

Abstract
Excessive accumulation of lipids in the liver is crucial in the pathogenesis of alcoholic steatohepatitis and may be partly mediated by impaired degradation of lipid droplets by autophagy. The E3 ubiquitin ligase SMAD‐specific E3 ubiquitin protein ligase 1 (SMURF1) regulates selective autophagy by ubiquitinating proteins on cargo destined for autophagic delivery to the lysosome for degradation. Here, we evaluated the role of SMURF1 in the regulation of hepatic lipid degradation in alcoholic steatohepatitis. In patients with severe alcoholic hepatitis, SMURF1 colocalized with lipid droplet membranes in liver explants. In a mouse model of alcoholic steatohepatitis, Smurf1 −/− mice fed an alcohol diet displayed increased hepatocyte accumulation of lipid droplets and triglycerides as well as more severe liver injury compared to wild‐type mice. The increased severity of liver steatosis in alcohol‐fed Smurf1 −/− mice was rescued by adeno‐associated virus (AAV) serotype 8‐mediated hepatic expression of wild‐type Smurf1 protein but not by mutant Smurf1 proteins either lacking the catalytically active cysteine 699 required for ubiquitin transfer or the N‐terminal C2 phospholipid membrane‐binding domain. Conclusion: Smurf1 plays a protective role in the pathogenesis of alcoholic steatohepatitis through a mechanism that requires both its ubiquitin‐ligase activity and C2 phospholipid‐binding domains. These findings have implications for understanding the roles of ubiquitin ligases in fatty liver disease.
Abbreviations
- AAV
adeno‐associated virus
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- AST
aspartate aminotransferase
- cDNA
complementary DNA
- H&E
hematoxylin and eosin
- HECT
homologous to the E6 activator protein carboxyl terminus
- mRNA
messenger RNA
- PBS
phosphate‐buffered saline
- PPAR‐γ
peroxisome proliferator‐activated receptor gamma
- SMURF1
SMAD‐specific E3 ubiquitin protein ligase‐1
The clinical spectrum of alcoholic liver disease includes alcoholic fatty liver, alcoholic steatohepatitis, cirrhosis, and hepatocellular cancer. Alcoholic fatty liver, also known as steatosis, is the earliest stage of alcoholic liver disease defined by the accumulation of lipid droplets in hepatocytes resulting from increased lipid synthesis and decreased lipid degradation (reviewed in Osna et al.1) More recently, defects in selective autophagy have also been shown to contribute to alcoholic fatty liver disease.2, 3
Autophagy protects cells from excessive stress by degrading cellular proteins and subcellular organelles and contributes to hepatocyte recovery from toxic effects of alcohol.4, 5 Following exposure to ethanol, hepatocytes respond by sequestering lipid droplets and mitochondria in autophagosomes, which then undergo degradation in lysosomes.4, 6 Pharmacologic or genetic inhibition of autophagy exacerbates ethanol‐induced liver injury in mice.6, 7 However, the detailed mechanisms by which autophagy protects against alcoholic liver disease are unknown.
The E3 ubiquitin ligase SMAD‐specific E3 ubiquitin protein ligase 1 (SMURF1) regulates selective autophagy by ubiquitinating proteins on cargo destined for autophagic delivery to the lysosome for degradation.8 Previous reports demonstrated that Smurf1 deficiency results in the development of spontaneous hepatic steatosis in aged mice8, 9 and aggravates hepatic steatosis in a mouse model of nonalcoholic fatty liver disease.9 The role of SMURF1 in the pathogenesis of alcoholic fatty liver disease is not known.
Here, we show that Smurf1 deficiency aggravates hepatic steatosis in a mouse model of alcoholic fatty liver disease. This protective effect of Smurf1 requires both its ubiquitin‐ligase activity and C2 phospholipid‐binding domains.
Method
Immunofluorescence of Human Liver Tissue
Archived liver explants from patients from Johns Hopkins School of Medicine who had acute alcoholic hepatitis were subjected to immunofluorescence analyses to detect colocalization of SMURF1 with the lipid droplet marker perilipin‐2 (PLIN2). Informed consent in writing was obtained from each patient, and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki, as reflected in a priori approval by the appropriate institutional review committee. No donor organs were obtained from executed prisoners or other institutionalized persons. Paraffin‐embedded 5‐μm tissue sections were deparaffinized in xylene and ethanol. Antigen retrieval was performed by boiling in 0.01 M citrate. Autofluorescence was quenched using the Trueblack lipofuscin autofluorescence quencher (catalogue number, 23007; Biotium, Inc.) as per the manufacturer’s protocol. Samples were blocked in 5% goat serum in phosphate‐buffered saline (PBS). Primary antibody against SMURF1 was obtained from Sigma (catalogue number, WH0057154M1), and primary antibody against PLIN2 was obtained from Abcam (catalogue number, 108323). Both primary antibodies were diluted 50‐fold in 5% goat serum in PBS and incubated with the samples overnight at 4°C. Secondary antibodies (goat anti‐mouse Alexa 488; catalogue number, A11029; and goat anti‐rabbit Alexa 568; catalogue number, A11036; Thermo Fisher Scientific) at 1:400 dilution were incubated with samples for 1 hour at room temperature. Nuclei were stained with Hoechst 33342 (catalogue number, 134406; Thermo Fisher Scientific) at a dilution of 1:2,500 for 30 minutes. Slides were mounted in Prolong diamond antifade mountant (catalogue number, P36961; Thermo Fisher Scientific). A Zeiss LSM880 Airyscan confocal microscope was used for obtaining Z‐stacks of imaging. Autoquant software (Media Cybernetics, Rockville, MD) was used for deconvolution analysis, and Imaris software (Oxford Instruments, Abingdon, United Kingdom) was used for final image processing.
Mouse Model of Alcoholic Fatty Liver Disease
Female C57BL/6 Smurf1 −/− mice10 or wild‐type littermate controls, aged 8‐12 weeks, were subjected to a two‐step alcohol‐exposure protocol, as described in Bertola et al.11 Briefly, this protocol consists of 10‐day feeding with a fresh liquid diet containing alcohol (Lieber‐DeCarli ‘82 Shake and Pour ethanol liquid diet; product number, F1258SP; Bio‐Serv) or with a control isocaloric diet (Lieber‐DeCarli ‘82 Shake and Pour control liquid diet; product number, F1259SP; Bio‐Serv). The liquid diet was made fresh and replenished daily between 7 pm and 8 pm. The 10‐day feeding was followed by a single intragastric administration of ethanol (5 g ethanol/kg body weight) or isocaloric maltose‐dextrin, administered between 7 am and 8 am. Mice were killed 9 hours later. Mice were housed with two animals per standard cage with wood shavings bedding and ad libitum access to water. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86‐23, revised 1985), and all animal studies were approved by the University of Texas Southwestern Institutional Animal Care and Use Committee.
AAV‐Mediated Smurf1 Expression in Mouse Livers
Murine Smurf1 complementary DNA (cDNA) was amplified from a mouse Smurf1 open reading frame clone (catalogue number, MG210325; Origene). The C699A mutation was introduced using the QuickChange II site‐directed mutagenesis kit (catalogue number, 200523; Agilent). Round‐the‐horn mutagenesis was used to obtain Smurf1 cDNA lacking the C2 domain. The cDNA sequences encoding the wild‐type, C699A, or ΔC2 Smurf1 proteins were cloned into the pLE282 plasmid containing the thyroid hormone‐binding globulin (TBG) promoter (obtained from Dr. Luke Engelking, University of Texas Southwestern Medical Center), using the Gibson assembly kit (catalogue number, E2611; New England Biolabs). AAV serotype 8 and the TBG promoter were chosen to ensure that transgene expression is specific to hepatocytes.12 AAV stock was produced at the Vector Core at the University of North Carolina at Chapel Hill.
AAV viral stock diluted in PBS was administered to female Smurf1 −/− mice at 3 × 1013 genomic copies per kilogram of body weight by tail vein injection in a total volume of 200 μL per mouse. The alcohol‐feeding protocol was initiated 7 days after the injection, as described above. At the conclusion of the experiment, messenger RNA (mRNA) from liver was extracted using the QIAGEN RNeasy Mini Kit (catalogue number, 74104) and reverse transcribed using the iScript master mix (catalogue number, 1708891; Bio‐Rad). Quantitative polymerase chain reaction was performed using the 5′ CAGCGACTCCGAAATCCTGA 3′ forward and 5′ GCCCAAGTTCATCGCAGTTC 3′ reverse primers.
Histopathological Analyses
Mouse liver sections were stained with hematoxylin and eosin (H&E) or Oil Red O (Molecular Pathology Core, Columbia University, New York, NY) and analyzed by microscopy.
Biochemical Analyses
Analysis of liver triglycerides and serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) was performed by the Metabolic Phenotyping Core Facility at the University of Texas Southwestern Medical Center.
Statistical Analysis
Statistical significance was determined using one‐way analysis of variance (ANOVA). Data are shown as mean ± SEM. P < 0.05 was considered significant. SPSS was used for statistical analysis.
Results
SMURF1 Associates With Hepatic LIPID Droplets in Patients With Alcoholic Hepatitis
The subcellular localization of SMURF1 within hepatocytes in human liver is unknown. In order to elucidate whether SMURF1 is present on lipid droplets, we used samples obtained from liver explants of patients with acute alcoholic hepatitis, a condition characterized by the presence of lipid droplets in hepatocytes.13 We found that SMURF1 colocalized with lipid droplet membranes in liver explants from these patients (Fig. 1). SMURF1 also colocalized with the smaller lipid droplets observed in the livers of healthy donors (Fig. 1). Thus, in human hepatocytes, SMURF1 colocalizes with lipid droplets, suggesting it may play a role in modulating their degradation.
Figure 1.
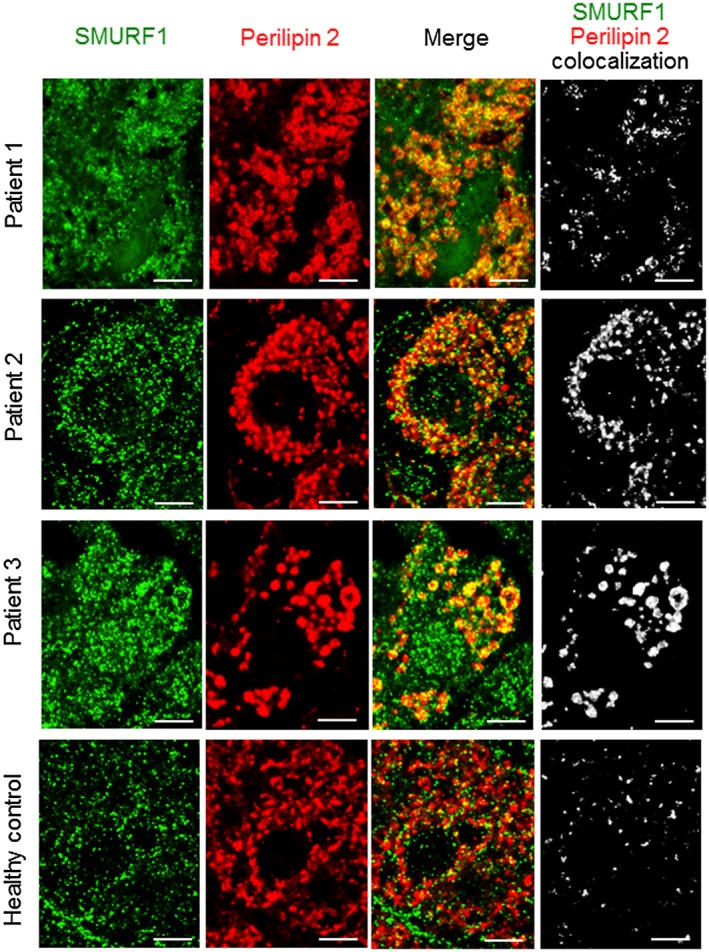
SMURF1 colocalizes with lipid droplets in human hepatocytes. Representative image of liver explants from 3 patients with severe alcoholic hepatitis and from 1 healthy donor. The white signal in the image on the right indicates regions of colocalization of SMURF1 (green) and perilipin 2 (red). Similar results were observed in liver explants from 5 independent patients. Scale bars, 5 μm.
SMURF1 is Required for LIPID Degradation in a Mouse Model of Alcoholic Fatty Liver Disease
To evaluate the role of Smurf1 in hepatic steatosis, we used a well‐established mouse model of alcoholic fatty liver disease, also known as the National Institute on Alcohol Abuse and Alcoholism protocol.11 This protocol consists of 10‐day feeding with a liquid diet containing alcohol, followed by a single intragastric administration of ethanol. As expected, wild‐type mice exposed to alcohol developed liver steatosis and injury (Fig. 2). Compared to wild‐type mice, alcohol‐fed Smurf1 −/− mice showed worsening of liver histopathology (Fig. 2A); more pronounced liver steatosis (as demonstrated by Oil Red O staining; Fig. 2A), increased liver:body weight ratio (Fig. 2B), and increased liver triglyceride content (Fig. 2C); and aggravation of liver injury, as demonstrated by increased serum levels of AST and ALT (Fig. 2D,E). These data indicated that Smurf1 plays a protective role in a mouse model of alcoholic fatty liver disease.
Figure 2.
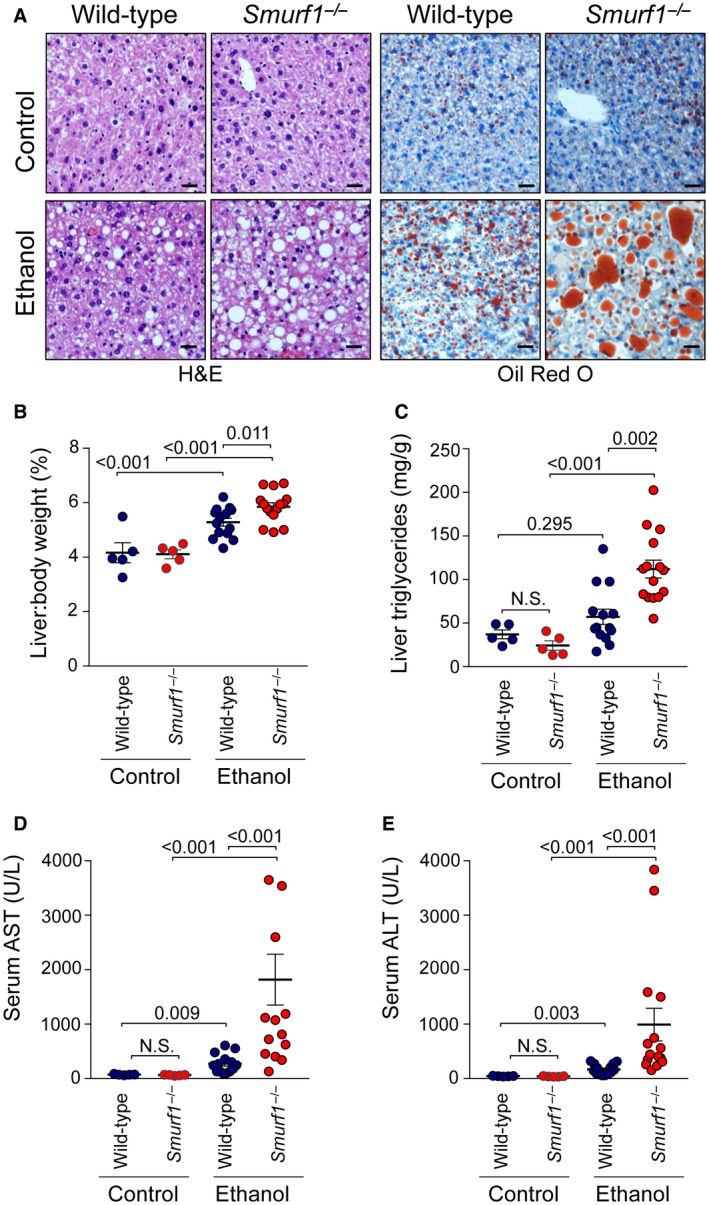
Smurf1 deficiency exacerbates ethanol‐induced hepatic steatosis and liver injury in mice. (A) Representative photomicrographs of hepatic injury and steatosis assessed by H&E and Oil Red O staining in livers of mice of the indicated genotype fed the control or ethanol diet. Quantitative assessment of hepatitis steatosis by measurement of (B) liver:body weight ratio and (C) hepatic triglyceride levels. Quantitative assessment of liver injury by measurement of serum (D) AST and (E) ALT. Combined data from two independent cohorts of animals are shown. Similar results were observed in each independent cohort. Pooled n = 5 (wild‐type, control diet); n = 5 (Smurf1 −/−, control diet); n = 14 (wild‐type, ethanol diet); n = 15 (Smurf1 −/−, ethanol diet). Horizontal lines represent mean and vertical lines represent SEM. Numbers in graphs denote P values. One‐way ANOVA was used for statistical analysis; AST and ALT values underwent logarithmic transformation prior to statistical analysis. Scale bars, 50 μm. Abbreviation: N.S., not significant.
Ubiquitin‐Ligase Activity and the C2 Domain of Smurf1 are Required for LIPID Degradation
Smurf1 is composed of an N‐terminal C2 domain involved in membrane binding, two central WW domains involved in protein–protein interactions, and a C‐terminal homologous to the E6‐activator protein carboxyl terminus (HECT) ubiquitin‐ligase domain (Fig. 3A).14 To determine which domains of Smurf1 are required for its protective effect in alcoholic steatohepatitis, we used AAV to overexpress wild‐type Smurf1 or Smurf1 mutants, including Smurf1 C699A, which has a loss‐of‐ubiquitin‐ligase function mutation in the HECT domain,15, 16 or Smurf1 ΔC2, which lacks amino acids 14‐137 and is defective in membrane localization but not ubiquitin‐ligase activity17 (Fig. 3B). Previous studies have demonstrated that the Smurf1 C2 domain is required for mitophagy8 and that both the C2 domain and C699 catalytically active site are required for selective bacterial autophagy.18
Figure 3.
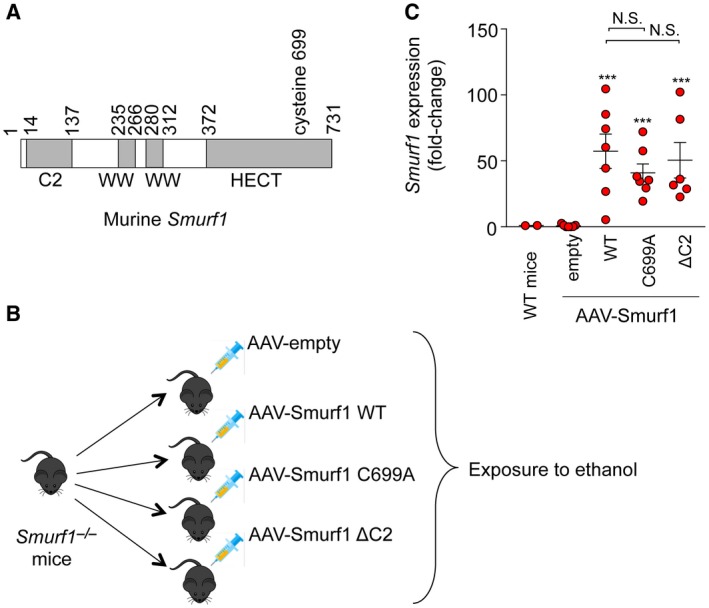
AAV‐Smurf1 overexpression in the livers of Smurf1 −/− mice. (A) Schematic representation of mouse Smurf1 protein. (B) Experimental design of studies to define functionally important domains of Smurf1 in protection against alcohol‐induced liver disease. (C) Expression of Smurf1 mRNA in the liver; n = 2 (wild‐type controls), n = 7 (empty AAV); n = 7 (wild‐type Smurf1); n = 7 (Smurf1 C699A); and n = 6 (Smurf1 ΔC2). Horizontal lines represent mean and vertical lines represent SEM. ***P < 0.001 versus empty AAV. One‐way ANOVA was used for statistical analysis; mRNA expression values were subjected to logarithmic transformation prior to statistical analysis. Abbreviations: N.S., not significant; WT, wild type.
Successful delivery of AAV‐Smurf1 to livers of Smurf1 −/− mice was demonstrated by high Smurf1 mRNA expression levels in recipients of AAV‐Smurf1, AAV‐Smurf1 C699A, and AAV‐Smurf1 ΔC2 compared to recipients of the empty AAV construct (Fig. 3C). Alcohol‐fed Smurf1 −/− mice expressing wild‐type AAV‐Smurf1 showed protection from liver steatosis and liver injury, as indicated by improved liver histology on H&E or Oil Red O staining (Fig. 4A) and by relative liver weight (Fig. 4B), liver triglycerides (Fig. 4C), and serum AST and ALT (Fig. 4D,E). In contrast, the protective effect of AAV‐Smurf1 expression in the livers of Smurf1 −/− was not observed in mice expressing AAV‐Smurf1 C699A and AAV‐Smurf1 ΔC2 as these livers developed a similar degree of hepatic steatosis and injury as mice expressing the empty AAV construct (Fig. 4A‐E). Thus, the protective role of Smurf1 in a mouse model of alcoholic fatty liver disease requires both its C2 phospholipid‐binding domain and ubiquitin‐ligase activity.
Figure 4.
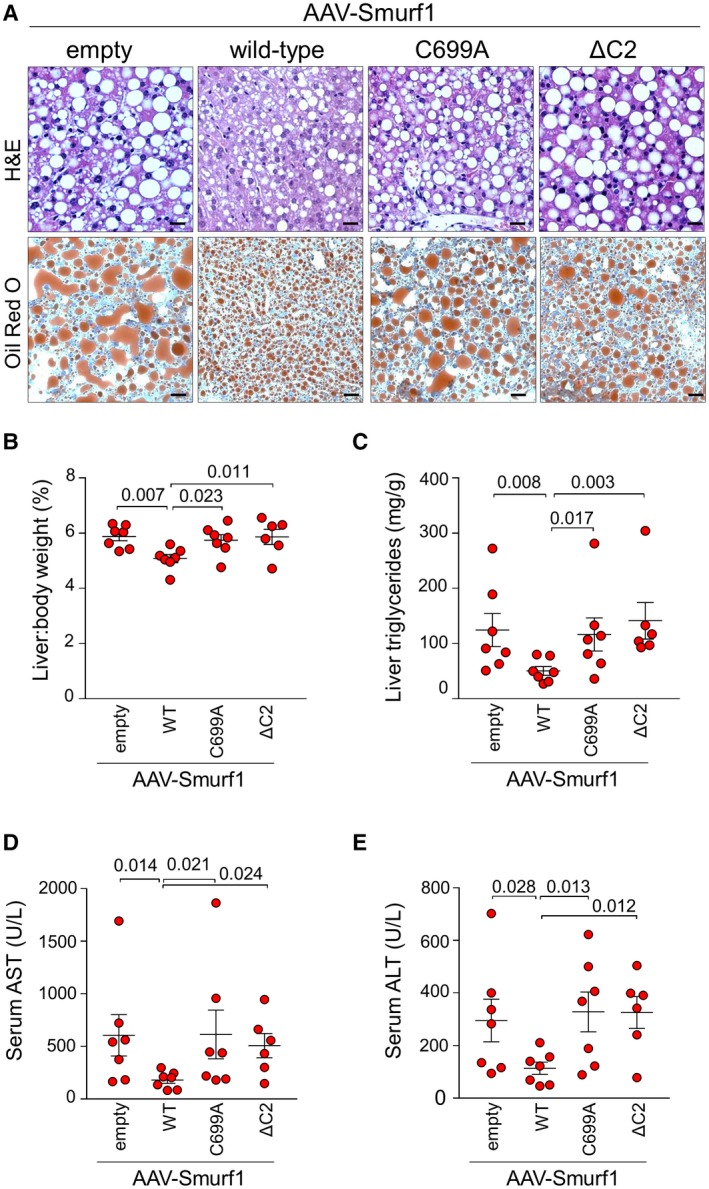
The protective function of Smurf1 in alcoholic steatohepatitis requires its ubiquitin‐ligase activity and C2 phospholipid‐binding domain. (A) Representative photomicrographs of liver injury and steatosis assessed by H&E and Oil Red O staining in Smurf1 −/− mice fed an ethanol diet and injected intravenously with empty AAV or AAV expressing wild‐type Smurf1, catalytically inactive Smurf1 (C699A), or Smurf1 lacking the C2 phospholipid‐binding domain (ΔC2). Quantitative assessment of hepatitis steatosis by measurement of (B) liver:body weight ratio and (C) hepatic triglyceride levels. Quantitative assessment of liver injury by measurement of serum (D) AST and (E) ALT; n = 7 (empty AAV); n = 7 (wild‐type Smurf1); n = 7 (Smurf1 C699A), and n = 6 (Smurf1 ΔC2). Horizontal lines represent mean and vertical lines represent SEM. Numbers in graphs denote P values. One‐way ANOVA was used for statistical analysis; AST, ALT, and liver triglyceride values underwent logarithmic transformation prior to statistical analysis. Scale bars, 50 μm. Abbreviation: WT, wild type.
Discussion
Alcoholic liver disease is defined by the presence of hepatic steatosis in humans and in animal models. In this study, we observed worsening liver steatosis in alcohol‐fed mice deficient in Smurf1, a ubiquitin ligase with a broad spectrum of targets,8, 9, 10, 18, 19 and demonstrated amelioration of steatosis following re‐introduction of wild‐type Smurf1 in the liver. Thus, Smurf1 plays a protective role in a mouse model of alcoholic fatty liver disease. Our experiments demonstrated that the role of Smurf1 is dependent on its ubiquitin‐ligase activity and on its N‐terminal C2 phospholipid membrane‐binding domain. This indicates that Smurf1 regulates hepatic steatosis either by ubiquitinating cytoplasmic cargo destined for autophagic degradation, by ubiquitinating proteins regulating autophagy or lipid metabolism, and/or by delivering autophagic substrates to the autophagosome. This concept is supported by published evidence that Smurf1 is a critical regulator of autophagic degradation of mitochondria and intracellular pathogens8, 18 and that it posttranslationally modifies ultraviolet radiation resistance‐associated gene protein (UVRAG), a protein involved in autophagosomal maturation,19 as well as peroxisome proliferator‐activated receptor gamma (PPARγ), a crucial transcriptional regulator of lipogenesis.9
Our observation that SMURF1 colocalizes with lipid droplets in human livers raises the question whether Smurf1 regulates degradation of lipid droplets by selective autophagy, a process known as lipophagy. Previous studies demonstrated that the autophagy protein microtubule‐associated protein 1A/1B‐light chain 3 (LC3) interacts with the phospholipid membrane of lipid droplets and targets lipid droplets for lysosomal degradation.20, 21 In a mouse model of alcoholic liver disease, lipid droplets colocalized with autophagosomes,5 and deletion of the autophagy protein autophagy‐related 7 (Atg7) in hepatocytes7 aggravated hepatic steatosis. Similarly, worsening of alcoholic fatty liver was observed in mice with genetic deficiency of Ras‐related protein 7 (Rab7), a small guanosine triphosphatase that facilitates recruitment of lysosomes to autophagosomes,22 or in mice with impaired lysosomal biogenesis due to genetic deficiency of transcription factor EB.3 Lipophagy is believed to be a selective process,20 but how the selection process of lipid droplet recognition by autophagy is accomplished is currently unknown. We hypothesize that by virtue of its ubiquitin‐ligase activity, SMURF1 tags specific proteins on lipid droplets for their recognition by autophagy receptors, resulting in lysosomal degradation of autophagocytosed lipid droplets. Alternatively, Smurf1 could regulate selective removal of lipid droplet membrane proteins, thus enabling lipid droplet remodeling and degradation by cytosolic lipases independently of autophagy. Specific proteins on lipid droplet membranes that undergo ubiquitination by SMURF1 are yet to be identified.
Smurf1 may also exert protective functions in alcoholic steatosis, at least in part, by promoting the autophagic clearance of damaged mitochondria (termed mitophagy). Liver tissues from patients with alcoholic liver disease have altered mitochondrial ultrastructure and impaired mitochondrial function, including decreased β‐oxidation. These mitochondrial changes in alcoholic liver disease are attributable to an increased reduced nicotinamide adenine dinucleotide/oxidized nicotinamide adenine dinucleotide ratio, increased levels of reactive oxygen species, damage to mitochondrial DNA, and impaired mitophagy (reviewed in Mansouri et al.23 and Galluzzi et al.24). Previously, Smurf1 was shown to be a critical regulator of mitophagy.8 Moreover, Smurf1 −/− mice accumulate abnormal mitochondria in hepatocytes, and elderly Smurf1 −/− mice develop spontaneous liver steatosis, with lipid droplets juxtaposed to the aberrant mitochondria in hepatocytes.8, 19 Therefore, it is possible that the worsening of hepatic steatosis in alcohol‐fed Smurf1 −/− mice observed in our study could be attributable to decreased lipolysis secondary to impaired mitophagy and accumulation of dysfunctional mitochondria.
In addition, in a mouse model of nonalcoholic fatty liver disease, Smurf1 was found to ubiquitinate PPARγ, a critical regulator of lipogenesis, and to suppress its transcriptional activity.9 Loss of Smurf1 up‐regulated PPARγ and its target genes involved in lipid synthesis and fatty acid uptake, and Smurf1−/− mice fed a high‐fat diet developed worsening of hepatic steatosis. The worsening of hepatic steatosis in Smurf1 −/− mice was reversed by pharmacological inhibition of PPARγ.9 Thus, in addition to impaired lipophagy and mitophagy, the worsening of hepatic steatosis in alcohol‐fed Smurf1 −/− mice observed in our study may be attributable to increased lipogenesis. Further studies are required to determine the precise cellular mechanisms by which Smurf1 exerts its protective effects in alcohol‐induced steatosis. The present study does not determine whether Smurf1 functions through lipophagy, mitophagy, PPARγ regulation, or other mechanisms.
In conclusion, this study demonstrates that Smurf1 plays a protective role in the pathogenesis of alcoholic steatohepatitis through a mechanism that requires both its ubiquitin‐ligase activity and C2 phospholipid‐binding domains. These findings have implications for understanding the roles of ubiquitin ligases in the pathogenesis of alcoholic fatty liver disease. Moreover, strategies to enhance the activity of Smurf1 may represent a new therapeutic target in the treatment of alcoholic liver disease.
Acknowledgment
We thank Ying E. Zhang (National Cancer Institute, National Institutes of Health, Bethesda, MD) for providing Smurf1 −/− mice; Luke Engelking for providing the pLE282 plasmid; and Zhongju Zhou, Lori Nguyen, and Liangcai Nie for technical assistance.
Supported by the National Institutes of Health (grants U19 AI142784‐01 to B.L. and T32 DK 7745‐22 to J.P.), Fondation Leducq (grant 15CBD04 to B.L.), and the Czech Society for Organ Transplantation (to D.E.).
Potential conflict of interest: Dr. Levine is a scientific cofounder of Casma Therapeutics, Inc. The other authors have nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Osna NA, Donohue TM Jr, Kharbanda KK. Alcoholic liver disease: pathogenesis and current management. Alcohol Res 2017;38:147‐161. [PMC free article] [PubMed] [Google Scholar]
- 2. Thomes PG, Trambly CS, Fox HS, Tuma DJ, Donohue TM Jr. Acute and chronic ethanol administration differentially modulate hepatic autophagy and transcription factor EB. Alcohol Clin Exp Res 2015;39:2354‐2363. [DOI] [PubMed] [Google Scholar]
- 3. Chao X, Wang S, Zhao K, Li Y, Williams JA, Li T, et al. Impaired TFEB‐mediated lysosome biogenesis and autophagy promote chronic ethanol‐induced liver injury and steatosis in mice. Gastroenterology 2018;155:865‐879.e812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ding WX, Li M, Yin XM. Selective taste of ethanol‐induced autophagy for mitochondria and lipid droplets. Autophagy 2011;7:248‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ding WX, Manley S, Ni HM. The emerging role of autophagy in alcoholic liver disease. Exp Biol Med (Maywood) 2011;236:546‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol‐induced hepatotoxicity and steatosis in mice. Gastroenterology 2010;139:1740‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan S, Zhou J, Chen X, Dong Z, Yin XM. Diverse consequences in liver injury in mice with different autophagy functional status treated with alcohol. Am J Pathol 2019;189:1744‐1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y, et al. Image‐based genome‐wide siRNA screen identifies selective autophagy factors. Nature 2011;480:113‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu K, Tang Y, Xu X, Dang H, Tang LY, Wang X, et al. Non‐proteolytic ubiquitin modification of PPARgamma by Smurf1 protects the liver from steatosis. PLoS Biol 2018;16:e3000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 2005;121:101‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen SJ, Sanmiguel J, Lock M, McMenamin D, Draper C, Limberis MP, et al. Biodistribution of AAV8 vectors expressing human low‐density lipoprotein receptor in a mouse model of homozygous familial hypercholesterolemia. Hum Gene Ther Clin Dev 2013;24:154‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009;360:2758‐2769. [DOI] [PubMed] [Google Scholar]
- 14. Cao Y, Zhang L. Pharmaceutical perspectives of HECT‐type ubiquitin ligase Smurf1. Curr Pharm Des 2013;19:3226‐3233. [DOI] [PubMed] [Google Scholar]
- 15. Yuan C, Qi J, Zhao X, Gao C. Smurf1 protein negatively regulates interferon‐gamma signaling through promoting STAT1 protein ubiquitination and degradation. J Biol Chem 2012;287:17006‐17015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheng PL, Lu H, Shelly M, Gao H, Poo MM. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron 2011;69:231‐243. [DOI] [PubMed] [Google Scholar]
- 17. Suzuki C, Murakami G, Fukuchi M, Shimanuki T, Shikauchi Y, Imamura T, et al. Smurf1 regulates the inhibitory activity of Smad7 by targeting Smad7 to the plasma membrane. J Biol Chem 2002;277:39919‐39925. [DOI] [PubMed] [Google Scholar]
- 18. Franco LH, Nair VR, Scharn CR, Xavier RJ, Torrealba JR, Shiloh MU, et al. The ubiquitin ligase Smurf1 functions in selective autophagy of Mycobacterium tuberculosis and anti‐tuberculous host defense. Cell Host Microbe 2017;21:59‐72. Erratum. In: Cell Host Microbe 2017;22:421‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng X, Jia Y, Zhang Y, Ma F, Zhu Y, Hong X, et al. Ubiquitination of UVRAG by SMURF1 promotes autophagosome maturation and inhibits hepatocellular carcinoma growth. Autophagy 2019;15:1130‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, et al. The MAP1‐LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun 2009;382:419‐423. [DOI] [PubMed] [Google Scholar]
- 22. Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015;61:1896‐1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mansouri A, Gattolliat CH, Asselah T. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology 2018;155:629‐647. [DOI] [PubMed] [Google Scholar]
- 24. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo‐San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J 2017;36:1811‐1836. [DOI] [PMC free article] [PubMed] [Google Scholar]