Tetu B, Mes-Masson AM, Bachvarov D. Global gene expression analysis of early response to chemotherapy treatment in ovarian cancer spheroids. BMC Genomics. 2008;9(1):99. doi: 10.1186/1471-2164-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baba T, Sakamoto Y, Kasamatsu A, Minakawa Y, Yokota S, Higo M, et al. Persephin: A potential key component in human oral cancer progression through the RET receptor tyrosine kinase-mitogen-activated protein kinase signaling pathway. Mol Carcinog. 2015;54(8):608–17. doi: 10.1002/mc.22127. [DOI] [PubMed] [Google Scholar]
- 82.Lee K, Byun K, Hong W, Chuang HY, Pack CG, Bayarsaikhan E, et al. Proteome-wide discovery of mislocated proteins in cancer. Genome Res. 2013;23(8):1283–94. doi: 10.1101/gr.155499.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thompson MP, Cooper ST, Parry BR, Tuckey JA. Increased expression of the mRNA for hormone-sensitive lipase in adipose tissue of cancer patients. Biochim Biophys Acta (BBA)-Mol Basis Dis. 1993;1180(3):236–42. doi: 10.1016/0925-4439(93)90044-2. [DOI] [PubMed] [Google Scholar]
- 84.Nath A, Chan C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci Rep. 2016;6:18669. doi: 10.1038/srep18669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22(2):271–82. doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin PC, Giannopoulou EG, Park K, Mosquera JM, Sboner A, Tewari AK, et al. Epigenomic alterations in localized and advanced prostate cancer. Neoplasia. 2013;15(4):IN2–5. doi: 10.1593/neo.122146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary text. Results of alternative analyses, including using the STRING protein interaction network and using alternative edge weights. (PDF 670 kb)
Data Availability Statement
Somatic mutation and survival data for TCGA BRCA samples are available at the NCI Genomic Data Commons (GDC) at https://gdc.cancer.gov/. Analysis scripts and generated data are available at https://www.cs.cmu.edu/~mruffalo/mut-edge-disrupt/.