

Abstract
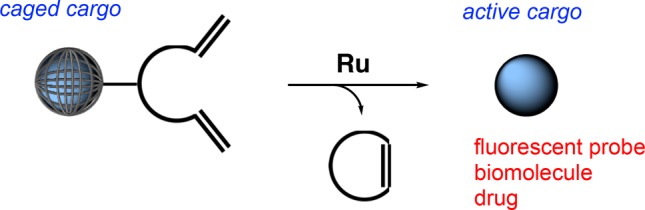
Bioorthogonal uncaging reactions offer versatile tools in chemical biology. In recent years, reactions have been developed to proceed efficiently under physiological conditions. We present herein an uncaging reaction that results from ring-closing metathesis (RCM). A caged molecule, tethered to a diolefinic substrate, is released via spontaneous 1,4-elimination following RCM. Using this strategy, which we term “close-to-release”, we show that drugs and fluorescent probes are uncaged with fast rates, including in the presence of mammalian cells or in the periplasm of Escherichia coli. We envision that this tool may find applications in chemical biology, bioengineering and medicine.
Bioorthogonal chemistry provides tools to scrutinize and modulate biological processes. Bertozzi pioneered the field by capitalizing on the Staudinger ligation,1 which paved the way to numerous bioorthogonal ligations.2 The possibility of triggering organic reactions in vivo provides tools for targeted therapy.3 Such reactions occur with high selectivity and turnover numbers (TONs) and proceed with fast kinetics under high dilution. There are several examples of dissociative reactions, mainly based on transition-metal catalysis and/or pericyclic reactions.3 The inverse-electron demand Diels–Alder reaction between tetrazine and trans-cyclooctene (TCO) is one of the fastest uncaging reactions.4 Versteegen et al. reported the cycloaddition between tetrazine and TCO bearing a carbamate to form 4,5-dihydropiridazine, which readily tautomerizes in aqueous media, Scheme 1a.5 This strategy, coined “click-to-release”, leads to the uncaging of the amine via the formation of the 1,4-tautomer. Antibody–drug conjugates are developed relying on the click-to-release approach to activate prodrugs in the extracellular space of tumors.6 In the context of metal-catalyzed biocompatible reactions, precious metals including ruthenium, gold and palladium occupy a place of choice. Indeed, these have been shown to catalyze the deallylation and depropargylation of caged substrates in cellulo and in vivo, Scheme 1b.7
Scheme 1. Selected Biocompatible Uncaging Reactions.
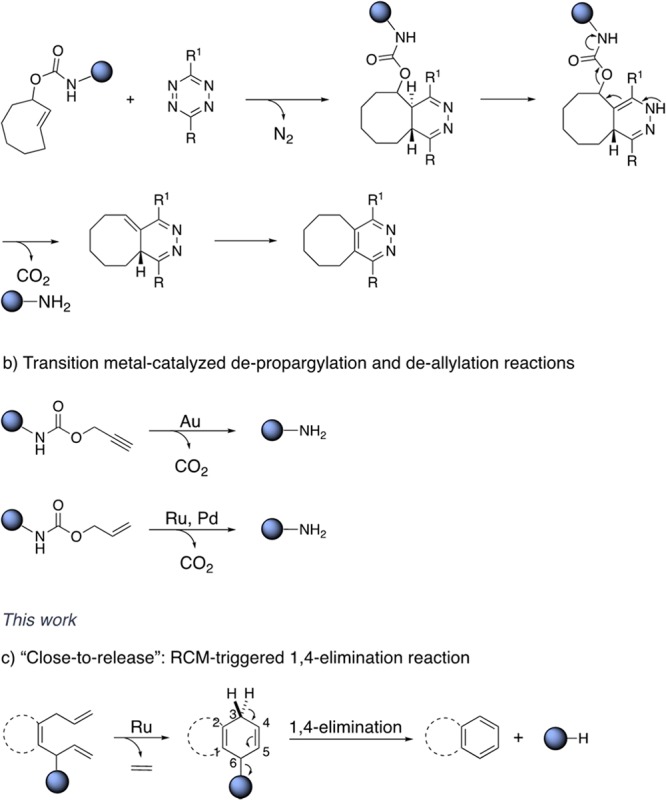
Recently, olefin metathesis has gained attention as a bioorthogonal tool in chemical biology.8 As Ru-based catalysts tolerate water and oxygen, a number of biocompatible protocols have been reported.9 Davis used olefin metathesis for bioorthogonal ligation via cross-metathesis. Schultz performed cross-metathesis of olefin-bearing proteins using the Hoveyda–Grubbs catalyst (HG-II).10 Our group reported the directed evolution of an artificial metathase in the periplasm of Escherichia coli.11 To expand on this work, we set out to develop a strategy that capitalizes on RCM, leading to the uncaging of a substrate. We contemplated the possibility that RCM may trigger the release of a cargo via an elimination reaction, driven by the generation of an aromatic moiety.
We were encouraged by reports of RCM leading to a transient 2,5-cyclohexadien-1-ol moiety, followed by spontaneous 1,4-elimination of water.12 We hypothesized that a leaving group may be subject to such a 1,4-elimination, Scheme 1c. This hypothesis was confirmed by subjecting the naphthalene precursor 1 to RCM in water. It led to good yields in the presence of various Ru-based metathesis catalysts. The HG-II performed best (see SI, Table S2), and was selected for all subsequent studies. The reactions were carried out in water or in buffered solutions at pH 6–7, with 10% acetone. Having identified that the 1,4-elimination of water proceeds under “near physiological” conditions (Table 1), we sought to equip the diolefinic substrate with a caged-cargo that could be released upon 1,4-elimination.
Table 1. Selected Results for the Aqueous RCM of Naphthalene Precursor 1.


[1] = 1.0 mM.
PBS buffer contains 50 mM MgCl2.
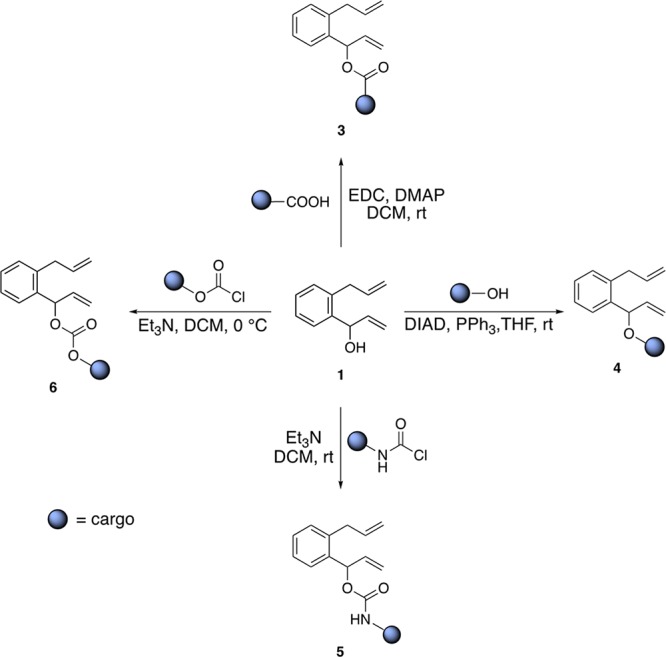
For this purpose, we substituted the hydroxy functionality by leaving groups amenable to release a cargo. Starting from the naphthalene precursor 1-(2-allylphenyl)prop-2-en-1-ol 1, four functional groups were evaluated, Scheme 2.
Scheme 2. Functionalization of the Naphthalene Precursor 1 for the Spontaneous Release of Cargoes.
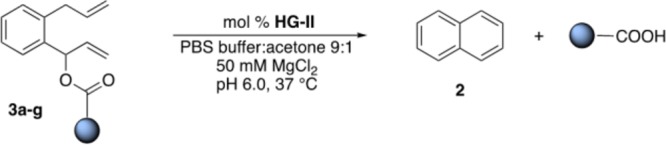
The results of the close-to-release uncaging of seven diolefinic substrates 3a–g bearing an ester moiety are collected in Table 2. In order to ensure that the release of the cargo occurs only as a result of the ring-closing event, the formation of both naphthalene and the cargo were monitored by NMR and gas chromatography mass spectrometry (GC–MS) (see SI, Figures S3 and S5).
Table 2. RCM-Triggered 1,4-Elimination of Carboxylic Acids.

Substrate concentration: 1.0 mM.
Substrate concentration: 2.5 mM in PBS buffer:acetone 3:1.
Reaction carried out at pH 7.4.
[3f] = 20 μM, 1% DMSO, pH 7.4.
These data highlight that both products are formed concurrently and in equimolar amounts, suggesting that the release of the acid-bearing cargo does not occur prior to the RCM event. We additionally monitored the stability of the ester substrates in the reaction media at 37 °C: no spontaneous hydrolysis was detected by GC–MS or NMR within 24 h (see SI, Figures S1 and S2). With the long-term goal of complementing the metabolism,13 we contemplated the possibility of uncaging nicotinic acid, a precursor of NAD(P)H, starting from substrate 3a. Gratifyingly, the RCM of substrate 3a led to the release of nicotinic acid with 34% yield (Table 2, entry 1). As the uncaging reaction required 5% or higher catalyst loading, we set out to improve the nucleofugacity by introducing electron-withdrawing groups on the aromatic cargo. Accordingly, halogen-substituted substrates 3b and 3c afforded 214 and 261 TONs at 0.2% catalyst loading, respectively (Table 2, entries 3 and 4). Introduction of p-trifluoroacetyl group 3d afforded 260 TONs using a 0.1% catalyst loading (Table 2, entry 5). With medical applications in mind, we evaluated the possibility of uncaging drugs as a result of RCM. The substrate 3e led to the release of valproic acid, an antineuroleptic drug, which gave the highest yield (64%) at pH 6 and 37% yield at physiological pH (Table 2, entries 6 and 7). The improved close-to-release activity observed at lower pH may be an asset for chemotherapy, leading to higher concentrations of drugs in acidic environments, typically present in hypoxic cancer cells.14 RCM of substrate 3f triggers the uncaging of Alofanib, a tyrosine-kinase inhibitor. The drug release was achieved with yields up to 43% albeit at high catalyst loading (Table 2, entry 9). Uncaging of myristic acid from 3g gave 235 TONs at 0.2% catalyst loading (Table 2, entry 10). The RCM of esters is characterized by fast rates: 80% of the observed 3-bromo-4-nitrobenzoic acid from substrate 3c is produced within 10 min (see SI, Figure S7).
Next, we tested the RCM of ether substrates 4a and 4b. Despite the fact that ether substrates might benefit from the allylic chalcogen effect,15 they proved more challenging than esters, which we trace-back to the poor nucleofugacity of alcoholates, Table 3.16 Accordingly, the uncaging efficiency of pentafluorophenol (pKa = 5.5) from derivative 4b was highest (Table 3, entries 3 and 4). Subjecting the pro-fluorogenic substrate 4a to RCM, led to the immediate appearance of fluorescence, resulting from the uncaging of umbelliferone (64 TONs, 32% yield at 0.5 mol % HG-II, Table 3, entry 2).
Table 3. RCM-Triggered 1,4-Elimination of Alcohols from Ethers 4a,b.


[4a] = 0.5 mM, 5% DMSO.
[4b] = 1.0 mM, 10% acetone.
Carbamates are frequently used as linkers to liberate amines.16Table 4 summarizes the results with carbamate substrates 5a–d. Decreasing the catalyst loading led to a marked increase in TONs. The RCM of substrate 5a, 5b and 5d gave respectively 49, 84 and 40 TONs at 0.2% (Table 4, entries 1, 3 and 6). Decreasing the catalyst loading to 0.05 mol %, (i.e., 0.5 μM HG-II) led to 97 TONs for the production of 4-chloro-3-(trifluoromethyl)aniline from substrate 5d (Table 4, entry 7). The fluorescent 4-methyl-7-aminocoumarin was uncaged from the pro-fluorogenic probe 5c at pH 7.4 in up to 40% yield (Table 4, entries 4 and 5). We also showed that carbonates, despite their hydrolytic propensity, lead to the release of alcohols. In PBS buffer at pH 6, GC–MS and NMR monitoring of the reaction using the activated p-nitrophenyl carbonate 6 (see SI, Figure S27), confirms that the release of the p-nitrophenol only occurs as a result of the RCM event: 44 TONs at 0.2% catalyst loading (Table 4, entries 8 and 9).
Table 4. RCM-Triggered 1,4-Elimination of Amines or Alcohols from Substrates 5 and 6.
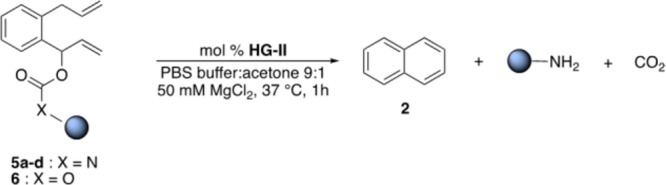

Substrate concentration: 1 mM.
[5b] = 2.5 mM, PBS buffer:acetone 3:1.
Reaction carried out for 16 h in PBS buffer containing 1% DMSO.
Despite the tolerance of Ru-based metathesis catalysts toward a variety of functional groups,17 these are irreversibly poisoned in the presence of a few equivalents of glutathione,11 nature’s ubiquitous redox buffer, present in mM concentrations in the cytoplasm of aerobic cells. To minimize the exposure of the soft-metal catalyst to thiols, we evaluated the viability of the close-to-release strategy under the following conditions: (i) in culture media, (ii) in the presence of E. coli cells and cell lysates, (iii) in the presence of HeLa cells and (iv) in the periplasm of E. coli. The results are collected in Table 5.
Table 5. Close-to-Release in Biological Media.

[3d] = 500 μM.
[4a] = 100 μM. All reactions included 1% or less DMSO, pH = 6–7.4.
Lyophilized E. coli cell lysate solution in PBS buffer.
Estimated uptake of biotinylated HG-II catalyst from 10 μM incubation.11
Substrates 3d and 4a were tested in biological media such as E. coli cells (or cell lysates) and Dulbecco Modified Eagle’s Medium (DMEM, solution with high concentrations of glucose, amino acids and vitamins). RCM of ester 3d afforded 69 TONs in DMEM at low catalyst loading (0.2%, corresponding to 1 μM, Table 5 entry 1). The pro-fluorogenic ether 4a afforded 32 TONs in both DMEM and PBS in the presence of E. coli cells, and 26 TONs in cell lysate at 1% catalyst loading, respectively (Table 5 entries 3 and 5). Next, we evaluated the uncaging of substrate 4a in media containing HeLa cells. Gratifyingly, we observed up to 50% uncaging of umbelliferone and up to 20 TONs within 90 min, (Table 5 entries 6 and 7, see SI Figures S22 and S23). The reaction in the presence of serum retained up to 64% of the activity compared to the reaction in DMEM, (Table 5, entry 8). The effect of substrate, products and catalyst on HeLa cell viability was evaluated using the MTT assay. After 24 h of incubation, the cells present excellent viability at the concentrations of catalyst and substrate (and the products umbelliferone and naphtalene) used in the activity assays (see SI, Figures S24 and S25). In the event that the generated naphthalene proved toxic, it may be replaced or decorated to minimize its harmfulness.
Finally, we tested the close-to-release uncaging strategy within the periplasm of E. coli. This cellular compartment contains significantly lower concentrations of glutathione which is mostly present in its disulfide form.18 As previously developed in our lab, we assembled in the periplasm of E. coli an artificial metathase based on the biotin–streptavidin technology (see SI, Figure S17).11,19 Upon addition of a biotinylated cofactor to E. coli cells harboring streptavidin in their periplasm, incubation with pro-fluorescent substrate 4a led to a marked increase in fluorescence, diagnostic of the release of umbelliferone (Table 5, entry 9, see SI, Figure S19).
This study demonstrates that ring-closing metathesis offers a versatile means to uncage carboxylic acids, alcohols and amines. We postulate that this represents an attractive addition to metal-catalyzed bioorthogonal reactions. The close-to-release strategy leads to the uncaging of metabolites, drugs and fluorescent probes under physiological conditions. Some of the most notable features include (i) low catalyst loading and concentrations, (ii) no inert atmosphere required, (iii) multiple turnovers in the periplasm of bacterial cells, in the presence of mammalian cells and in serum. This proof-of-concept study paves the way toward various applications such as metabolic engineering, in vivo imaging and prodrug activation, etc.
Acknowledgments
T.R.W. thanks the University of Basel, the NCCR Molecular Systems engineering, the SNF (grant 200020_182046) and the ERC (the DrEAM, advanced grant 694424). J.G.R. thanks the European Molecual Biology Organisation for a long-term fellowship (ALTF-194-2017). We thank Prof. Dr. Yaakov Benenson (d-bsse, ETHZ) for access to the mammalian cell culture facility
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b07193.
General information, experimental section, Figures S1–S30 and Tables S1–S19 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hang H. C.; Yu C.; Kato D. L.; Bertozzi C. R. A Metabolic Labeling Approach Toward Proteomic Analysis of Mucin-Type O-Linked Glycosylation. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 14846–14851. 10.1073/pnas.2335201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sletten E. M.; Bertozzi C. R. From Mechanism to Mouse: A Tale of Two Bioorthogonal Reactions. Acc. Chem. Res. 2011, 44, 666–676. 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lang K.; Chin J. W. Bioorthogonal Reactions for Labeling Proteins. ACS Chem. Biol. 2014, 9, 16–20. 10.1021/cb4009292. [DOI] [PubMed] [Google Scholar]; c Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]; d Das J. Aliphatic Diazirines as Photoaffinity Probes for Proteins: Recent Developments. Chem. Rev. 2011, 111, 4405–4417. 10.1021/cr1002722. [DOI] [PubMed] [Google Scholar]
- a Tu J.; Xu M.; Franzini R. M. Dissociative Bioorthogonal Reactions. ChemBioChem 2019, 20, 1615–1627. 10.1002/cbic.201800810. [DOI] [PubMed] [Google Scholar]; b Mejia Oneto J. M.; Khan I.; Seebald L. M.; Royzen M. In Vivo Bioorthogonal Chemistry Enables Local Hydrogel and Systemic Pro-Drug to Treat Soft Tissue Sarcoma. ACS Cent. Sci. 2016, 2, 476–482. 10.1021/acscentsci.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rossin R.; Renart Verkerk P.; van den Bosch S. M.; Vulders R. C. M.; Verel I.; Lub J.; Robillard M. S. In Vivo Chemistry for Pretargeted Tumor Imaging in Live Mice. Angew. Chem., Int. Ed. 2010, 49, 3375–3378. 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]; d Ji X.; Pan Z.; Yu B.; De La Cruz L. K.; Zheng Y.; Ke B.; Wang B. Click and Release: Bioorthogonal Approaches to “On-Demand” Activation of Prodrugs. Chem. Soc. Rev. 2019, 48, 1077–1094. 10.1039/C8CS00395E. [DOI] [PubMed] [Google Scholar]; e Li J.; Chen P. Development and Application of Bond Cleavage Reactions in Bioorthogonal Chemistry. Nat. Chem. Biol. 2016, 12, 129–137. 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]; f Neumann K.; Gambardella A.; Lilienkampf A.; Bradley M. Tetrazine-Mediated Bioorthogonal Prodrug-Prodrug Activation. Chem. Sci. 2018, 9, 7198–7203. 10.1039/C8SC02610F. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Devaraj N. K. The Future of Bioorthogonal Chemistry. ACS Cent. Sci. 2018, 4, 952–959. 10.1021/acscentsci.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Azoulay M.; Tuffin G.; Sallem W.; Florent J. C. A New Drug-Release Method Using the Staudinger Ligation. Bioorg. Med. Chem. Lett. 2006, 16, 3147–3149. 10.1016/j.bmcl.2006.03.073. [DOI] [PubMed] [Google Scholar]
- Fan X.; Ge Y.; Lin F.; Yang Y.; Zhang G.; Ngai W. S. C.; Lin Z.; Zheng S.; Wang J.; Zhao J.; Li J.; Chen P. R. Optimized Tetrazine Derivatives for Rapid Bioorthogonal Decaging in Living Cells. Angew. Chem., Int. Ed. 2016, 55, 14046–14050. 10.1002/anie.201608009. [DOI] [PubMed] [Google Scholar]
- Versteegen R. M.; Rossin R.; ten Hoeve W.; Janssen H. M.; Robillard M. S. Click to Release: Instantaneous Doxorubicin Elimination Upon Tetrazine Ligation. Angew. Chem., Int. Ed. 2013, 52, 14112–14116. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Peplow M. Click Chemistry Targets Antibody-Drug Conjugates for the Clinic. Nat. Biotechnol. 2019, 37, 835–837. 10.1038/d41587-019-00017-4. [DOI] [PubMed] [Google Scholar]
- a Sasmal P. K.; Streu C. N.; Meggers E. Metal Complex Catalysis in Living Biological Systems. Chem. Commun. 2013, 49, 1581–1587. 10.1039/C2CC37832A. [DOI] [PubMed] [Google Scholar]; b Völker T.; Dempwolff F.; Graumann P. L.; Meggers E. Progress Towards Bioorthogonal Catalysis with Organometallic Compounds. Angew. Chem., Int. Ed. 2014, 53, 10536–10540. 10.1002/anie.201404547. [DOI] [PubMed] [Google Scholar]; c Streu C.; Meggers E. Ruthenium-Induced Allylcarbamate Cleavage in Living Cells. Angew. Chem., Int. Ed. 2006, 45, 5645–5648. 10.1002/anie.200601752. [DOI] [PubMed] [Google Scholar]; d Yusop R. M.; Unciti-Broceta A.; Johansson E. M. V.; Sánchez-Martín R. M.; Bradley Palladium-Mediated Intracellular Chemistry. Nat. Chem. 2011, 3, 239–243. 10.1038/nchem.981. [DOI] [PubMed] [Google Scholar]; e Bray T. L.; Salji M.; Brombin A.; Pérez-López A. M.; Rubio-Ruiz B.; Galbraith L. C. A.; Patton E. E.; Leung H. Y.; Unciti-Broceta A. Bright Insights into Palladium-Triggered Local Chemotherapy. Chem. Sci. 2018, 9, 7354–7361. 10.1039/C8SC02291G. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Indrigo E.; Clavadetscher J.; Chankeshwara S. V.; Lilienkampf A.; Bradley M. Palladium-Mediated In Situ Synthesis of an Anticancer Agent. Chem. Commun. 2016, 52, 14212–14214. 10.1039/C6CC08666G. [DOI] [PubMed] [Google Scholar]; g Pérez-López A. M.; Rubio-Ruiz B.; Sebastián V.; Hamilton L.; Adam C.; Bray T. L.; Irusta S.; Brennan P. M.; Lloyd-Jones G. C.; Sieger D.; Santamaría J.; Unciti-Broceta A. Gold-Triggered Uncaging Chemistry in Living Systems. Angew. Chem., Int. Ed. 2017, 56, 12548–12552. 10.1002/anie.201705609. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Vidal C.; Tomás-Gamasa M.; Destito P.; López F.; Mascareñas J. L. Concurrent and Orthogonal Gold (I) and Ruthenium (II) Catalysis Inside Living Cells. Nat. Commun. 2018, 9, 1913–1921. 10.1038/s41467-018-04314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Rebelein J. G.; Ward T. R. In Vivo Catalyzed New-To-Nature Reactions. Curr. Opin. Biotechnol. 2018, 53, 106–114. 10.1016/j.copbio.2017.12.008. [DOI] [PubMed] [Google Scholar]
- a Binder J. B.; Raines R. T. Olefin Metathesis for Chemical Biology. Curr. Opin. Chem. Biol. 2008, 12, 767–773. 10.1016/j.cbpa.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lin Y. A.; Davis B. G.. Vignette: Extending the Application of Metathesis in Chemical Biology–The development of Site-Selective Peptide and Protein Modifications. Handbook of Metathesis Applications in Organic Synthesis, 2nd ed.; Grubbs R. H., O’Leary D. J., Eds.; Wiley-VCH: Weinheim, Germany, 2015; Vol. 2, Chapter (3), ; pp 295–309. [Google Scholar]; c Sabatino V.; Ward T. R. Aqueous Olefin Metathesis: Recent Developments and Applications. Beilstein J. Org. Chem. 2019, 15, 445–468. 10.3762/bjoc.15.39. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Isenegger P. G.; Davis B. G. Concepts of Catalysis in Site-Selective Protein Modifications. J. Am. Chem. Soc. 2019, 141, 8005–8013. 10.1021/jacs.8b13187. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Moellering R. E.; Cornejo M.; Davis T. N.; Del Bianco C.; Aster J. C.; Blacklow S. C.; Kung A. L.; Gilliland D. G.; Verdine G. L.; Bradner J. E. Direct Inhibition of the NOTCH Transcription Factor Complex. Nature 2009, 462, 182–188. 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Burtscher D.; Grela K. Aqueous Olefin Metathesis. Angew. Chem., Int. Ed. 2009, 48, 442–454. 10.1002/anie.200801451. [DOI] [PubMed] [Google Scholar]; b Jana A.; Grela K. Forged and Fashioned for Faithfulness–Ruthenium Olefin Metathesis Catalysts Bearing Ammonium Tags. Chem. Commun. 2018, 54, 122–139. 10.1039/C7CC06535C. [DOI] [PubMed] [Google Scholar]; c Tomasek J.; Schatz J. Olefin Metathesis in Aqueous Media. Green Chem. 2013, 15, 2317–2338. 10.1039/c3gc41042k. [DOI] [Google Scholar]; d Jordan J. P.; Grubbs R. H. Small-Molecule N-Heterocyclic-Carbene-Containing Olefin-Metathesis Catalysts for Use in Water. Angew. Chem., Int. Ed. 2007, 46, 5152–5155. 10.1002/anie.200701258. [DOI] [PubMed] [Google Scholar]; Neary W.; Isais T. A.; Kennemur J. Self-Immolative Bottlebrush Polypentenamers and their Macromolecular Metamorphosis. J. Am. Chem. Soc. 2019, 141, 14220. 10.1021/jacs.9b05560. [DOI] [PubMed] [Google Scholar]
- a Bhushan B.; Lin Y. A.; Bak M.; Phanumartwiwath A.; Yang N.; Bilyard M. K.; Tanaka T.; Hudson K. L.; Lercher L.; Stegmann M.; Mohammed S.; Davis B. G. Genetic Incorporation of Olefin Cross-Metathesis Reaction Tags for Protein Modification. J. Am. Chem. Soc. 2018, 140, 14599–14603. 10.1021/jacs.8b09433. [DOI] [PubMed] [Google Scholar]; b Lin Y. A.; Chalker J. M.; Floyd N.; Bernardes G. J. L.; Davis B. G. Allyl Sulphides are Privileged Substrates in Aqueous Cross-Metathesis: Application to Site-Selective Protein Modification. J. Am. Chem. Soc. 2008, 130, 9642–9643. 10.1021/ja8026168. [DOI] [PubMed] [Google Scholar]; c Ai H. W.; Shen W.; Brustad E.; Schultz P. G. Genetically Encoded Alkenes in Yeast. Angew. Chem., Int. Ed. 2010, 49, 935–937. 10.1002/anie.200905590. [DOI] [PubMed] [Google Scholar]; d Lin Y. A.; Chalker J. M.; Davis B. G. Olefin Cross-Metathesis on Proteins: Investigation of Chalcogen Effects and Guiding Principles in Metathesis Partner Selection. J. Am. Chem. Soc. 2010, 132, 16805–16811. 10.1021/ja104994d. [DOI] [PubMed] [Google Scholar]; e Chalker J. M.; Lin Y. A.; Boutureira O.; Davis B. G. Enabling Olefin Metathesis on Proteins: Chemical Methods for Installation of S-Allyl Cysteine. Chem. Commun. 2009, 3714–3716. 10.1039/b908004j. [DOI] [PubMed] [Google Scholar]
- Jeschek M.; Reuter R.; Heinisch T.; Trindler C.; Klehr J.; Panke S.; Ward T. R. Directed Evolution of Artificial Metalloenzymes for In Vivo Metathesis. Nature 2016, 537, 661–665. 10.1038/nature19114. [DOI] [PubMed] [Google Scholar]
- a Chen Y.; Dias H. V. R.; Lovely C. J. Synthesis of Fused Bicyclic Imidazoles by Ring-Closing Metathesis. Tetrahedron Lett. 2003, 44, 1379–1382. 10.1016/S0040-4039(02)02864-2. [DOI] [Google Scholar]; b Huang K.-S.; Wang E.-C.; Chen H.-M. Synthesis of Substituted Naphthalenes and Naphthols. J. Chin. Chem. Soc. 2004, 51, 585–605. 10.1002/jccs.200400090. [DOI] [Google Scholar]; c van Otterlo W. A. L.; de Koning C. B. Metathesis in The Synthesis of Aromatic Compounds. Chem. Rev. 2009, 109, 3743–3782. 10.1021/cr900178p. [DOI] [PubMed] [Google Scholar]; d Donohoe T. J.; Orr A. J.; Bingham M. Ring-Closing Metathesis as a Basis for the Construction of Aromatic Compounds. Angew. Chem., Int. Ed. 2006, 45, 2664–2670. 10.1002/anie.200503512. [DOI] [PubMed] [Google Scholar]
- Wallace S.; Balskus E. P. Opportunities for Merging Chemical and Biological Synthesis. Curr. Opin. Biotechnol. 2014, 30, 1–8. 10.1016/j.copbio.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Vander Heiden M. G.; Cantley L. C.; Thompson C. B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Anhäuser L.; Teders M.; Rentmeister A.; Glorius F. Bio-Additive-Based Screening: Toward Evaluation of the Biocompatibility of Chemical Reactions. Nat. Protoc. 2019, 14, 2599–2626. 10.1038/s41596-019-0190-2. [DOI] [PubMed] [Google Scholar]
- a Lin Y. A.; Davis B. G. The Allylic Chalcogen Effect in Olefin Metathesis. Beilstein J. Org. Chem. 2010, 6, 1219–1228. 10.3762/bjoc.6.140. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chalker J. M. Allyl Sulfides: Reactive Substrates for Olefin Metathesis. Aust. J. Chem. 2015, 68, 1801–1809. 10.1071/CH15311. [DOI] [Google Scholar]
- Alouane A.; Labruere R.; Le Saux T.; Schmidt F.; Jullien L. Self-Immolative Spacers: Kinetics, Aspects, Structure-Property Relationships, and Applications. Angew. Chem., Int. Ed. 2015, 54, 7492–7509. 10.1002/anie.201500088. [DOI] [PubMed] [Google Scholar]
- Ogba M.; Warner N. C.; O’Leary D. J.; Grubbs R. H. Recent Advance in Ruthenium-Based Olefin Metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. 10.1039/C8CS00027A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz D.; Beckwith J. Roles of Thiol-Redox Pathways in Bacteria. Annu. Rev. Microbiol. 2001, 55, 21–48. 10.1146/annurev.micro.55.1.21. [DOI] [PubMed] [Google Scholar]
- Deliz Liang A.; Serrano-Plana J.; Peterson R. L.; Ward T. R. Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Enzymatic Cascades and Directed Evolution. Acc. Chem. Res. 2019, 52, 585–595. 10.1021/acs.accounts.8b00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.