

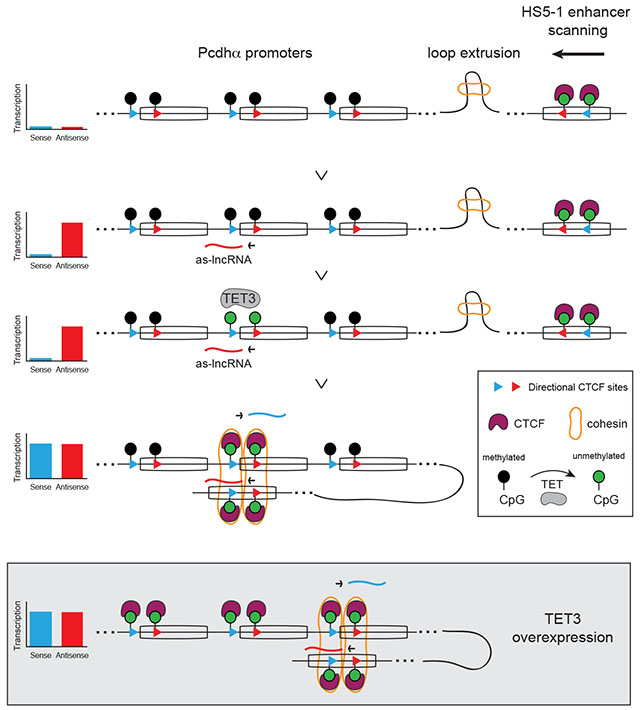
SUMMARY
Stochastic activation of clustered Protocadherin (Pcdh) α, β, and γ genes generates a cell-surface identity code in individual neurons that functions in neural circuit assembly. Here we show that Pcdhα gene choice involves the activation of an antisense promoter located in the first exon of each Pcdhα alternate gene. Transcription of an antisense long non-coding RNA (lncRNA) from this antisense promoter extends through the sense promoter, leading to DNA demethylation of the CTCF binding sites proximal to each promoter. Demethylation-dependent CTCF binding to both promoters facilitates Cohesin-mediated DNA looping with a distal enhancer (HS5-1), locking in the transcriptional state of the chosen Pcdhα gene. Uncoupling DNA demethylation from antisense transcription by Tet3 overexpression in mouse olfactory neurons promotes CTCF binding to all Pcdhα promoters, resulting in proximity-biased DNA looping of the HS5-1 enhancer. Thus, antisense transcription-mediated promoter demethylation functions as a mechanism for distance-independent promoter/enhancer DNA looping to ensure stochastic Pcdhα promoter choice.
Graphical Abstract
ETOC summary
Coupling transcription of a long noncoding RNA to DNA demethylation ensures stochastic promoter choice for clustered Protocadherin α genes, which is essential for the establishment of a neuronal surface identity code involved in circuit assembly.
INTRODUCTION
During brain development, individual neurons differentiate into distinct functional cell types, respond to a plethora of guidance molecules, and project into specific regions of the nervous system to form complex neural circuits. A key aspect of this process is the ability of neurites of individual neurons (axons and dendrites) to distinguish between themselves and neurites from other neurons (self vs. non-self) (Zipursky and Grueber, 2013). This process, known as self-avoidance, requires a unique combination of cell-surface homophilic recognition molecules that function as a molecular identity code (Zipursky and Grueber, 2013; Zipursky and Sanes, 2010). In mammals, this identity code is generated by random transcription of clustered Protocadherin (Pcdh) genes (Lefebvre et al., 2015; Mountoufaris et al., 2018) by means of a poorly understood mechanism of stochastic and combinatorial promoter choice (Esumi et al., 2005; Tasic et al., 2002; Wang et al., 2002; Wu and Maniatis, 1999). Pcdh genes have a unique genomic arrangement consisting of three closely linked gene clusters (α, β, and γ) that, together, span nearly 1 million base pairs (bp) of genomic DNA. The α and γ clusters are organized into variable (alternate and c-type) and constant regions, reminiscent of the organization of immunoglobin and T-cell receptor gene clusters (Wu and Maniatis, 1999) (Figure 1A, Pcdhα).
Figure 1: Transcription of sense and antisense RNA from Pcdhα alternative exons.

(A) Genomic organization of the human Pcdhα gene cluster. The stochastic expression of 13 alternate exons is driven by with their own promoter (arrows), and are equally likely to be activated. c1 and c2: c-type exons; 1-3: constant exons encoding the intracellular domain of Pcdh proteins. (B) Location and relative orientations (arrows) of the promoter and the exonic CBS sites (alternate exons) and the left and right CBS sites (HS5-1 enhancer). (C) Example of a Pcdhα promoter/HS5-1 enhancer complex bound to CTCF and Cohesin. (D) Sense (grey) and antisense (black) RNA (Total RNA, cRNA-Seq) at the Pcdhα cluster in SK-N-SH cells. CTCF, Rad21 and H3K4me3 (ChIP-Seq) relative to active exons (yellow). Pcdhαc2: active but not bound by CTCF or Rad21. Virtual 4C (cHi-C) on top (HS5-1 as a viewpoint). The x-axis: linear sequence of the genomic organization of the Pcdhα cluster. Numbers on the left-hand side of each track: minimum and maximum densities in reads per million.
Neuron-specific expression of individual Pcdhα genes requires long-range DNA looping between individual Pcdhα promoters and a transcriptional enhancer, called HS5-1 (hypersensitivity site 5-1) (Guo et al., 2012; 2015; Kehayova et al., 2011; Monahan et al., 2012; Ribich et al., 2006) (Figure 1A). Conserved transcriptional promoter sequences are located immediately proximal to every Pcdhα exon (Tasic et al., 2002) while the HS5-1 enhancer is located downstream of the constant exons, between the Pcdh α and the β clusters (Ribich et al., 2006) (Figure 1A, 1B and S1). These stochastic promoter/enhancer interactions occur independently on each of the two allelic chromosomes in diploid cells and require the binding of the CCCTC-binding protein (CTCF) and the Cohesin protein complex (Guo et al., 2012; Hirayama et al., 2012; Kehayova et al., 2011; Monahan et al., 2012) (Figure 1C). CTCF is an 11 zinc-finger (ZF) domain protein that, together with the Cohesin complex, plays a central role as an insulator of chromatin domains, and mediates genomewide promoter/enhancer interactions (Ghirlando and Felsenfeld, 2016; Ong and Corces, 2014). All Pcdhα alternate exons contain two CTCF binding sites (CBS): one in the promoter (pCBS) and one in the protein coding sequence of the first exon (eCBS) (Guo et al., 2012; Monahan et al., 2012) (Figure 1B). The two binding sites are separated by approximately 1000 bp, and similarly spaced CBS sites are located in the HS5-1 enhancer (L-CBS and R-CBS) (Guo et al., 2012; Monahan et al., 2012) (Figure 1B). Interestingly, the CTCF binding sites in Pcdhα promoters and the HS5-1 enhancer are in opposite relative orientations, and inversion of the HS5-1 enhancer results in a significant decrease in Pcdhα gene cluster expression, demonstrating the functional importance of this arrangement (Guo et al., 2015). This opposite relative orientation of promoter and enhancer CBS sites appears to be a general feature of eukaryotic chromosomes genome-wide (Guo et al., 2015; Rao et al., 2014), and has been proposed to play a critical role in promoting the spatial interaction between genes and transcriptional regulatory elements by a mechanism known as loop-extrusion (Fudenberg et al., 2016). In the context of the Pcdhα gene cluster, the loop-extrusion model predicts that the HS5-1 enhancer, bound by CTCF and the Cohesin proteins, scans the Pcdhα exons until it finds the exon bound by CTCF. However, this possibility has yet to be demonstrated.
A critical insight into the formation of Pcdhα promoter/enhancer complexes was provided by the observation that there is an inverse relationship between Pcdhα gene expression and DNA methylation of the Pcdhα promoters (Tasic et al., 2002; Toyoda et al., 2014). Specifically, the CTCF/Cohesin complex associates exclusively with transcriptionally active promoters, which are characterized by hypomethylation of the CBS sites and of the DNA sequences located between the two CBS sites (Guo et al., 2012). By contrast, CBS sites and the DNA between them are hypermethylated in inactive promoters, thus preventing CTCF/Cohesin binding (Guo et al., 2012). Although DNA methylation of the CTCF binding sites is likely to play an important role in the mechanism of stochastic Pcdhα promoter choice, the temporal relationship between promoter DNA methylation and promoter choice is not known. For example, it is not known whether promoter DNA methylation is the ground state upon which promoter choice operates, or whether all promoters are initially unmethylated and methylation of the inactive promoters occurs subsequent to stochastic promoter choice.
Here, we provide evidence that the ground state of Pcdhα promoter DNA is methylated and transcriptionally repressed in immature cells destined to become olfactory sensory neurons. Stochastic promoter demethylation occurs by a remarkable mechanism in which transcription of an antisense long noncoding RNA (lncRNA) is initiated from a promoter located within the downstream protein coding region of each Pcdhα exon. Transcription through the upstream sense promoter results in its demethylation, binding of CTCF and DNA looping to the HS5-1 enhancer. The binding of CTCF marks the promoter for engagement by the HS5-1 enhancer through DNA loop-extrusion (Fudenberg et al., 2016), thus eliminating enhancer/promoter proximity bias.
RESULTS
Transcription of sense and antisense RNA from clustered Pcdhα alternate exons
The mechanism of stochastic promoter choice in the Pcdhα gene cluster cannot be studied in vivo, as each neuron expresses a distinct repertoire of Pcdhα alternate exons. We therefore made use of the well-characterized human neuroblastoma cell line SK-N-SH, which stably expresses a distinct repertoire of Pcdhα isoforms through multiple cell divisions: α4, α8, α12, αc1, and αc2 (Guo et al., 2012) (Figure 1D). This stochastic pattern of expression in cell culture is indistinguishable from that observed in single neurons in vivo (Esumi et al., 2005; Mountoufaris et al., 2017). SK-N-SH cells thus provide a multicellular “avatar” for studying single cell expression of Pcdhα genes, and internal controls for exons that are transcriptionally silent.
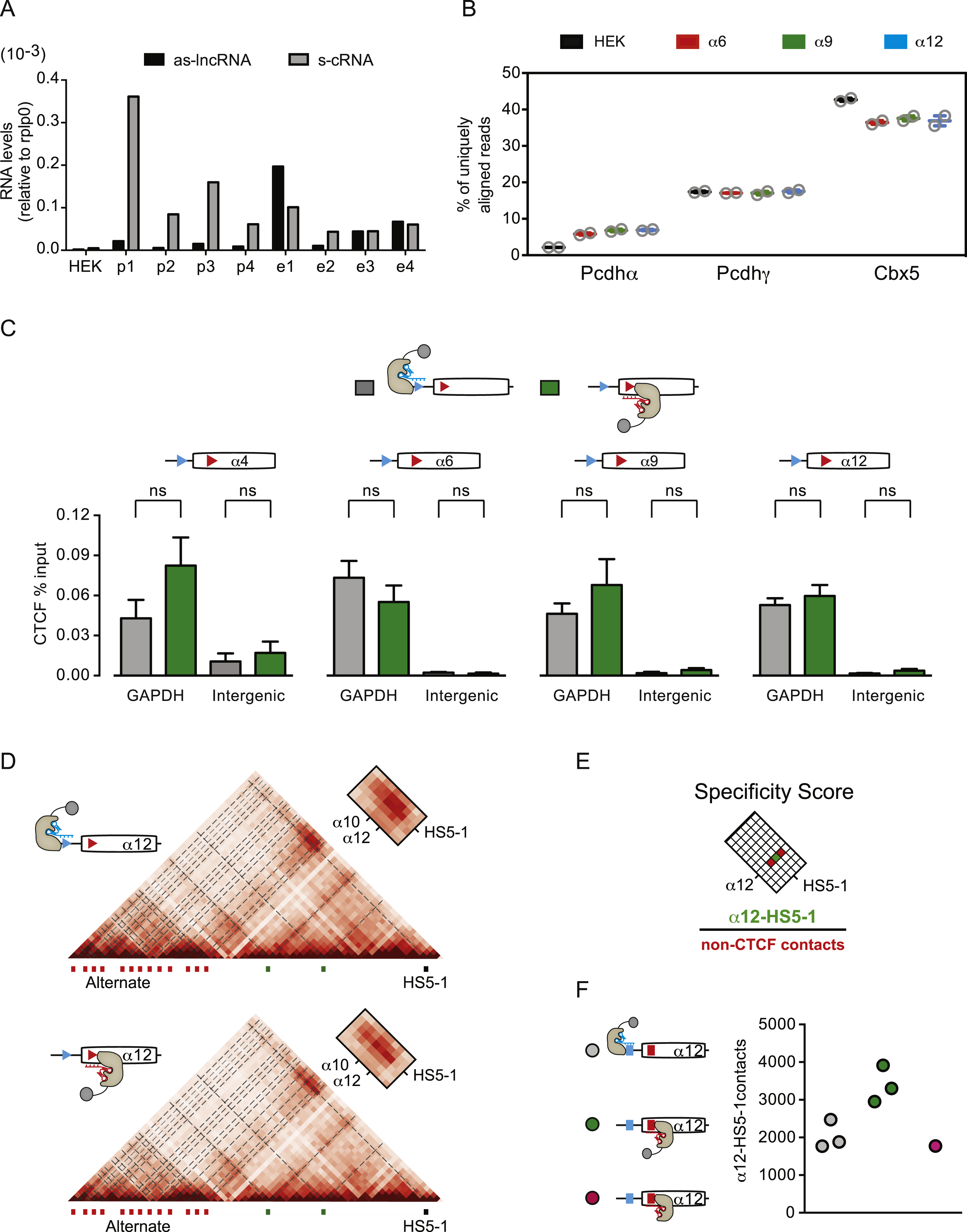
The low level of expression of Pcdh genes provides an additional challenge to the study of Pcdhα promoter choice. To optimize the analysis of Pcdh RNA precursors (pre-mRNA) and mature (mRNA) RNAs in SK-N-SH cells, we employed capture RNA-Sequencing (cRNA-Seq), and achieved a two order of magnitude enrichment of Pcdh RNA transcripts (Figure S1). Remarkably, this enrichment revealed a high level of antisense RNA transcription of the Pcdhα alternate exons, which contain dual CBSs in SK-N-SH cells (Figure 1D and S1B). By contrast, antisense RNA transcription was not detected within the two c-type exons, αc1 and αc2, which do not contain CBSs within their exons (Figure 1D). Antisense RNA was not observed in the Pcdh β or γ variable exons in SK-N-SH cells, which, like αc1 and αc2, do not contain exonic CBS sites (Figure S1B). We refer to the observed antisense RNA as as-lncRNA, as this high molecular weight RNA lacks open reading frames that encode protein. For clarity, we refer to the sense Pcdh coding RNA as s-cRNA (sense coding RNA).
Convergent promoters in both the Pcdhα alternative exons and HS5-1 enhancer
In order to characterize the nature of the antisense RNAs and to gain mechanistic insights into their function, we first localized their transcription start sites and the location of the promoter-paused RNAPII using Start-Seq (Nechaev et al., 2010). RNA isolated from stalled RNAPII at promoters are approximately 15-45 nucleotides long and contain a 5’ 7meG-cap (Figure 2A). Sequencing of these short RNAs revealed the position of paused RNAPII, thus acting as a proxy for the location of RNAPII-engaged promoters, and the transcriptional start site at a nucleotide-base resolution (Figure S2A). As expected, we observed promoter-proximal RNAPII at the pCBS-proximal promoter of the active Pcdh α4, α8, α12 and αc1 exons, and at the promoter of αc2 in SK-N-SH cells (Figure 2B). To our surprise, however, we also observed promoter-proximal RNAPII just upstream of the eCBS for α4, α8, and α12 in the antisense orientation (Figure 2B). Thus, sequences near the two CBSs of active Pcdhα genes act as convergent promoters, where antisense and sense RNA converge and partially overlap (Figure 2C, Pcdhα4 is shown). This is in contrast to the singular pCBS site in Pcdhαc1, which acts as a more canonical divergent promoter, where transcription of the antisense and sense RNA occurs in opposite directions and does not overlap (Figure 2C). Remarkably, Start-Seq analysis also identified a similar convergent promoter architecture of the two CBSs in the HS5-1 enhancer (Figure 2B and 2C). The position of TSS for Pcdh α4, α8 and α12 are shown in Figure 2D.
Figure 2: Convergent Pcdhα alternative exon promoters and the HS5-1 enhancer.

(A) Schematic diagram of Start-Seq. (B) Paused RNAPII (Start-Seq) relative to total RNA (cRNA-Seq), CTCF, H3K4me3 and H3K27ac (ChIP-Seq) in SK-N-SH cells. (C) Promoter architectures for Pcdhα4 (convergent), Pcdhαc1 (divergent) and the HS5-1 enhancer (convergent). (D) TSS of the as-lncRNA and s-cRNA from Pcdh α4, α8 and α12. (E) RNA splicing patterns of polyadenylated as-lncRNAs (cRNA-Seq) initiated from the Pcdhα4 and Pcdhα12 as indicated by the splice junctions in reads mapping to the as-lncRNAs, relative to CTCF and H3K4me3. Red triangles: antisense 5’ss described in (F). (F) Position, sequence and conservation of the antisense 5’ss located upstream of the pCBS (blue). CTCF in violet. Bar graph: distribution of the distance of the 5’ss from the pCBS.
For B and E, numbers on the left-hand side of each track: minimum and maximum densities in read per million. x-axis: linear sequence of the genomic organization of the Pcdhα cluster. Arrows in (E): position of transcription start sites by Start-Seq.
Mapping the location of the Pcdhα as-lncRNA promoters with respect to the as-lncRNAs revealed that these nuclear RNA precursors can be as long as 20 kb in length, and are spliced and polyadenylated with half-lives of the same order of magnitude as their respective s-cRNAs (Figure S1F). As an example, the as-lncRNA that initiates at the eCBS-proximal promoter of Pcdhα4 in SK-N-SH cells is transcribed through the pCBS-proximal promoter of Pcdhα4 and extends in the antisense direction all the way to the intergenic sequence between the Pcdh α1 and α2 exons (more than 20 kb) (Figure 2E). By contrast, the antisense RNA that initiates at the eCBS-promoter of Pcdhα12 extends to the Pcdhα11 exon (Figure 2E). In addition, we discovered the presence of a highly conserved 5’ splice site (5’ss), encoded in the antisense direction about 7 bp upstream of the pCBS core motif (Figure 2F). Usage of that 5’ss results in the most abundant polyadenylated as-lncRNA spliced isoform (Figure 2E). Remarkably, this site is absent from the pCBS of Pcdhαc1, as well as from the pCBS sites of the Pcdh β and γ clusters. These observations suggest that RNA splicing of this promoter-embedded 5’ splice site may be coupled to the activation of the pCBS promoter (See Discussion).
Antisense lncRNA and sense coding RNA are transcribed from the same active allele
The cRNA-Seq data obtained from SK-N-SH cells revealed a direct correlation between sense and antisense RNA transcription and transcriptionally active Pcdhα alternate exons. Because transcription of the Pcdhα alternate exons occurs independently on the two allelic chromosomes (Esumi et al., 2005), we sought to determine whether the as-lncRNA and the s-cRNA were transcribed from the same Pcdhα locus allele. To accomplish this, we used CRISPR-Cas9 gene editing to generate SK-N-SH cells heterozygous for the Pcdhα gene cluster, SK-N-SH-αhet (Figure 3A). We isolated two clones (SK-N-SH–αhet 1 and 2) expressing primarily α12, αc1 and αc2 from the remaining copy of the Pcdhα gene cluster (Figure 3B and 3C). Both clones showed expression of the as-lncRNA and s-cRNA from Pcdhα12 (Figure 3B and 3C), confirming that sense and antisense transcription originate from the same allele. Furthermore, chromatin immunoprecipitation sequencing studies (ChIP-Seq) for CTCF and Rad21, a subunit of the Cohesin complex, as well as capture in situ high-throughput chromosome conformation capture studies (cHi-C) performed in αhet-1 also demonstrated that the Pcdhα alternate exons, from which sense and antisense RNAs are transcribed, are bound by CTCF and Cohesin, and engaged in promoter/HS5-1 enhancer DNA looping (Figure 3C and 3D). We note that the αhet-1 and αhet-2 clones share a 16.7 kb deletion that truncates the Pcdhα8 exon and removes the Pcdh α9 and α10 exons (Figure 3C and 3D). This deletion was previously reported as a common feature of individuals from multiple populations of European and East Asian descent with no discernable phenotypic consequence (Noonan et al., 2003).
Figure 3: Antisense lncRNA transcription precedes sense cRNA transcription from the same active allele.

(A) Scissors: location of the gRNAs to generate SK-N-SH cells bearing a single copy of the Pcdhα gene cluster. PCR confirms the deletion. (B) Expression of Pcdh α4 and α12 relative to RPLPO in SK-N-SH-αhet 1 and 2 clonal cells compared to SK-N-SH-WT cells (RT-qPCR). (C) Total RNA (RNA-Seq) relative to Rad21, CTCF and H3K4me3 (ChIP-Seq) in SK-N-SH-αhet-1. (D) In situ cHi-C contact maps at 10kb resolution for SK-N-SH-αhet-1 (Left) and SK-N-SH-WT (Right) cells. Coordinates: 140,780,000-141,050,000, chr5 (hg38). (E) Schematic diagram of s4U-DRB-cRNA-Seq. (F) Nascent transcription at 20 minutes after release of RNAPII (s4U-DRB-cRNA-Seq) (G) Quantification of nascent transcription by RNAPII of the as-lncRNA and s-cRNA from Pcdh α4 (Left) and α12 (Right). The -s4U used as control. Errors (n=3) represent s.e.m. (H) Schematic diagram for the asynchronous activity of RNAPII.
Numbers on the left-hand side (C) and right side (F) of each track: minimum and maximum densities in read per million.
In contrast to SK-N-SH cells, a mixed population of primary neurons, each expressing a distinct repertoire of Pcdhα alternative exons, should collectively express as-lncRNAs from all the Pcdhα alternate exons, but not from Pcdh αc1 and αc2, or from the β or γ exons. As predicted, analysis of RNA from human primary neurons and from mouse mature olfactory sensory neurons (mOSNs) revealed lncRNA expression exclusively from all the Pcdhα alternate exons (Figure S2B and S2C). As in SK-N-SH cells, the as-lncRNA expressed in human and mouse primary neurons are spliced and polyadenylated (Figure 2E, S2B and S2C). However, in contrast to SK-N-SH cells, the levels of the as-lncRNAs in both human and mouse primary neurons appeared lower. We speculate that this difference could be a consequence of the mitotic (SK-N-SH) and the post-mitotic (primary neurons) state of the two cell types. We also note that an antisense lncRNA from the Pcdhα12 exon, similar to the one described and characterized above, was reported in human brain samples, but its significance was not understood (Lipovich et al., 2006).
The asymmetric nature of Pcdhα convergent promoters results in asynchronous sense and antisense RNA transcription
Antisense convergent transcription is a widespread phenomenon in the mammalian genome. Yet, its function, as well as the mechanism by which actively transcribing RNA polymerases translocate along a common stretch of DNA in opposite directions, remains unclear (see Discussion). To assess the activity of RNAPII at the pCBS-proximal and eCBS-proximal promoters, we analyzed transcription in SK-N-SH cells using s4UDRB-Seq (Fuchs et al., 2014). This method combines synchronization of RNAPII at promoters by 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) with incorporation of the nucleoside 4-thiouridine (s4U) during RNA synthesis (Figure 3E). Consistent with the Start-Seq data, we observed convergent elongating RNAPII from both pCBS- and eCBS-proximal promoters of α4, α8 and α12, and divergent RNAPII from the pCBS-proximal promoter of Pcdhαc1 (Figure 3F). We also observed convergent elongating RNAPII at the HS5-1 enhancer, consistent with the presence of convergent promoters as described above (Figure 3F). These data reveal a remarkable symmetry between the location of CTCF/Cohesin binding sites and sense and antisense transcription from the Pcdhα alternate promoters and the HS5-1 enhancer. However, in contrast to the sense and antisense RNA transcribed from Pcdhα alternate exons, both enhancer RNAs are not polyadenylated, and therefore appear to rapidly turnover over (Figure 1D, 1F and S2B).
Interestingly, quantification of nascent transcription of the antisense and sense RNAs assayed by s4UDRB-Seq revealed that, while RNAPII molecules at the Pcdhα active exons transcribe in a convergent manner, their activities appear asynchronous. That is, the as-lncRNA is transcribed earlier than the s-cRNA (Figure 3G and 3H). This asynchronous RNAPII activity reveals an intrinsic asymmetry in the activities of the two promoters, an observation consistent with the fact that the two promoters differ in their ability to bind to distinct classes of transcription factors (TF), as well as the fact that the two CBS sites, proximal to the sense and antisense promoters, differ in sequence and in their affinity for CTCF (Figure S2D).
Transcription of antisense lncRNAs triggers activation of Pcdhα sense promoters
To understand the functional significance of the asynchronous activity of the Pcdhα sense and antisense promoters, we designed a gain-of-function assay to uncouple transcription of the as-lncRNA from transcription of the sense coding Pcdhα mRNA in the context of the endogenous Pcdhα gene cluster. Specifically, we made use of a catalytic-inactive CRISPR-dCas9 protein fused to a tripartite transcriptional activator (dCas9-VPR) (Chavez et al., 2015) to selectively activate the pCBS-proximal or eCBS-proximal promoters of silent Pcdhα genes (Figure 4A). We chose HEK293T cells, as most Pcdhα genes are transcriptionally silent in this cell line, with the exception of Pcdh α10 and αc2. This property of HEK293T cells, together with the modularity of the CRISPR-dCas9 system, made it possible to selectively design guide RNAs for the transcriptional activation of Pcdh α4, α6, α9 and α12 (Figure S3). As expected, dCas9-VPR activation of the Pcdhα4 sense promoter resulted in robust synthesis of the Pcdhα4 s-cRNA (Figure 4B). Unexpectedly, activation of the Pcdhα4 antisense promoter not only led to high levels of antisense RNA transcription, but high levels of sense RNA transcription were also observed (Figure 4B). This pattern of sense and antisense RNA transcription did not depend upon the number of dCas9-VPR activators (1 vs. 4) nor on their position relative to the CBSs (Figure S4A). Most importantly, this pattern of transcription mirrored that of active exons observed in SK-N-SH cells (Figure 1D). As for the Pcdhα4, we also observed the same relationship between as-lncRNA and sense transcription for the Pcdh α6, α9, and α12 exons (Figure 4C and S4B).
Figure 4: Transcription of the antisense lncRNA triggers activation of sense promoters.

(A) Schematics of dCas9-VPR-mediated activation of pCBS- and eCBS-proximal promoters. (B) Activation of sense and antisense Pcdhα4 promoters by dCas9-VPR (RNA-Seq). (C) Activation of antisense Pcdh α6, α9, α12 promoters by dCas9-VPR (cRNA-Seq). Side boxes: zoom-in view of the convergent transcription. (D) Enrichment of H3K4me3 at the Pcdhα4 promoter (ChIP-qPCR). Errors (n=3) represent s.e.m. and statistical significance calculated with Student unpaired t-test.
For (B and C), the x-axis: the linear sequence of the genomic organization of the Pcdhα cluster. Arrows: initiation of transcription. Numbers on the left-hand side of each track: the minimum and maximum densities in read per million.
These observations suggest transcription of antisense RNA from the eCBS-proximal promoter activates the upstream cognate pCBS-proximal promoter to generate sense coding RNA. To test this possibility, we measured the levels of histone H3 lysine 4 trimethylation (H3K4me3), a histone post-translational modification that marks transcriptionally active promoters and is detected on DNA between the two CTCF-bound CBS sites in active Pcdhα genes (Figure 1D). As shown in Figure 4D, chromatin immunoprecipitation followed by quantitative PCR (ChIP-qPCR) resulted in an increase in H3K4me3 upon transcriptional activation of the antisense promoter by dCas9-VPR.
Antisense lncRNA transcription promotes CTCF binding and long-range promoter/enhancer DNA interactions
The expression of Pcdhα sense RNA transcripts requires the binding of CTCF and Cohesin to the pCBS and eCBS sites, and long-range DNA looping between active promoters and the HS5-1 enhancer (Guo et al., 2012; 2015). In ChIP-Seq experiments, we observed that both CBSs of Pcdh α4, α6, α9, and α12 in the HEK293T parental cell line used in this study are not bound to CTCF nor to the Cohesin subunit, Rad21 (Figure S3B). We therefore asked whether antisense transcription promotes the binding of CTCF to its binding sites in the activated exon. Consistent with the mechanistic coupling of promoter activation and CTCF/Cohesin binding (Guo et al., 2012; Monahan et al., 2012), we observed a statistically significant enrichment of CTCF occupancy at both the pCBS and eCBS sites upon dCas9-VPR activation of their antisense promoters relative to the activation of their sense promoters (Figure 5A). We note that the levels of CTCF binding at the activated Pcdhα promoters measured by ChIP-qPCR was lower than the one measured for a constitutive promoter such as GAPDH, but significantly higher than an intergenic DNA site (Figure S4C). We speculate that this lower CTCF enrichment is a consequence of the high degree of cell heterogeneity as a result of transient transfections of the dCas9-VPR constructs.
Figure 5: Antisense lncRNA transcription promotes CTCF binding and promoter/HS5-1 enhancer DNA interactions by DNA demethylation of the CBS sites.

(A) CTCF occupancy at the pCBS and the eCBS sites of Pcdh α4, α6, α9, α12 upon activation of the pCBS- (grey) and the eCBS-proximal (green) promoter by dCas9-VPR (ChIP-qPCR). (B) Left: Virtual 4C with Pcdhα12 promoter as a viewpoint for HEK293T cells activated with dCas9-VPR targeting the pCBS- (grey) or the eCBS-proximal promoter (green) of Pcdhα12. Black arrow: Specific interaction between the Pcdhα12 promoter and the HS5-1 enhancer. Right: Specificity scores for the interaction of the Pcdhα12 exon to the HS5-1 enhancer. (C) Relative levels of 5mC and 5hmC at the pCBS, eCBS and middle sequences of Pcdhα12 in HEK293T cells (white) and HEK293T cells with Pcdhα12 sense (grey) or antisense (green) promoters activated by dCas9-VPR.
Errors (n=3) represent s.e.m. and statistical significance calculated with Student unpaired t-test.
The binding of CTCF to the promoters and exons of dCas9-VPR-activated genes suggests the possibility that antisense transcription from the activated exon leads to CTCF/Cohesin-dependent long-range DNA looping between the active promoter and the HS5-1 enhancer. To address this hypothesis, we focused on the Pcdhα12 exon and performed three biologically independent in situ cHi-C experiments with HEK293T cells transfected with dCas9-VPR to activate either the Pcdhα12 pCBS-proximal or eCBS-proximal promoter (Figure S4D). To best discern newly formed long-range DNA contacts between the HS5-1 enhancer and the Pcdhα12 promoter, we calculated a specificity score indicating the signal-to-noise ratio of enhancer/promoter interaction in a 15 kb window at 5 kb resolution (Figure S4E). This analysis revealed a modest, but statistically significant, increase in specific DNA contacts between the Pcdhα12 promoter and the HS5-1 enhancer upon activation of the antisense Pcdhα12 promoter compared to the sense promoter (Figure 5B). Importantly, dCas9 without the transcriptional activator domain did not result in the formation of Pcdhα12/HS51 contacts (Figure S4F).
Antisense lncRNA transcription promotes DNA demethylation of Pcdhα promoters
The data presented thus far support a model in which antisense lncRNA transcription mediates the recruitment of CTCF to active Pcdhα alternate promoters. Given the observation that DNA methylation of the CBS sites blocks CTCF binding (Bell and Felsenfeld, 2000) and that both pCBS and eCBS sequences contain CpG dinucleotides, we reasoned that DNA demethylation could be the mechanism by which CTCF/Cohesin binds to the pCBS and eCBS following as-lncRNA transcription. To gain insight into the potential role of DNA methylation in the modularity of CTCF binding to both pCBS and eCBS sites, we obtained nucleotide resolution of the methylation of the CpG dinucleotides within the CBS sites by examining published ENCODE whole genome bisulfite sequencing (WGBS) data from SK-N-SH cells (Figure S5). Consistent with genome-wide studies (Wang et al., 2012), these data reveal how methylation at position C2 and C12 in the core CTCF motif can affect CTCF binding at both CBS sites (Figure S5C and S5D), and further suggest how additional methylation sites flanking C2 and C12 could also contribute to the regulation of CTCF binding to the pCBS and eCBS (Figure S5E). Quantification of the ENCODE WGBS data also revealed that the DNA sequence between the two Pcdhα CBS sites (“middle”) is hypormethylated in active exons (Figure S5F and S5G), consistent with previous reports on the relationship between methylation, CTCF binding and promoter activity (Guo et al., 2012; Kawaguchi et al., 2008; Tasic et al., 2002).
In mammals, 5-methylcytosine (5mC) modified CpG sequences are converted to unmodified cytosine (C) by the activity of TET deoxygenase enzymes, which mediate the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (Wu and Zhang, 2017). Thymine DNA glycosylase (TDG) then converts 5caC to C by a base excision repair mechanism (Wu and Zhang, 2017). 5hmC is a stable oxidation intermediate and its detection is a proxy for a pathway to active demethylation catalyzed by the TET proteins. Therefore, to directly test the possibility that transcription of the as-lncRNA leads to demethylation of CpG elements, we measured the levels of 5mC and 5hmC for the Pcdhα12 in HEK293T cells by Methylated DNA Immunoprecipitation (MeDIP) upon dCas9-VPR-mediated activation of its respective sense and antisense promoters. Consistent with our hypothesis, activation of the Pcdhα12 eCBS promoter resulted in a decrease of 5mC/5mhC levels at the pCBS, the eCBS and at the DNA sequence between the two CBS sites (Figure 5C). By contrast, activation of the Pcdhα12 pCBS-proximal promoter resulted in a statistically significant decrease of 5mC/5hmC levels only for the pCBS site (Figure 5C). To detect base-pair resolution of the changes occurring at the eCBS site, we performed bisulfite reactions followed by Sanger DNA sequencing, and observed a higher level of demethylation of all three CpG sites in the eCBS when antisense RNA is transcribed relative to when only sense transcription is initiated (Figure S5H).
Demethylation of Pcdhα promoters correlates with activation in vivo
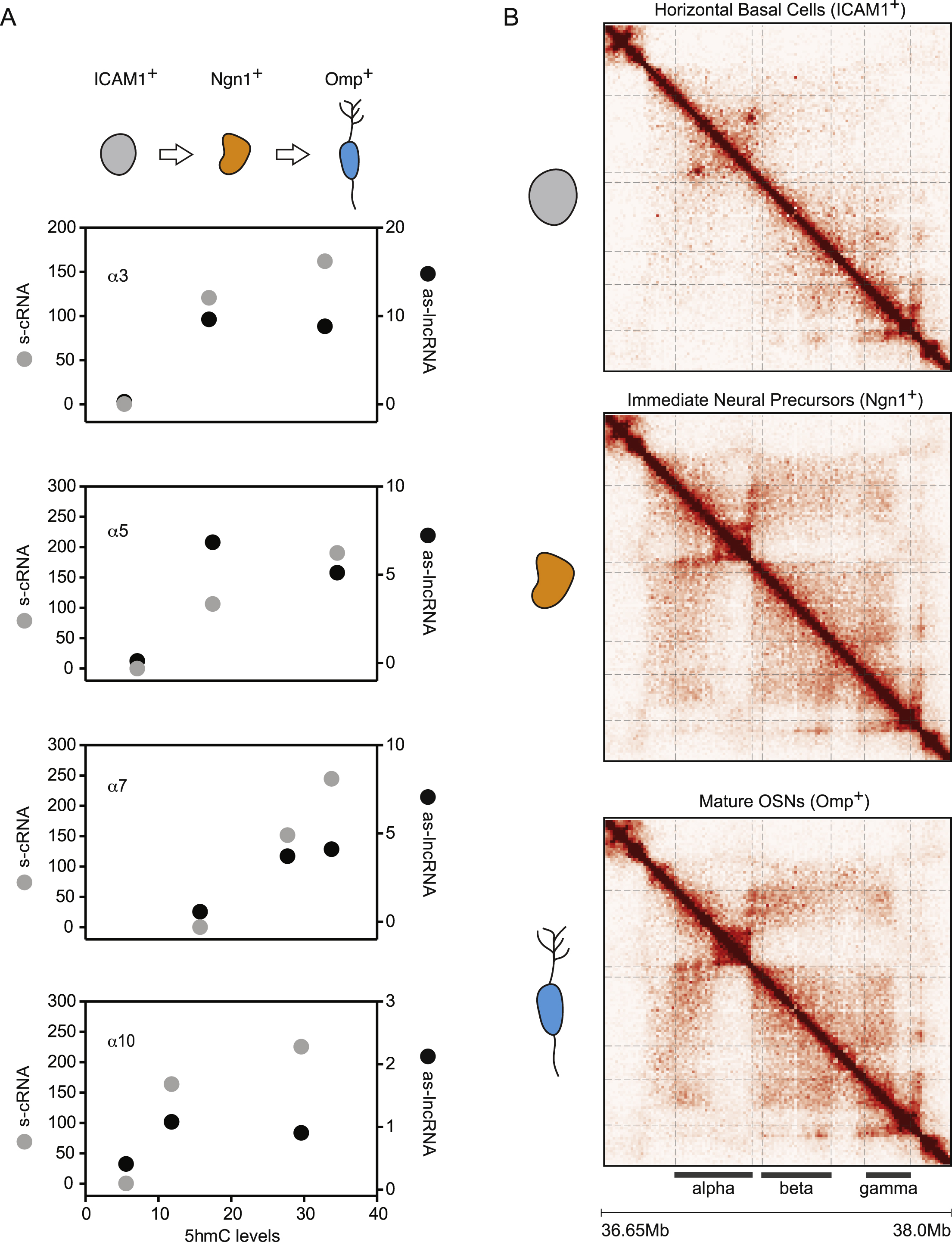
The data presented above suggest that the ground state of Pcdhα promoter DNA is methylated, and DNA demethylation, targeted by transcription of an antisense lncRNA, controls the binding of CTCF and Pcdhα sense promoter activation. To test this model in vivo, we made use of the mouse main olfactory sensory epithelium (mOE), as an in vivo developmental system to study the relationship between promoter DNA methylation and Pcdhα gene expression, as the Pcdh gene cluster is stochastically and combinatorially expressed in OSNs, and that Pcdhα genes play a fundamental role in OSN wiring (Hasegawa et al., 2008; Mountoufaris et al., 2017) (Figure 6A). We re-analyzed recently published data from a study of the levels of 5mC and 5hmC in the three cell types that represent discrete neurodevelopmental stages in the mOE: horizontal basal cells (ICAM1+), immediate neural precursors (Ngn1+) and mature olfactory sensory neurons (Omp+) (Figure 6A) (Colquitt et al., 2013). Horizontal basal cells are quiescent multipotent cells that produce all of the cell types present in the mOE; immediate neural precursors are post-mitotic cell precursors of olfactory sensory neurons, while olfactory sensory neurons are terminally differentiated primary sensory neurons. Consistent with our model, we found that the Pcdhα alternate exons and their promoters are enriched in 5mC in iCAM1+ cells, indicating that the pre-neuronal ground state of all Pcdhα alternate promoter DNA is methylated and repressed (Figure 6B and 6C). However, with the development of olfactory sensory neurons (ICAM1+ → Ngn1+ → Omp+), we observed an increase of 5hmC in the Pcdhα alternate promoters and exons (Figure 6B and 6D). To determine whether conversion of 5mC to 5hmC is accompanied by activation of Pcdhα promoters, we performed RNA-Seq experiments in ICAM1+, Ngn1+ and Omp+ cells. Consistent with our hypothesis, conversion of 5mC to 5hmC correlates with the expression of both antisense long noncoding and sense coding Pcdhα RNAs (Figure 6E, 6F and S6A). Finally, we determined whether Pcdhα expression is accompanied by the formation of long-range DNA contacts between the Pcdhα promoters and the HS5-1 enhancer in vivo, and performed in situ Hi-C experiments in ICAM1+, Ngn1+ and Omp+ cells (Figure S6B). We observed a strong increase in alternate promoters/HS5-1 enhancer interaction during neuronal differentiation of the mOE (Figure 6G). These data, collectively, provide in vivo support of our observations made in human cell lines.
Figure 6: DNA demethylation at Pcdhα promoters correlates with their activation in vivo.

(A) Top: Schematics of the maturation of the mouse main olfactory epithelium (OE) and the assembly of a functional neural circuit (glomerulus). Bottom: Schematics of stochastic Pcdhα promoter choice in individual mOSNs. (B) 5mC (Black) and 5hmC (Green) profiles of the Pcdhα alternate promoters and exons in ICAM+, Ngn1+ and Omp+ cells. x-axis: linear sequence of the mouse Pcdhα cluster. Numbers on the left-hand side of each track: minimum and maximum read densities in read per million. (C-F) Average of cumulative RPM values for the Pcdhα alternate promoters/exons for 5mC (C), 5hmC (D), as-lncRNAs (E) and s-cRNAs (F). (G) Average of cumulative in situ Hi-C contacts for the Pcdhα alternate promoters/exons. For (C-G), data in Box and whiskers. Error bars: minimum and maximal values and statistical significance calculated with one-way ANOVA.
Stochastic DNA demethylation ensures random Pcdhα promoter choice by the CTCF/Cohesin complex via DNA loop-extrusion
Analysis of the Hi-C data from Ngn1+ and Omp+ cells revealed architectural “stripes” along the Pcdhα gene cluster (Figure S6B and 7A), a feature that has been associated with the activity of the Cohesin complex in the assembly of promoter/enhancer complexes during DNA loop-extrusion (Vian et al., 2018). A prediction of the DNA loop-extrusion model for the assembly of a Pcdhα promoter/enhancer complex is that uncoupling CTCF binding to Pcdhα promoters from DNA looping to the HS5-1 enhancer by the Cohesin complex should result in an overall loss of expression of all Pcdhα exons. To test this possibility, we conditionally deleted the Cohesin subunit, Rad21, in mouse olfactory sensory neurons (Figure S7A) using OMPiresCre. With this driver, Rad21 is deleted in post-mitotic, fully differentiated, OSNs in which Pcdhα promoter choice has already occurred (Figure 6C–G and S7B). However, upon deletion of Rad21, a loss of long-range DNA contacts between the Pcdhα promoters and the HS5-1 enhancer was observed (Figure 7A and 7B). More importantly, loss of DNA contacts correlated with a significant loss of expression of all Pcdhα exons as determined by RNA-Seq (Figure 7C). Thus, continuous Cohesin activity appears to be required for the maintenance of DNA looping in the Pcdhα cluster, even in the absence of cell division.
Figure 7: Stochastic DNA demethylation ensures random Pcdhα promoter choice via DNA loop-extrusion.

(A) Hi-C contacts maps at 10kb resolution for the Pcdhα cluster in wild-type (Left) and Rad21 KO (Right) mOSNs; max: 100 reads per billion Hi-C contacts. (B and C) Average HiC contacts of the HS5-1 enhancer with the individual Pcdhα promoters (B) and average RPM values of s-cRNA for individual Pcdhα exons (C) in mOSNs (Blue) and mOSNs Rad21 KO (Black). (D) Left: 5hmC (MeDIP-Seq) and CTCF (ChIP-Seq) profiles in mOSNs (Blue) and mOSNs upon Tet3 overexpression (Red). Right: Quantification of CTCF binding. (E) Hi-C contact maps at 10kb resolution for the Pcdhα cluster in mOSNs overexpressing Tet3; max: 100 reads per billion Hi-C contacts. (F and G) Average HiC contacts of the HS5-1 enhancer with the individual Pcdhα promoters (F) and average RPM values of s-cRNA for individual Pcdhα exons (G) mOSNs overexpressing Tet3. (H) Model for how coupling of as-lncRNA transcription and DNA demethylation ensures stochastic and HS5-1 distance-independent choice of a Pcdhα promoter. Overexpression of Tet3 results in non-random and HS5-1 distance-biased Pcdhα promoter choice.
These data suggest that stochastic antisense transcription ensures random demethylation of Pcdhα promoters to ensure an HS5-1-distant-independent assembly of a CTCF/Cohesin-mediated enhancer/promoter complex by DNA loop-extrusion. A prediction of this model is that uncoupling DNA demethylation from antisense lncRNA transcription would result in a non-random looping of Pcdhα promoters to the HS5-1 enhancer. We tested this possibility by overexpressing Tet3 in OSNs (Figure S7A). Tet3 is the most highly expressed Tet protein in OSNs, and has been shown to associate with the Pcdhα promoters in differentiated neuronal precursor cells (Li et al., 2016). Overexpression of Tet3 resulted in strong demethylation of Pcdhα promoters, as indicated by a large increase in 5hmC levels(Figure 7D and S7C) and by an increase of CTCF binding to CBS sites genome-wide (Figure S7D), and to all Pcdhα exons, irrespective of the transcription state of their cognate as-lncRNAs (Figure 7D and S7E). To address the function of uncoupling as-lncRNA transcription from stochastic DNA demethylation, we performed Hi-C and RNA-Seq on mOSNs overexpressing Tet3. Remarkably, despite the fact that all Pcdhα exons are bound by CTCF, and that the expression of the as-lncRNAs is maintained (Figure 7D and S7E), overexpression of Tet3 resulted in a strong bias in Pcdhα promoter/HS5-1 enhancer contacts, specifically biased towards the Pcdhα12 promoter (Figure 7E and 7F) and a concomitant bias in Pcdhα12 expression relative to all other Pcdhα exons, as determined by RNA-Seq (Figure 7G). Thus, CTCF bound to the CBS sites of Pcdhα12 created a “roadblock” for Cohesin, preventing the HS5-1 enhancer from engaging upstream Pcdhα promoters.
DISCUSSION
Stochastic, combinatorial expression of individual Pcdh protein isoforms in Purkinje (Esumi et al., 2005) and olfactory sensory neurons (Mountoufaris et al., 2017) generates distinct combinations of Protocadherin isoforms that function as a cell-surface identity code for individual neurons (Mountoufaris et al., 2018). This conclusion has been confirmed more broadly through single cell RNA sequencing studies in a variety of neuronal cell types (Tasic et al., 2018). Here we identify a mechanism by which Pcdhα alternate exon promoters are stochastically activated in individual neurons, and propose a model that may apply more broadly to regulate enhancer/promoter interactions and gene expression in vertebrates.
Insights into the mechanism of stochastic Pcdhα promoter choice
We provide evidence that stochastic activation of individual Pcdhα alternate promoters requires mechanistic coupling between transcription of a large multiply-spliced, polyadenylated antisense lncRNA and DNA demethylation of the Pcdhα promoters and CTCF binding sites (Figure 7H). Transcription of this lncRNA, initiated at the eCBS-proximal promoter, leads to the demethylation, de-repression and activation of Pcdhα proximal sense strand promoters. This step occurs coordinately with CTCF binding to its CBS sites located proximal to both promoters, and the formation of CTCF/Cohesin-dependent long-range DNA looping between the demethylated promoter and the HS5-1 enhancer. These observations are consistent with a promoter scanning mechanism in which the HS5-1 enhancer, bound by CTCF and Cohesin, translocates to the most enhancer-proximal demethylated and CTCF-bound promoter by DNA loop-extrusion, leading to the stochastic production of a specific Pcdhα mRNA (Figure 7H). Remarkably, the as-lncRNA initiated at a Pcdhα eCBS-proximal promoter transcribes through its cognate pCBS-proximal promoter and extends through upstream sense promoters. However, the only sense promoter that is activated in this process is the sense promoter immediately proximal to the antisense promoter. We speculate that this proximal specificity is a consequence of functional coupling between transcription and RNA processing mediated by the carboxy-terminal (CTD) of the RNAPII, the cap-binding complex and the spliceosome (Maniatis and Reed, 2002). In support of this hypothesis, we identified a highly conserved and active 5’ss immediately upstream of each pCBS site in the Pcdhα alternate exons (Figure 2E and 2F). Thus, the spliceosome may be recruited to the vicinity of the sense promoter by transcriptional read-through. While functional coupling between Tet-mediated DNA demethylation, CTCF and the spliceosome has been reported elsewhere (Marina and Oberdoerffer, 2016), additional studies will be required to test this hypothesis in the context of Pcdhα promoters.
A fundamental question raised by our model is how antisense promoters are stochastically activated in individual neurons during development. Given the observation that the ground state of the Pcdhα gene cluster is inactive and marked by 5mC in horizontal basal cells in the mouse olfactory epithelium, we speculate that activation of eCBS-proximal promoters in the Pcdhα gene cluster is regulated by transcription factors capable of binding methylated DNA. Finally, we note that the mechanism of stochastic promoter choice in the Pcdhα gene cluster must differ from the mechanism of promoter choice in the Pcdh β and γ gene clusters. Specifically, the β and γ gene clusters do not have CTCF binding sites within the alternate exons, and we have not detected as-lncRNAs in either the Pcdh β or γ gene clusters. Additional studies will be required to understand the mechanisms that underlie stochastic promoter choice in these Pcdh gene clusters.
The molecular logic of convergent promoters
Convergent transcription, such as the example described for the Pcdhα alternate exons, can produce long and stable antisense noncoding RNAs that overlap with the sense coding RNA (Brown et al., 2018). Interestingly, genes that are activated by antisense convergent RNA are characterized by an overall low level of expression of sense and antisense RNAs and, a unique chromatin signature that facilitates their transcription (Brown et al., 2018). We speculate that, at least in the case described here, low levels of RNA expression, together with differences in the chromatin environment in the two convergent promoters, permits the two convergent RNAPII to productively translocate along DNA without significant interference.
The example of convergent transcription described here also suggests a model in which noncoding antisense RNA transcription couples RNAPII activity to a DNA deoxygenase TET enzyme activity. We note that there are precedents for a transcription-dependent mechanism of transcriptional activation coupled to DNA demethylation. Specifically, transcription of the tumor suppressor gene, TCF21, is activated by the antisense lncRNA, TARID, whose transcription is initiated at an intronic promoter sequence located within the TCF21 gene (Arab et al., 2014). Transcription of TARID leads to the formation of promoter-associated R-loops. These DNA-RNA hybrids are recognized by the growth arrest and DNA damage protein 45A, GADD45A, which, in turns, mediates the recruitment of TET1 to drive TET-mediated DNA demethylation and activation of the TCF21 sense strand promoter (Arab et al., 2019). It is therefore possible that the same, or similar, mechanism is used for stochastic choice of Pcdhα promoters.
A general mechanism for stochastic promoter activation
We used the differentiating mouse olfactory epithelium as an in vivo model system for studying stochastic Pcdhα gene activation. We were therefore struck by the similarities in regulatory logic between Pcdhα and olfactory receptor (OR) promoter choice. In both cases, the ground state of the stochastically chosen promoters is repressed and inaccessible to transcriptional activator proteins. In the case of the Pcdhα gene cluster, this repression is mediated predominantly by DNA methylation (Tasic et al., 2002; Toyoda et al., 2014), while OR genes are repressed by the assembly of constitutive heterochromatin (Magklara et al., 2011). In both of these cases, however, repressive DNA or histone modifications are replaced by activating marks, concomitantly with selective binding of transcription factors that promote DNA looping between promoters and distant enhancers. As all the Pcdhα genes are clustered on a single chromosome, stochastic Pcdhα promoter choice is accomplished in cis via DNA looping to the enhancer. This mechanism of promoter choice differs from OR promoter choice, which has been shown to require the formation of a multi-chromosomal, multi-enhancer hub that activates only one out of 2800 OR alleles distributed throughout the genome (Markenscoff-Papadimitriou et al., 2014; Monahan et al., 2019). Most likely, reliance on cis versus trans interactions also explains why Pcdhα and OR genes require distinct mechanisms to achieve transcriptional stochasticity. In the case of Pcdhα genes, CTCF and Cohesin are critical for stochastic enhancer/promoter interactions. The proposed loop-extrusion mechanism allows the HS5-1 enhancer to scan the gene cluster locally for the most proximal promoter bound by CTCF. In contrast, OR enhancers cannot deploy loop-extrusion mechanisms to activate OR transcription because this process cannot accommodate trans chromosomal interactions, which may explain the absence of CTCF and Cohesin binding sites in OR enhancers and promoters (Monahan et al., 2019). Consequently, as Pcdhα choice relies on stable CTCF promoter binding, DNA demethylation provides an effective mechanism for stochastic promoter activation. An important consequence of this mechanism is that, since antisense transcription and DNA demethylation are coupled and appear to occur in a stochastic fashion, DNA loop-extrusion will not create a bias toward the selection of the Pcdhα promoter most proximal to the enhancer (Pcdhα13 and Pcdhα12 in human and mouse, respectively). Rather, DNA loop-extrusion identifies the promoter bound to CTCF, providing an elegant mechanism to overcome selection biases driven by genomic proximity. In fact, we have shown that such a bias occurs if as-lncRNA transcription and DNA demethylation are uncoupled. Finally, our experiments highlight another important property of the loop-extrusion-mediated promoter/enhancer complex mechanism: the dynamic nature of enhancer promoter interactions that requires continuous Cohesin expression even in post-mitotic cells. This observation is reminiscent of the cell-division-independent role of Cohesin in the expression of the T-cell receptor α locus (Seitan et al., 2011). Continual maintenance of promoter enhancer interactions is further highlighted by the striking observation that demethylation of all the Pcdhα promoters, after one is chosen, results in bias towards the HS5-1-proximal alternate promoters. These observations suggest that if Pcdhα promoter choice is stable for the life of OSNs, then a mechanism must be in place to prevent demethylation of the non-chosen promoters.
It remains to be seen if the proposed mechanism of stochastic Pcdhα choice is applicable to other clustered gene families where stochastic gene expression occurs. An interesting example of promoter stochasticity is the process of V(D)J recombination, whereby Cohesin-mediated loop-extrusion appears to bias RAG-mediated recombination of the variable Vh exons that are most proximal to the iEμ enhancer (Jain et al., 2018). However, even in this system, there is a set of Vh exons that recombine in a distance-independent fashion, which could be accomplished by similar molecular mechanisms as the ones described here, ensuring optimal diversity in the generation of immunoglubulins.
STAR METHODS
CONTACT FOR REAGENT AND RESORCE SHARING
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tom Maniatis (tm2472@cumc.columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and Cell culture
SK-N-SH cells (XX female) were purchased from ATCC and cultured in RPMI-1640 supplemented with 10% (vol/vol) FBS, 1X GlutaMax, 1mM sodium pyruvate, 1X non-essential amino acids, and 1% penicillin-streptomycin. HEK293T cells (XX female) were purchased from ATCC and cultured in DMEM supplemented with 10% (vol/vol) FBS, 1X GlutaMax, 1mM sodium pyruvate, 1X non-essential amino acids, and 1% penicillin-streptomycin. Cells were maintained at 37°C in a 5% (vol/vol) CO 2 incubator.
Generation of a CRISPR-inducible SK-N-SH cell line (SK-N-SH-iCas9)
CRISPR-inducible SK-N-SH cells were generated as previously described for Human pluripotent stem cells (hPSCs) (Zhu et al., 2014) with the following differences: (1) the Puro-Cas9 donor plasmid was substituted with a GFP-Cas9 donor plasmid and (2) the Neo-M2rtTA donor plasmid was substituted with a mCherry-M2rtTA donor plasmid. Dual color cells were sorted by flow cytometry and genotyped by PCR and further karyotyped.
Generation of SK-N-SH heterozygous for the Pcdhα cluster (SK-N-SH-αhet)
SK-N-SH-iCas9 cells were plated at 50% density in a 6-well dish, dox-induced (at a concentration of 2 mg/mL) for 48 hours (refresh Media with 1X RPMI with Dox for every day of induction). On days 3 and 5, the cells were transfected with 1 μg (total) of sgRNAs. On day 6, the GFP/mCherry positive and DAPI negative were single cells sorted on plates pre-coated with MEF feeder cells. The cells were allowed to grow for a month until visible colonies were observed, replica plated and genotyped by PCR. We isolated two clones (1 and 2) and named this cell line as SK-N-SH-αhet. Deletion of one copy of the Pcdhα cluster in the SK-N-SH-αhet1 clone was further confirmed by Sanger DNA sequencing and further karyotyped.
Animals
Mice were treated in compliance with the rules and regulations of IACUC under protocol number AC-AAAO3902. All experiments were performed on primary FACS-sorted cells from dissected main olfactory epithelium from animals (both male and female) of age between 4 to 12 weeks. HBC cells were sorted from keratin5-creER;rt-gfp mice, INP cells were sorted from the brightest GFP populations of ngn1-GFP mice, OSNs were sorted from omp-IRES-GFP mice. Rad21 conditional knockout mOSNs was achieved by crossing Rad21 conditional allele mice (Seitan et al., 2011) to OMP-ires-Cre mice (Omptm1(cre)Jae). Recombined cells were purified by including a Cre-inducible tdTomato allele (ROSA26-tdtomato, Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J ) in the cross and selecting tdTomato positive cells by FACS. Overexpression of Tet3 in mOSNs was achieved by crossing tetotet3-IRES-GFP to omptta mice to obtain tetotet3-IRES-GFP;omptta mice. Control mice were achieved by crossing tetoGFP to omptta mice to obtain tetoGFP;omptta mice. GFP positive cells were sorted by FACS for both tetotet3-IRES-GFP;omptta and tetoGFP;omptta mice. In the text and the figures, we refer to the Rad21 conditional knockout in mOSNs as Rad21 KO and the Tet3 overexpression in mOSNs as Tet3 overexpression.
METHODS DETAILS
Fluorescence activated cell sorting of HBCs, INPs and mOSNs
Cells were dissociated into a single-cell suspension by incubating freshly dissected main olfactory epithelium with papain for 40 minutes at 37°C according to the Worthington Papain Dissociation System. Following dissociation and filtering for three times through a 35 μm cell strainer, cells were resuspended in 1X PBS with 5% FBS. For in situ Hi-C and ChiP-Seq experiments, upon dissociation, cells were fixed with 1% formaldehyde for 10 minutes at room temperature. Formaldehyde was quenched by adding glycine to a final concentration of 0.125 M for 5 minutes at room temperature. Cells were then washed with 1X cold PBS and resuspended in 1X PBS with 5% FBS. Fluorescent cells were then sorted on a BD Aria II or Influx cell sorter.
Transfections of plasmids into HEK293T cells
One day prior to lipid-mediated transfection, HEK293T cells were seeded in a 6-well plate at a density of about 2 million cells per well. For plasmid DNA transfections, 3 μg of total DNA was added to 125 μL of Opti-MEM containing 5 μL of P300 reagent, followed by an addition 125 μL of Opti-MEM containing 7.5 μL of Lipofectamine 3000 per well. The two solutions were mixed and incubated at room temperature for 5 minutes and the solution was added dropwise to cells. Plates were then incubated at 37°C for 48 or 72 hours in a 5% CO2 incubator. After incubation, cells were harvested in 1 mL of TRIzol.
RNA isolation and sequencing
RNA was isolated using TRIzol. Cell lysate was extracted with bromo-chloropropane and RNA was precipitated with 100% isopropanol supplemented with 10 μg of glycoblue for 10 min at room temperature and then pelleted at 16,000 x g for 30 min at 4C. The RNA pellet was washed once with 75% ethanol and then resuspended in RNase-free water to a maximal concentration of 200ng/μl. Genomic DNA contaminants were removed by Turbo DNase. Removal of Turbo DNase was performed by phenol:chloroform extraction and RNA was precipitated as described above and resuspended in RNase-free water and stored at −80C. Sequencing libraries for total RNA and polyadenylated RNA from SK-N-SH cells and human neurons were made using the NEBNext Ultra II Directional RNA Library Prep Kit. Sequencing libraries for total RNA from HEK293T cells and the SK-N-SH-αhet clones were made using the SMARTer Stranded Total RNA-Seq Pico input mammalian RNA kit. The quality of all the libraries was assessed by bioanalyzer and quantified using a combination of bioanalyzer and qubit. Libraries were sequenced on a NEXT-Seq 500/550.
Design of the myBaits Capture Library
To overcome the low level of Pcdh expression in both primary neurons and SK-N-SH cells, we made use of an RNA-based enrichment strategy to capture pre-processed and mature RNA species. We refer to this approach as Capture RNA-Sequencing (cRNA-Seq) (see also Figure S1 for a schematic of the myBaits enrichment procedure).
myBaits targeted capture kits were designed and purchase from MYcroarray (Arbor Biosciences, http://www.arborbiosci.com). A total of 16,357 biotinylated RNA probes covering about 90.42% of the Pcdh α (chr5: 140159476-140429082, hg19) and γ (chr5:140705658-140911381, hg19) clusters were synthesized. We also designed baits for the CBX5 locus (chr12:54624724-54673956, hg19) to serve as a positive control for our enrichment protocol. Baits were design satisfying at least one of the following conditions:
- No blast hit with a Tm above 60°C
- No more than 2 hits at 62.5-65°C or 10 hits in the same interval and at least one neighbor candidate being rejected
- No more than 2 hits at 65-67.5°C and 10 hits at 62.5-65°C and two neighbor candidates on at least one side being rejected
- No more than a single hit at or above 70°C and no more than 1 hit at 65-67.5°C and 2 hits at 62.5-65°C and two neighbor candidates on at least one side being rejected
Sequencing libraries from RNA-Seq or HiC-Seq were multiplexed at the desired ratio and captured using the myBaits Capture Library protocol for 18 hours at 65°C. Captured libraries were eluted in RNase-free water and further amplified. The quality of captured libraries was assessed by bioanalyzer and quantified using a combination of bioanalyzer and qubit. Libraries were sequenced on a NEXT-Seq 500/550.
RNAPII pausing
Start-Seq experiments were previously described (Nechaev et al., 2010) with the following changes: (1) about 10 million SK-N-SH cells were used for each replicate experiment, (2) the 2 μl of RNA 5’ Pyrophosphohydrolase, RppH, (NEB M0356S, 5 U/μl) was used in conjunction with ThermoPol Buffer (NEB B9004) to remove the 5’cap to the short-RNAs for 1 hr at 37°C, (3) RNA-Seq libraries were prepared with the NEXTflex small RNA kit v3. Start-RNA libraries were sequenced using single-end 75-nt cycles on an Illumina NextSeq 500/550 instrument. The location of promoter-proximal RNAPII and the transcriptional start sites (TSS) were determined by analysis of the full-length reads.
RNAPII elongation
SK-N-SH cells were treated with 100 μM of 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) or DMSO for 6 hours to block phosphorylation of the carboxy-terminal domain (CTD) of RNAPII, which is required to release paused RNAPII from promoters in the transition from initiation to productive elongation. DRB inhibition is reversible, and upon removal from the cell culture media, a wave of newly transcriptionally elongating RNAPII leads to the incorporation of 4-thiouridine (s4U) into newly synthesized RNAs. s4U is rapidly incorporated into living cells without the need of cell lysis or nuclear isolation. Given the thiol-specific reactivity of s4U, s4U-labeled nascent RNA can be covalently and reversibly captured and sequenced. s4UDRB experiments were performed as previously described (Fuchs et al., 2014) with the following changes: 1 mM s4U was added to media 20 min before cells were harvested. After 6h, DRB and s4U-containing media was removed and replaced with s4U-containing media, and cells were harvested with TRIzol after 0, 8, or 20 min after DRB removal. Cells were flash frozen and stored at −80°C. A no DRB and a no s 4U controls were also performed.
Total RNA was purified and s4U-RNA was enriched using MTS-biotin chemistry (Duffy et al., 2015). Briefly, cells were lysed in TRIzol, extracted once with chloroform and the nucleic acids were precipitated with isopropanol. DNA was removed with Turbo DNase. DNase protein was removed by phenol:chloroform:isoamylalcohol extraction, and the RNA was isolated using isopropanol precipitation. RNA was sheared to ~200 bp by adding shearing buffer (150 mM Tris-HCl pH 8.3, 225 mM KCl, 9 mM MgCl2) and heating to 94 °C for 4 min, followed by quenching on ice with EDTA. Sheared RNA was purified using a modified protocol with the RNeasy Mini Kit (Qiagen). To biotinylate the s4U-RNA, 150 μg sheared RNA was incubated with 60 μg MTS-biotin in biotinylation buffer (150 μL total volume) for 30 min. Excess biotin was removed via chloroform extraction using Phase-Lock Gel Tubes. RNA was precipitated with a 1:10 volume of 3 M NaOAc and an equal volume of isopropanol and centrifuged at 20,000 x g for 20 min. The pellet was washed with an equal volume of 75% ethanol. Purified RNA was dissolved in 200 μl RNase-free water. Biotinylated RNA was separated from non-labeled RNA using glycogen-blocked Dynabeads Streptavidin C1 Beads (Invitrogen). Beads (200 μl were added to each sample and incubated for 15 min at room temperature, then washed three times with high salt wash buffer (1 ml each, 100 mM Tris-HCl (pH 7.4), 10 mM EDTA, 1 M NaCl, and 0.1% Tween-20). In order to improve the stringency of the washes, an additional three washes with buffer TE (10 mM Tris pH 7.4, 1 mM EDTA) at 55 °C were performed. s 4U-RNA was eluted from Dynabeads with 200 μl freshly prepared elution buffer (10 mM DTT, 100 mM NaCl, 10 mM Tris pH 7.4, 1 mM EDTA) and incubated for 15 min. Enriched RNA was purified by ethanol precipitation and re-biotinylated as above. Excess biotin was removed via chloroform extraction using Phase-Lock Gel Tubes and RNA was purified by RNeasy Mini Kit. s4U-RNA was enriched on streptavidin beads as above and beads were washed three times with high salt wash buffer. s4U-RNA was eluted as above and spiked with 200 pg Schizosaccharomyces pombe total RNA. 10 ng total RNA from input and enriched RNA samples was used for library preparation with the SMARTer Stranded Total RNA-seq Kit Pico Input Mammalian (Clontech) according to the manufacturer’s instructions. Input and enriched samples were multiplexed with Illumina barcodes and sequenced using paired-end 2 × 75-nt cycles on an Illumina NextSeq 500/550 instrument.
RNA half-life
SK-N-SH cells were treated with 100 μM DRB for 0, 15, 30, 60, 120, 240, 480, 960 minutes to inhibit transcription. Total RNA was purified as described above and levels of antisense lncRNA and sense cRNA were measured by qPCR.
Chromatin Immunoprecipitation (ChIP-Seq and ChIP-qPCR)
The following antibodies were used for chromatin immunoprecipitation studies: CTCF (donated by Victor Lobanenkov), Rad21 (Abcam ab992), Histone H3 Lysine 4 tri-methyl (ThermoFisher PA5-27029), Histone H3 Lysine 27 acetylation (Abcam ab4729), FLAG (Sigma F1804). With the exception of ChIP-Seq experiments for CTCF performed in mOSNs where ~1 million sorted cells were used per IP, about 5 million cells were used. Cells were crosslinked with 1% formaldehyde for 10 minutes at room temperature. Formaldehyde was quenched by adding glycine to a final concentration of 0.125 M for 5 minutes at room temperature. Cells were then washed with 1X cold PBS with protein inhibitors twice and pelleted. Cell pellets were stored at −80C till use. Cells were lysed in lysis buffer (50 mM Tris pH 7.5, 140 mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100) for 10 minutes. Nuclei were span for 10 minutes at 1000g and resuspended in the sonication buffer (10 mM Tris pH 7.5, 0.5% SDS) as 56 nuclei per 300 μl sonication buffer. Chromatin was sheared by Bioruptor for 30 cycles at cycling condition 30/30 (ON/OFF time in seconds). Following a spin at 13,000g for 10 minutes to remove debris, the sheared chromatin was diluted such as the final binding buffer concentration was 15 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS) and incubated for 2 hours with dynabeads G pre-equilibrated in the binding buffer for pre-clearing of the chromatin. Post-cleared chromatin was then incubated with the specific antibody overnight (1 μg of antibody was used per 56 nuclei). The next day, dynabeads G were added to the chromatin-antibody mix for 2 hours. A total of four washes with 1X wash buffer (100 mM Tris pH 7.5, 500 mM LiCl, 1% NP-40, 1% sodium deoxycholate) and one wash with TE buffer (10 mM Tris pH 7.5, 1 mM EDTA) were performed. The elution was performed at 65°C for 1 hour in the elution buffer (1% SDS, 250 mM NaCl, 2 mM DTT). All steps, with the exception of the elution, were performed at 4°C. All buffers, with the exception of the TE and elution buffer contained 1X protease inhibitors. The eluted chromatin was reverse-crosslinked overnight at 65°C and the DNA was purified with the Zymo DNA kit.
Libraries for ChIP-Seq were prepared using the NEBNext Ultra II DNA Library Prep Kit. The quality of the libraries was assessed by bioanalyzer and quantified using a combination of bioanalyzer and qubit. Libraries were sequenced on a NEXT-Seq 500/550.
In situ Chromatin Capture Conformation (Hi-C)
HEK293T cells transfected with dCas9-VPR-GFP plasmids were fixed with 1% formaldehyde and GFP-positive cells were FACS-sorted. About 500,000 cells (SK-N-SH or HEK293T) were lysed and intact nuclei were processed through an in situ Hi-C protocol as previously described with a few modifications (Rao et al., 2014). Briefly, cells were lysed with 50 mM Tris pH 7.5 0.5% Igepal, 0.25% Sodium-deoxychloate, 0.1% SDS, 150 mM NaCl, and protease inhibitors. Pelleted intact nuclei were then resuspended in 0.5% SDS and incubated for 20 minutes at 65°C for nuclear permeabilization . After quenching with 1.1% Triton-X for 10 minutes at 37°C, nuclei were digested with 6 U/ μl of Dpnll in 1x Dpnll buffer overnight at 37°C. Following initial digestion, a second Dpnll digestion was performed at 37°C for 2 hours. Dpnll was heat-inactivated at 65°C for 20 minutes. For the 1.5hr fill-in at 37°C, biotinylated dGTP was used instead of dATP to increase ligation efficiency. Ligation was performed at 25°C for 4 hours. Nuclei were then pelleted and sonicated in 10 mM Tris pH 7.5, 1 mM EDTA, 0.25% SDS on a Covaris S220 for 16 minutes with 2% duty cycle, 105 intensity, 200 cycles per burst, 1.8-1.85 W, and max temperature of 6°C. DNA was reverse cross-linked overnight at 65°C with proteinase K and RNAse A.
Reverse cross-linked DNA was purified with 2x Ampure beads following the standard protocol. Biotinylated fragments were enriched using Dynabeads MyOne Strepavidin T1 beads. The biotinylated DNA fragments were prepared for next-generation sequencing on the beads by using the Nugen Ovation Ultralow kit protocol with some modifications. Following end repair, magnetic beads were washed twice at 55°C with 0.05% Tween, 1 M NaCl in Tris/EDTA pH 7.5. Residual detergent was removed by washing the beads twice in 10 mM Tris pH 7.5. End repair buffers were replenished to original concentrations, but the enzyme and enhancer was omitted before adapter ligation. Following adaptor ligation, beads underwent five washes with 0.05% Tween, 1 M NaCl in Tris/EDTA pH 7.5 at 55°C and two washes with 10mM Tris pH 7.5. DNA was amplified by 10 cycles of PCR, irrespective of starting material. Beads were reclaimed and amplified unbiotinylated DNA fragments were purified with 0.8x Ampure beads. Quality and concentration of libraries were assessed by Agilent Bioanalyzer and Qubit. In situ Hi-C libraries from SK-N-SH and HEK293T cells were size-selected and enriched as described above using the myBaits Capture Library protocol described above and sequenced paired-end on NextSeq 500 (2x75bp).
Methylated DNA Immunoprecipitation (MeDIP)
The following antibodies were used: 5-Methylcytosine (5-mC) antibody (Active Motif 39649) and 5-Hydroxymethylcytosine (5-hmC) antibody (Active Motif 39791).
HEK293T cells were transfected with the appropriate set of dCas9 plasmids and incubated at 37°C for 72 hours in a 5% CO2 incubator. Genomic DNA was extracted using the PureLink Genomic DNA Mini Kit (Invitrogen). A total of 2 μg of DNA was diluted into 300 ml TE sonication buffer (10 mM Tris pH 7.5, 1 mM EDTA). Genomic DNA was sheared by Bioruptor for 18 cycles at cycling condition 30/90 (ON/OFF time in seconds). The sheared DNA was diluted to a final IP buffer of 15 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 and incubated overnight with 1 μg of antibody. The next day, a mixture of dynabeads A and G were added to the DNA-antibody mix for 2 hours. A total of three washes with 1X IP buffer were performed. The elution was performed at 55°C for 3 hours with rigorous shaking in the elution buffer (1% SDS, 250 mM NaCl). All steps, with the exception of the elution, were performed at 4°C. The eluted DNA was purified with the Zymo DNA kit.
Bisulfite DNA Reactions
Bisulfite DNA reactions were performed using the TrueMethyl oxBS module, Nugen, following the steps indicated by the protocol. Primers were designed using the MethPrimer. PCR products were cloned and sequenced (at least 15 clones per condition). Data were analyzed using QUMA (http://quma.cdb.riken.jp).
Immunofluorescence
The MOE was dissected from 14-week old Rad21 KO (Rad21-fl/fl;OMP-cre) mice and littermate controls (Rad21-fl/fl). Tissue was embedded in OCT and then coronal cryosections were collected at a thickness 12 mM. Tissue sections were air dried on slides for 10 minutes and then fixed with cold 4 % PFA for 10 minutes. After fixation, slides were washed with PBST (PBS with 0.1 % Triton X-100) and then stained with primary antibody for Rad21 (1:1000 dilution, Abcam Cat# ab42522, RRID: AB_945133) in PBST-DS overnight at 4°C. Slides were then washed, stained with DAPI (2.5 μg/mL) and the secondary antibody (Donkey anti-rabbit IgG conjugated to Alexa-488, diluted 1:1000, Thermo Fisher Scientific Cat# A-21206, RRID:AB_2535792) in PBST-DS for 1 hour, washed, and then mounted with Vectashield. Confocal images were collected with a Zeiss LSM 700 and image processing was carried out with ImageJ (NIH).
Bioinformatic Analysis of Sequencing Data
For RNA-Seq experiments, raw FASTQ files were aligned with either Tophat or STAR using hg19 or mm10 reference genomes. When libraries were made following the SMARTer Stranded Total RNA-Seq, the initial 4 base pairs of both paired reads were trimmed prior to alignment.
For ChiP-Seq experiments, raw FASTQ files were aligned using Bowtie2 using hg19 reference genome upon adapter sequences removal using CutAdapt. Uniquely aligning reads were selected using Samtools and reads with alignment quality below 30 (-q 30) were removed. The HOMER software package was used to generate signal tracks.
For in situ Hi-C experiments, raw FASTQ files were processed through use of the Juicer Tools Version 1.76 pipeline with one modification. Reads were aligned to hg38 using BWA 0.7.17 mem algorithm and specifying the −5 option implemented specifically for in situ Hi-C data. For captured Hi-C libraries, contact matrices were normalized to 2kb resolution by first reporting counts as reads per billion Hi-C contacts, then by normalizing with the Knight Ruiz (KR) matrix balancing algorithm focused on the alpha Pcdh cluster (chr5:140780000-141046000; hg38). For uncaptured libraries (mm10 Hi-C), matrices were KR normalized genome wide.
For generating a contact matrix, scales were set to a minimum of 0 reads and a maximum of 2*(mean normalized reads) in order to report a relative enrichment of contacts.
DNaseI and ChIP data for H3K4me3, CTCF, Rad21, ELF1, GABP, TCF12, MAX, YY1 in SK-N-SH cells were obtained from the ENCODE data matrix.
For Start-Seq experiments, raw FASTQ files were aligned using Bowtie2. TSS peaks were determined using Homer and the most abundant TSS reported in Figure 2.
In situ Hi-C data for INP and OSN cells were obtained from (Monahan et al., 2019).
CRISPR gRNA design
All guide RNA (gRNAs) were designed as truncated 18mer long sequences to increase their binding specificity using the CRISPR design web tool (http://crispr.mit.edu). With the exception of the Pcdhα9, where a total of two gRNAs were used to activate either the pCBS-proximal or the eCBS-proximal promoters, we used four gRNAs for the activation of the pCBS-proximal and eCBS-proximal promoters of Pcdh α4, α6, α12.
In vitro transcription of gRNAs
The gRNAs were transcribed using the MEGAshortscript T7 Transcription Kit by Life Technologies (AM1354M), purified by phenol-chloroform and transfected in the SK-N-SH-iCas9 cells by RNAimax lipofectamine reagent.
QUANTIFICATION AND STATISTICS
The statistical tests used in this study are indicated in the respective figure legends. In general, data with single independent experiments were analyzed by Student unpaired t-test to determine statistical significant effects (p < 0.05). Data with multiple independent experiments were analyzed by one-way ANOVA to determine statistical significant effects (p < 0.05).
DATA AND SOFTWARE AVAILABILITY
The data discussed in this work have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE115862.
Supplementary Material
Figure S1, Related to Figure 1: RNA-Sequencing and Capture RNA-Sequencing.
(A) Schematic diagram of Capture RNA-Sequencing (cRNA-Seq). The white, pink and blue bars indicate RNA from the Pcdh α, β and γ gene clusters, respectively. The brown bars indicate RNA from the rest of the genome. (B) RNA-Seq and cRNA-Seq from total RNA isolated from SK-N-SH cells. Red bar: myBaits for the Pcdh α and γ clusters. (C) Enrichment of Pcdh α and γ RNAs by cRNA-Seq. The CBX5 gene was used as a positive control for our capture procedure as we developed myBaits probes to enrich for RNA molecules expressed from the CBX5 locus as well. (D) Sense and antisense RNA reads sequenced by either RNA-Seq or cRNA-Seq. (E) Expression values (RPKM) for as-lncRNA and s-cRNA expressed from Pcdhα4 and Pcdhα12 in SK-N-SH cells. (F) Half-lives (minutes) for as-lncRNA and s-cRNA expressed from Pcdhα4 and Pcdhα12 in SK-N-SH cells.
{kind=link}
Figure S2, Related to Figure 2: Expression of convergent Pcdhα antisense and sense RNA in human and mouse primary neurons
(A) Start-Seq signal from two biological replicate experiments in SK-N-SH cells ranked by decreasing read density relative to known transcriptional start sites (TSS) genome-wide. (B) Polyadenylated (PolyA) RNA and Total RNA from human primary neurons (cRNA-Seq). (C) Polyadenylated (PolyA) RNA and Total RNA from mouse olfactory sensory neurons (RNA-Seq). (D) DNaseI hypersensitivity and ChiP-Seq data for distinct transcription factors associated with the active exons by the pCBS-proximal and eCBS-proximal promoters in SK-N-SH cells. Specifically, TFs belonging to the ETS family bind to the pCBS-proximal promoter, while TFs belonging to the bHLH family bind to the eCBS-proximal promoter. It is interesting to note that both of these classes of TFs are implicated in regulating genes involved in neuronal development and differentiation, such as members of the cell-adhesion protein family (Hollenhorst et al., 2011).
For (B) and (C), the x-axis represents the linear sequence of the genomic organization of the human (B) and mouse (C) Pcdhα gene cluster and the numbers on the left-hand side of each track represent the minimum and maximum densities in read per million.
{kind=link}
Figure S3, Related to Figure 3: Recruitment of dCas9-VPR to Pcdhα sense and antisense promoters
(A) Location of the gRNAs used to activate Pcdh α4, α6, α9 and α12, relative to their respective pCBS and the eCBS sites. (B) dCas9-VPR is recruited at the Pcdh α4 and α12 pCBS-proximal and eCBS-proximal promoters. H3K4me3, Rad21 and CTCF (ChIP-Seq) from parental HEK293T cells. The x-axis represents the linear sequence of the genomic organization of the Pcdhα human cluster and the numbers on the left-hand side of each track represent the minimum and maximum densities in read per million. (B) Zoom-in of the dCas9-VPR ChIP-Seq tracks from (B) for Pcdh α4 (Left) and Pcdh α12 (Right).
{kind=link}
Figure S4, Related to Figure 4: Functional outcomes of the activation of sense and antisense promoters by dCas9-VPR
(A) Activation of the pCBS-proximal and eCBS-proximal Pcdhα4 promoters by a single dCas9-VPR protein. (B) Percent of uniquely aligned reads from cRNA-Seq for the Pcdh α and γ gene cluster and the CBX5 locus for HEK293T cells (black) and HEK293T cells transfected with gRNA activating the eCBS-proximal promoter of α6 (Red), α9 (green) and α12 (blue). The primary data are shown in Figure 4C. (B) Percent input of CTCF occupancy, as determined by ChIP-qPCR, at the GAPDH promoter (positive control) and at an intergenic DNA region (negative control) for the experiments shown in Figure 5A where the pCBS-proximal (grey) and the eCBS-proximal (green) promoters of Pcdh α4, α6, α9, α12 are activated by dCas9-VPR. Errors (n=3) represent s.e.m. and statistical significance was calculated with a Student unpaired t-test. (D) In situ cHi-C cumulative maps for the pCBS-proximal promoter activation (Top) and eCBS-proximal promoter activation (Bottom) of Pcdhα12. (E) Schematics of the specificity score used to calculate novel specific contacts between the CTCF binding sites of Pcdhα12 and the HS5-1 enhancer. The specificity score indicates the signal-to-noise ratio of the interaction in a 15 kb window at 5 kb resolution. (F) cHi-C contacts between the Pcdhα12 promoter and the HS5-1 enhancer: pCBS-proximal promoter activation (grey); eCBS-proximal promoter activation (green); recruitment of dCas9 (without the VPR activator) to the eCBS-proximal promoter (pink). Y-axis indicated total cHiC contacts.
{kind=link}
Figure S5, Related to Figure 5: Antisense lncRNA transcription mediates DNA demethylation of Pcdhα promoters
(A and B) Top: Schematics of the pCBS and the eCBS relative to the 11 Zinc fingers of the CTCF protein. Module 2 and 3 represent the core CBS motif. Bottom: DNA Logo for the human Pcdhα pCBS and eCBS sites. CTCF binding to the core CBS motif is significantly affected by DNA methylation at position 2 and 12. (C and D) Nucleotide resolution of the percent CpG DNA methylation of the pCBS and eCBS of Pcdh α4, α12 (ON exons) and α3, α13 (OFF exons) in SK-N-SH cells as determined by whole-genome bisulfite sequencing (WGBS). (E) Average of percentage of CpG methylation at the pCBS (Top) and eCBS (Bottom) of active (ON) and inactive Pcdhα exons (OFF) assayed by WGBS relative to CTCF occupancy of those sites assayed by ChIP-Seq in SK-N-SH cells. (F) Percent CpG DNA methylation of the DNA sequence between the two CBSs (middle) in Pcdh α4, α12 (ON) and α3, α13 (OFF) in SK-N-SH cells. (G) Average of percent CpG methylation of the DNA sequence between the two CBSs of active (ON) and inactive Pcdhα exons (OFF). (H) Nucleotide resolution of the percent CpG DNA methylation of the eCBS of Pcdhα12 in HEK293T cells upon sense promoter activation (Top) and antisense promoter activation (Bottom) assayed by bisulfite sequencing.
{kind=link}
Figure S6, Related to Figure 6: DNA demethylation correlates with Pcdhα expression in vivo
(A) Changes in 5hmC (x-axis) relative to the expression of the s-cRNA (left y-axis, grey) and the as-lncRNA (right x-axis, black) during the maturation of olfactory sensory neurons. Data for Pcdh α3, α5, α7 and α10 are shown. (B) In situ Hi-C contact maps at 10kb resolution for horizontal basal cells (ICAM1+, Top), immediate neural precursors (Ngn1+, Middle) and mature olfactory sensory neurons (Omp+, Bottom).
{kind=link}
Figure S7, Related to Figure 7: Rad21 knockout and Tet3 overexpression in mature olfactory sensory neurons
(A) Log2 fold change for Rad21 from Rad21fl/fl;OMPcre mice and Tet3 from tetotet3iresGFP;omptta relative to mOSNs from control mice. (B) Rad21 immunofluorescence (green) in MOE sections from 14-week-old control (Rad21-fl/fl) and Rad21 KO (Rad21 fl/fl;OMPcre) mice. Nuclei are stained with DAPI (magenta). Rad21 is lost from mOSNs but retained in apical sustentactular cells and basal immature cells. Scale bar = 20 μm. (C) Average of cumulative RPM values for the Pcdhα alternate promoters/exons for 5hmC for horizontal basal cells (HBC), immediate neural precursors (INP), and control or Tet3 overexpressing mature olfactory sensory neurons (mOSN). (D) CTCF profiles in mOSNs (Left) and mOSNs overexpressing Tet3 (Right) as measured by ChIP-Seq. (E) RNA-Seq profiles for s-cRNA (grey) and as-lncRNA (black) in mOSNs and mOSNs upon Tet3 overexpression. The x-axis represents the linear sequence of the genomic organization of the mouse Pcdhα cluster and the numbers on the left-hand side of each track represent the minimum and maximum densities in read per million.
{kind=link}
Supplemental Data Table 3, Related to Figures 3, 6 and 7: Table of sequence processing steps for the Hi-C experiments
Supplemental Data Table 1, Related to Figures 3 and 5: Table of the primers used in this study
Supplemental Data Table 2, Related to Figures 3, 4 and 5: Table of the guide RNAs (gRNAs) used in this study
Highlights.
A conserved antisense promoter is located within each of the Pcdhα alternate exons
Antisense lncRNA transcription leads to DNA demethylation of promoters and CBSs
CTCF/Cohesin drive the assembly of Pcdhα promoter/enhancer complex via loop-extrusion
Coupling lncRNA transcription to DNA demethylation ensures stochastic promoter choice
ACKNOWLEDGEMENTS
We thank Drs. Richard Axel, Charles Zuker, Max Gottesman, David Hirsh, Germano Cecere, George Mountoufaris, Enrico Cannavo and members of the Maniatis, Lomvardas and Simon labs for critical discussions and suggestions on the manuscript. D.C. and T.M. would like to thank Dr. Karen Adelman for advice with the Start-Seq experiment, Dr. Ye Zhang and Dr. Ben Barres for the generous gift of human brain neurons, Dr. Victor Lobanenkov and Dr. Elena Pugacheva for the CTCF monoclonal antibody and, Ira Schieren for assistance with flow cytometry. This work was supported by a Helen Hay Whitney Postdoctoral Fellowship (D.C. and R.D.), an NIH F32 Postdoctoral Fellowship GM108474 (K.M.), an NIH F31 Predoctoral Fellowship DC016785 (A.H.), an NIH Path to Independence Award K99/R00 K99GM121815 (D.C.), NIH New Innovator Award DP2 HD083992-01 (M.D.S.), NIH grants 1R01MH108579 and 5R01NS088476 (T.M.), and NIH grant R01DC013560 and HHMI Faculty Scholar (SL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Arab K, Karaulanov E, Musheev M, Trnka P, Schaefer A, Grummt I, and Niehrs C (2019). GADD45A binds R-loops and recruits TET1 to CpG island promoters. Nat Genet 51, 217–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arab K, Park YJ, Lindroth AM, Schafer A, Oakes C, Weichenhan D, Lukanova A, Lundin E, Risch A, Meister M, et al. (2014). Long noncoding RNA TARID directs demethylation and activation of the tumor suppressor TCF21 via GADD45A. Mol. Cell 55, 604–614. [DOI] [PubMed] [Google Scholar]
- Bell AC, and Felsenfeld G (2000). Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485. [DOI] [PubMed] [Google Scholar]
- Brown T, Howe FS, Murray SC, Wouters M, Lorenz P, Seward E, Rata S, Angel A, and Mellor J (2018). Antisense transcription-dependent chromatin signature modulates sense transcript dynamics. Mol. Syst. Biol 14, e8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E, Lin S, Kiani S, Guzman CD, Wiegand DJ, et al. (2015). Highly efficient Cas9-mediated transcriptional programming. Nat Meth 12, 326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquitt BM, Allen WE, Barnea G, and Lomvardas S (2013). Alteration of genic 5-hydroxymethylcytosine patterning in olfactory neurons correlates with changes in gene expression and cell identity. Proc. Natl. Acad. Sci. U.S.a 110, 14682–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, and Simon MD (2015). Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol. Cell 59, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esumi S, Kakazu N, Taguchi Y, Hirayama T, Sasaki A, Hirabayashi T, Koide T, Kitsukawa T, Hamada S, and Yagi T (2005). Monoallelic yet combinatorial expression of variable exons of the protocadherin-alpha gene cluster in single neurons. Nat Genet 37, 171–176. [DOI] [PubMed] [Google Scholar]
- Fuchs G, Voichek Y, Benjamin S, Gilad S, Amit I, and Oren M (2014). 4sUDRB-seq: measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome Biol 15, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell Reports 15, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirlando R, and Felsenfeld G (2016). CTCF: making the right connections. Genes & Development 30, 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Maniatis T, Monahan K, Myers RM, Monahan K, Wu H, Gertz J, Varley KE, Li W, Myers RM, et al. (2012). CTCF/cohesin-mediated DNA looping is required for protocadherin promoter choice. Proceedings of the National Academy of Sciences 109, 21081–21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, et al. (2015). CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. 162, 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa S, Hamada S, Kumode Y, Esumi S, Katori S, Fukuda E, Uchiyama Y, Hirabayashi T, Mombaerts P, and Yagi T (2008). The protocadherin-alpha family is involved in axonal coalescence of olfactory sensory neurons into glomeruli of the olfactory bulb in mouse. Mol. Cell. Neurosci 38, 66–79. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Tarusawa E, Yoshimura Y, Galjart N, and Yagi T (2012). CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Reports 2, 345–357. [DOI] [PubMed] [Google Scholar]
- Hollenhorst PC, McIntosh LP, and Graves BJ (2011). Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem 80, 437–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Ba Z, Zhang Y, Dai H-Q, and Alt FW (2018). CTCF-Binding Elements Mediate Accessibility of RAG Substrates During Chromatin Scanning. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi M, Toyama T, Kaneko R, Hirayama T, Kawamura Y, and Yagi T (2008). Relationship between DNA methylation states and transcription of individual isoforms encoded by the protocadherin-alpha gene cluster. Journal of Biological Chemistry 283, 12064–12075. [DOI] [PubMed] [Google Scholar]
- Kehayova P, Monahan K, Chen W, and Maniatis T (2011). Regulatory elements required for the activation and repression of the protocadherin-alpha gene cluster. Proceedings of the National Academy of Sciences 108, 17195–17200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JL, Sanes JR, and Kay JN (2015). Development of Dendritic Form and Function. Annu. Rev. Cell Dev. Biol 31, 741–777. [DOI] [PubMed] [Google Scholar]
- Li X, Yue X, Pastor WA, Lin L, Georges R, Chavez L, Evans SM, and Rao A (2016). Tet proteins influence the balance between neuroectodermal and mesodermal fate choice by inhibiting Wnt signaling. Proc. Natl. Acad. Sci. U.S.a 113, E8267–E8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipovich L, Vanisri RR, Kong SL, Lin C-Y, and Liu ET (2006). Primate-specific endogenous cis-antisense transcription in the human 5q31 protocadherin gene cluster. J. Mol. Evol 62, 73–88. [DOI] [PubMed] [Google Scholar]
- Magklara A, Yen A, Colquitt BM, Clowney EJ, Magklara A, Markenscoff-Papadimitriou E, Evans ZA, Kheradpour P, Mountoufaris G, Carey C, et al. (2011). An epigenetic signature for monoallelic olfactory receptor expression. Cell 145, 555–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, and Reed R (2002). An extensive network of coupling among gene expression machines. Nature 416, 499–506. [DOI] [PubMed] [Google Scholar]
- Marina RJ, and Oberdoerffer S (2016). Epigenomics meets splicing through the TETs and CTCF. Cc 15, 1397–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markenscoff-Papadimitriou E, Allen WE, Colquitt BM, Goh T, Murphy KK, Monahan K, Mosley CP, Ahituv N, and Lomvardas S (2014). Enhancer interaction networks as a means for singular olfactory receptor expression. Cell 159, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan K, Horta A, and Lomvardas S (2019). LHX2- and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature 565, 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan K, Rudnick ND, Kehayova PD, Pauli F, Newberry KM, Myers RM, and Maniatis T (2012). Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of protocadherin-α gene expression. Proc. Natl. Acad. Sci. U.S.a 109, 9125–9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountoufaris G, Canzio D, Nwakeze CL, Chen WV, and Maniatis T (2018). Writing, Reading, and Translating the Clustered Protocadherin Cell Surface Recognition Code for Neural Circuit Assembly. Annu. Rev. Cell Dev. Biol 34, 471–493. [DOI] [PubMed] [Google Scholar]
- Mountoufaris G, Chen WV, Hirabayashi Y, O’Keeffe S, Chevee M, Nwakeze CL, Polleux F, and Maniatis T (2017). Multicluster Pcdh diversity is required for mouse olfactory neural circuit assembly. Science 356, 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Fargo DC, Santos dos G, Liu L, Gao Y, and Adelman K (2010). Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 327, 335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan JP, Li J, Nguyen L, Caoile C, Dickson M, Grimwood J, Schmutz J, Feldman MW, and Myers RM (2003). Extensive linkage disequilibrium, a common 16.7-kilobase deletion, and evidence of balancing selection in the human protocadherin alpha cluster. Am. J. Hum. Genet 72, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong C-T, and Corces VG (2014). CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribich S, Tasic B, and Maniatis T (2006). Identification of long-range regulatory elements in the protocadherin-alpha gene cluster. Proceedings of the National Academy of Sciences 103, 19719–19724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitan VC, Hao B, Tachibana-Konwalski K, Lavagnolli T, Mira-Bontenbal H, Brown KE, Teng G, Carroll T, Terry A, Horan K, et al. (2011). A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature 476, 467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Nabholz CE, Baldwin KK, Kim Y, Rueckert EH, Ribich SA, Cramer P, Wu Q, Axel R, and Maniatis T (2002). Promoter choice determines splice site selection in protocadherin alpha and gamma pre-mRNA splicing. Mol. Cell 10, 21–33. [DOI] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Smith KA, Graybuck L, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 229542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda S, Okano M, Tarusawa E, Kawaguchi M, Hirabayashi M, Kobayashi T, Toyama T, Oda M, Nakauchi H, Yoshimura Y, et al. (2014). Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron 82, 94–108. [DOI] [PubMed] [Google Scholar]
- Vian L, Pekowska A, Rao SSP, Kieffer-Kwon K-R, Jung S, Baranello L, Huang SC, Khattabi El L., Dose M, Pruett N, et al. (2018). The Energetics and Physiological Impact of Cohesin Extrusion. Cell 173, 1165–1178.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. (2012). Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 22, 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su H, and Bradley A (2002). Molecular mechanisms governing Pcdh-gamma gene expression: evidence for a multiple promoter and cis-alternative splicing model. Genes & Development 16, 1890–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, and Maniatis T (1999). A striking organization of a large family of human neural cadherin-like cell adhesion genes. 97, 779–790. [DOI] [PubMed] [Google Scholar]
- Wu X, and Zhang Y (2017). TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18, 517–534. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Gonzalez F, and Huangfu D (2014). The iCRISPR platform for rapid genome editing in human pluripotent stem cells. Meth. Enzymol 546, 215–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipursky SL, and Grueber WB (2013). The molecular basis of self-avoidance. Annu. Rev. Neurosci 36, 547–568. [DOI] [PubMed] [Google Scholar]
- Zipursky SL, and Sanes JR (2010). Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. 143, 343–353. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, Related to Figure 1: RNA-Sequencing and Capture RNA-Sequencing.
(A) Schematic diagram of Capture RNA-Sequencing (cRNA-Seq). The white, pink and blue bars indicate RNA from the Pcdh α, β and γ gene clusters, respectively. The brown bars indicate RNA from the rest of the genome. (B) RNA-Seq and cRNA-Seq from total RNA isolated from SK-N-SH cells. Red bar: myBaits for the Pcdh α and γ clusters. (C) Enrichment of Pcdh α and γ RNAs by cRNA-Seq. The CBX5 gene was used as a positive control for our capture procedure as we developed myBaits probes to enrich for RNA molecules expressed from the CBX5 locus as well. (D) Sense and antisense RNA reads sequenced by either RNA-Seq or cRNA-Seq. (E) Expression values (RPKM) for as-lncRNA and s-cRNA expressed from Pcdhα4 and Pcdhα12 in SK-N-SH cells. (F) Half-lives (minutes) for as-lncRNA and s-cRNA expressed from Pcdhα4 and Pcdhα12 in SK-N-SH cells.
Figure S2, Related to Figure 2: Expression of convergent Pcdhα antisense and sense RNA in human and mouse primary neurons
(A) Start-Seq signal from two biological replicate experiments in SK-N-SH cells ranked by decreasing read density relative to known transcriptional start sites (TSS) genome-wide. (B) Polyadenylated (PolyA) RNA and Total RNA from human primary neurons (cRNA-Seq). (C) Polyadenylated (PolyA) RNA and Total RNA from mouse olfactory sensory neurons (RNA-Seq). (D) DNaseI hypersensitivity and ChiP-Seq data for distinct transcription factors associated with the active exons by the pCBS-proximal and eCBS-proximal promoters in SK-N-SH cells. Specifically, TFs belonging to the ETS family bind to the pCBS-proximal promoter, while TFs belonging to the bHLH family bind to the eCBS-proximal promoter. It is interesting to note that both of these classes of TFs are implicated in regulating genes involved in neuronal development and differentiation, such as members of the cell-adhesion protein family (Hollenhorst et al., 2011).
For (B) and (C), the x-axis represents the linear sequence of the genomic organization of the human (B) and mouse (C) Pcdhα gene cluster and the numbers on the left-hand side of each track represent the minimum and maximum densities in read per million.
Figure S3, Related to Figure 3: Recruitment of dCas9-VPR to Pcdhα sense and antisense promoters
(A) Location of the gRNAs used to activate Pcdh α4, α6, α9 and α12, relative to their respective pCBS and the eCBS sites. (B) dCas9-VPR is recruited at the Pcdh α4 and α12 pCBS-proximal and eCBS-proximal promoters. H3K4me3, Rad21 and CTCF (ChIP-Seq) from parental HEK293T cells. The x-axis represents the linear sequence of the genomic organization of the Pcdhα human cluster and the numbers on the left-hand side of each track represent the minimum and maximum densities in read per million. (B) Zoom-in of the dCas9-VPR ChIP-Seq tracks from (B) for Pcdh α4 (Left) and Pcdh α12 (Right).
Figure S4, Related to Figure 4: Functional outcomes of the activation of sense and antisense promoters by dCas9-VPR
(A) Activation of the pCBS-proximal and eCBS-proximal Pcdhα4 promoters by a single dCas9-VPR protein. (B) Percent of uniquely aligned reads from cRNA-Seq for the Pcdh α and γ gene cluster and the CBX5 locus for HEK293T cells (black) and HEK293T cells transfected with gRNA activating the eCBS-proximal promoter of α6 (Red), α9 (green) and α12 (blue). The primary data are shown in Figure 4C. (B) Percent input of CTCF occupancy, as determined by ChIP-qPCR, at the GAPDH promoter (positive control) and at an intergenic DNA region (negative control) for the experiments shown in Figure 5A where the pCBS-proximal (grey) and the eCBS-proximal (green) promoters of Pcdh α4, α6, α9, α12 are activated by dCas9-VPR. Errors (n=3) represent s.e.m. and statistical significance was calculated with a Student unpaired t-test. (D) In situ cHi-C cumulative maps for the pCBS-proximal promoter activation (Top) and eCBS-proximal promoter activation (Bottom) of Pcdhα12. (E) Schematics of the specificity score used to calculate novel specific contacts between the CTCF binding sites of Pcdhα12 and the HS5-1 enhancer. The specificity score indicates the signal-to-noise ratio of the interaction in a 15 kb window at 5 kb resolution. (F) cHi-C contacts between the Pcdhα12 promoter and the HS5-1 enhancer: pCBS-proximal promoter activation (grey); eCBS-proximal promoter activation (green); recruitment of dCas9 (without the VPR activator) to the eCBS-proximal promoter (pink). Y-axis indicated total cHiC contacts.
Figure S5, Related to Figure 5: Antisense lncRNA transcription mediates DNA demethylation of Pcdhα promoters
(A and B) Top: Schematics of the pCBS and the eCBS relative to the 11 Zinc fingers of the CTCF protein. Module 2 and 3 represent the core CBS motif. Bottom: DNA Logo for the human Pcdhα pCBS and eCBS sites. CTCF binding to the core CBS motif is significantly affected by DNA methylation at position 2 and 12. (C and D) Nucleotide resolution of the percent CpG DNA methylation of the pCBS and eCBS of Pcdh α4, α12 (ON exons) and α3, α13 (OFF exons) in SK-N-SH cells as determined by whole-genome bisulfite sequencing (WGBS). (E) Average of percentage of CpG methylation at the pCBS (Top) and eCBS (Bottom) of active (ON) and inactive Pcdhα exons (OFF) assayed by WGBS relative to CTCF occupancy of those sites assayed by ChIP-Seq in SK-N-SH cells. (F) Percent CpG DNA methylation of the DNA sequence between the two CBSs (middle) in Pcdh α4, α12 (ON) and α3, α13 (OFF) in SK-N-SH cells. (G) Average of percent CpG methylation of the DNA sequence between the two CBSs of active (ON) and inactive Pcdhα exons (OFF). (H) Nucleotide resolution of the percent CpG DNA methylation of the eCBS of Pcdhα12 in HEK293T cells upon sense promoter activation (Top) and antisense promoter activation (Bottom) assayed by bisulfite sequencing.
Figure S6, Related to Figure 6: DNA demethylation correlates with Pcdhα expression in vivo
(A) Changes in 5hmC (x-axis) relative to the expression of the s-cRNA (left y-axis, grey) and the as-lncRNA (right x-axis, black) during the maturation of olfactory sensory neurons. Data for Pcdh α3, α5, α7 and α10 are shown. (B) In situ Hi-C contact maps at 10kb resolution for horizontal basal cells (ICAM1+, Top), immediate neural precursors (Ngn1+, Middle) and mature olfactory sensory neurons (Omp+, Bottom).
Figure S7, Related to Figure 7: Rad21 knockout and Tet3 overexpression in mature olfactory sensory neurons
(A) Log2 fold change for Rad21 from Rad21fl/fl;OMPcre mice and Tet3 from tetotet3iresGFP;omptta relative to mOSNs from control mice. (B) Rad21 immunofluorescence (green) in MOE sections from 14-week-old control (Rad21-fl/fl) and Rad21 KO (Rad21 fl/fl;OMPcre) mice. Nuclei are stained with DAPI (magenta). Rad21 is lost from mOSNs but retained in apical sustentactular cells and basal immature cells. Scale bar = 20 μm. (C) Average of cumulative RPM values for the Pcdhα alternate promoters/exons for 5hmC for horizontal basal cells (HBC), immediate neural precursors (INP), and control or Tet3 overexpressing mature olfactory sensory neurons (mOSN). (D) CTCF profiles in mOSNs (Left) and mOSNs overexpressing Tet3 (Right) as measured by ChIP-Seq. (E) RNA-Seq profiles for s-cRNA (grey) and as-lncRNA (black) in mOSNs and mOSNs upon Tet3 overexpression. The x-axis represents the linear sequence of the genomic organization of the mouse Pcdhα cluster and the numbers on the left-hand side of each track represent the minimum and maximum densities in read per million.
Supplemental Data Table 3, Related to Figures 3, 6 and 7: Table of sequence processing steps for the Hi-C experiments
Supplemental Data Table 1, Related to Figures 3 and 5: Table of the primers used in this study
Supplemental Data Table 2, Related to Figures 3, 4 and 5: Table of the guide RNAs (gRNAs) used in this study
Data Availability Statement
The data discussed in this work have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE115862.