

Abstract
Acetaminophen (APAP)‐induced liver injury is the most common cause of acute liver failure (ALF) in the Western world. APAP toxicity progresses to multiorgan dysfunction and thus has broader whole‐body implications. Importantly, greater 30‐day mortality has been observed in liver transplant recipients following ALF due to APAP‐related versus non‐APAP‐related causes. Reasons for this discrepancy have yet to be determined. Extrahepatic toxicities of APAP overdose may represent underappreciated and unaddressed comorbidities within this patient population. In the present study, rapid induction of apoptosis following APAP overdose was observed in the intestine, an organ that greatly influences the physiology of the liver. Strikingly, apoptotic cells appeared to be strictly restricted to the intestinal crypts. The use of leucine‐rich repeat‐containing G protein–coupled receptor 5 (LGR5) reporter mice confirmed that the LGR5‐positive (+) crypt base stem cells were disproportionately affected by APAP‐induced cell death. Although the apoptotic cells were cleared within 24 hours after APAP treatment, potentially long‐lived consequences on the intestine due to APAP exposure were indicated by prolonged deficits in gut barrier function. Moreover, small intestinal cell death was found to be independent of tumor necrosis factor receptor signaling and may represent a direct toxic insult to the intestine by exposure to high concentrations of APAP. Conclusion: APAP induces intestinal injury through a regulated process of apoptotic cell death that disproportionately affects LGR5+ stem cells. This work advances our understanding of the consequences of APAP toxicity in a novel organ that was not previously considered as a significant site of injury and thus presents potential new considerations for patient management.
Abbreviations
- +
positive
- ALF
acute liver failure
- ALT
alanine aminotransferase
- APAP
acetaminophen
- creERT2
Cre recombinase estrogen receptor 2 fusion protein
- EGFP
enhanced green fluorescent protein
- ELISA
enzyme‐linked immunosorbent assay
- FITC
fluorescein isothiocyanate
- GFP
green fluorescent protein
- H&E
hematoxylin and eosin
- IgG
immunoglobulin G
- IHC
immunohistochemistry
- IP
intraperitoneal
- IRES
internal ribosome entry site
- LGR5
leucine‐rich repeat‐containing G protein–coupled receptor 5
- PARP
poly (adenosine diphosphate ribose) polymerase
- PBS
phosphate‐buffered saline
- PUR
puromycin
- RT
room temperature
- tm1Imx
targeted mutation 1 Immunex Research and Development Corporation
- TNF
tumor necrosis factor
- TNFR
tumor necrosis factor receptor
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
- WT
wild type
Acetaminophen (APAP) is an extensively used analgesic both in over‐the‐counter and prescription medication formulations due to its generally favorable side effect profile in comparison to nonsteroidal anti‐inflammatory drugs and opiates. At therapeutic doses, APAP is primarily metabolized by glucuronidation and sulfonation pathways and therefore is safely tolerated by most individuals.1 However, clearance of excessive amounts of APAP relies on cytochrome‐mediated oxidative metabolism primarily by hepatocytes, which involves generation of the highly reactive and toxic metabolite N‐acetyl‐p‐benzoquinione imine (NAPQI).1 Thus, at high doses, APAP causes significant liver injury and necrosis due to overwhelming oxidative stress, protein adduct formation, and cellular malfunction.1, 2
APAP overdose as a result of either a “therapeutic misadventure” or intentional self‐harm represents the leading cause of acute liver failure (ALF) in the United States, Canada, and the United Kingdom.3 Despite posing a widespread clinical issue, there is a lack of effective therapies for APAP‐induced hepatotoxicity to date. Currently, the standard of care is supplementation with N‐acetylcysteine (NAC), which limits hepatic accumulation of NAPQI by replenishing antioxidant glutathione stores. Although it is readily available, NAC supplementation is often started too late because the clinical symptoms of APAP‐induced liver failure often do not manifest until the peak of liver injury has been reached.1
Although some patients are able to survive the toxic insult and eventually recover due to the natural regenerative capacity of liver tissue, emergent liver transplantation is indicated for approximately 20% to 30% of patients due to excessive liver injury.1, 4 Even for those fortunate enough to procure a transplantable liver on urgent notice, transplant recipients who had developed ALF due to APAP toxicity versus ALF from non‐APAP‐related causes were found to suffer from a lower 30‐day survival.5 Explanations for this discrepancy remain unknown, but poorer medical compliance and greater adverse events related to psychologic comorbidities were speculated causes.
Undoubtedly, the major life‐threatening concern for APAP overdose is widespread liver cell death that rapidly progresses to ALF. However, APAP toxicity can also result in eventual multiorgan dysfunction. Thus, there are likely overlooked pathological effects of APAP overdose on target organs other than the liver that potentially contribute to the increased morbidity and mortality observed in APAP‐induced ALF transplant recipients. For example, a few studies have highlighted this possibility by identifying injury and inflammation in the brain, lung, and kidney following APAP overdose.6, 7, 8 The intestine is an organ with a particularly important role in both regulating and itself being modulated by liver disease due to the close physiological connections through portal circulation and hepatic biliary output.9 Critically, the intestine must maintain a tightly regulated and efficient barrier to entry for the countless species of bacteria, fungi, and viruses that comprise the gut microbiome. As has now been well established, loss of intestinal barrier function is a major contributor to various modes of liver injury and potentially other disorders,9 including those considered to be of neurologic origin, such as Parkinson’s disease.10 Importantly, although a few studies have focused on potential contributions of the gut microbiota on APAP‐induced liver injury,11, 12 APAP‐induced intestinal damage has not been fully explored.13, 14
In this study, we found that, in addition to the well‐characterized liver injury following APAP intoxication, damage also occurs within the intestine. Using a murine model of APAP hepatotoxicity, we demonstrate that intestinal apoptotic cell death occurs in the acute phases following APAP intoxication independent of tumor necrosis factor (TNF) receptor (TNFR) signaling. Specifically, our findings indicate that death of intestinal cells is an early event following APAP intoxication, is strictly limited to the crypts, and predominantly affects epithelial stem cell populations. However, we found that hepatocyte cell death occurred by an apoptosis‐independent mechanism, a finding that is consistent with previous reports.15 Together, these observations indicate that APAP intoxication can induce differential modes of injury across the intestine and liver and suggest that intestinal injury may be a neglected complication suffered by patients during APAP‐induced ALF with potential implications on overall outcome.
Materials and Methods
Mice
We purchased 6‐ to 8‐week‐old male C57BL6/J wild‐type (WT) and heterozygous leucine‐rich repeat‐containing G protein–coupled receptor 5–enhanced green fluorescent protein–internal ribosome entry site–Cre recombinase estrogen receptor 2 fusion protein (LGR5‐EGFP‐IRES‐creERT2) reporter mice from the Jackson Laboratory (Bar Harbor, ME). Age‐matched and sex‐matched B6.129S‐Tnfr superfamily, member 1a targeted mutation 1 Immunex Research and Development Corporation (sf1a)tm1Imx Tnfrsf1btm1Imx/J mice were originally purchased from the Jackson Laboratory but were bred and maintained in‐house. All animals were housed in specific pathogen‐free housing in compliance with the Emory Institutional Animal Care and Use Committee and National Institutes of Health guidelines.
APAP Treatment
Mice were fasted for 12 hours before treatment with 500 mg/kg APAP (Sigma‐Aldrich, St Louis, MO) or an equivalent volume of phosphate‐buffered saline (PBS) vehicle by intraperitoneal (IP) injection. APAP solution was prepared fresh on the day of injection by heating 25 mg/mL APAP in PBS at 75°C until fully dissolved (approximately 5 minutes). APAP solutions were sterile filtered and cooled to 37°C. APAP and PBS control solutions were kept in a 37°C water bath during injections to prevent precipitation. Food was returned to mice immediately after treatment, and heating pads were provided in cages to prevent hypothermia. Animals were euthanized 4 or 24 hours after treatment by CO2 inhalation.
Following euthanasia, blood was collected by cardiac puncture. Livers were gently perfused with ice‐cold sterile PBS through the portal vein and harvested for further analysis. Ileum tissue (approximated as the most distal third of the small intestine) was excised and trimmed of associated mesenteric fat. Ileum samples were opened by a longitudinal cut, and fecal contents were removed by gently shaking the tissue while immersed in cold sterile PBS. Portions of the liver and ileum were frozen on dry ice and placed in long‐term storage at −80°C.
In vivo intestinal permeability assay
To assess intestinal permeability at 4 hours after APAP treatment, mice were restricted from access to food and water for 12 hours before APAP injection. At the time of injection, mice were also orally gavaged with 1 g of 4‐kDa fluorescein isothiocyanate (FITC)–dextran (Sigma‐Aldrich) per kilogram of body weight. FITC–dextran solution was dissolved at 160 mg/mL in PBS. Food and water were returned to mice immediately following gavage, and 4 hours later mice were euthanized and blood was collected. To assess intestinal permeability at 24 hours after APAP treatment, mice were restricted from access to food but had free access to water for 12 hours before APAP injection. After injection, food was returned to the mice. Mice were restricted from access to water starting 8 hours after APAP treatment. The animals were then gavaged with FITC–dextran as described above at 20 hours after APAP treatment. Water was returned to the animals until 4 hours later (at 24 hours after APAP injection) when mice were euthanized and blood was collected.
Blood samples were protected from light the entire time following harvest. Blood was incubated at room temperature (RT) for at least 1 hour to clot, and serum was separated by high‐speed centrifugation for 10 minutes. Serum aliquots were diluted 1:1 with PBS and analyzed in duplicate for fluorescence readings at 488/530 nm. FITC–dextran concentration in serum samples was determined according to standard curve values generated from serially diluting (in PBS) the FITC–dextran solution used at the time of oral gavage. All serum samples were normalized to control mouse serum without FITC–dextran, whereas standard curve wells were normalized to PBS alone.
Tissue Homogenization and Western Blot Analysis
Small portions of frozen animal tissue were lysed in a modification of Abcam’s intestinal lysis buffer recipe consisting of 150 mM NaCl, 50 mM Tris‐HCl pH 7.4, 1% Triton X‐100, 1% sodium deoxycholic acid, and 0.1% sodium dodecyl sulfate (SDS) reconstituted with 1X protease inhibitor cocktail (Sigma‐Aldrich). Tissues were homogenized either by a handheld rotary homogenizer (#985370; BioSpec Products, Bartlesville, OK) or with Lysing Matrix D ceramic beads in a FastPrep24 5G grinder (MP Biomedicals, Santa Ana, CA) used at the preregistered settings specified for the respective tissues. All samples within each experimental set were homogenized by the same method to ensure equivalent comparison of protein contents. Following homogenization, samples were cleared of solid debris by centrifugation in a tabletop centrifuge at maximum speed for 20 minutes at 4°C. Controls for apoptotic activation markers were generated by acquiring whole‐cell lysates from Raw 264.7 macrophages (ATCC, Manassas, VA) incubated with media only or 10 μg/mL puromycin (PUR) for approximately 18 hours. Protein concentration of lysates was quantified by DC protein assay (BioRad, Hercules, CA). Equal amounts of protein were diluted in 1X sample loading buffer containing 5% β‐mercaptoethanol and heated at 95°C for 5‐10 minutes. Following reduction, 25‐30 μg of each protein sample was used for SDS–polyacrylamide gel electrophoresis and western blot analysis. Blots were incubated overnight at 4°C in primary antibody solutions at the following concentrations in 5% bovine serum albumin/trishydroxymethylaminomethane‐buffered saline Tween 20 (TBST): rabbit anti‐poly (adenosine diphosphate ribose) polymerase (PARP) 1:1,000 (#9532; Cell Signaling Technology, Danvers, MA), mouse anti‐β‐actin 1:5,000 (#A2228; Sigma‐Aldrich), mouse anti‐β‐tubulin 1:5,000 (#T8328; Sigma‐Aldrich), rabbit anti‐caspase‐9 1:1,000 (#NBP2‐67362; Novus Biologicals, Centennial, CO), rabbit anti‐cleaved caspase‐8 1:1,000 (#8592; Cell Signaling Technology). Secondary antibody solutions consisting of goat anti‐rabbit immunoglobulin (IgG) or goat anti‐mouse IgG (Jackson ImmunoResearch, West Grove, PA) were applied for 1 hour at RT both at a 1:10,000 dilution in 5% milk/TBST. Western blots were developed using Clarity Western ECL blotting substrate (BioRad) and read using a ChemiDoc XRS+ System and Image Lab software v6.0.1 (BioRad).
Histology and Immunohistochemistry
At the time animals were euthanized, mouse ileums were cleaned as described above and excess fluid was absorbed by placing ileums on paper towels. Ileums were adhered to a flat toothpick, gently rolled along their length from proximal to distal end, and then transferred into 10% phosphate‐buffered formalin for fixation overnight. Harvested livers were cut into small sections and also fixed by overnight incubation in 10% formalin. Tissues were transferred into 70% ethanol for long‐term storage and later paraffin embedded for sectioning. Sections 10 μm thick were used for all staining procedures. Hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC) analysis of cleaved caspase‐3 (#9961, 1:200; Cell Signaling Technology) were performed by the Pathology Core, Yerkes National Primate Research Center, Emory University School of Medicine. Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) staining was done using a commercial kit (#11684817910; Roche, Indianapolis, IN), and slides were counterstained with hematoxylin.
Tissue Cryosections and Immunofluorescence
Ileum tissue was harvested as described above and immediately frozen in VWR frozen section compound (VWR, Radnor, PA) for sectioning. Frozen ileum sections were fixed in 4% paraformaldehyde in PBS for 15 minutes at RT and stained with rabbit anti‐cleaved caspase‐3 (1:400; Cell Signaling Technology) and biotinylated mouse anti‐GFP tag (#MA515256BTIN, 1:500; Thermo Fisher Scientific, Waltham, MA). Goat anti‐rabbit IgG (heavy [H]+light [L] chains)‐AlexaFluor 594 (1:500; Jackson ImmunoResearch) and FITC–conjugated streptavidin (BD Biosciences, San Jose, CA) were used for secondary staining. Staining, mounting, and imaging of slides were done as described.16
Serologic Measurements
Serum levels of alanine aminotransferase (ALT) were measured by the Pathology Core, Yerkes National Primate Research Center, Emory University School of Medicine. Quantification of TNFα in serum was determined using a mouse TNFα enzyme‐linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Serum samples were diluted 1:1 with reagent diluent for TNFα ELISA analysis. Cytokine array analysis of serum harvested from mice 4 hours after APAP or PBS treatment was conducted according to the manufacturer’s instructions (#ab133993; Abcam, Cambridge, United Kingdom).
Statistical Analysis
Data are presented as sample means ± SEM. The two‐tailed Student t test was performed using GraphPad Prism 7.0a software (GraphPad Software, La Jolla, CA) with statistical significance considered as P < 0.05.
Results
Acute APAP Toxicity Rapidly Increases Intestinal Permeability and Apoptosis of Crypt Cells in Small Intestine
Several recent studies in diverse models of injury, such as sepsis and radiation injury, have demonstrated that intestinal injury occurs within remarkably quick time frames after insult.17, 18 Thus, we sought to better understand the early pathological events that occur following APAP intoxication. To this end, we treated fasted mice with a toxic dose of APAP and harvested liver and intestinal tissue 4 hours later for analysis. This acute 4‐hour time point was selected based off the likely presence of significant early liver injury. Consistent with previous observations, serum ALT levels were significantly elevated in APAP‐treated animals versus controls (Fig. 1A; 8,391 ± 2,375 vs. 105.7 ± 5.69 U/L, respectively). Animals were concurrently administered FITC–dextran at the time of APAP treatment to determine whether early changes in gut permeability occurred during APAP toxicity. Surprisingly, we observed a roughly 2.5‐fold increase in serum recovery of FITC–dextran by 4 hours after APAP treatment compared to controls (Fig. 1B; 2.36 ± 0.24 vs. 0.89 ± 0.13 μg/mL, respectively). These data indicate that APAP impacted intestinal physiology within the same time frame as induction of liver injury.
Figure 1.
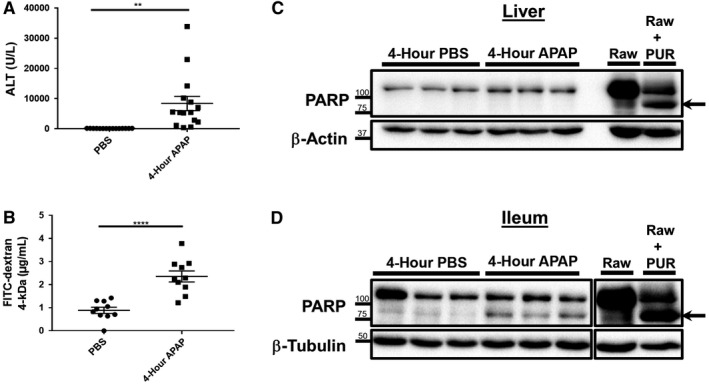
Increased intestinal permeability and enhanced PARP cleavage is detected within the ileum during the acute response to APAP overdose. Six‐ to eight‐week‐old male C57BL/6J mice were fasted and restricted from water for 12 hours. Mice were then treated with 500 mg/kg APAP by IP injection and 1 g/kg 4‐kDa FITC–dextran by oral gavage. Water and food were returned to the animals until they were euthanized 4 hours later. Serum was analyzed for (A) levels of ALT and (B) recovery of FITC–dextran. Data are represented as means ± SEM and are representative of 10‐15 mice per group from at least three independent experiments. Statistical analyses were made using the Student t test; **P < 0.01, ****P < 0.0001. Total protein lysates (25‐30 μg) of (C) liver and (D) distal ileum tissue were analyzed by western blot for PARP cleavage. Lysates of Raw 264.7 macrophages treated with or without 10 μg/mL PUR for 18 hours were included as controls. Arrows indicate cleaved PARP. Breaks in the depicted ileum image represent nonadjacent wells on the same gel. Western blot images are representative of 10‐12 mice per group from at least three independent experiments.
We postulated that the enhanced gut permeability that occurred in response to APAP intoxication was due to intestinal injury. Therefore, we next investigated protein tissue lysates for cleavage of PARP as a marker for apoptotic cell death.19 Despite the high serum ALT levels that were consistent with severe liver injury, western blots of liver tissue revealed the absence of PARP cleavage above trace detection levels (Fig. 1C). However, whole ileum lysates unexpectedly revealed a significant increase of cleaved‐PARP signal in APAP‐treated mice when compared to the baseline amounts detected in controls (Fig. 1D). Because there was no consequent reduction in the expression levels of total PARP within ileum lysates, this potentially suggested that the increase in PARP cleavage arose from a relatively small population of cells. Thus, in order to clarify these differential observations between the liver and ileum, we next examined histopathological tissue sections by H&E staining. As expected, in comparison to PBS‐treated mice, liver sections from APAP‐treated animals displayed significant areas of liver cell death occurring primarily around the central vein (Fig. 2A). Corroborating our western blot analyses, intestinal sections revealed the appearance of small condensed vesicles consistent with the appearance of apoptotic bodies that were present only within the crypts of APAP‐treated but not control mice (Fig. 2A). To further confirm whether these vesicles represent apoptotic cells, we performed IHC for cleaved/activated caspase‐3, the effector molecule and well‐verified marker for apoptosis.15 Consistent with the lack of detectable PARP cleavage by western blot, both control and APAP‐treated liver tissues displayed an absence of caspase‐3 staining (Fig. 2B). However, ample positive cleaved caspase‐3 staining was observed within the intestinal crypts of APAP‐treated mice, supporting the hypothesis that the vesicles observed by H&E staining were derived from apoptotic cells (Fig. 2B).
Figure 2.
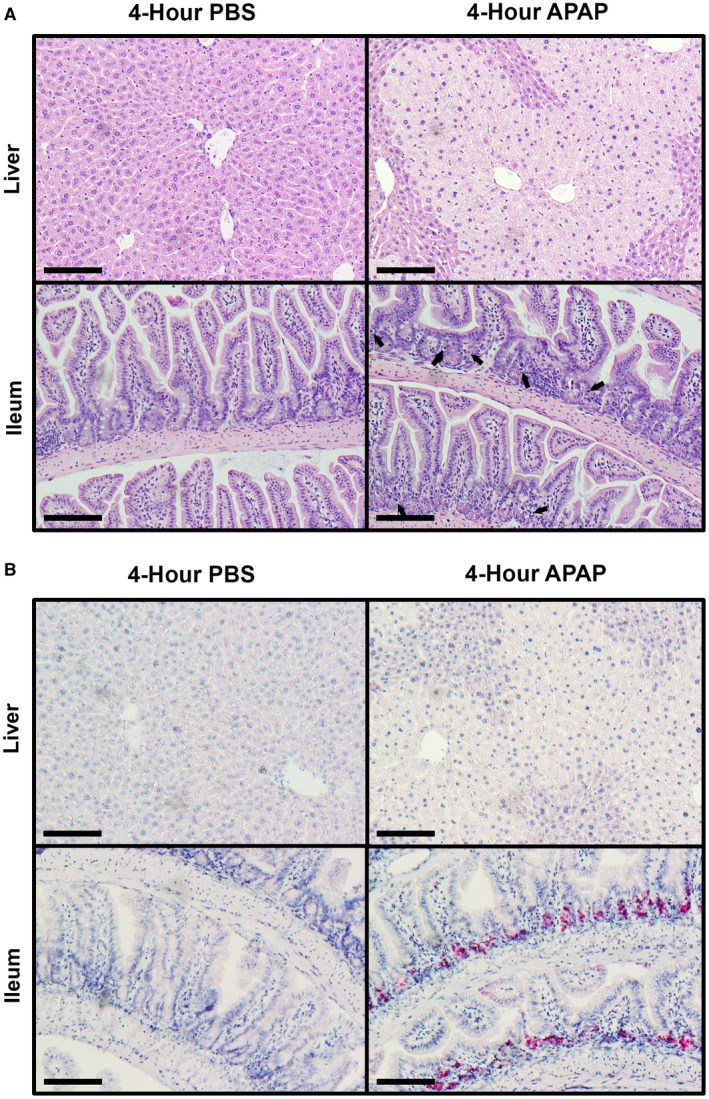
Apoptosis is induced in the ileum but not liver during the acute response to APAP intoxication. Six‐ to eight‐week‐old male C57BL/6J mice were fasted for 12 hours and treated with 500 mg/kg APAP by IP injection. Food was returned to the animals, and they were euthanized 4 hours later. Formalin‐fixed paraffin‐embedded sections of liver and distal ileum tissue were analyzed by (A) H&E staining and (B) IHC staining for cleaved caspase‐3. All provided images were taken at magnification ×200 and are representative of at least three mice per group. Scale bars represent 150 μm; arrows indicate apoptotic bodies.
APAP‐Induced Intestinal Apoptosis Occurs Early Following the Onset of Intoxication
The appearance of apoptotic cells within the intestine following acute APAP overdose was strictly limited to cells within the crypts (Fig. 2A,B). Because the intestinal crypts are home to various populations of adult stem cells that are responsible for regulating the physiological turnover of enterocytes,20 we questioned what changes may occur to the intestine within a longer period of time after APAP treatment. Therefore, we similarly assessed gut barrier function and investigated PARP cleavage in liver and ileum lysates at 24 hours after intoxication. In comparison to control animals, ALT levels remained significantly elevated in APAP‐treated mice (Fig. 3A; 24,276 ± 2,593 vs. 96.67 ± 2.64 U/L, respectively). Serum FITC–dextran recovery was also significantly higher in APAP‐treated animals compared to controls (Fig. 3B; 2.62 ± 0.71 vs. 0.60 ± 0.08 μg/mL, respectively). PARP cleavage was still undetected in liver lysates, suggesting that apoptosis was not playing a significant role in APAP‐induced liver cell death and injury (Fig. 3C). However, although an increase in PARP cleavage was still observed at 24 hours in APAP‐treated mice ileums versus controls, the effect was less pronounced compared to results seen at 4 hours (Figs. 1D, 3D). Therefore, in order to determine whether there were any changes at the tissue structure level, we again examined histopathological sections at 24 hours after APAP treatment. By this time, livers demonstrated extensive regions of late necrotic death. However, only sparse apoptotic bodies remained within ileum sections without any gross alteration of crypt–villus structure (Fig. 4A). Cleaved caspase‐3 staining was similarly sparse within ileum sections at 24 hours after APAP treatment (Fig. 4B). As was the case during the earlier phases of APAP hepatotoxicity, liver sections were negative for cleaved caspase‐3 staining at 24 hours. Moreover, a 48‐hour time course experiment indicated that apoptotic cells in the small intestine crypts did not appear until sometime between 1 and 4 hours after APAP intoxication (Supporting Fig. S1). Together, these data suggest that death of APAP‐sensitive intestinal cell populations occurs early, but not immediately, after overdose and that clearance of these apoptotic cells is largely complete by 24 hours. Further, apoptosis does not appear to be a significant contributor to direct APAP hepatotoxicity.
Figure 3.
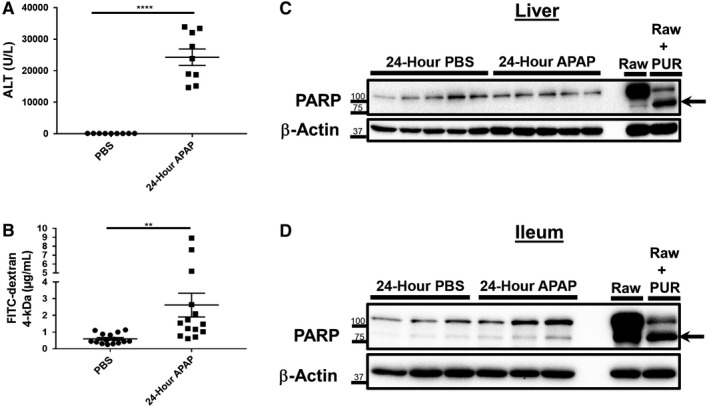
Intestinal barrier deficits persist at 24 hours after APAP overdose despite a reduction in ileum PARP cleavage. Six‐ to eight‐week‐old male C57BL/6J mice were fasted for 12 hours and treated with 500 mg/kg APAP by IP injection. Food was returned to the animals, and they were restricted from water access starting from 8 hours after APAP injection. After an additional 12 hours, mice were gavaged with 1 g/kg 4‐kDa FITC–dextran. Water was then returned to the animals until they were euthanized 4 hours later (a total of 24 hours after APAP treatment). Serum was analyzed for levels of (A) ALT and (B) recovery of FITC–dextran. Data are represented as means ± SEM and n = 9‐15 mice per group from at least three independent experiments. Statistical analyses were made by the Student t test; **P < 0.01, ****P < 0.0001. Total protein lysates (25‐30 μg) of (C) liver and (D) distal ileum tissue were analyzed by western blot for PARP cleavage. Lysates of Raw 264.7 macrophages treated with or without 10 μg/mL PUR for 18 hours were included as controls. Arrows indicate cleaved PARP. Western blot images are representative of 10 to 15 mice per group from at least three independent experiments.
Figure 4.
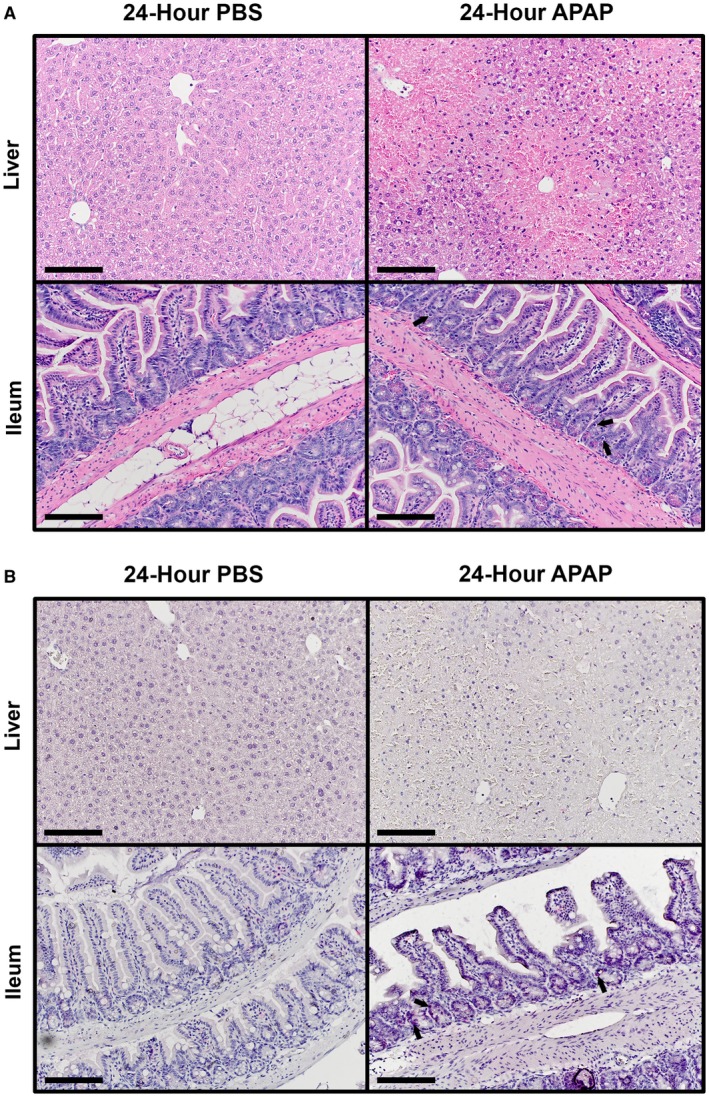
Intestinal apoptotic bodies are cleared within 24 hours after APAP overdose. Six‐ to eight‐week‐old male C57BL/6J mice were fasted for 12 hours and treated with 500 mg/kg APAP by IP injection. Food was returned to the animals, and they were euthanized 24 hours later. Formalin‐fixed paraffin‐embedded sections of liver and distal ileum tissue were analyzed by (A) H&E staining and (B) IHC staining for cleaved caspase‐3. All provided images were taken at magnification ×200 and are representative of at least three mice per group. Scale bars represent 150 μm; arrows indicate (A) apoptotic bodies and (B) cells with positive staining for cleaved caspase‐3.
APAP‐Induced Small Intestinal Apoptosis Occurs Independent of TNFR Signaling
The appearance of apoptotic bodies within the ileum was observed alongside significant liver injury (Figs. 1A, 2B). It was unclear whether the intestinal injury occurred as a result of the systemic release of soluble mediators from the damaged liver tissue or due to a direct mechanism localized at the intestine. For instance, studies have demonstrated that APAP‐induced liver injury results in the release of inflammatory and cytotoxic cytokines, such as TNFα and high‐mobility group box 1 (HMGB1), into the serum.14, 21 TNFα itself has been shown to play a role in inducing intestinal injury and increasing intestinal permeability in various disease models.22, 23, 24 For these reasons, we first focused on investigating potential roles of TNFα signaling in APAP‐induced intestinal toxicity. As reported,21 we detected greater concentrations of TNFα in the serum of mice 4 hours after APAP treatment in comparison to PBS‐treated controls (Fig. 5A; 64.75 ± 22.88 vs. 0.43 ± 0.43 pg/mL TNFα, respectively). However, some variability was noted in this observation because TNFα remained undetectable in the serum from approximately 35% of APAP‐treated mice. In order to directly investigate a potential role of TNFα signaling in mediating APAP‐induced intestinal crypt cell death either by systemic or localized mechanisms, we employed knockout mice for both the TNFRα chain and TNFRβ chain (TNFR−/−). TNFR−/− mice were found to suffer similar levels of liver necrosis and enhanced intestinal permeability within the acute response to APAP intoxication (Supporting Fig. S2). PARP cleavage was absent in both APAP‐treated WT and TNFR−/− mice, as anticipated (Fig. 5B). However, ileum lysates demonstrated that an increase in PARP cleavage occurred even in the absence of TNFR signaling (Fig. 5C). Furthermore, the appearance of apoptotic bodies by H&E, IHC staining for cleaved/activated caspase‐3, and direct staining of apoptotic cells by TUNEL assay was identical between APAP‐treated WT and TNFR−/− ileum sample sections (Fig. 5D). Because our observations to this point were restricted to the small intestine, we additionally investigated whether the colonic epithelium was injured during APAP intoxication. Cleaved/activated caspase‐3 was detected within the colon 4 hours after APAP treatment. However, unlike the small intestine, the colonic epithelium of TNFR−/− mice demonstrated significantly less cleaved caspase‐3 staining than WT animals (Supporting Fig. S3). These results indicate that the apoptotic response of small intestinal crypt cells, but not colonocytes, that is induced during APAP intoxication occurs independently from TNFα signaling.
Figure 5.
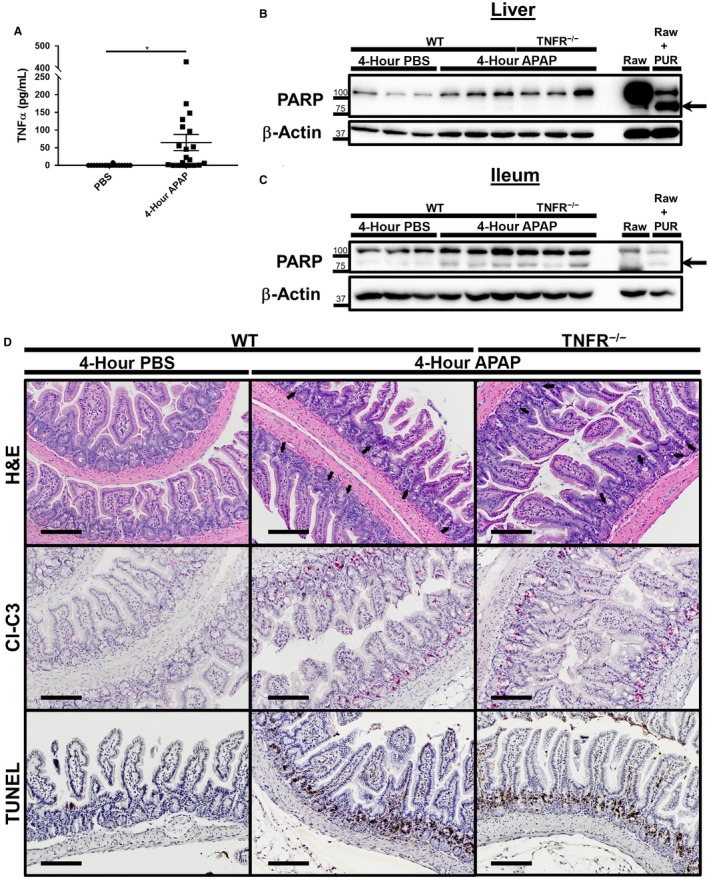
Apoptotic death of intestinal crypt cells following APAP overdose occurs independent of TNFR signaling. Six‐ to eight‐week‐old male C57BL/6J and age‐matched TNFR−/− male mice were fasted for 12 hours and treated with 500 mg/kg APAP by IP injection. Food was returned to the animals, and they were euthanized 4 hours later. Serum was analyzed for (A) concentration of TNFα by sandwich ELISA. Concentrations are represented as means ± SEM and are representative of 18‐20 mice per group from at least six independent experiments. Statistical analyses were made using the Student t test; ***P < 0.001. Total protein lysates (25‐30 μg) of (B) liver and (C) distal ileum tissue were also analyzed by western blot for PARP cleavage. Lysates of Raw 264.7 macrophages treated with or without 10 μg/mL PUR for 18 hours were included as controls. Arrows indicate cleaved PARP. Western blot images are representative of 10‐11 mice per group from at least three independent experiments. Formalin‐fixed paraffin‐embedded sections of distal ileum tissue were analyzed by (D) H&E staining, IHC staining for cleaved caspase‐3, and TUNEL assay. All provided images were taken at magnification ×200 and are representative of at least three mice per group. Scale bars represent 150 μm; arrows indicate apoptotic bodies. Abbreviation: CL‐C3, cleaved caspase 3.
Small Intestinal Crypt Stem Cells are Disproportionately Susceptible to APAP‐Induced Cell Death
The small intestines of both WT and TNFR−/− mice suffered widespread apoptosis 4 hours after APAP treatment, yet all cell death remained restricted to the intestinal crypts. The intestinal crypt base houses the epithelial stem cell populations, most notably the rapidly dividing LGR5‐positive (+) crypt base columnar cells.20 LGR5+ cells and other subsets of small intestinal crypt cells are highly susceptible to cytotoxic stressors, such as radiation and numerous chemotherapeutics.25, 26, 27 Therefore, we hypothesized that the APAP‐induced death we observed was disproportionately affecting LGR5+ stem cell populations. To test this hypothesis, we treated heterozygous LGR5‐EGFP‐IRES‐creERT2 reporter mice with APAP or PBS and harvested their ileums 4 hours later. Immunofluorescence analysis of fresh frozen ileum revealed diffuse expression of GFP in the intestinal crypts of control animals that was reduced in APAP‐treated mice (Fig. 6). Consistent with IHC staining, apoptotic bodies were detected in the intestinal crypts of APAP‐treated animals that had strong staining for cleaved caspase‐3. Merging of these two channels demonstrated that the majority of these apoptotic bodies colocalized with strong condensed GFP+ signals (Fig. 6). Together, these data demonstrate that intestinal toxicity due to high‐dose APAP exposure disproportionately induced death of LGR5+ stem cells.
Figure 6.
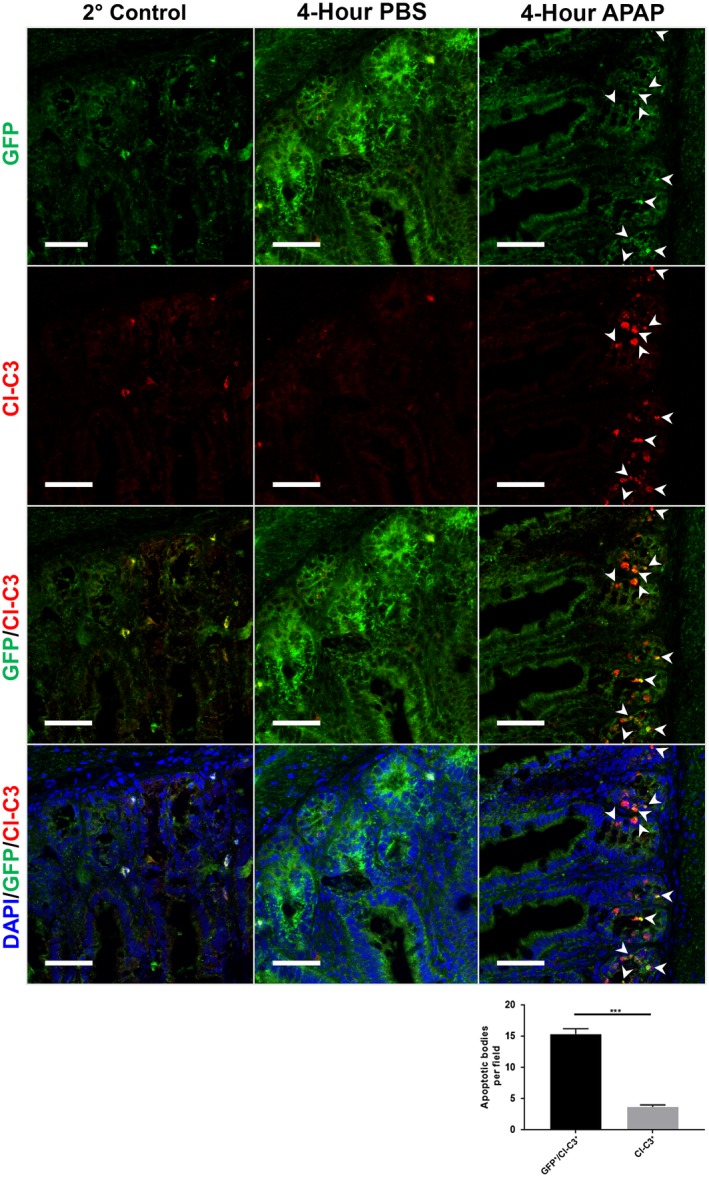
APAP‐induced intestinal apoptosis disproportionately affects LGR5+ stem cells. Six‐ to eight‐week‐old male heterozygous LGR5‐EGFP‐IRES‐creERT2 reporter mice were fasted for 12 hours and treated with 500 mg/kg APAP by IP injection. Food was returned to the animals, and they were euthanized 4 hours later. Frozen ileum sections were stained for expression of GFP and cleaved caspase‐3. All slides were counterstained with DAPI and analyzed by confocal immunofluorescence microscopy. All images except the 2° control are representative of n = 3 mice per group and are magnification ×400. Scale bars represent 75 μm; white arrowheads indicate sites of colocalization of GFP with cleaved caspase‐3. Quantification graph indicates analysis of GFP‐expressing (GFP+/Cl‐C3+) and GFP‐nonexpressing (Cl‐C3+) apoptotic blebs from three APAP‐treated animals; ***P < 0.001. Abbreviations: 2° control, secondary antibody control; CL‐C3, cleaved caspase‐3; DAPI, 4′,6‐diamidino‐2‐phenylindole.
Discussion
Widespread liver cell death that rapidly progresses to ALF is without question the definitive threat posed by APAP toxicity. However, the specific kinetic progression and mechanistic nature of APAP liver injury remains incompletely characterized. For instance, a somewhat controversial topic is the relative contributions of the various forms of cell death (e.g., apoptosis, necrosis, pyroptosis).2, 28 Even to this day, publications report conflicting evidence either in favor of or against a role of hepatic apoptosis in APAP hepatotoxicity.15, 29, 30 Although this topic was not the focus of the current study, our data provide the strongest support to the argument that direct hepatic injury due to APAP toxicity is primarily driven by necrotic cell death. Instead, we surprisingly found that widespread apoptosis occurred throughout the crypts of the small intestine rapidly after APAP overdose. Interestingly, this observation confirms the speculations made by Possamai et al.13 that serum markers of apoptotic injury measured in human patient samples were derived from dying gut cells. Although a few studies have demonstrated potential toxicity of APAP on intestinal cell lines and organoids in vitro,31, 32, 33 to our knowledge, this study provides the first direct evidence that APAP induces regulated cell death to intestinal cells in vivo.
Our observations highlight the fact that APAP toxicity is a whole‐body issue with pathological effects beyond liver injury. Previous reports have indicated APAP induces inflammation and injury of the kidney, lung, and brain.6, 7, 8 For this study, we focused on the gut due to the critical role the intestinal physiology and the microbiome play in regulating chronic conditions of the liver9 and potentially other organs.10 Currently, the role that intestinal barrier function and intestinal microbiota play in APAP‐induced liver injury is somewhat unclear. On one hand, germ‐free mice were found to be equally susceptible to APAP liver injury as conventionally housed animals.12 On the other hand, antibiotic‐treated mice were found to be protected from APAP hepatotoxicity, which was linked to the contribution of the bacterial metabolite 1‐phenyl‐1,2‐propanedione enhancing liver injury.11 However, because liver injury is the immediate and acute concern with APAP toxicity, an overlooked and unexplained observation is that APAP ALF transplant recipients have been found to have higher short‐term mortality rates than patients who have undergone transplantation for other etiologies of ALF.5 Although the causes for this observation are currently unclear, greater noncompliance to treatment regimens among APAP ALF transplant recipients was one speculated cause. However, it is entirely possible that this increased short‐term mortality may in fact be in part caused by other toxic implications of APAP overdose beyond those directly on the liver that are left unaddressed by liver transplantation alone. To this end, we hypothesize that these toxic insults on target organs in addition to the liver are comorbid events with health implications that will become more important to consider as management of APAP ALF improves in the future.
In our present observations, APAP‐induced small intestinal cell death was strictly limited to populations of cells found within the crypts, which is a pattern of cell death that is strikingly similar to that induced by radiation exposure, cancer chemotherapy, and some forms of sepsis.18, 26, 34 This topographic location along the crypt–villus structure is enriched in various intestinal epithelial stem cell populations, such as rapidly dividing LGR5+ cells and quiescent B lymphoma Mo‐MLV insertion region1 homolog expressing (BMI‐1+) cells. Indeed, use of LGR5‐EGFP‐IRES‐creERT2 reporter mice demonstrated that the majority of these dying cells were LGR5+ cells. However, it is currently unclear what percentage of LGR5+ cells survive APAP toxicity and whether the apoptotic bodies that did not colocalize with the GFP reporter tag represent late remnants of LGR5+ cells or death of small subsets of LGR5– cellular populations. Regardless, the specificity of APAP‐induced death to intestinal stem cells implies that APAP intestinal injury is likely to have longer lasting effects on overall intestinal physiology.20, 35 Although this remains to be fully determined, our observations at least indicate that enhanced gut permeability is also triggered at an early stage of APAP toxicity but persists even after apoptotic cells are cleared from the crypts.
Unfortunately, the direct mechanism of APAP‐induced apoptotic death remains unclear. Although we originally hypothesized that the intestinal injury occurred as a secondary result of massive hepatic necrosis, our evidence suggests that these events are likely independent from each other. Our initial focus for testing this hypothesis was TNFα, which has been shown both in the present work and by previous studies to be rapidly elevated in the serum following APAP overdose.21 However, a significant subset of animals did not have elevated serum TNFα levels at a time point at which intestinal apoptosis was widespread. Furthermore, intestinal apoptosis was highly prominent before the peak of liver injury, as suggested by our ALT and histologic analyses. By 24 hours after APAP overdose, a time by which liver injury had further progressed, apoptosis of intestinal stem cells was no longer present. Therefore, it seems most likely that APAP induces a direct toxic insult localized to the intestine itself. Furthermore, although TNFR−/− signaling may contribute to colonic injury, it does not play a significant role in APAP‐induced injury to the small intestine. Whether other members of the death receptor family, such as Fas/FasL, play a role in APAP‐induced intestinal injury remains to be determined. It is also distinctly possible that APAP may induce cytotoxic stress‐related intrinsic cell death in intestinal stem cells because the gut does express cytochrome P450 2E1 (CYP2E1), the predominant metabolic enzyme for production of APAP‐related reactive oxygen species.2, 36, 37, 38 Elucidating the relative roles of intrinsic versus extrinsically induced cell death will have to be a focus of future work on extrahepatic APAP toxicity, including intestinal injury.
In conclusion, we provide the observation that APAP overdose induces rapid apoptosis of intestinal stem cells, predominantly of the LGR5+ population at the base of the crypts. The pattern of this intestinal cell death mimics that observed by radiation‐induced injury and has potential long‐term consequences. The massive acute loss of LGR5+ stem cells is possibly a unique aspect of APAP ALF that distinguishes this patient population from those suffering ALF from other causes. Thus, it is possible that comorbid intestinal dysfunction contributes to the greater mortality observed after transplantation for ALF caused by APAP versus non‐APAP causes. Characterizing the mechanism of this death and the potential contributions APAP‐induced intestinal injury have on overall morbidity and mortality will be an important advancement toward improving the management of patients suffering from APAP toxicity and ALF.
Supporting information
Acknowledgment
We thank Dr. Pratyusha Mandal for the helpful scientific discussions and technical support in guiding this work; Krystel Chopyk for assistance in final preparation of the figures; and Dr. Brian Evavold, Dr. Joshy Jacob, and Dr. Malú Tansey as critical sources of feedback and scientific critique. We are grateful to the Pathology Cores of the Emory Vaccine Center and the veterinary and animal care staff of Yerkes National Primate Research Center, Emory University School of Medicine, for their assistance. This research project was supported in part by the Emory University Integrated Cellular Imaging Microscopy Core.
Supported by National Institutes of Health (grants R01AI124680, R01AI126890, and R01AI136533 to A.G.; R01DK044234 to M.J.C.); Office of Research Infrastructure Programs/Office of the Director (P51OD011132 to A.G.; formerly National Center For Research Resources number P51RR000165 to the Yerkes National Primate Research Center); National Institute of Alcohol Abuse and Alcoholism, Ruth L. Kirschtein National Research Service Award Individual Predoctoral Fellowship (F31AA024960 to D.M.C.); National Institute of General Medical Sciences Institutional Research and Academic Career Development Award (2K12GM000680‐16 to J.D.S.); National Institute of Diabetes and Digestive and the Kidney Diseases Mentored Career Development Award (K01DK109025 to M.T.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Potential conflict of interest: Nothing to report.
See Editorial on Page 1421
References
Author names in bold designate shared co‐first authorship.
- 1. Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N. Acetaminophen‐induced hepatotoxicity: a comprehensive update. J Clin Transl Hepatol 2016;4:131‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iorga A, Dara L, Kaplowitz N. Drug‐induced liver injury: cascade of events leading to cell death, apoptosis or necrosis. Int J Mol Sci 2017;18:1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bernal W, Wendon J. Acute liver failure. N Engl J Med 2013;369:2525‐2534. [DOI] [PubMed] [Google Scholar]
- 4. Bunchorntavakul C, Reddy KR. Acetaminophen‐related hepatotoxicity. Clin Liver Dis 2013;17:587‐607. [DOI] [PubMed] [Google Scholar]
- 5. Cooper SC, Aldridge RC, Shah T, Webb K, Nightingale P, Paris S, et al. Outcomes of liver transplantation for paracetamol (acetaminophen)‐induced hepatic failure. Liver Transpl 2009;15:1351‐1357. [DOI] [PubMed] [Google Scholar]
- 6. Neff SB, Neff TA, Kunkel SL, Hogaboam CM. Alterations in cytokine/chemokine expression during organ‐to‐organ communication established via acetaminophen‐induced toxicity. Exp Mol Pathol 2003;75:187‐193. [DOI] [PubMed] [Google Scholar]
- 7. da Silva MH, da Rosa EJ, de Carvalho NR, Dobrachinski F, da Rocha JB, Mauriz JL, et al. Acute brain damage induced by acetaminophen in mice: effect of diphenyl diselenide on oxidative stress and mitochondrial dysfunction. Neurotox Res 2012;21:334‐344. [DOI] [PubMed] [Google Scholar]
- 8. Kennon‐McGill S, McGill MR. Extrahepatic toxicity of acetaminophen: critical evaluation of the evidence and proposed mechanisms. J Clin Transl Res 2018;3:297‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut‐liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 2018;15:397‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell 2016;167:1469‐1480.e1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gong S, Lan T, Zeng L, Luo H, Yang X, Li N, et al. Gut microbiota mediates diurnal variation of acetaminophen induced acute liver injury in mice. J Hepatol 2018;69:51‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Possamai LA, McPhail MJ, Khamri W, Wu B, Concas D, Harrison M, et al. The role of intestinal microbiota in murine models of acetaminophen‐induced hepatotoxicity. Liver Int 2015;35:764‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Possamai LA, McPhail MJ, Quaglia A, Zingarelli V, Abeles RD, Tidswell R, et al. Character and temporal evolution of apoptosis in acetaminophen‐induced acute liver failure*. Crit Care Med 2013;41:2543‐2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang R, Zou X, Tenhunen J, Zhu S, Kajander H, Koskinen ML, et al. HMGB1 neutralization is associated with bacterial translocation during acetaminophen hepatotoxicity. BMC Gastroenterol 2014;14:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaeschke H, Duan L, Akakpo JY, Farhood A, Ramachandran A. The role of apoptosis in acetaminophen hepatotoxicity. Food Chem Toxicol 2018;118:709‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chopyk DM, Kumar P, Raeman R, Liu Y, Smith T, Anania FA. Dysregulation of junctional adhesion molecule‐A contributes to ethanol‐induced barrier disruption in intestinal epithelial cell monolayers. Physiol Rep 2017;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, DeLaney A, et al. Caspase‐8 collaborates with caspase‐11 to drive tissue damage and execution of endotoxic shock. Immunity 2018;49:42‐55.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qiu W, Leibowitz B, Zhang L, Yu J. Growth factors protect intestinal stem cells from radiation‐induced apoptosis by suppressing PUMA through the PI3K/AKT/p53 axis. Oncogene 2010;29:1622‐1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35:495‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barker N, van Oudenaarden A, Clevers H. Identifying the stem cell of the intestinal crypt: strategies and pitfalls. Cell Stem Cell 2012;11:452‐460. [DOI] [PubMed] [Google Scholar]
- 21. Simpson KJ, Lukacs NW, McGregor AH, Harrison DJ, Strieter RM, Kunkel SL. Inhibition of tumour necrosis factor alpha does not prevent experimental paracetamol‐induced hepatic necrosis. J Pathol 2000;190:489‐494. [DOI] [PubMed] [Google Scholar]
- 22. Van Hauwermeiren F, Armaka M, Karagianni N, Kranidioti K, Vandenbroucke RE, Loges S, et al. Safe TNF‐based antitumor therapy following p55TNFR reduction in intestinal epithelium. J Clin Invest 2013;123:2590‐2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schulzke JD, Bojarski C, Zeissig S, Heller F, Gitter AH, Fromm M. Disrupted barrier function through epithelial cell apoptosis. Ann N Y Acad Sci 2006;1072:288‐299. [DOI] [PubMed] [Google Scholar]
- 24. Chen P, Starkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis‐induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 2015;61:883‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Metcalfe C, Kljavin NM, Ybarra R, de Sauvage FJ. Lgr5+ stem cells are indispensable for radiation‐induced intestinal regeneration. Cell Stem Cell 2014;14:149‐159. [DOI] [PubMed] [Google Scholar]
- 26. Yu J. Intestinal stem cell injury and protection during cancer therapy. Transl Cancer Res 2013;2:384‐396. [PMC free article] [PubMed] [Google Scholar]
- 27. Ijiri K, Potten CS. Further studies on the response of intestinal crypt cells of different hierarchical status to eighteen different cytotoxic agents. Br J Cancer 1987;55:113‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krenkel O, Mossanen JC, Tacke F. Immune mechanisms in acetaminophen‐induced acute liver failure. Hepatobiliary Surg Nutr 2014;3:331‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Du K, Ramachandran A, Weemhoff JL, Woolbright BL, Jaeschke AH, Chao X, et al. Mito‐tempo protects against acute liver injury but induces limited secondary apoptosis during the late phase of acetaminophen hepatotoxicity. Arch Toxicol 2019;93:163‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li X, Lin J, Lin Y, Huang Z, Pan Y, Cui P, et al. Hydrogen sulfide protects against acetaminophen‐induced acute liver injury by inhibiting apoptosis via the JNK/MAPK signaling pathway. J Cell Biochem 2019;120:4385‐4397. [DOI] [PubMed] [Google Scholar]
- 31. Li AP, Alam N, Amaral K, Ho MD, Loretz C, Mitchell W, et al. Cryopreserved human intestinal mucosal epithelium: a novel in vitro experimental system for the evaluation of enteric drug metabolism, cytochrome P450 induction, and enterotoxicity. Drug Metab Dispos 2018;46:1562‐1571. [DOI] [PubMed] [Google Scholar]
- 32. Schafer C, Schroder KR, Hoglinger O, Tollabimazraehno S, Lornejad‐Schafer MR. Acetaminophen changes intestinal epithelial cell membrane properties, subsequently affecting absorption processes. Cell Physiol Biochem 2013;32:431‐447. [DOI] [PubMed] [Google Scholar]
- 33. Bundscherer AC, Malsy M, Gruber MA, Graf BM, Sinner B. Acetaminophen and metamizole induce apoptosis in HT 29 and SW 480 colon carcinoma cell lines in vitro. Anticancer Res 2018;38:745‐751. [DOI] [PubMed] [Google Scholar]
- 34. Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot DM 2nd, Buchman TG, et al. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia‐induced sepsis. JAMA 2002;287:1716‐1721. [DOI] [PubMed] [Google Scholar]
- 35. Bull‐Otterson L, Feng W, Kirpich I, Wang Y, Qin X, Liu Y, et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS ONE 2013;8:e53028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bergheim I, Bode C, Parlesak A. Distribution of cytochrome P450 2C, 2E1, 3A4, and 3A5 in human colon mucosa. BMC Clin Pharmacol 2005;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Forsyth CB, Voigt RM, Shaikh M, Tang Y, Cederbaum AI, Turek FW, et al. Role for intestinal CYP2E1 in alcohol‐induced circadian gene‐mediated intestinal hyperpermeability. Am J Physiol Gastrointest Liver Physiol 2013;305:G185‐G195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue‐selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol 2003;43:149‐173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials