

SUMMARY
Argonaute proteins loaded with microRNAs (miRNAs) or small interfering RNAs (siRNAs) form the RNA-Induced Silencing Complex (RISC), which represses target RNA expression. Predicting the biological targets, specificity, and efficiency of both miRNAs and siRNAs has been hamstrung by an incomplete understanding of the sequence determinants of RISC binding and cleavage. We applied high-throughput methods to measure the association kinetics, equilibrium binding energies, and single-turnover cleavage rates of RISC. We find that RISC readily tolerates insertions of up to seven nucleotides in its target opposite the central region of the guide. Our data uncover specific guide:target mismatches that enhance the rate of target cleavage, suggesting novel siRNA design strategies. Using these data, we derive quantitative models for RISC binding and target cleavage and show that our in vitro measurements and models predict knockdown in an engineered cellular system.
Graphical Abstract
eTOC blurb
By high-throughput, quantitative characterization of binding and cleavage for >40,000 distinct RISC targets, Becker et al. reveal principles of miRNA regulation and siRNA function. These data enable construction of quantitative models of binding and cleavage and are used to explain mRNA knockdown in cells.
INTRODUCTION
In eukaryotes, Argonaute (AGO) proteins loaded with ~21–23 nt RNA guides form the RNA-induced silencing complex (RISC). In the RNA interference (RNAi) pathway, small interfering RNAs (siRNAs) direct RISC to bind and cleave extensively complementary RNA targets at the phosphodiester bond opposite guide positions g10 and g11 (Hammond et al., 2000; Zamore et al., 2000; Elbashir et al., 2001). Synthetic siRNAs have become a mainstay of molecular biological research, and the first siRNA drug was approved in 2018 (Wittrup and Lieberman, 2015; Chakraborty et al., 2017; Dowdy, 2017; Adams et al., 2018). In contrast, animal microRNAs (miRNAs) bind targets through their seed sequence—guide bases g2–g7—and accelerate target degradation (Baek et al., 2008; Selbach et al., 2008; Guo et al., 2010) or inhibit mRNA translation (Doench, 2003; Doench and Sharp, 2004; Hendrickson et al., 2009; Bazzini et al., 2012).
AGO proteins assign distinct functions to regions of the small RNA (Lewis et al., 2003; Krek et al., 2005; Wee et al., 2012; Salomon et al., 2015). By pre-organizing guide nucleotides g2–g5 into the seed sequence (Wang et al., 2008; Elkayam et al., 2012; Schirle and MacRae, 2012; Schirle et al., 2014), AGOs reduce the entropic penalty of target binding and accelerate RNA hybridization to near-diffusion-limited rates (Parker et al., 2009; Salomon et al., 2015).
The sequence determinants of RISC binding and cleavage have been identified chiefly from low-throughput, quantitative biochemical assays (Wee et al., 2012; Deerberg et al., 2013; Jo et al., 2015; Salomon et al., 2015). These methods sample a small portion of the sequence space occupied by RISC targets. Conversely, high-throughput sequencing methods that rely on crosslinking miRNAs to their targets (Chi et al., 2009; Helwak et al., 2013; Clark et al., 2014) cannot measure the effect of target sequence on binding kinetics. The lack of high-throughput data precludes accurate prediction of RISC targets from fundamental biophysical principles, frustrating efforts to predict the identity and extent of repression of miRNA targets and limiting the design of specific and potent siRNAs.
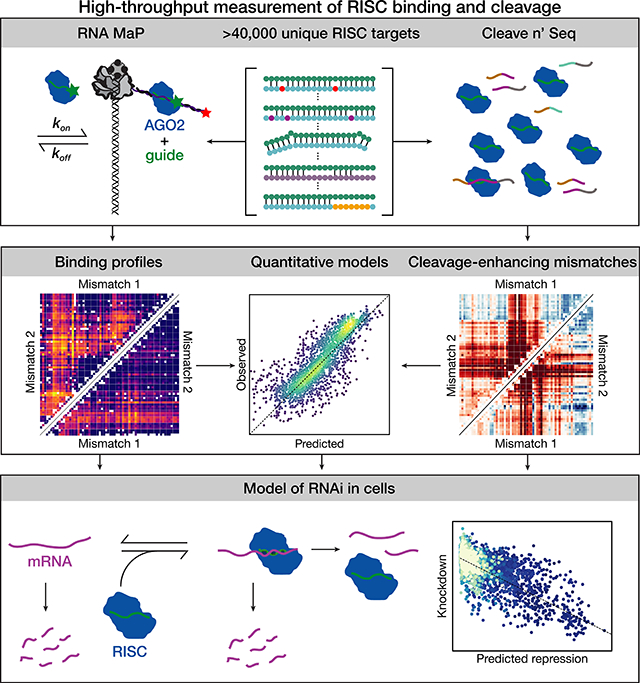
Here, we report RISC binding affinity, association kinetics, and rates of cleavage for ~20,000 target variants of two miRNAs: let-7a and miR-21. We use target RNA libraries comprising both canonical and noncanonical targets, to probe the effects of mismatches, guide and target bulges, and local secondary structure. Our data highlight the distinct binding determinants for let-7a and miR-21: let-7a binds mainly via its seed, whereas miR-21 binding requires both seed and 3′ supplemental pairing. Remarkably, RISC binding tolerates as many as seven nucleotides inserted into the target across from the central region of the guide. Finally, we describe specific mismatches flanking the cleavage site and at the 3′ end of the guide that enhance target cleavage. Our quantitative, high-throughput biophysical measurements allow us to construct predictive models for target binding and cleavage, and we use these models to explain knockdown of thousands of miR-21 targets in living cells.
RESULTS
High-Throughput Measurement of RISC Binding
To define the sequence determinants of RISC binding, we designed libraries comprising ~20,000 distinct RNA targets per guide, including all singly and doubly mismatched targets; a subset of targets with ≥3 mismatches; targets containing insertions (≤7 nucleotides) and deletions; as well as targets predicted by TargetScan, Diana-microT, miRanda-mirSVR and PicTar2, and targets identified by CLASH (Figure 1B and Figure S1A) (Krek et al., 2005; Betel et al., 2010; Reczko et al., 2012; Helwak et al., 2013; Khorshid et al., 2013; Agarwal et al., 2015). Target libraries were sequenced on an Illumina MiSeq and transcribed in situ to generate clusters of RNA tethered to DNA templates of known sequence (Figure 1A) (Buenrostro et al., 2014; She et al., 2017; Denny et al., 2018). To eliminate potential secondary structures and cryptic binding sites, DNA oligonucleotides were annealed to the RNA flanking the target sequence. We measured binding kinetics using catalytically inactive D669A mutant AGO2 (Liu et al., 2004) and cleavage using wild-type AGO2 loaded with 3′ Alexa555-labeled let-7a, a seed-driven miRNA, or miR-21, a miRNA that requires 3′ supplemental pairing for its highest affinity binding (Salomon et al., 2015). RISC was continuously flowed into the MiSeq chip at 37°C at multiple concentrations, enab ling determination of the association rate (kon; Figure 1C) and dissociation constant (KD; Figure 1D) for tens of thousands of target RNAs. All experiments included a large number of replicate clusters for each target sequence (~50/target), enabling accurate measurement and error estimation for all reported values (STAR Methods). By constraining the maximum fluorescence values to an empirical distribution defined by high affinity variants, we were able to estimate KD values between 10 pM and 10 nM. Variants with KD values near the detection limit were defined by fewer points and typically had larger confidence intervals on their affinities. To test whether KD estimates near the detection limit were robust, we progressively removed the highest and lowest concentrations from our KD curves and found that even when KD values were defined by a single point, they were fit to nearly identical KD values as were originally determined using all concentrations (STAR Methods and Figure S1C).
Figure 1. High-throughput Characterization of RISC Binding to in situ Transcribed RNA.

(A) Schematic of RISC binding to RNA targets in a sequenced flow cell.
(B) Summary of the let-7a target library. The number of targets in each class is indicated by the sum of targets with an affinity <10 pM (dark blue), affinities ranging between 10 pM and 10 nM (light blue), and targets with affinity > 10 nM (gray). The number of targets for which association was measured is shown in orange.
(C) A representative set of RISC association data for a single target. Error bars correspond to the 95% confidence interval on the median fluorescence. The plot to the right shows the relationship between RISC concentration and observed rate, from which the association rate was determined.
(D) Representative binding isotherms for four RISC targets (shown in corresponding color in schematic) containing different degrees of complementarity to the guide (in gray).
See also Figure S1.
RISC Association Proceeds via Binding of the Seed Region
Consistent with previous results, guide:target complementarity within the seed determines the rate of RISC target binding in our assay (Figures 2A–2D and Figure S2A) (Wee et al., 2012; Chandradoss et al., 2015; Salomon et al., 2015). For both guides, mismatches with seed nucleotides g2–g5 most slowed association rates; these nucleotides are preorganized into a helical geometry and are accessible for the initial target search (Figure 2B) (Schirle et al., 2014; Salomon et al., 2015). Mismatches at seed positions g7 and g8 and mismatches outside the seed generally slowed the association rate of RISC with target <2-fold (Figures 2A–2D and Figure S2A). Target libraries also contained RNA with stretches of mismatches starting at every target position and ending at every other position: provided the target was fully complementary to the let-7a or miR-21 seed, target association rate was unaffected by as many as 13 contiguous mismatches (Figure 2C).
Figure 2. Sequence Determinants of AGO2 Association Kinetics.

(A) Association rates for miR-21 (upper left) and let-7a (lower right) loaded RISC binding to single and double mismatched targets. To find the rate corresponding to a particular double mismatch, identify the first mismatch on the horizontal axis, and the second mismatch on the vertical axis. The intersection indicates the double mismatched target. Axes are labeled with the 3′ end of the target (5′ end of the guide) starting at position 1. Gray crosses indicate missing data. Colors are centered on the association rate of the perfectly complementary (PC) target (white) with blue representing faster and red slower. Color bar is displayed in panel C.
(B) Association rates for tandem double mismatches mapped onto the AGO2 crystal structure (PDB ID: 4W5N).
(C) Association rates for miR-21 (upper left) and let-7a (lower right) targets containing stretches of complementary nucleotide mismatches (e.g., A to U). Examples are shown for mismatch stretches 2–4 and 5–9 on the right of the panel. For the 2–4 mismatches, the corresponding targets in the heatmap are located at the intersection of 2 on the ‘beginning complement mismatch’ axis and 4 on the ‘ending complement mismatch’ axis. Colors are scaled as in panel A.
(D) Change in association rates for tandem triple mismatches of miR-21 targets relative to a PC target (dotted line). Each boxplot includes the 27 triple substitutions for the three indicated target bases.
(E) Change in association rates for perfect complement miR-21 targets with increasingly long hairpins bound to either the seed (blue) or non-seed (orange) end of the target sequence relative to a PC target with no flanking complementarity (dotted line). For each length of complementarity to the target sequence, there are up to five corresponding stem loops of different lengths.
(F) Change in association rates for miR-21 targets containing 1–3 insertions of each base relative to a PC target (dotted line).
(G) Change in association rates for miR-21 targets containing single and double deletions relative to a PC target (dotted line).
See also Figure S2.
Secondary structure likely affects miRNA activity by competing with miRNA binding (Kedde et al., 2010). Among our targets, non-seed mismatches primarily slowed RISC association by sequestering the target site in a stable secondary structure. We used RNAfold (Lorenz et al., 2011) to predict the structure of every target sequence in each library: for targets where mismatches outside the seed slowed association >2-fold, 67% of let-7a targets (407) and 63% of miR-21 targets (1,281) had predicted internal secondary structures more stable than −1.5 kcal·mol−1 (Figure S2B). For example, a miR-21 target bearing t12C (3.7-fold decrease) or t12G (3.1-fold decrease) instead of t12U formed a hairpin that slowed target binding as much as a seed mismatch (Figure S2C). t1G substitutions also slowed miR-21 RISC target finding ~5-fold by stabilizing an RNA hairpin (ΔGRNAfold = −4.74 kcal·mol−1) that occludes the target seed (Figure S2C). let-7a target sequences formed fewer internal structures, likely because the fully complementary let-7a target sequence contains no guanine nucleotides. The observed effects of secondary structure are consistent with previous results showing that structures sequestering the seed-match reduce RISC cleavage more than structures sequestering the 5′ region of the binding site (Ameres et al., 2007). Our libraries also included targets with progressively longer hairpins at either end of the target, enabling systematic investigation of the effects of secondary structure. In contrast to structures sequestering the seed-complementary target nucleotides, structures that sequestered non-seed pairing regions of the target did not affect RISC association (Figure 2E). Thus, RNA structure in the seed sequence can modulate RISC association kinetics.
Insertions and Deletions Minimally Affect RISC Association Rates
Effects of target insertions on RISC association have not been systematically studied. By measuring RISC association to targets containing 1–7 nucleotide insertions, we found that association was primarily affected when these perturbations were within the seed-pairing region (Figure 2F and Figure S2D); insertions between t3 and t4 had the largest effects (≤8-fold). Targets with one or two deletions are predicted to require a bulge in the guide strand to accommodate flanking base pairs. We found that only single deletions of nucleotide t3 for let-7a and nucleotides t3, t4, or t12 for miR-21 slowed target finding >2-fold (Figure 2G and S2E). Removing two consecutive nucleotides from fully complementary targets only decreased the miR-21 RISC association rate when the deletions were within the seed (>6-fold reduction) (Figure 2G). In general, insertions were better tolerated than deletions at the same target position. For example, for miR-21, inserting three adenosine, cytosine, or uridine nucleotides between t3 and t4 slowed the association rate less than deleting nucleotide t3. These data suggest that target bulges in the seed-pairing region, which are predicted to face the solvent, are more readily accommodated than unpaired seed nucleotides in the guide, which face the protein and have their mobility restricted by a network of contacts between the miRNA backbone and AGO2.
Seed Complementarity in concert with 3′ Supplemental Pairing is Needed for High-Affinity Binding by Some miRNAs
Recruitment of deadenylases and other mRNA degradation factors by RISC likely depends on RISC occupancy at one or more target sites within the mRNA. To identify which target bases determine the fraction of target RNA bound by RISC, we measured RISC binding affinity to mismatched targets. For let-7a, position 3 and 4 mismatches led to the largest changes in affinity. In contrast, for miR-21, mismatches throughout the seed or in the 3′ supplemental region had similar effects on RISC affinity (Figures 3A–3B). let-7a targets containing mismatches at the same positions as miR-21 targets often bound with higher affinity (Figures 3A–3C). Both the reliance of miR-21 on supplemental pairing and the overall lower affinities of miR-21 RISC for its targets are likely consequences of the lower GC content of the miR-21 seed sequence.
Figure 3. Target Sequence Contributions to AGO2 Binding Energies.

(A) Binding energies for miR-21 (upper left) and let-7a (lower right) loaded RISC binding to single and double mismatched targets. Axes are labeled with the 3′ end of the target (5′ end of the guide) starting at position 1. White boxes represent missing data. Color bar is displayed in panel C.
(B) Effect of tandem triple substitutions in the target sequence on miR-21 (top) and let-7a (bottom) binding affinity. Dashed lines indicate the limits of detection and the numbers above and below the line indicate the number of targets in each group that fell beyond those limits.
(C) Binding energies for miR-21 (upper left) and let-7a (lower right) targets containing different length stretches of complementary nucleotide mismatches (e.g., A to U).
(D) Binding affinities for targets containing progressively more complementarity to RISC.
(E) Binding affinities for RISC loaded with miR-21 (top) or let-7a (bottom) to targets with 1–7 nucleotides insertions. Dashed lines indicate the limits of detection and points below the line bound with higher affinity than the detection limit.
See also Figure S3.
In lieu of predicting RISC affinity for each potential target, many miRNA target prediction algorithms define canonical site types, which are thought to possess variable efficacies. For both let-7a and miR-21, 8mer sites (targets with complementarity at positions 2–8 and a t1A) bound with the highest affinity on average, followed by 7merm-8 sites (targets with complementarity at positions 2–8), 7mer-A1 sites (targets with complementarity at positions 2–7 and a t1A), and 6mer sites (targets with complementarity at positions 2–7; Figure S3C). We observed a range of affinities to predicted targets containing the same seed types. Some of this variance could be explained by predicted RNA secondary structure (R2 = 0.38 for 8mer seed targets; Figure S3E). We removed targets predicted to have internal structures with ΔGRNAfold < −2 kcal·mol−1, and replotted the affinity distributions (Figure S3D). Sequence context still strongly affected binding affinity: the KD values for let-7a (<10 pM to >10 nM) and miR-21 (37 pM to >10 nM) spanned a ~1000-fold range, with the most favorable contexts having KD values comparable to those reported previously (Wee et al., 2012; Salomon et al., 2015). Comparing site types, we found that a t1A increased binding affinity by an average of 1 kcal·mol−1; human AGO2 contains a pocket between the MID and L2 domains that recognizes an unpaired adenosine through hydrogen bonding between Ser561 and the N6 amine of the adenine (Schirle et al., 2014; Schirle et al., 2015). All miR-21 site types bound with lower affinity than the corresponding let-7a site (Figure S3D), again likely a consequence of the greater let-7a seed GC content.
To test whether these differences in affinity reflect differences in mRNA regulation, we compared the median binding affinities for each seed type to published RNA-seq data collected following transfection of a let-7a decoy into 3T3 cells (Werfel et al., 2017). Considering only RNAs containing a single canonical binding site in their 3′ UTR and binning RNAs by seed type revealed a strong correlation (R2 = 0.99) between binding affinity and the mean log2 change in target abundance for each seed type (Figure S3F), suggesting that binding affinity is a key factor in mRNA repression by microRNAs. For canonical let-7a targets we observed <1.5-fold differences in median association rates (Figure S2F), indicating that the difference in affinity and thus regulation at these sites results from increased dwell times rather than decreased association rates.
Central Pairing can Reduce RISC Binding Affinity
Guide pairing to target bases t9 and t10 has been shown to reduce RISC affinity to seed-matched targets, leading to the proposal that pairing at these central positions requires an unfavorable conformational change (Schirle et al., 2014; Salomon et al., 2015). Additionally, natural miRNA targets rarely engage in central pairing (Khorshid et al., 2013; Grosswendt et al., 2014). To further characterize this phenomenon, we examined the contributions of central bases to binding by looking at all stretches of contiguous mismatches (Figures 3C–3D). For seed-matched miR-21 targets, complementarity to each additional base pair from t10 to t13 decreased binding affinity (Figures 3C–3D). This pattern was also observed for let-7a, but only for complementarity to t10 and t11 (Figure 3D). This indicates that complementarity at central positions can reduce binding affinity for most biological targets, which have little or no 3′ pairing. Interestingly, central pairing increased the binding affinity of many targets containing little or no seed complementarity (Figure 3C and Figures S3A–S3B), suggesting that the conformation associated with central pairing may specifically destabilize seed binding. However, even for seedless miR-21 targets, targets base paired from t10–t21 bound with lower affinity than targets paired from t11–t21 (Figure 3C).
AGO2 can Tolerate Seven Nucleotide Target Insertions without Substantial Decreases In Binding Affinity
RISC has been proposed to first find and bind to the seed-complementary region of a target, then loop out intervening, non-complementary target sequences in order to pair with complementary 3′ supplementary target bases (Schirle et al., 2014; Bartel, 2018; Sheu-Gruttadauria et al., 2019). Our libraries included 452 miR-21 and 463 let-7a of 560 possible targets bearing 1–7 nucleotide insertions allowing us to test whether long stretches of RNA can be looped out to connect complementary regions between the guide and target. Only 258 of let-7a and 156 of miR-21 bulged targets detectably reduced RISC binding affinity (Figure 3E). Most (53% for miR-21 and 45% for let-7a) of these insertions disrupted seed binding. For miR-21, only 9 of 95 (17 were not measured) target insertions between t8 and t12 detectably reduced binding affinity, demonstrating that long stretches of RNA can loop out to allow for pairing of the guide to target nucleotides more distant from each other. Functional biological targets containing large bulges have been described for miR-122, which binds the Hepatitis C viral RNA at a site predicted to contain a large central hairpin (Machlin et al., 2011; Luna et al., 2015). Our data suggest that this binding mode may be more common than previously appreciated: four target bases complementary to the 3′ supplementary region are predicted to occur by chance every 256 nucleotides.
Mechanisms for High-Affinity Binding to Noncanonical Targets
Although the seed sequence is the primary specificity determinant for target binding, several classes of “noncanonical” targets with incomplete seed sequences have been proposed to support RISC binding (Shin et al., 2010; Chi et al., 2012). Our target library included 513 miR-21 and 1162 let-7a noncanonical targets predicted by different algorithms or identified by CLASH. These putative noncanonical targets include 3′-compensatory and centered sites, as well as sites containing a single G:U wobble in a 6mer seed (Betel et al., 2010). The vast majority (95% for miR-21; 89% for let-7a) did not bind at the concentrations measured (Figure S3C). The two highest affinity let-7a noncanonical targets formed G:U pairs with the let-7a seed that were bolstered by 3′ supplemental pairing (Figure S3G). After removing targets predicted to form stable structures (ΔGRNAfold < −2 kcal·mol−1), only 18.7% (104) of the 556 let-7a and 5% (11) of the 228 miR-21 putative noncanonical sites bound with a KD <10 nM (ΔG < −11.3 kcal·mol−1; Figure S3D). Thus, most noncanonical targets identified by prediction algorithms or CLASH correspond to low affinity binding sites unlikely to be substantially occupied in vivo.
Our libraries included many nucleation bulge sites, sequences in which a nucleotide inserted between t5 and t6 can base pair with g6. The best studied let-7a nucleation bulge site, UAACCUC (Chi et al., 2012), occurred in 32 of the predicted targets in our let-7a library. let-7a RISC bound these targets weakly: the median affinity was 9.04 nM (ΔG = −11.4 kcal·mol−1), and binding to 15 sites was below the limit of detection (KD >10 nM). Just three UAACCUC sites, which were buttressed by 5–7 additional non-seed base pairs, bound with an affinity < 2 nM (ΔG < −12.3 kcal·mol−1; Figure S3H), an affinity similar to a 6-mer site (−12.1 kcal·mol−1).
Our libraries included targets with different extents of complementarity but lacking a canonical seed match. Some of these targets are similar to centered sites, which contain 11–12 bases of contiguous central complementarity (Shin et al., 2010). The binding affinities of these targets demonstrate that the length of contiguous complementarity needed for high-affinity binding depends on both sequence and position within the guide. For example, let-7a RISC bound targets with uninterrupted complementarity from t5–t17 (KD = 2.5 nM; ΔG = −12.2 kcal·mol−1) or t5–t16 (KD = 3.5 or 4.1 nM; ΔG = −12.0 or −11.9 kcal·mol−1 for two variants), affinities similar to a 6mer seed match. In contrast, targets complementary to let-7a from t5–t15 bound with affinity (KD >10 nM; ΔG > −11.3 kcal·mol−1) weaker than a 6mer seed match. For miR-21, but not let-7a, targets containing complementarity from t11–t21 bound more tightly than either 7mer-m8 or 7mer-A1 sites (Figure 3C). Our data suggest that centered sites or extensively complementary 3′ only sites could be functional, but the length and position of complementarity required likely depends on the distribution of GC content within the miRNA.
Imperfect seed complementarity has been proposed to render RISC binding dependent on 3′ compensatory pairing (Bartel, 2009). Supporting this view, many targets both imperfectly matching the seed and bearing additional two or three mismatches outside the seed failed to detectably bind RISC (Figure S3A–S3B). Thus, target sites without complete seed complementarity bind only when they contain extensive distal complementarity.
High-Throughput Measurement of RISC Cleavage Rate
siRNAs are typically designed to be fully complementary to their target. This design paradigm has been challenged by evidence that AGO cleavage activity can be enhanced by specific guide:target mismatches (Tang et al., 2003; Haley and Zamore, 2004; Ameres et al., 2007). Moreover, mismatches can allow siRNAs to discriminate between targets that differ by a single nucleotide (Dykxhoorn et al., 2006; Schwarz et al., 2006; Pfister et al., 2009). However, identifying mismatches that improve siRNA efficacy or specificity currently requires testing large numbers of individual siRNAs.
We developed RISC Cleave-’n-Seq (RISC-CNS) to enable high-throughput measurements of RISC cleavage rates and rapidly identify favorable guide:target mismatches for an individual siRNA sequence (Figure 4A and Figures S1A–S1B). RISC-CNS begins by incubating a library of RNA targets with a 10-fold molar excess of RISC to achieve single-turnover conditions. Cleavage is measured after various times by reverse transcribing and sequencing the targets remaining uncut. Normalized sequencing data was fit to single exponential curves—which yielded essentially the same values as a model incorporating association and dissociation rates (Figures S4B–S4C, STAR methods)—to determine cleavage rates for 22,607 let-7a and 7,841 miR-21 targets (Figure S4A).
Figure 4. RISC Cleave ’n-Seq (CNS) Enables High-throughput Measurement of Single Turnover Cleavage Kinetics.

(A) Method to determine single turnover cleavage rates for RISC targets.
(B) Cleavage rates for miR-21 (upper left) and let-7a (lower right) targets with single and double substitutions. Deep red represents targets for which no detectable cleavage was observed. Targets colored in blue were cleaved faster than the fully complementary target.
(C) Cleavage rates of miR-21 (upper left) and let-7a (lower right) targets containing different length stretches of complementary nucleotide mismatches (e.g., A to U). Color bar as in (B).
(D) Cleavage rates for miR-21 (top) and let-7a (bottom) targets containing three consecutive substitutions. The black dotted line represents the cleavage rate of the fully complementary RNA target, whereas the gray dotted line indicates the cleavage rate detection limit. The numbers at the bottom of the plot represent the number of targets in each group for which no cleavage was observed.
See also Figure S4.
Central Target Mismatches can both Inhibit or Enhance Cleavage
RISC cleaves its RNA target at the phosphodiester bond linking target nucleotides t10 and t11 (Elbashir et al 2001), and central base pairing (g9–g12) is required for efficient target cleavage (Haley and Zamore, 2004; Ameres et al., 2007; Wee et al., 2012) because it moves the scissile phosphate into the catalytic site (Ma et al., 2005; Parker et al., 2005). RISC-CNS revealed that for otherwise fully complementary targets, mismatches at t10 and t11 caused the greatest reduction in target cleavage rate (Figure 4B). For all possible let-7a triple mismatches and 21 of 27 miR-21 triple mismatches at t9–t11, cleavage was undetectable (>500-fold kcleave decrease; Figure 4D). For both guides, a target mismatch produced by changing t10U to t10C was better tolerated than other t10 base substitutions, likely because substitution of another pyrimidine at the cleavage site is less disruptive to the helical geometry required for cleavage (Figure 4B). Moreover, mismatches at t13, a position not usually considered part of the central region, actually perturbed cleavage more than t12 mismatches.
Surprisingly, some mismatches near the cleavage site enhanced cleavage. The rate of cleavage for a target bearing t12A mismatched with the let-7a g12G (0.14 s−1) was 2.5-fold faster than the fully complementary t12C target (0.055 s−1) (Figure 4B). The let-7a target bearing a t12A mismatch had the fastest cleavage rate of any single mismatch (Figure 4B, let-7a diagonal), and the cleavage rates of 37 of the 60 doubly mismatched targets containing a t12A mismatch were faster than the fully complementary target (Figure 4B). Counterintuitively, the t12 mismatches that most weakened the affinity of let-7a RISC (Figure 3A) showed the greatest enhancement in cleavage rates (Figure 4B): changing t12C to a t12A (0.14 s−1) increased the cleavage rate while a 12G substitution (0.018 s−1) and a t12U substitution (0.003 s−1) decreased the cleavage rate relative to the fully complementary let-7a target (0.055 s−1). miR-21 RISC showed the same trend: t12U>A (0.027 s−1) or t12U>G (0.019 s−1) slowed the rate of cleavage less than t12U>C (0.013 s−1) relative to the fully complementary t12A target (0.087 s−1; Figure 4B). Interestingly, the majority of seed mismatches had small effects on single turnover cleavage rates (Figure 4B), and some seed mismatches accelerated cleavage (t8G or t7C mismatches in miR-21 and t5 mismatches in let-7a). This finding is not without precedent: certain seed mismatches can enhance cleavage by the zebrafish homolog of AGO2 (Chen et al., 2017). Thus, an siRNA fully complementary to its target is unlikely to be optimal, perhaps because specific mismatches reduce strain in the RNA duplex and enable the RISC:target complex to more fully populate the catalytically competent conformation.
Target Mismatches to the Guide 3′ End Accelerate Single-Turnover Cleavage
Target:guide mismatches at the 3′ end of the guide (g17–g21) increase the rate of multiple turnover target cleavage, a phenomenon hypothesized to reflect faster release of the cleaved products (Tang et al., 2003; Haley and Zamore, 2004; Wee et al., 2012; Salomon et al., 2015). In our experiments, such mismatches also increased the rate of single-turnover cleavage, suggesting that unpairing the guide 3′ end lowers the barrier to RISC adopting a cleavage-competent conformation (Figures 4B–4D). Remarkably, single, double, and triple mismatches from t15–t21 for miR-21 and t16–t21 for let-7a increased the single-turnover cleavage rate (Figures 4B–4D). Even when nucleotides t15–t21 (miR-21) or t16–t21 (let-7a) were all simultaneously mismatched with the guide, the cleavage rate increased (Figure 4C).
Guide and Target Bulges Have Different Effects on Cleavage Kinetics
In the context of a fully complementary sequence, single insertions and deletions (indels) had similar effects on RISC association rate and binding affinity (Figures S5A and S5B). In contrast, insertions or deletions at the same target position had markedly different effects on the single turnover cleavage rate for both guides (Figure 5A). Single insertions that disrupted central pairing resulted in nearly undetectable target cleavage (<0.0002 s−1 for let-7a and <0.001 s−1 for miR-21), insertions in the seed slightly lowered the cleavage rate, and insertions opposite the distal 3′ end of the guide enhanced cleavage. Interestingly, target deletions between positions t4 and t8 resulted in cleavage rates that were >30-fold slower than either of the flanking insertions. Unlike target insertions flanking t11, targets bearing t11 deletions were readily cleaved.
Figure 5. Target Insertions and Deletions Result in Out of Phase Trends for Cleavage Rates.

(A) Cleavage rates for miR-21 (left) and let-7a (right) single insertions (blue dots) and single deletions (orange dots). Indels that correspond to multiple target positions are plotted in all possible target positions. The cleavage rate of the fully complementary target is indicated by the dotted line. Targets for which no cleavage was detected are plotted below the solid black line. Orange line, all single deletions; blue line, mean of the single insertions.
(B) let-7a cleavage rates were mapped onto the RNA components of the AGO2 crystal structure (PDB ID: 4W5O). Target insertions were mapped onto the 9mer RNA target such that the mean of all insertions between t1 and t2 are mapped onto t1. Single deletion cleavage rates were mapped onto the guide strand of the structure. Cleavage rates near the wild-type rate are colored white, while immeasurably slow cleavage rates are colored deep red. The first frame shows both the guide and target strands as they enter the central cleft of the protein, while the second frame shows only the guide strand. The third frame shows the guide strand as it exits the central cleft of the protein.
See also Figure S5.
Mapping the let-7a perturbations onto the AGO2 RISC crystal structure (Schirle et al., 2014) suggests an explanation for the distinct effects of indels on RISC function (Figure 5B). Because AGO2 constrains the seed nucleotides of its guide RNA, looping out an extra guide base to accommodate a target deletion is sterically prohibited. This restriction in guide geometry may force the extra base to be stacked into the duplex, as was observed in crystal structures of DNA target-bound TtAgo with DNA guide bulges (Sheng et al., 2017). Accommodating the extra guide base in the RNA duplex likely distorts the cleavage site, preventing efficient cleavage. By base g10, the guide backbone has begun to exit the central cleft of AGO and is facing solvent, suggesting that the extra guide base can loop out of the duplex with less disruption of the cleavage site. By contrast, the target bases pairing to the seed region have a solvent-facing backbone and unpaired bases are readily looped out, but as the target strand passes through the central cleft of the protein, the target backbone begins to abut the PAZ domain of the protein and becomes sterically constrained. The significant effects on cleavage activity of helical imperfections well outside the cleavage site highlights the structural sensitivity of AGO2.
Models for RISC Binding Affinity and Cleavage Kinetics
To predict the binding affinity and cleavage rates of any miR-21 or let-7a RISC target, we modeled binding and cleavage separately for each guide. An alignment algorithm enabled prediction of the binding register for each target (Figures 6A and S6A). To model binding affinity, we included one energy parameter for each base at each position (84 parameters). Because the effect of indels depends primarily on their position—seed, central, or 3′ supplemental—and whether they perturb the guide or target strand, we included bulge opening and extension penalties for each of these regions for each strand (12 parameters). We also included a base-pairing initiation term and a term to account for internal RNA structure. This linear energetic model predicts 61% of the variance in binding affinity for let-7a and 55% for miR-21 RISC (Figure 6B); more complex models performed marginally better (Figure S6C).
Figure 6. Predictive Models for AGO2 Binding Affinity and Cleavage Kinetics.

(A) Schematic of alignment of guide and target sequences to identify bound orientation.
(B) Comparison of binding affinity predicted by let-7a and miR-21 specific models to observed binding affinities.
(C) Comparison of cleavage rates predicted by let-7a and miR-21 specific models, or by a general cleavage model to observed cleavage rates. The color of the points represents the density of points at that position, with yellow being the densest and purple being the least dense.
(D) Parameters obtained from fitting miR-21 cleavage model or a general cleavage model.
See also Figure S6.
Next, we sought to define an appropriate set of features for our cleavage model. Because double-mismatch cleavage rates are predicted well by single-mismatch cleavage rates (Figure S6B), we employed a linear model consisting of parameters for each mismatched base at each position (3 nucleotides × 21 positions = 63 parameters). Insertions of different nucleotides generally had similar effects, allowing us to use single parameters for any base insertion at any given position (Figure S4G). From positions t1–t11, increasing the extent of target bulges led to a decreasing cleavage rate, whereas increasing the extent of target bulges from position t12–t21 had no effect on the cleavage rate and, in some cases, increased the cleavage rate (Figure S4G). As a result, target bulge parameters scaled with bulge length for bases t1–t11, but not for positions t12–t21 (20 parameters). For guide bulges, effects were generally additive, and multiple guide bulges typically abolished cleavage activity (Figures S4E–S4F). To account for this, we included guide bulge penalties for each position from t2–t20 (19 parameters). A model containing these 102 parameters was trained on targets containing single and double mismatches and single indels for both let-7a (1,766) and miR-21 (2,084). We then predicted the cleavage rates of targets containing three mismatches or multiple indels (2,361 for let-7a and 2,765 for miR-21). This model fit well to the data collected on targets with ≤2 mismatches, insertions, or deletions and quantitatively predicted with high accuracy (R2 = 0.71 for let-7a and 0.72 for miR-21) the cleavage rates of targets containing >2 perturbations (Figure 6C).
Given that we could accurately predict the cleavage rates for targets of each guide, and that we observed similar qualitative behaviors for cleavage by RISC when loaded with either let-7a or miR-21, we constructed a generalizable model to predict the cleavage rate of any RISC complex. This model included parameters for transitions or transversions at each position, along with the bulge parameters described above. We fit this 81-parameter model to the combined let-7a and miR-21 training data. The model accurately predicted cleavage rates of targets (5,126) containing triple mismatches or multiple indels (R2 = 0.66; Figure 6C). The fit model parameters reflect the physical constraints on RISC cleavage (Figure 6D). For example, transversions perturbed cleavage more than transitions at positions t6–t11, suggesting that the greater helical perturbations introduced by transversions at positions t6–t11 may propagate to the cleavage site (Figure 6D). Supporting this view, guide bulges 5′ to the cleavage site were also more disruptive (Figure 5). Conversely, at positions t12–t19, transversions were often cleaved at faster rates and likely increase the ability of the RISC ternary complex to obtain a cleavable conformation relative to more readily accommodated transversions.
RISC Kinetic Parameters Predict Knockdown in Cells
To determine how well in vitro biochemical parameters predict siRNA efficacy in cells, we deployed a cellular system for measuring the change in abundance of thousands of miR-21 targets. Using CRISPR-Cas9, we deleted the entire pri-miR-21 hairpin from HEK-293 Flp-In T-REx cells (Figure S7A). We cloned a subset of the miR-21 target library (the 6,327 sequences used in RISC-CNS) into the 3′ UTR of an eGFP reporter plasmid and stably integrated this library into the miR-21−/− cell line. We then transfected six concentrations of a miR-21 siRNA, isolated RNA from cells after 48 h, and sequenced uncleaved RNA targets. After normalizing for sequencing depth, we calculated the change in steady-state target abundance as the fraction of counts from each miR-21 transfection relative to the counts from the mock transfected miR-21 knockout line, which were highly reproducible between replicates (Figures 7A–7B).
Figure 7. Binding Affinity and Cleavage Rate Affect Knockdown in Cells.

(A) Scheme used to measure change in abundance of miR-21 targets.
(B) Comparison of normalized counts obtained from replicate miR-21 siRNA transfection experiments at the same concentration. Points are colored by density, with yellow being the densest and blue being the least dense.
(C) Biochemical model for predicting siRNA knockdown from measured kon and kcleave, and predicted koff of each target. Sample shown is from the 100 nM miR-21 transfection. Individual targets are colored by RISC-CNS measured cleavage rate. Red dot, perfectly complementary target. Dotted line has slope of −1 and intercept of 0.
(D) Knockdown of targets bearing single mismatches at each miR-21 siRNA concentration transfected.
(E) Knockdown of targets with single insertions (blue dots) or deletions (orange dots) following 100 nM transfection. Indels that correspond to multiple target positions are plotted in all possible target positions. Dotted line, target fully complementary to the siRNA. Orange line, all single deletions; blue line, mean of the single insertions.
(F) siRNA-directed (100 nM) reduction in abundance for all tandem, doubly mismatched targets. Dotted line, target fully complementary to the siRNA.
See also Figure S7.
We derived a biochemical model to predict the change in target abundance at steady-state based on each target’s association, dissociation, and cleavage rate, as well as the free RISC concentration, the basal mRNA decay rate, and the miRNA-accelerated decay rate (Figure S7B). Because we were unable to measure dissociation rates for extremely high affinity (KD<10 pM) targets, we used dissociation rates estimated by multiplying model predicted affinities by measured association rates (koff = KD × kon). The free RISC concentration was fit as a constant for all targets for each miR-21 transfection concentration. Because all reporter constructs had essentially the same 3′ UTR length and sequence composition, the basal mRNA decay rate and miRNA-accelerated decay rate were assumed to be constant for all targets, and these parameters were fit globally. Unlike RISC-CNS cleavage rates, the flanking context of targets significantly influenced target knockdown in cells (Figure S7E), likely due to the greater length of flanking sequence, which may increase competing secondary structure formation or binding of cellular proteins. For this reason, only targets containing five adenosines flanking the target region were used in model fitting and subsequent analyses (4,483 sequences). While this does not eliminate differential effects of structure or other RNA binding proteins on the targets examined, it does reduce their likelihood of confounding comparative analyses. Many variants with RISC-CNS cleavage rates >10-fold faster than their estimated dissociation rates showed little change in abundance in cells, suggesting that the cleavage rate is slower in cells than the cleavage rate measured in vitro or that the dissociation rate in cells is much faster than the dissociation rate measured in vitro. An increase in the dissociation rate could reflect differences in ionic environment or the activity of RNA helicases or other RNA-binding proteins. To account for this discrepancy, we fit a single dissociation-rate scaling term for all targets across all treatment conditions. This model performed well for each of the three highest miR-21 transfection conditions (R2 = 0.59, 0.56, 0.55; Figure 7C and Figure S7C).
Next, we examined the effect of single-nucleotide mismatches on knockdown in cells (Figure 7D and Figure S7F). In agreement with RISC-CNS results (Figure 4B), t13 mismatches resulted in less target reduction than most seed mismatches, highlighting the importance of t13 pairing for efficient target cleavage. Mismatches from t17–t21 resulted in target reduction equal to or greater than that observed for the perfectly complementary (PC) target, in agreement with our finding that mispairing at these positions enhances the rate of target cleavage. Many targets containing t8G substitutions also exhibited greater knock-down than the PC target. Similarly, the effect of target indels was predicted by RISC-CNS: deletions in the seed-matching region yielded little or no target knockdown in cells, while single target insertions between t2 and t9 resulted in similar or slightly less knockdown than the PC target (Figure 7E). While single-nucleotide insertions in the seed-matching sequence of the target caused only a modest reduction (<2-fold on average) in siRNA efficacy relative to the PC target, yet, as RISC-CNS predicted, the insertion of two or more bases in the seed binding region reduced siRNA efficacy (Figure S7D). Insertion of 1–3 nucleotides at target positions t11–t15 reduced RISC activity a similar amount (<2-fold) regardless of the length of the insertion, consistent with the RISC-CNS findings (Figure S4G).
Finally, our library of targets included all 180 possible tandem double mismatches (Figure 7F). As predicted by RISC-CNS, target mismatches to both t20 and t21 enhanced target cleavage. Targets bearing tandem mismatches in the cleavage site, particularly t9t10, were better RISC substrates than targets with tandem mismatches in either the seed or 3′ supplemental region. Yet such t9t10 mismatched targets—which bind RISC with high affinity (KD <10 pM)—were not cleaved in RISC-CNS experiments. These targets are thus likely substrates for miRNA-mediated transcript destabilization rather than cleavage (Hutvágner and Zamore, 2002; Zeng et al., 2002; Doench, 2003).
A limitation of this experimental and modeling approach is that we are unable to account for certain target-specific effects, such as target-directed miRNA degradation (TDMD) (Ameres et al., 2010). We anticipate that further characterization of this process for a diversity of targets would enable more accurate modeling of knockdown by RISC in future work.
DISCUSSION
Thermodynamic Binding Specificity of miRNAs
Accurate prediction of miRNA-mediated mRNA repression requires a quantitative description of RISC binding affinities for a range of targets. Although non-seed binding sites have been proposed to comprise a substantial fraction of miRNA-binding sites (Loeb et al., 2012; Helwak et al., 2013; Grosswendt et al., 2014), our data show that the majority of sites lacking canonical seed-matched sequences bind RISC with affinities below our detection limit, suggesting that it is unlikely that these sites function in vivo. Our data suggest that whenever pairing to the seed is interrupted by multiple substitutions or bulged nucleotides on the guide, then a long, contiguous stretch of 3′ complementarity is needed to maintain a physiologically relevant binding affinity.
Large target bulges distal to the seed-matching sequence are well tolerated by RISC, suggesting that RISC can readily loop out large stretches of target RNA to add 3′ supplemental complementarity. Finally, our data demonstrate that the specificity landscapes of different miRNAs can be highly sequence dependent. Since miR-21 RISC seed binding is weaker, 3′ pairing is required to achieve an affinity comparable to let-7a RISC for a seed-matched target. This is likely driven by the low GC content of the miR-21 seed: miR-21 8mer targets are bound ~1.5 kcal·mol−1 (~11-fold) weaker than let-7a (Figures S3C–S3D), and a similar difference was observed for 7mer-m8 sites. Thus, miRNAs such as miR-21 must reach higher than typical steady-state levels or bind to sites with additional 3′ complementarity to produce the same functional effects as most other miRNAs.
A Physical Model for RISC Binding and Cleavage
Several stepwise models of RISC activity have been proposed (Ameres et al., 2007; Mayr and Bartel, 2009; Schirle et al., 2014; Salomon et al., 2015; Bartel, 2018). Our results provide a detailed, biochemical perspective into key steps on this pathway. RISC searches for candidate targets by pre-organizing seed nucleotides g2–g5 into a solvent-accessible helix (Wang et al., 2008). Within this region, pairing to guide bases g3 and g4, are particularly essential for productive binding. Upon binding, the AGO2 α-helix 7 shifts, exposing the distal end of the seed, allowing target pairing to propagate to guide bases g6–g8 (Schirle et al., 2014).
During this initial phase of binding, AGO2 undergoes a conformational change, repositioning the 3′ supplemental region of the guide into a near A-form helix. This region is now primed to act as a second nucleation site. Skipping central guide pairing may explain why even large target insertions here are well tolerated by RISC. With binding anchored at two positions, pairing to highly complementary targets can progress into the central region. A cleavage-competent conformation requires pairing at central bases t9–t11, but pairing at positions t8 and t12 is often dispensable, and occasionally detrimental. Continuous base stacking through the cleavage site is thus not required, and single-stranded character here can facilitate cleavage in some cases. It has been suggested that base pairing downstream of the 3′ supplemental region helps to pull the 3′ end of the guide from the PAZ domain (Tomari and Zamore, 2005), facilitating helix winding and enhancing cleavage. Our data argue against this idea. Pairing past guide base 16 actually slows the single-turnover cleavage rate, suggesting that central and 3′ supplemental pairing may be sufficient to extricate the 3′ end of the guide from the PAZ domain, and unpairing at the 3′ end may relieve some conformational strain at the cleavage site.
The finding that complementarity past guide base g16 is unnecessary for efficient cleavage by mammalian AGO2 is consistent with findings for other AGO and PIWI proteins. Mismatches from g17–g21 have little effect on cleavage by Drosophila Ago2 (Wee et al., 2012). Similarly, the PIWI proteins Aubergine and Ago3, whose guides range from 23–27 nt, are predicted to cleave targets containing complementarity from g2–g16 and g2–g14, respectively (Wang et al., 2014). TtAgo, a DNA-guided DNA endonuclease from the eubacterium Thermus thermophilus, reaches maximal cleavage rate with targets paired only from g2–g16 (Wang et al., 2009). Thus, the requirement for g2–g16 complementarity for target cleavage is conserved among Argonautes separated by >2 billion years of evolution.
Design Principles for Specific and Effective siRNAs
siRNAs are widely used tools in biomedical research, and RNAi-based therapeutics are now used in humans. Mismatches at the 3′ end of the guide have been shown to increase the rate of multiple-turnover target cleavage, which was proposed to result from increased product release (Tang et al., 2003; Haley and Zamore, 2004), and to enhance knockdown of target RNAs in reporter assays, which was explained by a decrease in TDMD (De et al., 2013). Our data identify many mismatches that enhance target cleavage by two guide RNAs. Consistently, mismatches at the 3′ end of the guide accelerated single-turnover cleavage rates. In cells, 3′ mismatches enhanced target knockdown relative to a fully complementary target. In principle, this effect could result from increased single-turnover cleavage rates, increased product release, decreased TDMD, or a combination of all three. Regardless, when designing siRNAs, incorporating 3′ mismatches, particularly from g18–g21, will likely enhance target knockdown.
Designing highly specific siRNAs can be achieved by tuning affinity or cleavage rate. Our cleavage experiments in cells show that both strategies can discriminate between targets with single nucleotide differences. For example, discriminating between a fully complementary miR-21 target and one with a single mismatch was most effective when the mismatch was at position 3 or position 13 (Figure 7D). The effect of a seed mismatch at position 3 is likely due to decreased affinity (Figure 3A), while, at position 13, the effect primarily reflects a decrease in cleavage rate (Figure 4B). This information can be used to design highly specific siRNAs that can discriminate between sequences containing SNPs. Indeed, previous work has suggested that siRNA mismatches at positions 3 and 13 may enable discrimination between SOD1 RNAs containing a single mismatch (Schwarz et al., 2006). However, whether binding or cleavage perturbations enable better single-nucleotide discrimination will likely depend on the affinity and concentration of the siRNA. For higher affinity siRNAs, mismatches affecting binding may be less useful on their own since dissociation may be much slower than cleavage even when a mismatch is introduced. Maximizing siRNA specificity may also require mismatches at multiple positions, even when the goal is to discriminate between SNPs, because the greatest specificity is predicted to occur when the dissociation rate is similar to or greater than the cleavage rate (Pfister et al., 2009; Bisaria et al., 2016).
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, William Greenleaf (wjg@stanford.edu).
METHOD DETAILS
Library Design
Target libraries for let-7a and miR-21 loaded RISC were designed to include all single mismatches, all double mismatches, a subset of triple mismatches, all single target insertions and deletions, all target insertions of 2–7 identical nucleotides, pairs of 2–5 consecutive transitions or transversions, four way combinations of two consecutive transitions or transversions (eight total mismatches), stretches of mismatches to the complement target base of all lengths throughout the target sequence, the top 1,000 predicted targets from four algorithms (TargetScan, Diana-microT, miRanda-mirSVR, and PicTar2), and targets identified with the CLASH experimental method. Each designed target was placed within context sequence that typically consisted of five flanking adenosine nucleotides on the 5′ and 3′ ends of the target. The predicted targets were included with the 5 flanking nucleotides present around the actual target sequences. We also included targets identified from CLIP experiments in mice (Chi et al., 2009), but the mm9 coordinates were lifted over to hg19 to identify the corresponding human targets, which were included in the library. Since these lifted targets were not experimentally determined, they were not used in comparing predicted targets (Figure S3) but were included to add more sequence diversity for model fitting. The perfectly complementary target was also placed in 225 distinct five nucleotide contexts, and the single mismatches were placed in four five nucleotide contexts to test for the effects of the flanking sequence. The perfectly complementary target was also placed in sequence contexts longer than five nucleotides that were designed to form RNA secondary structure with the target region. An overview of the library designs is shown in Figure S1 and the full list of sequences is available in Table S1.
Assembly and Sequencing of Library
Target libraries were synthesized by Custom Array (Bothell, WA) such that each variant was flanked by common 5′ and 3′ priming sequences. Predicted target variants were ordered with an alternate 3′ priming sequence so that these variants could be separated from the rest of the library. Ordered sequences ranged from 73 bp to 129 bp, and sequences shorter than the longest variant had random sequence appended until all variants were the same length. The first miR-21 library was ordered as a 12,000 oligonucleotide synthesis and contained 6,327 unique variants. The let-7a and second miR-21 libraries were ordered as part of two separate 92,000 oligonucleotide syntheses and contained 22,641 and 12,768 unique variants respectively.
Synthesized libraries were assembled into full constructs compatible with Illumina sequencing and with generation of RNA on chip (Figure S1B). The assembly reactions were carried out in a 20 μl volume of 1× NEBNext Master Mix (NEB, M0541) with ~10 nM of synthesized library, 10 nM of “T7A1 promoter and stall sequence”, 50 nM of “Illumina Adapter (P5) and T7A1 promoter oligo”, 50 nM of either “Illumina Adapter (P7) and designed library R2 sequence” or “Illumina Adapter (P7) and predicted target library R2 sequence”, and 250 nM of both Illumina Adapter (P5) and Illumina Adapter (P7) (oligonucleotide sequences available in Table S2). SYBR green was added at a final concentration of 0.6× to assembly reactions so that assembly progress could be monitored. Reactions were loaded into a QuantStudio qPCR thermocycler and went through cycles of 98°C for 10 s, 63°C for 30 s, and 72°C for 30 s until the SYBR green signal of a reaction began to plateau, after which the reaction was paused and assembly reaction was removed. Assembly reactions ran between 14 and 19 cycles. Completed assemblies were purified using a QIAquick PCR purification kit, and a portion of the purified product was visualized on an agarose gel to confirm specific assembly of the intended product.
Assembled libraries were diluted and quantified against a standard library of PhiX (Illumina, Hayward, CA). PhiX standard was prepared by diluting stock PhiX to 200 pM in water and then serially diluting by 2-fold eight times, resulting in a standard curve that spanned 200 pM to 1.56 pM. Diluted libraries and the PhiX standard were amplified in qPCR reactions containing 500 nM primers (Illumina Adapter Sequences P5 and P7; Table S2) in 1× NEBNext Master Mix (NEB, M0541) with 0.6× SYBR green. Reactions were cycled at 98°C for 10 s, 63°C for 30 s, and 72 °C for 30 s for a total of 20 cycles. Standards were run in duplicate, and all library samples were run in triplicate. Quantified libraries were then sequenced on an Illumina MiSeq instrument using custom read 1 and read 2 primers that flanked the variable region of each library sequence (Table S2). Libraries typically represented 10–20% of the total sequencing chip, with the rest of the chip comprised of high-complexity genomic libraries. Libraries were sequenced in two steps using paired end sequencing with 76-bp reads. Because library variable regions were all shorter than this read length, all variants were fully sequenced in both directions in each sequencing run.
Processing Sequencing Data
Following sequencing, tile and x, y coordinates of each cluster were extracted. Clusters were deemed library members based on aligning a segment of the read 2 sequence (either 5′-AGA TCG GAA GAG CGG TTC AG-3′ or 5′-CGG ACG CGG GAA GAC AGA AT-3′). Fiducial marks were identified by aligning the exact fiducial sequence (5′-TAG CCA GCC TGA TAA GTA ACA CCA CCA CTG-3′). Fiducial marks and library members identified in this manner were used for registering tiles prior to experiments and for registering sequencing data to images during image processing. Because all library members were shorter than the read sequence, each variant was fully sequenced twice during sequencing. Only clusters that exactly matched a known library sequence in both reads were fit in downstream data analysis for determination of kon and KD.
RISC Purification
S100 extract was generated from SV40 large T-antigen immortalized AGO2−/− MEFs that stably overexpress mouse AGO2 (O’Carroll et al., 2007). Cell extract was essentially prepared as described (Dignam et al. 1983). Briefly, the cell pellet was washed three times in ice-cold PBS and once in Buffer A (10 mM HEPES-KOH (pH 7.9), 10 mM potassium acetate, 1.5 mM magnesium acetate, 0.01% w/v CHAPS, 0.5 mM DTT, 1 mM AEBSF, hydrochloride, 0.3 μM Aprotinin, 40 μM Bestatin, hydrochloride, 10 μM E-64, 10 μM Leupeptin hemisulfate). The supernatant was removed, and 0.11 cell pellet volumes of Buffer B (300 mM HEPES-KOH (pH 7.9), 1.4 M potassium acetate, 30 mM magnesium acetate, 0.01% w/v CHAPS, 0.5 mM DTT, 1 mM AEBSF, hydrochloride, 0.3 μM Aprotinin, 40 μM Bestatin, hydrochloride, 10 μM E-64, 10 μM Leupeptin, hemisulfate) was added, followed by centrifugation at 100,000 × g for 20 min at 4°C. Ice-cold 80% (w/v) glycerol was then added to achieve a 20% (w/v) final glycerol concentration, followed by gentle inversion to mix. S100 was aliquoted, frozen in liquid nitrogen, and stored at ‒80°C.
To load AGO2-RISC, 30 nM duplex siRNA with a 3′ Alexa Fluor 555 (Life Technologies) labeled guide strand was incubated in S100 extract for 1.5 h at 37°C in 15 mM HEPES-KOH (pH 7.9), 100 mM potassium acetate, 5 mM magnesium acetate, 5 mM DTT, 1 mM ATP, 25 mM creatine phosphate, 30 μg·mL−1 creatine kinase. RISC was purified as described (Flores-Jasso et al., 2013). Briefly, the assembled AGO2-RISC was incubated overnight at 4°C with a biotinyl ated, 2′-O-methyl capture oligonucleotide linked to streptavidin paramagnetic beads (Dynabeads MyOne Streptavidin T1, Life Technologies). RISC was eluted with a competitor oligonucleotide for 2 h at room temperature. Excess competitor oligonucleotide was removed by incubating the eluate with streptavidin paramagnetic beads (Dynabeads MyOne Streptavidin T1, Life Technologies) for 15 min at room temperature. The RISC was concentrated, and the potassium acetate concentration was adjusted to 100 mM (f.c.) by centrifugal ultrafiltration (Amicon Ultra-centrifugal filter, 10K MWCO, EMD Millipore, Billerica, MA). The concentration of active, purified RISC was measured by pre-steady-state target cleavage assays at 23°C in the presenc e of 100 nM 32P-radiolabeled target RNA. The concentration of catalytically inactive, purified RISC was measured by fluorescence with Typhoon FLA-7000 (GE Healthcare) following denaturing polyacrylamide gel electrophoresis.
Imaging Station Setup
A custom instrument that enables biochemical measurements to be made in a MiSeq flow cell was constructed as described in (She et al., 2017). The camera, lasers, Z-stage, XY–stage, syringe pump, and objective lens used in the instrument were salvaged from an Illumina GAIIx. These parts were combined with a fluidics adaptor designed to interface with Illumina MiSeq chips, a temperature control system, and laser control electronics to enable real time biochemical measurements in MiSeq flow cells. Imaging was performed using either a 400 ms exposure time at 150 mW fiber input power of a 660 nm laser and a 664 nm long pass filter (Semrock) or with a 600 ms exposure time at 150 mW input power of a 530 nm laser and a 590 nm center wave length and 104 nm guaranteed minimum 93% bandwidth band pass filter (Semrock).
Generation of RNA on the Sequencing Flow Cell
MiSeq flow cells containing sequenced libraries were loaded into the custom imaging station for in situ RNA generation (Buenrostro et al., 2014, She et al., 2017). All steps were executed using custom xml scripts to control the imaging station’s pump, stage movement, Peltier heater, lasers, and camera. Unless otherwise stated, all wash volumes were 100 μl and flowed at 100 μl·min−1.
Regeneration of Double-Stranded DNA
For the first experiment after sequencing, DNA not covalently attached to the flow cell surface was removed by heating the flow cell to 55°C and washing with 100% (v/v) formamide. The flow cell was then heated to 60°C an d incubated in Cleavage buffer (80 mM Tris-HCl (pH 8.0), 80 mM NaCl, 0.05% v/v Tween 20, 100 mM TCEP) for 10 min to remove residual fluorescence from sequencing reversible terminators.
Cy3-labeled fiducial mark oligonucleotides and 5′ biotinylated roadblock oligonucleotides (Table S2) were hybridized to the distal end of library ssDNA molecules in multiple phases. First, the flow cell was incubated in Hybridization buffer (5× SSC buffer (ThermoFisher 15557036), 5 mM EDTA, 0.05% v/v Tween 20) containing 500 nM of each oligonucleotide for 12 min at 60°C, followed by 12 min at 40°C. The flow cell was washed in Annealing buffer (1× SSC, 5 mM EDTA, 0.05% v/v Tween 20), and then incubated in Annealing buffer containing 500 nM of each oligonucleotide for 8 min at 40°C. Following oligon ucleotide hybridization, the temperature was lowered to 37°C and the flow cell w as washed with Klenow buffer (1× NEB buffer 2 (NEB B7002S), 0.25 mM of each dNTP, 0.01% v/v Tween 20). The hybridized oligonucleotides were extended into dsDNA by adding one line volume (65 μl) of Klenow buffer containing 0.2 U/μl Klenow fragment (3′→5′ exo‐minus (NEB M0212)) and pumping 9 μl of Klenow buffer every 5 min for a total of 30 min. Following dsDNA generation, the flow cell was washed with Hybridization buffer.
Because the success of RNA generation was determined by annealing of a labeled stall oligonucleotide to the nascent RNA molecule, it was necessary to block this DNA sequence in the event that dsDNA generation was less than 100% efficient. Blocking of the ssDNA stall sequence was achieved by incubating the flow cell in Hybridization buffer containing 500 nM unlabeled stall oligonucleotide for 10 min, washing with annealing buffer, and then incubating the flow cell in Annealing buffer containing 500 nM unlabeled stall oligonucleotide for 10 min. After another Annealing buffer wash, the flow cell was incubated in Annealing buffer containing 500 nM of labeled stall oligonucleotide for 10 min. The flow cell was imaged after this step to serve as a baseline image for RNA generation.
RNA Generation
After dsDNA generation, the flow cell was incubated for 5 min in 1 μM streptavidin (PROzyme, SA10) in Annealing buffer. The streptavidin binds the biotinylated oligonucleotides used for dsDNA generation and stalls E. coli RNA polymerase holoenzyme (RNAP; NEB M0551) during RNA generation. After washing with Annealing buffer, the flow cell was incubated for 5 min in 5 μM biotin (ThermoFisher B20656) in Annealing buffer to saturate the remaining streptavidin binding sites. The flow cell was washed again with Annealing buffer, and then washed with Initiation buffer (2.5 μM each of ATP, GTP, and UTP in R-reaction buffer (20 mM Tris-HCl (pH 7.5), 7 mM MgCl2, 20 mM NaCl, 0.1 mM EDTA, 1.5% glycerol, 0.01% v/v Tween 20, 0.5 mM DTT)). One line volume (65 μl) of Initiation buffer containing 0.06 U/μl of RNAP was applied to the flow cell, after which 9 μl of Initiation buffer was pumped every 100 s for a total of 10 min. Because the Initiation buffer lacks CTP, RNAP is allowed to initiate transcription on dsDNA molecules containing the T7A1 sequence, but then stalls part way through transcribing the stall sequence. Unbound RNAP was then removed from the flow cell with an Initiation buffer wash. RNAP was extended by adding Extension buffer (10 mM NTPs in R-reaction buffer) containing 500 nM each of labeled stall DNA oligonucleotide and R2 DNA blocking oligonucleotides (Table S2) and incubating for 5 min. The labeled stall oligonucleotide binds to the 5′ end of the newly transcribed RNA molecule and serves the dual purpose of blocking this common sequence while also allowing for assessment of RNA generation efficiency. The R2 oligonucleotides serve to block the 3′ common sequence of each RNA molecule, leaving only the variable target sequences single stranded. To ensure efficient blocking, the flow cell is incubated in 500 nM of each oligonucleotide in Blocking buffer (1× SSC, 7 mM MgCl2, 0.05% v/v Tween 20) for an additional 10 min. Finally, the flow cell was washed with AGO2 Sample buffer (30 mM HEPES-KOH (pH 7.3), 120 mM potassium acetate, 3.5 mM magnesium acetate, 1 mM DTT, 50 μg/mL BSA, 10 μg/mL yeast tRNAs, 0.05% v/v Tween 20).
Measurement of association rates and equilibrium dissociation constants on chip
After RNA was transcribed in the MiSeq flow cell, AGO2 loaded with a labeled guide was introduced at various concentrations to measure association kinetics. For let-7a, association was measured at 63 pM, 125 pM, 250 pM, and 500 pM for the entire library. For miR-21, association was measured at 25 pM, 188 pM, 375 pM, and 1 nM for the second part of the library, and at 50 pM, 125 pM, 250 pM, and 500 pM for the initial library.
Tiles were imaged continuously during the first 20 min of association, with each tile being imaged approximately every 90 s. For association experiments lasting longer than 20 min, additional images were taken at log spaced intervals. By collecting association data at multiple concentrations, we were able to fit association constants and were able to use the fraction bound at the end of each association to construct equilibrium binding curves.
After each association experiment, the chip was washed with 500 μl Wash buffer (10 mM Tris-HCl (pH 8.0), 5 mM EDTA, 0.05% v/v Tween 20), and then all protein, RNA and non-covalently attached DNA was stripped by heating the chip to 55°C and flowing 100% formamide. RNA was regenerated for each subsequent experiment.
Measurement of Cleavage Rates
Transcription of Library
To construct the target libraries for the cleavage experiments, a T7 promoter was added by PCR to the DNA oligonucleotide-pool library designed for the array experiment. RNA target libraries were transcribed with T7 RNA Polymerase for 3 h using the following conditions: 16 mM MgCl2, 2 mM Spermidine, 40 mM Tris-HCl (pH 7.5), 0.01% Triton X-100, 2 mM each dNTP, and 40 mM DTT. The resulting products were treated with DNase-I and purified using Qiagen RNeasy Mini columns. For let-7a the full designed library and the library of predicted targets in the short sequence context was used for cleavage experiments. However, for miR-21, only the initial designed library (6,327 variants), containing the less degenerate sequences for which cleavage is more relevant, was used for the cleavage experiments.
Cleavage Experimental Protocol
Cleavage assays were performed in cleavage buffer (30 mM HEPES-KOH (pH 7.3), 120 mM potassium acetate, 3.5 mM MgCl2, 1 mM DTT, and 0.1% v/v Tween-20). Prior to the reactions, DNA blocking oligonucleotides were annealed to the target RNA primer sequences to prevent structure formation or interaction between the primer sequence and the protein by adding 1.25× excess blocking oligonucleotides to the RNA in cleavage buffer without MgCl2. The resulting mixture was heated to 70°C and cool ed slowly to room temperature (10 min) to anneal the oligonucleotides to the RNA. After annealing the oligonucleotides, the RNA target libraries were diluted to the reaction concentrations and MgCl2 concentration was adjusted to 3.5 mM. For each reaction, the RNA target library concentration was set to 10% of the protein concentration to ensure that there would be minimal depletion of protein. The miR-21 reactions were performed at 8 nM RISC and the let-7a reactions were performed at 4 nM RISC. High concentrations of RISC were used to limit the effects of association on the observed cleavage rate such that for the vast majority of target variants the rate measured would reflect the single turnover cleavage rate constant. Reactions were initiated by mixing the protein and target libraries at 37°C and incubating for log spaced amounts of time ranging from 15 s to 32 min. Additionally, one reaction was immediately quenched after mixing the components and a no protein control went through the same procedure. The reactions were quenched at −80°C and once all reactions were complete, they we re immediately placed at 95°C to denature the protein and prevent any additional cleavage in the downstream library generation steps. The reactions were then treated with DNase-I to remove the blocking oligonucleotides and the resulting RNA was reverse transcribed with superscript IV reverse transcriptase. The resulting cDNA was barcoded for each time point using NEBNext 2× high-fidelity master mix and 250 nM of each timepoint barcode. PCR progress was monitored by including 0.6× SYBR Green in the reaction and stopped when the SYBR Green signal began to plateau to minimize the total number of PCR cycles to prevent introduction of bias at this step. The resulting libraries were purified using Qiagen QIAquick PCR Purification columns and quantified for sequencing with qPCR (see Assembly and Sequencing of Library Above).
Sequencing of Cleavage Libraries
Paired end sequencing (2 × 36) of the resulting libraries was performed with 75 bp High Output Next Seq kits on a NextSeq500. Custom read 1, 2, and index primers were spiked into the run to sequence the cleavage libraries.
Cell Culture
HEK-293 Flp-In T-REx cells (Invitrogen) were cultured in DMEM with 10% FBS, GlutaMAX, and penicillin-streptomycin. Cells were maintained in a humidified CO2 incubator at 37°C and examined regularly to ensure absence of mycoplasma contamination.
Generation of miR-21 Knockout Cell Line
Cas9-gRNA ribonucleoprotein complexes containing two tracrRNA:crRNAs flanking the miR-21 hairpin (5′-TGA TAA GCT ACC CGA CAA GGT GG-3′; 5′-CGA TGG GCT GTC TGA CAT TTT GG-3′) were transfected into HEK-293 Flp-In T-REx cells according to the Alt-R CRISPR-Cas9 user guide (IDT), except that RNAiMAX was replaced with Lipofectamine 3000 (Invitrogen). Transfected cells were incubated for 48 h, after which single cells were sorted into 96-well plates. After 3 weeks, viable clones were genotyped using primers that flanked the miR-21 hairpin (5′-TCA AAT CCT GCC TGA CTG TCT G-3′ and 5′-CCA GAG TTT CTG ATT ATA AAC AAT GAT GC-3′). Homozygous edited clones were further expanded and deletion of the miR-21 hairpin was confirmed by amplification and electrophoresis of the miR-21 locus and by a TaqMan RT-qPCR miRNA assay specific for mature miR-21 (Applied Biosystems).
Preparation of miR-21 Library piggyBac Reporter Constructs
All oligonucleotides used to construct the miR-21 plasmid library are reported in Table S2. The CMV promoter, eGFP coding sequence, and SV40 poly(A) signal sequence were amplified from existing plasmids in the lab. Each of these components was amplified using primers containing homology arms to neighboring segments, and an EcoRI site was added upstream of the CMV promoter and a XhoI site was added downstream of the SV40 poly(A) sequence. The promoter, gene, and poly(A) signal sequence were assembled using NEBuilder HiFi DNA Assembly master mix with equimolar mixing of components. After assembly, the full gene was amplified further using only the outermost primers.
The PB-U6insert-EF1puro backbone was amplified such that the U6 promoter was removed and an EcoRI site was added upstream of the EF1 promoter and an XhoI site was added inside of the 5′ piggyBac right (3′) inverted repeat. The reporter gene was inserted into the amplified PB-EF1puro backbone to create the PB-CMV-GFP-EF1puro plasmid, wherein the CMV and EF1 promoters faced in opposite directions.
To prepare the miR-21 target sequences for cloning into the PB-CMV-GFPEF1puro plasmid, the fully assembled version 1 miR-21 (6,327 variants) array library was used as a template. The variable target region of the library was amplified 15 cycles using primers that introduced restriction sites on each end of the target. The library was then cloned 61 bases downstream of the GFP stop codon and 93 bases upstream of the SV40 poly(A) signal sequence.
Stable Transfection of miR-21 Target Library
miR-21 knockout cells were grown to 90% confluency in a 6-well tissue culture plate. 200 ng of purified miR-21 target plasmid library was co-transfected with or without 200 ng Super piggyBac Transposase Expression Vector (SBI) using lipofectamine 3000 (Invitrogen) according to manufacturer’s instructions. After 24 h, transfected cells were passaged into a 10-cm tissue culture plates. After another 24 h, culture media was replaced with culture media containing 2 μg·mL−1 Puromycin. Media was replaced every 3 days until the negative control cells (those without Transposase expression vector co-transfection) were all dead.
Knockdown in Cells
miR-21 knockout cells containing the miR-21 target library were plated in six-well plates at ~300,000 cells per well. After 24 h, cells were transfected with variable miR-21 siRNA (Dharmacon) concentrations (100, 20, 4, 0.8, 0.16, or 0.032 nM) using lipofectamine 3000 (Invitrogen) according to manufacturer’s instructions. Cells were incubated for 48 h, after which RNA was isolated from each well using a Quick-RNA MiniPrep kit (Zymo). On-column DNase I treatment was performed for all samples according to manufacturer’s recommendation. RNA was then reverse-transcribed using superscript IV reverse transcriptase and an RT primer specific to the region immediately 3′ to the variable region of the miR-21 target reporter constructs. The resulting cDNA was barcoded and prepared for sequencing as described for the in vitro cleavage libraries. Paired-end sequencing was performed as described above for in vitro cleavage libraries.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Processing and Image Fitting
To map sequencing data to array experimental images, the previously extracted tile and coordinate information was cross-correlated to images iteratively. This process resulted in cluster coordinates being mapped to images at sub-pixel resolution as previously described (She et al., 2017; Denny et al., 2018). After coordinate mapping, each cluster was fit to a two-dimensional Gaussian to quantify fluorescence.
Association Curve Fitting
Following quantification of cluster intensity at each time point, association rates were fit for each variant. As imaging all DNA clusters required 18 images to be taken, the time for each image was set as the median time for the 18 images taken in that round of imaging. To account for variability between illumination and focus in each imaging cycle, the fluorescence intensity at each timepoint was normalized by dividing by the median fluorescence intensity of a fiducial mark (a fluorescent DNA oligonucleotide hybridized directly to single stranded DNA) that otherwise should have constant fluorescence intensity during the experiment. Association rates were determined by fitting the following single exponential to the median fluorescence of all clusters representing a single molecular variant at each timepoint:
where fintensity is the fluorescence intensity, feq is the fluorescence intensity at infinite time, fo is the fluorescence intensity at time 0, and kobs is the observed rate. Least-squares fitting here and for the equilibrium and cleavage fitting below was carried out using the python package lmfit.
Error in the measurement of the observed rates was estimated by bootstrapping the clusters representing each molecular variant. All clusters representing a single variant were sampled with replacement and the median fluorescence of the resampled clusters was fit to the above equation. This was repeated 1,000 times to generate 95% confidence intervals on the observed rate constant fits.
After computing the observed association rates, the observed rates for each variant were fit to the following equation to compute the association rate:
where kobs is the observed rate, kon is the association constant, koff is the dissociation constant, and [RISC] is the concentration of loaded AGO2.
Equilibrium Binding Curve Fitting
Initial Fitting of Single Clusters
The maximum fluorescence values determined by fitting the association experiments at each concentration were used to fit equilibrium dissociation constants. Using fit feq values, which represent the equilibrium binding at infinite time, ensured that the values used to fit equilibrium binding curves represented the amount of binding at equilibrium for each concentration. Since we found that the perfectly complementary target was fully bound at all concentrations that we performed experiments at, we normalized the feq values for all variants to the feq value of the perfectly complementary sequence at a given concentration (feq,norm = feq/feq,Perfect Complement). This allowed us to account for differences between experiments related to RNA production, illumination, and fiducial mark signal. The equilibrium fluorescence values at each concentration were fit to the following equation to determine the dissociation constant:
where feq,norm is the normalized equilibrium fluorescence intensity, fmax is the normalized fluorescence intensity when the target is fully bound, fmin is the normalized fluorescence intensity of the unbound target, [RISC] is the concentration of loaded AGO2, and KD is the dissociation constant. Since the fmin was very low for all variants it was constrained to be between 0 and 2 percent of the fully bound signal for the perfectly complementary target. The fraction bound at each concentration corresponds to the following:
Determination of fmax Distribution
After fitting this equation for all variants, it was necessary to account for uncertainty in the true fmax value for variants that were not fully bound at the highest experimental concentration. To do this we estimated the distribution of fmax values across all variants. This distribution was estimated by selecting all variants with KD values less than 30 pM, which should be fully saturated at the highest experimental concentration. The feq,norm values for all of these variants at the highest concentration was then used as the fmax distribution.
Bootstrapping to Estimate KD and Error
To estimate the KD and error for each molecular variant we first determined if the fmax distribution needed to be enforced. In cases where the maximum fluorescence achieved at any concentration exceeded the lower limit of the 95% confidence interval of the fmax distribution or the single cluster fit resulted in a KD more than 8-fold below the highest concentration, the fmax value was allowed to float during fitting. For these variants, the feq,norm values at each concentration were sampled from the feq values determined when bootstrapping the association rate fits. This was repeated 100 times to generate 95% confidence intervals for the equilibrium constant. The variants that did not reach a significant fluorescence level at any concentration and were not high enough affinity to reach near saturation at the highest concentration were also fit by sampling the feq,norm values at each concentration from the feq,norm values determined when bootstrapping the association rate fits. However, rather than allowing the fmax value to float, a value was selected from the fmax distribution determined with the high-affinity targets (above), and the fmax was constrained to this value during fitting. The final equilibrium constant was set to the median of these 100 fits and the 95% confidence interval was defined from all the fit values.
Validation of KD Limits of Detection
Despite only measuring binding at four concentrations, we were able to resolve differences in binding affinity between 10 pM and 10 nM. This was possible due to a few experimental features. First, binding was measured to ~50 independent clusters in parallel for each target in our library, a number that far exceeds the number of replicates performed in most biochemical studies. This allowed a high-confidence measure of binding signal at each concentration. Second, we were able to accurately quantify the expected fluorescence for saturated binding (i.e. fmax) using strong binders on the chip. Given an accurate measure of expected signal at saturation, as well as an accurate measure of the fraction bound at concentrations with non-negligible (>5% bound) and non-saturating (<86% bound) binding, we could fit a standard binding curve to these points constrained to saturate at our estimate fmax. This fit thus required a single free parameter, allowing estimation of KD even for targets where only a single concentration exhibited greater than 5% of maximum binding signal (the amount of binding expected for a 10 nM KD at 500 pM).
To provide evidence that this method accurately estimates the dissociation constant when only 5–10% binding was observed at a single concentration, we progressively down sampled the points defining the let-7a KD curves and refit the dissociation constants. This allowed us to compare dissociation constants defined by ~5% binding at the highest concentration to dissociation constants fit with all four points. When the highest point is removed, leaving a range of 63 pM–250 pM, the dissociation constants are highly correlated with those calculated using the full 63–500 pM concentration range (Figure S1C). Notably, the variants that only have 5–10% binding at 250 pM (lighter shaded region in plot), are fit to very similar dissociation constants, with an RMSE of 0.17 kcal/mol (an average of a 1.3–fold difference in the dissociation constant). Similarly, when the two highest concentration points are removed, we again observed good agreement between KD fits, and the points with only 5%–10% binding at 125 pM are fit to similar KD values, (RMSE of 0.26 kcal/mol), as when three points with greater than 5% bound are included (Figure S1C). Finally, when we use only the lowest concentration point to compute the dissociation constants, we can compare dissociation constants fit with 4 points with >5% binding to those fit with a single point with 5% binding (indicated by light shaded region in Figure S1C). The targets with only 5–10% binding at 63 pM are fit to KD values that deviate from the four-point fits by an average of 0.39 kcal/mol (~2–fold average difference in KD), although there is some bias towards higher affinity KD values. This bias may reflect a systematic deviation in the amount of binding at specific points, which would likely lead to a small shift in the KD calculated when this is the only point used, when compared to the KD calculated from multiple points. Interestingly, even the variants with less than 5% binding at a single down sampled point are still fit to dissociation constants that correlate well with their 4-point dissociation constant (regions to the right of the shaded regions in the top row of Figure S1C), indicating that our restricted dissociation constant estimation range (i.e >10 nM; <5% bound at 500 pM) is fairly conservative.
For targets with dissociation constants below our lowest concentration (50 pM), we were able to directly observe the amount of saturating binding (fmax) for each target. As a result, for any concentrations with sub-saturating binding, we could calculate the fraction bound at that concentration, and the corresponding dissociation constant. To show that we can compute dissociation constants from a single point with <86% binding, we again sub-sampled the data. Removing the lowest 1, 2, or 3 concentrations all resulted in average differences of less than 1.5–fold when the dissociation constants computed from a single concentration with less than 86% binding were compared to those computed from multiple concentrations with <86% binding (light shaded region in Figure S1C). This analysis demonstrates that even when KD values were defined by a single point, they were fit to nearly identical KD values as were originally determined using all concentrations.
Cleavage Rate Fitting
Sequencing data was first converted to counts for sequences in the designed library that had the same sequence in both read1 and read2. A set of highly degenerate normalization sequences was used to normalize the counts and account for any nonspecific RNA degradation or differences in sequencing depth at each time point using the following formula:
where countsi is the number of raw sequencing counts for a given variant at timepoint i, countsi,normalized is the number of normalized counts for a given variant at timepoint i, median(normalization counts)0 is the median number of raw sequencing counts across all normalization sequences at timepoint 0, and median(normalization counts)i is the median number of raw sequencing counts across all normalization sequences at timepoint i. For miR-21, the normalization sequences included 10 sequences with long stretches of central and seed mismatches. For let-7a, since the library tested for cleavage was much larger, all sequences with nucleotides t7–t11 mismatched were used as normalization sequences. Following normalization, the counts for each variant were fit to the following single exponential equation to determine the cleavage rate:
where countsmax is the counts at time 0, countsmin is the counts at infinite time, and kcleave is the single turnover cleavage rate. Variants for which the mean of the final 3 time points was greater than the mean of the first two time points or the overall change in counts was less than 15% of the median number of counts were defined as non-cleavers (kcleave < 0.0002 s−1) due to the insufficient loss of signal throughout the experiment. The cleavage data was also fit to an alternative model that incorporated both binding and cleavage. The following model:
was fit to the counts data using the relative association rates measured in the RNA array experiments and the dissociation rates determined from the association rates and dissociation constants measured in the RNA array experiments. In theory, cleavage rates for variants with slow association rates (e.g., seed mismatches) or exceptionally fast cleavage rates may deviate from the single exponential approximation. Using the above equations, we simulated the potential effects of using a single exponential fit to estimate the cleavage rate for variants with different cleavage rates, association rates, and dissociation rates, and showed that, for targets with slow association rates and fast cleavage rates, it is possible to underestimate the cleavage rate when using a single exponential fit (Figure S4B). However, when we fit our data to this model, we found that the exponential approximation yields essentially the same values as a model incorporating the measured relative association rates and dissociation rate (Figure S4C), so the values for the simpler, single-exponential, model were used.
Like in the array binding experiments, five nucleotides of RNA sequence flanking each side of the target site are accessible in RISC-CNS. This allows measuring the effect of 225 different five-nucleotide flanking contexts on cleavage rates of a target fully complementary to let-7a or miR-21. Flanking sequences had only modest effects on cleavage rate (kcleave, mean ± S.D. of 0.077 ± 0.024 s−1 for miR-21 and 0.037 ± 0.013 s−1 for let-7a), suggesting that the rates measured by RISC-CNS are generally insensitive to local secondary structure or biases from PCR amplification or high-throughput sequencing (Figure S4D).
RNA-Seq Data Analysis
The raw counts table that included RNA-Seq with (“AL7_Inp_rep1”, “AL7_Inp_rep2”) and without (“AC_Inp_rep1”,”AC_Inp_rep2”a) a let-7a decoy was downloaded from ArrayExpress (E-MTAB-5386). Log2 fold change between the control and let-7a decoy experiments was computed with DESeq2. Only genes containing an average of 10 counts or more in these 4 samples were used for downstream analysis.
Comparison of Seed Type Binding Affinity
3′ UTRs for the Gencode transcripts identified as representative transcripts in TargetScan were downloaded for mm10. We scanned through each 3′ UTR and counted the number of 6mer, 7mer-A1, 7mer-m8, and 8mer seed sequences. We selected transcripts containing only a single instance of a canonical seed site and compared the median log2 change for each of class of seed types to the median affinity measured for each canonical seed type.
Model Fitting
Alignment of Sequences
A dynamic programming approach was used to align the target and guide sequences. The following parameters were defined for the dynamic programming: a parameter for each nucleotide at positions t1–t21 (84 total) that are referred to as Epn where p is the guide position that the target is bound to (t1–t21) and n is the nucleotide (A, C, G, or U); parameters for opening target and guide bulges in the seed, central, and 3′ supplemental region (6 total); parameters for extension of target and guide bulges in the seed, central, and 3′ supplemental region (6 total); and a parameter for initiation of pairing following a bulge or mismatch. The parameters used were inspired by findings in binding experiments.
Four matrices (Ntarget × Nguide) were initialized to track the cases where the final subproblem ends with a match (M), a mismatch (N), a target bulge (T), and a guide bulge (G). For all matrices, the rows i represent the position in the target and the columns j represent the position in the guide. Row 1 of the match matrix is then initialized as follows:
Where t1 is the target nucleotide at position 1. Column 1 of the match matrix was initialized as:
The following recursions were then used to populate the four matrices:
If ti,gj complementary:
otherwise:
These recursions allowed us to identify the best register ending with a match at ti:gj, a mismatch at ti:gj, and a guide or target bulge at ti:gj. Following population of the four matrices, the minimum value in the matched matrix was selected as the most likely binding register. From the minimum entry in the matrix, a traceback to identify the steps taken to get to that point was performed, enabling to reconstruction of the optimal binding register.
Model for AGO2 Binding Affinity
To fit a model predicting RISC binding model, all miR-21 and let-7a sequences were aligned with the above method. Features were defined for each base at each position (21 positions × 4 bases/position = 84 parameters), for opening target and guide bulges in the seed, central, and 3′ supplemental region (2 strands × 3 regions = 6 parameters), for extension of target and guide bulges in the seed, central, and 3′ supplemental region (2 strands × 3 regions = 6 parameters), and for initiation of pairing following a bulge or mismatch. One additional feature used to account for RNA secondary structure was also included. This feature was calculated as the difference in the energy of the ensemble of RNA secondary structures formed when the seed region was involved in structure and when the seed region was constrained to be unstructured. These RNA structure predictions were made using the following commands in RNAfold. For the case of no constraint (no region forced to not form structure): RNAfold -T 37 C -p0 --noPS -i inputfile.fa. and for the case when a constraint was included: RNAfold -T 37 C -p0 --noPS -C -i inputfile.fa. Each of these commands were provided with a fasta file containing the RNA targets and, for the case where constraints were included they were indicated as:
UUUUUACUAUACAACCUCCUACCUCAUUUUU
..................xxxxxxxx.....
For fitting, data was filtered to only include RNA targets for which we could quantitatively measure a binding constant (10 nM > KD > 10 pM) and only sequences of length 39 nucleotides or less. Testing and training sets of equal size were randomly selected from the filtered data. All fitting was done using scikit-learn module in Python 2.7. The model was fit with Ridge regression to prevent parameters from being fit to large, non-physical values. All fits were performed with an intercept, which represents the intrinsic affinity of the protein for any nucleic acid strand.
Fitting of Cleavage Model
For miR-21 RISC, cleavage data was only collected on the original library (7,675 unique sequences; see assembly and sequencing of library above) that included primarily single mismatches, double mismatches, triple mismatches, insertions of different lengths, single and double deletions, combinatorial insertions, and structured and context variants. We filtered out the structured and context variants when doing the model fitting since this would have introduced many occurrences of the perfect complement target. We filtered the let-7 cleavage data to include the same classes of variants as the miR-21 data. This enabled comparison of the performance of the two models, as well as building and testing of a general model with similar data from both guides. Additionally, we are primarily interested in predicting cleavage for highly complementary sequences, and most of the remainder of the library probes questions relevant to miRNA binding but, since many of these targets have large numbers of mismatches, little cleavage activity is observed. Prior to fitting, the data was filtered to remove targets that we did not measure a cleavage rate for kcleave < 0.0001 s−1 and targets that had a poor goodness of fit (R2 < 0.6).
To fit the cleavage model, we first aligned all miR-21 and let-7a target sequences. After alignment we defined features for each mismatch at each position and for guide and target bulges at each positions. We performed a constrained fit when fitting the models for let-7a and miR-21 specific cleavage. The mismatch penalties were constrained to be no more than 2 natural logs below and 1 natural log above the observed single mismatch penalties during fitting. The guide and target bulge penalties were constrained to be no more than 1.5 natural logs below and 0.5 natural log above the observed single bulge penalties during fitting. The model was then fit to the single mismatches, double mismatches, single position target insertions, and single deletions using the lmfit module in Python 2.7. Following fitting of models for let-7 and miR-21, a general model was fit to both datasets. This model included the same bulge parameters as the guide specific models, but only included position specific parameters for transitions and transversions since the base depends on the microRNA/siRNA. This model was fit to all single and double mismatched targets and single insertions and deletions of let-7a and miR-21 and tested on triple mismatched targets and targets with multiple insertions and deletions for both sequences. Fitting of this model was performed with ridge regression in scikit-learn in Python 2.7.
Analysis of siRNA Efficacy in Cells
Sequence data was converted to counts and normalized as described above for in vitro cleavage data. The change in the abundance of each target sequence i for each condition j was calculated to be:
Where normalized countsi,0 is the normalized count of target i in the mock transfected cells.
Biochemical Model Derivation and Fitting
We aimed to predict mRNA steady state knockdown with a kinetic model of RISC activity. This approach has the benefit of not requiring any assumptions about the concentration of target RNA relative to the Km of the interaction (the free ligand approximation)—a significant limitation of many classical biochemical models of enzyme activity. For a given miR-21 target, we considered the following molecular species and rates:
[mRNA], concentration of unbound target mRNA,
[RISC], concentration of unbound, miR-21-loaded RISC,
[RISC:mRNA], concentration of loaded RISC bound to target mRNA,
[RISC:cutRNA], concentration of loaded RISC bound to cut mRNA,
ktrans, mRNA transcription rate,
kdegrade, basal mRNA degradation rate,
kon, association rate of RISC for target mRNA,
koff, dissociation rate of RISC from target mRNA,
kdecay, rate of miRNA accelerated mRNA decay, not through direct cleavage,
kcleave, single-turnover cleavage rate for RISC on target mRNA,
krelease, rate of product release.
We considered four rate equations describing RISC activity:
| 1) |
| 2) |
| 3) |
| 4) |
The quantity measured in the in cell knockdown assay is the total uncleaved RNA, or [RISC:mRNA] + [mRNA]. By setting each of the above equations to 0 and solving the system of equations, it can be shown that:
| 5) |
The maximum possible mRNA concentration [mRNAmax] was assumed to occur in the absence of any miR-21 siRNA:
| 6) |
Therefore, the change in the abundance of a target mRNA is given by:
| 7) |
This equation contains three unknown parameters: kdecay, kdegrade, and [RISC]. Because all targets were placed in a nearly identical gene context, we assumed that kdecay and kdegrade are constant across all targets and all miR-21 transfections. The free RISC concentration [RISC] was constrained to be at most the transfected miR-21 concentration, and was fit for each transfection condition. We observed that many targets had little knockdown in cells despite having in vitro cleavage rates >10-fold faster than their corresponding dissociation rates. We surmised that the cellular dissociation rates might be significantly faster than the measured in vitro rates. To account for this, we added a dissociation rate scaling term C, which was fit as a constant across all targets and all transfection conditions. Alternatively, if we scaled the cleavage rate rather than the dissociation rate the model performed similarly, suggesting that it is difficult to know whether cleavage or dissociation is most different in cells.
| 8) |
This model was fit using experimentally measured association rates and cleavage rates for each target. Dissociation rates were inferred from model predicted affinities. To limit differential effects of structure or other RNA binding proteins on the targets examined, only targets containing five adenosines flanking the targets region and that had values for all of the required parameters were used in model fitting and subsequent analyses (4,483 sequences).
DATA AND SOFTWARE AVAILABILITY
The kinetic and thermodynamic measurements generated in this paper are available for download as a supplemental table (Table S1) and on Mendeley Data (http://dx.doi.org/10.17632/fzh7pfpmmb.1). Sequencing data have been deposited in the NCBI Sequence Read Archive with SRA accession PRJNA512481. Custom software for determination of association rates, cleavage rates, and dissociation constants is available on GitHub.
Supplementary Material
Table S1. Kinetic and Thermodynamic Measurements for Each Target in this Study, Related to Figures 1–7 and S1–S7
Table S2. Oligonucleotides Used in this Study, Related to STAR Methods
Highlights.
Binding energies, association and cleavage rates measured for >40,000 RISC targets
AGO2 tolerates large insertions in the target opposite the central region of the guide
Some guide:target mismatches enhance the single-turnover RISC cleavage rate
In vitro measured biochemical parameters explain knockdown in cells
ACKNOWLEDGMENTS
This work was supported in part by NIH grants GM65236 and P01HD078253 to P.D.Z. and R01GM111990 to W.J.G. W.J.G. acknowledges support as a Chan-Zuckerberg Investigator. B.O.R. and W.R.B. were supported in part by the Stanford MSTP training grant (T32GM007365). W.R.B. was supported in part by the SIGF affiliated with ChEM-H. The HEK-293 Flp-In T-REx cell line was a gift from the Riedel-Kruse lab.
Footnotes
DECLARATION OF INTERESTS
P.D.Z. is a member of the scientific advisory boards of Alnylam Pharmaceuticals, Voyager Therapeutics, and ProQR. He is also a consultant for The RNA Medicines Company. The authors will file a provisional patent application on the work described.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang C-C, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, et al. (2018). Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med 379, 11–21. [DOI] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam J-W, and Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Martinez J, and Schroeder R (2007). Molecular basis for target RNA recognition and cleavage by human RISC. Cell 130, 101–112. [DOI] [PubMed] [Google Scholar]
- Ameres SL, Horwich MD, Hung J-H, Xu J, Ghildiyal M, Weng Z, and Zamore PD (2010). Target RNA-directed trimming and tailing of small silencing RNAs. Science 328, 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villén J, Shin C, Camargo FD, Gygi SP, and Bartel DP (2008). The impact of microRNAs on protein output. Nature 455, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2018). Metazoan MicroRNAs. Cell 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, and Giraldez AJ (2012). Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D, Koppal A, Agius P, Sander C, and Leslie C (2010). Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 11, R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaria N, Jarmoskaite I, and Herschlag D (2016). Specificity Principles in RNA-Guided Targeting. [DOI] [PMC free article] [PubMed]
- Buenrostro JD, Araya CL, Chircus LM, Layton CJ, Chang HY, Snyder MP, and Greenleaf WJ (2014). Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nat. Biotechnol 32, 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty C, Sharma AR, Sharma G, Doss CGP, and Lee S-S (2017). Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 8, 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandradoss SD, Schirle NT, Szczepaniak M, MacRae IJ, and Joo C (2015). A Dynamic Search Process Underlies MicroRNA Targeting. Cell 162, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GR, Sive H, and Bartel DP (2017). A Seed Mismatch Enhances Argonaute2-Catalyzed Cleavage and Partially Rescues Severely Impaired Cleavage Found in Fish. Mol. Cell 68, 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, and Darnell RB (2009). Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460, 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, and Darnell RB (2012). An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol 19, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PM, Loher P, Quann K, Brody J, Londin ER, and Rigoutsos I (2014). Argonaute CLIP-Seq reveals miRNA targetome diversity across tissue types. Sci. Rep 4, 5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De N, Young L, Lau P, Meisner N, Morrissey DV, and MacRae IJ (2013). Highly Complementary Target RNAs Promote Release of Guide RNAs from Human Argonaute2. Mol. Cell. 50, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deerberg A, Willkomm S, and Restle T (2013). Minimal mechanistic model of siRNA-dependent target RNA slicing by recombinant human Argonaute 2 protein. Proc. Natl. Acad. Sci. U. S. A 110, 17850–17855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny SK, Bisaria N, Yesselman JD, Das R, Herschlag D, and Greenleaf WJ (2018). High-Throughput Investigation of Diverse Junction Elements in RNA Tertiary Folding. Cell 174, 377–390.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, and Roeder RG (1983). Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG (2003). siRNAs can function as miRNAs. Genes Dev. 17, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, and Sharp PA (2004). Specificity of microRNA target selection in translational repression. Genes Dev. 18, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdy SF (2017). Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol 35, 222–229. [DOI] [PubMed] [Google Scholar]
- Dykxhoorn DM, Palliser D, and Lieberman J (2006). The silent treatment: siRNAs as small molecule drugs. Gene Ther. 13, 541–552. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, and Tuschl T (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Elkayam E, Kuhn C-D, Tocilj A, Haase AD, Greene EM, Hannon GJ, and Joshua-Tor L (2012). The structure of human argonaute-2 in complex with miR-20a. Cell 150, 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosswendt S, Filipchyk A, Manzano M, Klironomos F, Schilling M, Herzog M, Gottwein E, and Rajewsky N (2014). Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol. Cell 54, 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, and Bartel DP (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley B, and Zamore PD (2004). Kinetic analysis of the RNAi enzyme complex. Nat. Struct. Mol. Biol 11, 599–606. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Bernstein E, Beach D, and Hannon GJ (2000). An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404, 293–296. [DOI] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, and Tollervey D (2013). Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, and Brown PO (2009). Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol. 7, e1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvágner G, and Zamore PD (2002). A microRNA in a multiple-turnover RNAi enzyme complex. Science 297, 2056–2060. [DOI] [PubMed] [Google Scholar]
- Jo MH, Shin S, Jung S-R, Kim E, Song J-J, and Hohng S (2015). Human Argonaute 2 Has Diverse Reaction Pathways on Target RNAs. Mol. Cell 59, 117–124. [DOI] [PubMed] [Google Scholar]
- Kedde M, van Kouwenhove M, Zwart W, Oude JA, Elkon R, and Agami R (2010). A Pumilio-induced RNA structure switch in p27–3′ UTR controls miR-221 and miR-222 accessibility. Nature Cell Biology 12, 1014–1020. [DOI] [PubMed] [Google Scholar]
- Khorshid M, Hausser J, Zavolan M, and van Nimwegen E (2013). A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat. Methods 10, 253–255. [DOI] [PubMed] [Google Scholar]
- Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. (2005). Combinatorial microRNA target predictions. Nat. Genet 37, 495–500. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Shih I-H, Jones-Rhoades MW, Bartel DP, and Burge CB (2003). Prediction of mammalian microRNA targets. Cell 115, 787–798. [DOI] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song J, Hammond SM, Leemor J, and Hannon GJ (2004). Argonaute2 Is the Catalytic Engine of Mammalian RNAi. Science 305, 1437–1441. [DOI] [PubMed] [Google Scholar]
- Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, and Rudensky AY (2012). Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol. Cell 48, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Siederdissen C.H. zu, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol. Biol 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna JM, Scheel TKH, Danino T, Shaw KS, Mele A, Fak JJ, Nishiuchi E, Takacs CN, Catanese MT, de Jong YP, et al. (2015). Hepatitis C virus RNA functionally sequesters miR-122. Cell 160, 1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J-B, Yuan Y-R, Meister G, Pei Y, Tuschl T, and Patel DJ (2005). Structural basis for 5′-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature 434, 666–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machlin ES, Sarnow P, and Sagan SM (2011). Masking the 5’ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proceedings of the National Academy of Sciences 108, 3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, and Bartel DP (2009). Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carroll D, Mecklenbrauker I, Das PP, Santana A, Koenig U, Enright AJ, Miska EA, and Tarakhovsky A (2007). A Slicer-independent role for Argonaute 2 in hematopoiesis and the microRNA pathway. Genes Dev. 21, 1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Parizotto EA, Wang M, Roe SM, and Barford D (2009). Enhancement of the seed-target recognition step in RNA silencing by a PIWI/MID domain protein. Mol. Cell 33, 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Mark Roe S, and Barford D (2005). Structural insights into mRNA recognition from a PIWI domain–siRNA guide complex. Nature 434, 663–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister EL, Kennington L, Straubhaar J, Wagh S, Liu W, DiFiglia M, Landwehrmeyer B, Vonsattel J-P, Zamore PD, and Aronin N (2009). Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr. Biol. 19, 774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczko M, Maragkakis M, Alexiou P, Grosse I, and Hatzigeorgiou AG (2012). Functional microRNA targets in protein coding sequences. Bioinformatics 28, 771–776. [DOI] [PubMed] [Google Scholar]
- Salomon WE, Jolly SM, Moore MJ, Zamore PD, and Serebrov V (2015). Single-Molecule Imaging Reveals that Argonaute Reshapes the Binding Properties of Its Nucleic Acid Guides. Cell 162, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, and MacRae IJ (2012). The crystal structure of human Argonaute2. Science 336, 1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, Sheu-Gruttadauria J, and MacRae IJ (2014). Structural basis for microRNA targeting. Science 346, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, Sheu-Gruttadauria J, Chandradoss SD, Joo C, and MacRae IJ (2015). Water-mediated recognition of t1-adenosine anchors Argonaute2 to microRNA targets. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, Ding H, Kennington L, Moore JT, Schelter J, Burchard J, Linsley PS, Aronin N, Xu Z, and Zamore PD (2006). Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet. 2, e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, and Rajewsky N (2008). Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63. [DOI] [PubMed] [Google Scholar]
- She R, Chakravarty AK, Layton CJ, Chircus LM, Andreasson JOL, Damaraju N, McMahon PL, Buenrostro JD, Jarosz DF, and Greenleaf WJ (2017). Comprehensive and quantitative mapping of RNA–protein interactions across a transcribed eukaryotic genome. Proceedings of the National Academy of Sciences 114, 3619–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng G, Gogakos T, Wang J, Zhao H, Serganov A, Juranek S, Tuschl T, Patel DJ, and Wang Y (2017). Structure/cleavage-based insights into helical perturbations at bulge sites within T. thermophilus Argonaute silencing complexes. Nucleic Acids Res. 45, 9149–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu-Gruttadauria J, Xiao Y, Gebert LF, and MacRae IJ (2019). Beyond the seed: structural basis for supplementary microRNA targeting by human Argonaute2. EMBO J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Nam J-W, Farh KK-H, Chiang HR, Shkumatava A, and Bartel DP (2010). Expanding the microRNA targeting code: functional sites with centered pairing. Mol. Cell 38, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Reinhart BJ, Bartel DP, and Zamore PD (2003). A biochemical framework for RNA silencing in plants. Genes Dev. 17, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomari Y, and Zamore PD (2005). Perspective: machines for RNAi. Genes Dev. 19, 517–529. [DOI] [PubMed] [Google Scholar]
- Wang W, Yoshikawa M, Han BW, Izumi N, Tomari Y, Weng Z, and Zamore PD (2014). The Initial Uridine of Primary piRNAs Does Not Create the Tenth Adenine that Is the Hallmark of Secondary piRNAs. Mol. Cell 56, 708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Juranek S, Li H, Sheng G, Tuschl T, and Patel DJ (2008). Structure of an Argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 456, 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Juranek S, Li H, Sheng G, Wardle GS, Tuschl T, and Patel DJ (2009). Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature 461, 754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee LM, Flores-Jasso CF, Salomon WE, and Zamore PD (2012). Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell 151, 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werfel S, Leierseder S, Ruprecht B, Kuster B, and Engelhardt S (2017). Preferential microRNA targeting revealed by in vivo competitive binding and differential Argonaute immunoprecipitation. Nucleic Acids Res. 45, 10218–10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittrup A, and Lieberman J (2015). Knocking down disease: a progress report on siRNA therapeutics. Nat. Rev. Genet 16, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Tuschl T, Sharp PA, and Bartel DP (2000). RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101, 25–33. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Wagner EJ, and Cullen BR (2002). Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol. Cell 9, 1327–1333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Kinetic and Thermodynamic Measurements for Each Target in this Study, Related to Figures 1–7 and S1–S7
Table S2. Oligonucleotides Used in this Study, Related to STAR Methods
Data Availability Statement
The kinetic and thermodynamic measurements generated in this paper are available for download as a supplemental table (Table S1) and on Mendeley Data (http://dx.doi.org/10.17632/fzh7pfpmmb.1). Sequencing data have been deposited in the NCBI Sequence Read Archive with SRA accession PRJNA512481. Custom software for determination of association rates, cleavage rates, and dissociation constants is available on GitHub.