

Abstract
Ursodeoxycholic acid (UDCA) is commonly used to treat several liver disorders in adults and children, including primary sclerosing cholangitis (PSC) for which it is not U.S. Food and Drug Administration approved. UDCA treatment has an uncertain impact on disease outcomes and has been reported in high doses to be associated with worse outcome in adults with PSC. In this context, controlled withdrawal and reintroduction of UDCA in children with PSC were studied. Prior to study initiation, participants were required to have alanine aminotransferase (ALT) and gamma‐glutamyl transpeptidase (GGT) <2 times the upper limit of normal on stable UDCA dosing. The study included four phases: I (stable dosing), II (50% UDCA reduction), III (UDCA discontinuation), IV (UDCA reintroduction), with a primary endpoint of change in ALT and GGT between phases I and III. We enrolled 27 participants (22 completed) between March 2011 and June 2016. Changes in mean ALT and GGT between phases I and III were ALT, +29.5 IU/L (P = 0.105) and GGT, +60.4 IU/L (P = 0.003). In 7 participants, ALT and GGT ≤29 IU/L did not rise above 29 IU/L (null response group). Eight participants had increases of ALT or GGT >100 IU/L (flare group). None developed elevated bilirubin. All flares responded to UDCA reinstitution. Serum GGT, interleukin‐8, and tumor necrosis factor α levels were higher in the flare group at baseline. Liver biochemistries increased in children with PSC during controlled UDCA withdrawal; one third increased above 100 IU/L and one third remained normal during UDCA withdrawal. Conclusion: The impact of prolonged UDCA use in childhood PSC and the significance of a biochemical flare are unclear. Further studies of the natural history and treatment of pediatric PSC and UDCA use are needed.
Abbreviations
- AIH
autoimmune hepatitis
- ALP
alkaline phosphatase
- ALT
alanine aminotranferase
- ANOVA
analysis of variance
- AST
aspartate aminotransferase
- DB
direct bilirubin
- GGT
gamma‐glutamyl transpeptidase
- IBD
inflammatory bowel disease
- IL
interleukin
- INR
international normalized ratio
- PSC
primary sclerosing cholangitis
- PT
prothrombin time
- SAE
serious adverse event
- SDF
severe disease flare
- TNF
tumor necrosis factor
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
- UPLC
ultra‐high‐performance liquid chromatography
Primary sclerosing cholangitis (PSC) is a chronic hepatobiliary disease of unknown etiology that causes chronic inflammation and obliterative fibrosis of the intrahepatic and/or extrahepatic biliary tree, leading to cholestasis, stricturing, hepatic fibrosis, and ultimately to cirrhosis and end‐stage liver disease.1 In adults and children, PSC leads to significant morbidity and mortality, and 2% of pediatric liver transplants in the United States are for PSC.2, 3 Unfortunately, “recurrent” disease occurs in up to 10% of children who undergo transplantation.4, 5 The prognosis of PSC is related to the mode of presentation, which correlates with stage of disease. In a large international cohort, complications of portal hypertension developed in one third of children with PSC within 10 years of diagnosis.3 Cholangiocarcinoma, while quite rare in childhood, has been described in adolescents with PSC.3
There are many controversial areas in pediatric PSC, including optimal diagnostic criteria, predictors of outcome, and therapeutic approaches.6 Diagnostic criteria have not been well‐delineated in pediatric PSC but are suggested to include biochemical features of biliary disease (e.g., elevated gamma‐glutamyl transpeptidase [GGT]), histologic findings consistent with cholangiocyte‐directed injury, and/or cholangiographic evidence of biliary injury. None of these features have been the subject of a rigorous analysis of reliability and reproducibility in children. Overlap of PSC with autoimmune hepatitis (AIH), present in 30%‐50% of pediatric patients with PSC, also referred to as autoimmune sclerosing cholangitis, may be an important factor in disease progression and severity.7, 8 Given the progressive nature of pediatric PSC, there is significant biomedical relevance in identifying prognostic biomarkers of future progression and developing evidence‐based therapies that can slow evolution to end‐stage liver disease.
Effective therapeutic approaches to the management of pediatric PSC remain uncertain given the lack of reliable treatment data. Immunosuppression is presumed to be important for children with autoimmune overlap.7, 9 Ursodeoxycholic acid (UDCA) has been a mainstay of therapy in pediatrics, although quality of evidence supporting its long‐term use and benefits is lacking.10, 11 Vancomycin has also been suggested to have therapeutic value in PSC in relatively small studies, and the overall experience with this drug is significantly less than with UDCA.12, 13 Because of the paucity of data, clinicians, patients, and families are eager to employ any therapeutic regimen that may slow the progression of the disease. In this context, anecdotal approaches are often implemented to emphasize the critical need to avoid potential harm of unproven therapies.
The finding of potential toxicity of “high‐dose” UDCA therapy in adults with PSC has led to a vexing problem in recent approaches to disease management.14 In light of this unexpected finding, the American Association for the Study of Liver Diseases recommended against the use of UDCA in adults with PSC.15 This recommendation was not extended to children but raised questions about the safety of a therapy in children that seems efficacious, at least from a biochemical perspective.7, 16 Withdrawal of UDCA therapy in adults with PSC resulted in statistically significant worsening of liver biochemistries 3 months after withdrawal.17 In that study, consecutive adult patients with PSC were enrolled regardless of their response to UDCA therapy. In 2010, shortly after the report of potential UDCA toxicity, the present pediatric UDCA withdrawal study was initiated (Withdrawal of Ursodeoxycholic Acid Therapy in Pediatric Primary Sclerosing Cholangitis [WUPPSC, NCT01088607]). Clinically, study investigators had observed biochemical disease flares in adolescents who were nonadherent to UDCA for PSC; this improved with reinitiation of UDCA treatment. As such, WUPPSC was designed to assess the potential utility of UDCA as a therapy in pediatric PSC. The study focused on participants who were biochemically responsive to UDCA, as reflected by the requirement of alanine aminotransferase (ALT) and GGT less than 2 times the upper limit of normal (ULN) at study entry, thereby potentially enhancing the ability to identify a therapeutic effect. Unlike the adult study,17 the design involved both a staged withdrawal of therapy and a monitored reinstitution of UDCA in pediatric patients with PSC with mild and stable disease. The results of this novel investigation are reported.
Participants and Methods
Participants
Twenty‐seven previously diagnosed male and female participants with PSC, all less than 21 years of age without ethnic or racial restrictions, were recruited from 12 participating pediatric centers between March 2011 and June 2016. Diagnostic criteria for PSC in this study included two of the following three features: 1) serum GGT increased more than 50% above the ULN for age; 2) endoscopic retrograde cholangiopancreatography, percutaneous transhepatic cholangiogram, or magnetic resonance cholangiopancreatography findings of intrahepatic and/or extrahepatic bile duct irregularities consistent with PSC; and 3) liver biopsy abnormalities consistent with chronic biliary injury. Patients with PSC/AIH overlap who met the criteria for PSC plus had liver histologic features of AIH were included.8
In addition to the diagnosis of PSC, participants were also required to have the following: 1) biochemically quiescent liver disease defined by an ALT and GGT less than 2.0 times ULN measured on two separate occasions greater than 2 weeks apart; 2) prior and ongoing UDCA therapy at a dose of greater than 13 mg/kg/day or 600 mg/day for more than 6 months by commercially available pill form (compounded liquid preparations were avoided due to bioavailability concerns); 3) ability to swallow pills; and 4) quiescent inflammatory bowel disease (IBD) as reflected by revised Pediatric Ulcerative Colitis Activity Index18 or Short Pediatric Crohn's Disease Activity Index19 scores (see Supporting Materials). Major exclusion criteria included (see also Supporting Materials for details) any other primary liver disease, advanced liver disease and cirrhosis with decompensation, any evidence of portal hypertension with palpable spleen and hypersplenism being used as surrogate markers, previous injury/surgery of the biliary tract, and any systemic disease that secondarily involved the biliary system.
Protocol
The study protocol consisted of four phases (Supporting Fig. S1). During phase I (4 weeks), participants were maintained on UDCA at their baseline clinically prescribed dosing and were monitored for stability. In phase II (4 weeks), the UDCA dose was reduced by 50% and then stopped completely in phase III (8 weeks) with continued monitoring. Dosing was restarted in phase IV (8 weeks) at a standardized dose of as close to but not to exceed 20 mg/kg/day in two divided doses. Baseline and endpoint visits occurred at weeks 0, 16, and 24, with face‐to‐face surveillance visits at weeks 4, 8, 12, and 20. Participants were maintained on all other clinically prescribed medications for their liver disease and IBD, such as immunosuppressives, during the course of the study. Surveillance visits included review of any new symptoms, study adherence by pill count, adverse events, and collection/review of the following laboratory tests: ALT, aspartate aminotransferase (AST), total bilirubin (TB), direct bilirubin (DB), alkaline phosphatase (ALP), GGT, albumin, and prothrombin time/international normalized ratio (PT/INR). Endpoint evaluations included full history, physical examination, monitoring for adverse events, and collection of plasma and serum for inflammatory biomarkers, bile acid analysis, ALT, AST, TB, DB, ALP, GGT, albumin, PT/INR, platelet count, white blood cell count with differential, and hematocrit/hemoglobin.
Participants were monitored for significant biochemical worsening of their liver disease, referred to as a significant disease flare (SDF) and defined by a rise in ALT or GGT to greater than 10 times ULN, rise in DB to greater than 5 times entry level, or clinical manifestations of acute liver failure or decompensated cirrhosis. If an SDF was detected, the participant was immediately placed back on UDCA at the dose prescribed by the study protocol as above, moved to phase IV of the study, and the event was reported as a serious adverse event (SAE). Also, if the ALT or GGT was greater than 5 times ULN at any laboratory draw, a 2‐week follow‐up surveillance blood draw could be ordered at the discretion of the site investigator.
We hypothesized that participants would exhibit a biochemical flare during UDCA withdrawal that would resolve when placed back on the drug. The primary endpoints for the study were the change in ALT and GGT at the end of phase III compared to the beginning of the baseline phase I period (drug withdrawal). The secondary endpoints were the change in ALT and GGT at the end of phase IV compared to the end of phase III (drug reinstitution) and changes in inflammatory cytokines at the end of phase III compared to either baseline (withdrawal) or to the end of phase IV (reinstitution).
Laboratory Testing
All blood samples for chemistries, hematology, and coagulation testing were collected at study sites and shipped to a central laboratory (Q2 Solutions, Valencia, CA) for automated analysis. Results were reported to centers within 48 hours, then entered into the secure online study database at the Data Coordinating Center (Emmes Corporation, Rockville, MD).
Inflammatory Cytokine Measurement
Baseline and endpoint plasma samples were shipped to the Clinical Laboratory Improvement Amendments‐approved Pediatric Clinical and Translational Research Center, University of Colorado, for cytokine measurement using Luminex multi‐analyte technology (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA). Analysis included interleukin (IL)‐1β, IL‐2, IL‐4, IL‐5, IL‐6, IL‐8, IL‐10, IL‐15, tumor necrosis factor (TNF)‐α, interferon (IFN)‐γ, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), macrophage inflammatory protein (MIP)‐1β, and eotaxin. Individual cytokine analyses were performed in duplicate, and the background signal (saline only) was subtracted for each readout. Results are shown as mean of duplicate samples.
Bile Acid Determination
Bile acid analysis to determine UDCA levels was carried out on baseline and endpoint plasma samples. Whole blood was collected in heparin tubes, and plasma was isolated and stored at −80° C. UDCA was quantified by an ultra‐high‐performance liquid chromatography–tandem mass spectrometry (UPLC/MS‐MS) assay20 with modifications. Bile acids were first isolated from plasma with a Strata‐X C18 solid‐phase extraction column (#8B‐S100‐TAK; Phenomenex). Standard solutions of ursodeoxycholate (C1020, CAS# 128‐13‐2; Steraloids) were used to generate a standard curve (0‐2.5 µM) to determine UDCA concentration. Separation was achieved using UPLC (Waters Corporation) followed by MS‐MS (TQD Tandem Mass Spectrometer; Waters Corporation) with a Waters Cortecs C18 + UPLC column (#186007367; Waters). Analyte separation and elution were achieved using a 10 mM ammonium acetate, 0.15% ammonium hydroxide aqueous mobile phase and a methanol/10 mM ammonium acetate 0.15% ammonium hydroxide elution phase. We used d4‐ursodeoxycholate (#D3819; CDN, Inc.) as an internal control in each sample.
Ethical Considerations
Informed consent in writing was obtained for each participant, and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected by a priori approval by the institutional review committees of each participating center. The study was monitored by a nonconflicted external data and safety monitoring board.
Statistical Analysis
All participants who completed the study were included in the final statistical analyses. All analyses were performed for all such participants for the group as a whole. Further ad hoc analyses included stratification of the participants according to the following three subgroup categories defined based on the UDCA response of ALT and GGT parameters: null, ALT, and GGT persistently ≤29 IU/L (i.e., normal); flare, ALT, and/or GGT >100 IU/L during phase II and/or III; indeterminant, ALT, and/or GGT >29 IU/L and <100 IU/L during phase II and/or III.
Participants’ demographics and other baseline characteristics (i.e., disease diagnosis groups, time since diagnosis, UDCA dosing, IBD diagnosis, immunosuppressive medication, all clinical laboratory and cytokine parameters) were summarized in aggregate and by UDCA response subgroup categories using descriptive statistics, including mean and SD for continuous outcomes and frequencies and percentages for categorical outcomes. Differences in participants’ characteristics and laboratory values at baseline among the groups were tested using analysis of variance (ANOVA) for continuous measures and chi‐square or Fisher exact test if expected cell sizes were less than five for categorical measures. A pairwise comparison between the groups was done if the overall test comprising all the subgroups showed any significantly different results.
To assess the primary endpoint of the study, ALT and GGT values at the end of phase III were compared with those at the beginning of the baseline phase I in aggregate and by post hoc analysis of UDCA response subgroups using paired t test with no adjustment for multiplicity given the exploratory nature of the analysis. A similar method was used for secondary endpoint analyses to provide statistical comparison between the end of phase III and end of phase IV values. These analyses were replicated for a more conservative estimate of phase III and phase IV data, where maximum of ALT and GGT values across the visits within these phases were considered for the analysis. Spaghetti plots for ALT and GGT by UDCA response subgroups for all the visits were generated to review participants’ responses to UDCA treatment and withdrawal across all the visits and phases of the study.
For all the statistical analyses above, P < 0.05 was considered significant. All the analyses were performed in SAS version 9.4 (SAS Institute Inc., Cary, NC).
Results
Study Participants
Participant demographics and clinical characteristics in aggregate and by UDCA response group are shown in Table 1. Most of the baseline characteristics were similar among treatment response groups. A majority were male participants (77.3%), with a mean age of 13.9 + 3.3 (mean + SD) years at entry into the study and 12.0 + 3.7 years at diagnosis. A slight majority (54.5%) had AIH/PSC overlap, 86.4% had IBD (63.2% ulcerative colitis, 15.8% Crohn's disease, and 21.0% indeterminant), and 40.9% were on immunosuppressive medication at entry to the study. Two diagnostic criteria for PSC were met by 81.8% of participants, and 18.2% met all three (Supporting Table S1). Baseline laboratory values are shown in Table 2. All had quiescent PSC as defined by GGT and ALT < 2.0 times ULN. None of the participants had laboratory evidence of hypersplenism or decompensated liver disease.
Table 1.
Baseline Demographics and Physical Characteristics by UDCA Group: Primary Analysis Population
| UDCA Response Group | Total (N = 22) | P Value* (Overall) | P Value† | |||
|---|---|---|---|---|---|---|
| Null (n = 7) | Flare (n = 8) | Indeterminant (n = 7) | ||||
| Age at diagnosis (years) | ||||||
| n | 7 | 8 | 7 | 22 | 0.101 | |
| Mean (SD) | 9.7 (2.2) | 12.4 (4.4) | 13.9 (3.2) | 12.0 (3.7) | ||
| Age at start of phase I (years) | ||||||
| n | 7 | 8 | 7 | 22 | 0.157 | |
| Mean (SD) | 12.6 (2.1) | 13.4 (3.8) | 15.9 (3.3) | 13.9 (3.3) | ||
| Sex | ||||||
| Male | 5 (71.4%) | 6 (75.0%) | 6 (85.7%) | 17 (77.3%) | >0.999 | |
| Female | 2 (28.6%) | 2 (25.0%) | 1 (14.3%) | 5 (22.7%) | ||
| Ethnicity | ||||||
| Hispanic or Latino | 1 (14.3%) | 1 (12.5%) | 1 (14.3%) | 3 (13.6%) | 0.906 | |
| Not Hispanic or Latino | 6 (85.7%) | 7 (87.5%) | 5 (71.4%) | 18 (81.8%) | ||
| Not reported | 0 | 0 | 1 (14.3%) | 1 (4.5%) | ||
| Race | ||||||
| Black or African American | 0 | 0 | 1 (14.3%) | 1 (4.5%) | 0.299 | |
| White or Caucasian | 6 (85.7%) | 7 (87.5%) | 3 (42.9%) | 16 (72.7%) | ||
| Not reported | 1 (14.3%) | 1 (12.5%) | 3 (42.9%) | 5 (22.7%) | ||
| Diagnosis group | ||||||
| PSC | 2 (28.6%) | 3 (37.5%) | 5 (71.4%) | 10 (45.5%) | 0.316 | |
| PSC/AIH overlap | 5 (71.4%) | 5 (62.5%) | 2 (28.6%) | 12 (54.5%) | ||
| IBD diagnosis at study entry | ||||||
| Present | 6 (85.7%) | 7 (87.5%) | 6 (85.7%) | 19 (86.4%) | >0.999 | |
| Absent | 1 (14.3%) | 1 (12.5%) | 1 (14.3%) | 3 (13.6%) | ||
| On immumosuppressive medication | ||||||
| Yes | 3 (42.9%) | 4 (50.0%) | 2 (28.6%) | 9 (40.9%) | 0.862 | |
| No | 4 (57.1%) | 4 (50.0%) | 5 (71.4%) | 13 (59.1%) | ||
| Immunosuppressive medication type | ||||||
| Corticosteroids | 1 (14.3%) | 0 | 1 (14.3%) | 2 (9.1%) | 0.644 | |
| Others | 2 (28.6%) | 4 (50.0%) | 1 (14.3%) | 7 (31.8%) | ||
| N/A | 4 (57.1%) | 4 (50.0%) | 5 (71.4%) | 13 (59.1%) | ||
| Height z score | ||||||
| n | 7 | 7 | 7 | 21 | 0.018 | 0.016 (null vs. flare) |
| Mean (SD) | −0.5 (1.0) | 0.6 (0.4) | −0.2 (0.5) | −0.0 (0.8) | 0.432 (null vs. indeterminant) | |
| 0.008 (flare vs. indeterminant) | ||||||
| Weight z score | ||||||
| n | 7 | 7 | 7 | 21 | 0.301 | |
| Mean (SD) | −0.3 (0.9) | 0.4 (0.5) | 0.0 (1.0) | 0.0 (0.8) | ||
| BMI z score | ||||||
| n | 7 | 7 | 7 | 21 | 0.991 | |
| Mean (SD) | 0.006 (0.6) | 0.022 (0.6) | −0.033 (1.1) | −0.001 (0.7) | ||
| Time since diagnosis (years) | ||||||
| n | 7 | 8 | 7 | 22 | 0.063 | |
| Mean (SD) | 3.0 (1.6) | 1.3 (1.2) | 1.9 (1.2) | 2.0 (1.5) | ||
| UDCA dose per kg | ||||||
| n | 6 | 7 | 7 | 20 | 0.34 | |
| Mean (SD) | 13.0 (2.5) | 14.8 (3.4) | 11.8 (4.6) | 13.2 (3.7) | ||
| UDCA dose ranges (mg/kg/day) | ||||||
| >30 | 1 (14.3%) | 0 | 0 | 1 (4.5%) | 0.383 | |
| 20‐30 | 0 | 1 (12.5%) | 0 | 1 (4.5%) | ||
| 13‐20 | 1 (14.3%) | 4 (50.0%) | 2 (28.6%) | 7 (31.8%) | ||
| <13 | 5 (71.4%) | 3 (37.5%) | 5 (71.4%) | 13 (59.1%) | ||
Overall P value computed using ANOVA for continuous data and chi‐square or Fisher exact test for categorical data to test any difference between UDCA subgroups.
P value for pairwise comparisons computed using two‐sample t test or Wilcoxon sign rank test for continuous data and chi‐square or Fisher exact test for categorical data between each pair of UDCA subgroups. Pairwise comparisons shown only if overall P value is significant.
Abbreviations: BMI, body mass index; N/A, not applicable.
Table 2.
Summary of Clinical Laboratory Results at Baseline by UDCA Response Group
| UDCA Response Group | Total (N = 22) | P Value* (Overall) | P Value† | |||
|---|---|---|---|---|---|---|
| Null (n = 7) | Flare (n = 8) | Indeterminant (n = 7) | ||||
| Hemoglobin (g/dL) | ||||||
| n | 7 | 8 | 7 | 22 | 0.169 | |
| Mean (SD) | 12.9 (0.7) | 12.9 (1.0) | 14.0 (1.8) | 13.3 (1.3) | ||
| White blood cell count (103/μL) | ||||||
| n | 7 | 8 | 7 | 22 | 0.771 | |
| Mean (SD) | 6.0 (1.9) | 5.5 (2.4) | 6.2 (1.1) | 5.9 (1.8) | ||
| Platelets (103/μL) | ||||||
| n | 7 | 8 | 7 | 22 | 0.433 | |
| Mean (SD) | 265.1 (57.0) | 273.9 (122.0) | 218.7 (49.4) | 253.5 (84.8) | ||
| ALT (IU/L) | ||||||
| n | 7 | 8 | 7 | 22 | 0.289 | |
| Mean (SD) | 15.3 (6.0) | 26.8 (19.5) | 21.1 (10.3) | 21.3 (13.8) | ||
| AST (IU/L) | ||||||
| n | 7 | 8 | 7 | 22 | 0.639 | |
| Mean (SD) | 25.4 (5.7) | 30.3 (10.7) | 28.4 (11.6) | 28.1 (9.5) | ||
| ALP (IU/L) | ||||||
| n | 7 | 8 | 7 | 22 | 0.921 | |
| Mean (SD) | 262.6 (86.5) | 262.0 (154.1) | 237.7 (141.0) | 254.5 (126.0) | ||
| GGT (IU/L) | ||||||
| n | 7 | 8 | 7 | 22 | 0.004 | 0.018 (null vs. flare) |
| Mean (SD) | 12.4 (2.4) | 44.9 (25.4) | 22.9 (11.8) | 27.5 (21.3) | 0.046 (null vs. indeterminant) | |
| 0.056 (flare vs. indeterminant) | ||||||
| Total bilirubin (mg/dL) | ||||||
| n | 7 | 8 | 7 | 22 | 0.563 | |
| Mean (SD) | 0.9 (1.4) | 0.5 (0.3) | 0.7 (0.3) | 0.7 (0.8) | ||
| Direct/conjugated bilirubin (mg/dL) | ||||||
| n | 7 | 7 | 7 | 21 | 0.813 | |
| Mean (SD) | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.1) | ||
| Albumin (g/dL) | ||||||
| n | 7 | 8 | 7 | 22 | 0.02 | 0.008 (null vs. flare) |
| Mean (SD) | 4.7 (0.2) | 4.2 (0.3) | 4.5 (0.3) | 4.5 (0.3) | 0.335 (null vs. indeterminant) | |
| 0.127 (flare vs. indeterminant) | ||||||
| Prothrombin time (seconds) | ||||||
| n | 7 | 7 | 7 | 21 | 0.704 | |
| Mean (SD) | 11.9 (1.9) | 11.5 (0.6) | 12.0 (1.0) | 11.8 (1.2) | ||
| INR | ||||||
| n | 7 | 7 | 7 | 21 | 0.439 | |
| Mean (SD) | 1.1 (0.1) | 1.1 (0.1) | 1.2 (0.1) | 1.2 (0.1) | ||
Overall P values computed using ANOVA.
P values for pairwise comparisons computed using t test or Wilcoxon sign rank test. Pairwise comparisons shown only if overall P value is significant.
Study Course
Of the total of 27 participants recruited for the study, 22 completed all phases of the study and constituted the primary analysis group (Supporting Fig. S2). There were five early withdrawals: one due to an ulcerative colitis flare, two due to study protocol nonadherence, one with an out of range baseline laboratory value, and one withdrawal reason was not disclosed by the participant. There were three SAEs reported, including two SDF events and one IBD flare. Both participants with SDF were placed back on UDCA and moved to phase IV, with subsequent remission of their flare.
GGT and ALT levels in the three response groups during the course of the study are depicted in Fig. 1. Seven participants had baseline levels of ALT and GGT ≤29 IU/L that did not rise above 29 IU/L during the 24 weeks of the study (null response group, Fig. 1A,B). Eight participants had increases of ALT or GGT >100 IU/L on either reduced or no UDCA therapy (flare response group, Fig. 1C,D). Seven participants had intermediate responses (indeterminant response group, Fig. 1E,F). All flares responded to UDCA reinstitution by the end of the study. One subject flared on UDCA reintroduction (Fig. 1E,F). Follow‐up after the study demonstrated resolution of this flare with reduction of ALT to normal and GGT to less than 2 times ULN within 7 weeks with continued UDCA therapy. There were two SDFs (one during phase II and one during phase III) (Fig. 1C,D, red arrows). Both participants were placed back on UDCA and moved to phase IV, with subsequent flare resolution. None developed elevated bilirubin, and there were no significant increases in ALP levels. At study baseline, height z scores were different in null versus flare participants (P = 0.016; Table 1) and flare versus indeterminant groups (P = 0.008; Table 1). The diagnosis of isolated PSC versus AIH/PSC overlap, use of immunosuppressives, or presence of IBD did not impact flare or null status (Table 1). Interestingly, the null group had baseline GGT levels significantly lower than the flare and indeterminant groups and albumin levels higher than the flare group (Table 2), suggesting that these two baseline laboratory values may be predictive of a disease flare.
Figure 1.
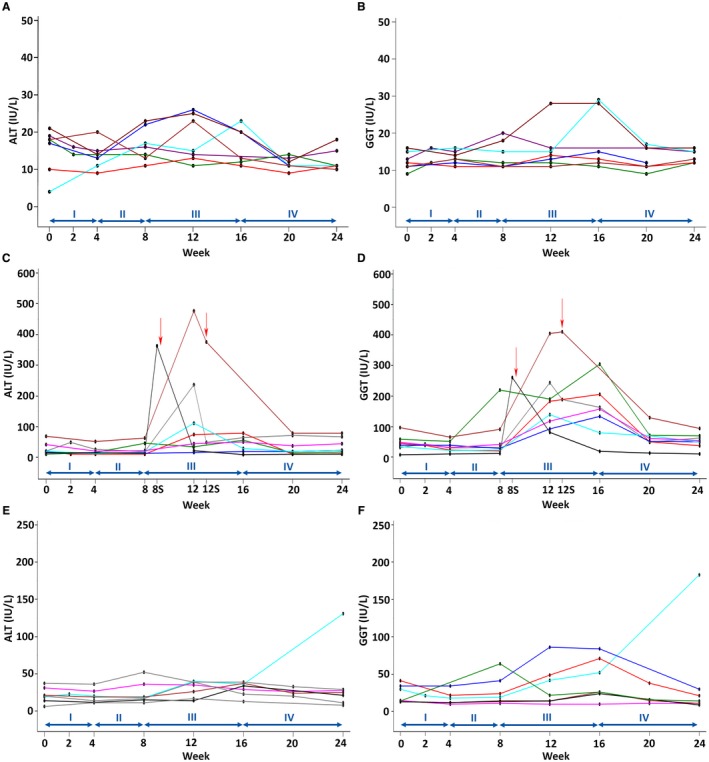
Serum ALT and GGT levels in study participant response groups. (A,B) Null group; (C,D) flare group; (E,F) indeterminant group. The y axis shows serum levels, and the x axis shows study week and phase. Colored lines depict individual participants. In the flare group (C,D), visits 8S and 12S are off‐study visits and red arrows indicate time of early reinstitution of UDCA due to SDF in 2 participants.
Study Endpoint Analysis
Aggregate clinical laboratory results by study phase for the primary analysis population are summarized in Supporting Table S2. Significant changes occurred in GGT and ALT, as depicted in Fig. 2A, which shows results of the analysis of change in ALT and GGT from study baseline to end of phase III for all participants as well as the three response groups. Although the increase in ALT within the indeterminant group was significant, as shown, the aggregate mean change across all groups did not reach statistical significance. For GGT, the average increases across all response groups as well as within the flare and indeterminant groups were statistically significant. The changes in ALT and GGT from baseline to maximum values in phases II and III are shown in Fig. 2B. The average increase in ALT across all groups as well as the increases within the flare and indeterminant groups reached statistical significance. For GGT, the average increases across all response groups and within the null, flare, and indeterminant groups were statistically significant.
Figure 2.
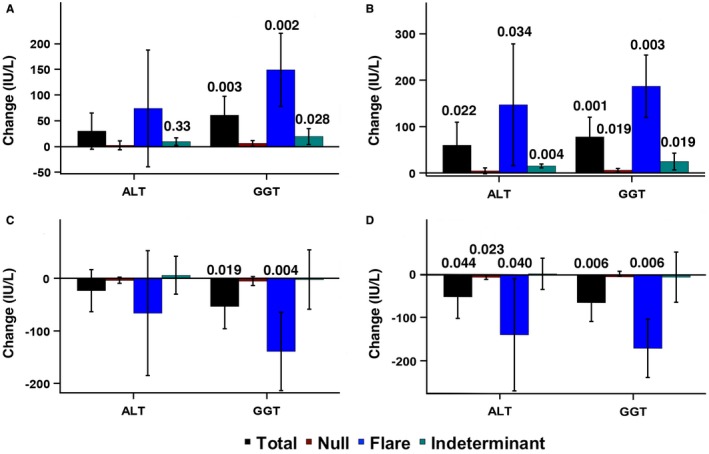
Serum ALT and GGT levels for study endpoints. (A) Results of the analysis of change in serum ALT and GGT levels from study baseline to end of phase III for total participants and response groups. (B) Change in serum ALT and GGT levels from baseline to maximum values in phases II and III. (C) Change in serum ALT and GGT levels from the end of phase III to the end of phase IV. (D) Change in serum ALT and GGT levels from the maximum value in phases II and III to the minimum value in phase IV. Data in all panels represent mean + SD. P values shown at the top of bars for test of within‐group change between the two time points were calculated using paired t test.
The change in ALT and GGT from the end of phase III to the end of phase IV is shown in Fig. 2C. The mean change in ALT across all response groups as well as within each group did not reach statistical significance. However, in the case of GGT, the mean change among all response groups was significant as was the within‐group change for the flare response group, as shown. The changes in ALT and GGT from the maximum value in phases II and III to the minimum value in phase IV are shown in Fig. 2D. The mean change in ALT across all groups and the decreases within the null and flare groups reached statistical significance. For GGT, the average decrease across all response groups as well as within the flare group, as shown, was statistically significant.
Inflammatory Cytokines
Inflammatory cytokine levels (IL‐1β, IL‐2, IL‐4, IL‐5, IL‐6, IL‐8, IL‐10, IL‐15, TNF‐α, IFN‐γ, GM‐CSF, MIP‐1β, and eotaxin) showed no clear pattern of change over time between study phases, although the limited numbers of samples due to loss in storage may have precluded identification of some changes (Supporting Fig. S3). Baseline IL‐8 levels were significantly higher in the flare group compared to the indeterminant group (Fig. 3A), and baseline levels of TNF‐α were significantly higher in the flare and indeterminant groups compared to the null group (Fig. 3B).
Figure 3.
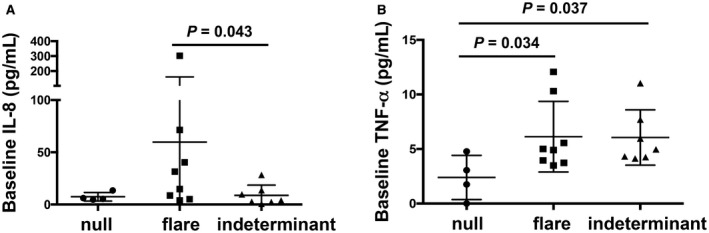
Baseline plasma levels from 4 null, 8 flare, and 7 indeterminant participants. (A) IL‐8 and (B) TNF‐α treatment response groups. Null, circle; flare, square; indeterminant, triangle. Each symbol represents an individual participant. Horizontal lines depict mean and SD for each group.
Bile Acid Analysis
Serum UDCA levels in 13 participants at baseline, end of phase III, and end of phase IV are shown in Fig. 4. With UDCA withdrawal, 11 participants exhibited a decline in serum UDCA to very low levels (<0.4 µM), with the exception of 1 individual whose level actually increased from 4.3 µM to 13.1 µM and another who showed a decline from a high baseline level (26.7 µM) to a still elevated level (7.6 µM) off UDCA. With UDCA reinstitution, there was a variable response, ranging from minimal increases to return to near baseline levels. The same 2 participants who had anomalous responses to withdrawal both unexpectedly exhibited a decline in UDCA from end of phase III to end of phase IV levels. Interestingly, both of these individuals belonged to the null response group.
Figure 4.
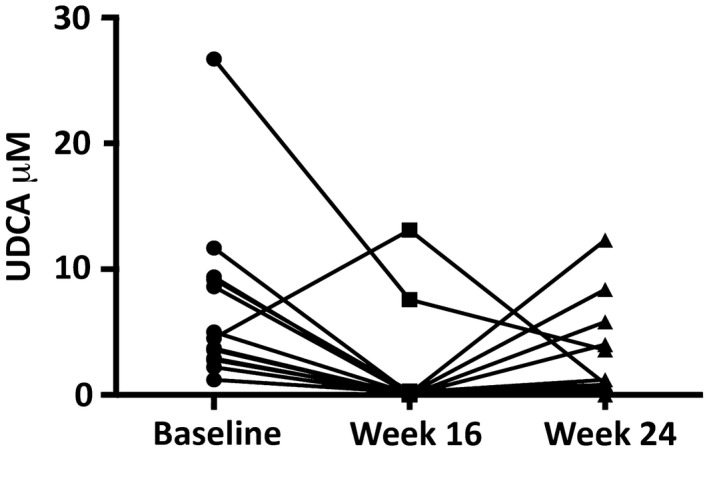
Plasma UDCA levels in 13 study participants at baseline (circle) and at 16 (square) and 24 (triangle) weeks. Each line represents an individual participant.
Discussion
UDCA is a hydrophilic bile acid first isolated from Ursus maritimus (polar bear).21 It is a minor bile acid in humans, and oral dosing enriches the bile acid pool up to 40%‐60%, depending on the dose used.22 It is currently U.S. Food and Drug Administration approved for use in primary biliary cholangitis and for cholesterol gallstone dissolution in adults. However, it has been widely used off label in adults and children with other liver diseases, including PSC. Potential therapeutic mechanisms include increase in the hydrophilicity index of the bile acid pool, stimulation of choleresis, cytoprotection, and immunomodulatory and anti‐inflammatory effects.23, 24, 25, 26 The role of UDCA in the treatment of PSC is controversial, both in adults and children. In a prospective, blinded, placebo‐controlled trial in adults, high‐dose UDCA therapy was shown to result in worse long‐term outcomes and increased SAEs compared to controls, despite short‐term improvements in liver biochemistries.14 Similar studies have not been performed in children, although short‐term biochemical improvement has been reported.7, 16 Consequences of UDCA withdrawal in children with PSC are important for informing parents who may wish to discontinue the drug based on the adult study and to inform clinical trial design of UDCA studies with a control arm or any new PSC medications (e.g., vancomycin, farnesoid X receptor agonists) that would optimally be performed off UDCA.13
During our study, we found that average liver biochemistries increased modestly overall with controlled UDCA withdrawal in children with quiescent PSC. Further analysis after stratification into treatment response groups showed that one third had normal ALT and GGT that remained normal in the absence of UDCA therapy (null group), one third increased above 100 IU/L (flare group), and the rest had intermediate responses (indeterminant group). None developed elevated bilirubin or significant increases in ALP levels, and all flares responded to the reinstitution of UDCA therapy. Diagnosis of PSC versus AIH/PSC overlap, use of immunosuppressive medications, or presence of IBD did not impact response status.
In this study, we observed that baseline levels of plasma IL‐8 and TNF‐α were higher in the flare group compared to null and/or indeterminant groups. There have been few studies of serum biomarkers reported in adult patients with PSC and none previously reported in children with PSC. A recent study on bile and serum biomarkers in adults with PSC revealed that IL‐8 along with other biomarkers was significantly increased in PSC compared to controls. Moreover, serum IL‐8 levels independently predicted transplant‐free survival.27 Another adult study analyzed cytokine levels before and after UDCA treatment in patients with PSC.28 Similar to our findings, elevation in TNF‐α and IL‐8 were reported but none were significantly affected by UDCA therapy. Due to the small number of patient samples available for cytokine analyses, broad conclusions regarding cytokines as potential biomarkers of disease severity cannot be made. However, the findings do suggest that a large multicenter study of immunophenotyping in pediatric PSC is warranted to provide insight into biomarkers of disease severity, immunopathogenesis, and potential therapeutic targets.
There are several potential reasons for the variability we observed in response to UDCA withdrawal. Eight weeks off of UDCA may not be sufficient for some patients to generate a biliary inflammatory response sufficient to elevate serum biochemistries. Higher mean serum GGT and lower serum albumin at baseline were present in the flare group compared to the null group and may be predictive of a flare. Also, despite remaining in the “normal” range throughout the study, GGT levels rose significantly in the null group during or after UDCA withdrawal. These observations suggest that even when these tests are within the normal limits, patients may have low‐grade indolent inflammation and be primed for a flare following UDCA withdrawal. This is also supported by the baseline cytokine findings for IL‐8 and TNF‐α in our study. However, in order to determine if these cytokines will be useful as biomarkers for predicting a flare during or after UDCA withdrawal, future investigations are necessary.29
While we measured serum UDCA levels, our methodology did not assess for UDCA conjugates, leading to potential underestimation of the total serum UDCA levels. However, our data do support that there was withdrawal of medication during phase III in the majority of participants. The variable increase in UDCA levels with reinstitution of the drug may be explained by variability in the time required to re‐establish prewithdrawal levels as well as our measurement excluding the conjugated species. Alternatively, some parents may have decided not to restart the medication, although all flare participants responded to UDCA reinstitution. Interestingly, only 1 participant in the indeterminant group flared during phase IV. The high withdrawal levels in the 2 participants noted with anomalous levels probably, at least partially, explains the lack of a flare and inclusion in the null response group.
Limitations of our study included a relatively small sample size as recruitment was difficult for several reasons. In children, PSC is less common than in adults, with an incidence of 0.23 cases per 100,000 person‐years.30 Because of safety considerations in a vulnerable population, inclusion and exclusion criteria were relatively strict and may have introduced bias for selection of participants more responsive to UDCA or with milder disease. Recruitment was also challenging because the study involved withdrawing a drug that had resulted in an initial improvement in liver tests and parents are more likely to participate in a trial testing a new drug. Study retention was also problematic with 5 of 27 participants lost from the study due to early withdrawal for various reasons (Supporting Fig. S2). Finally, the period of time our participants were on a reduced dose or off UDCA was relatively brief (12 weeks).
The results of this study may have important implications for pediatric PSC therapy. The fact that biochemical flares occurred when the drug was reduced or withdrawn strongly suggests that UDCA was having at least a biochemical therapeutic effect in a significant number of study participants. Long‐term “hard” endpoint trials for drug efficacy are extremely difficult to perform in children. In adults, ALP has been used as a biomarker for disease severity for most PSC trials, with data showing that patients who respond to UDCA therapy, based on significant improvement or normalization of ALP, have a better prognosis and disease outcome.31 Thus, there may be a subgroup of adult patients with PSC who are UDCA responders and perhaps should be treated.32 In children, a significant contribution from bone to serum ALP precludes its use as a biliary biomarker, and GGT is therefore used, as in our study, despite its own inherent limitations.33 A recent retrospective analysis of a large international cohort of pediatric patients with PSC found that 46% of patients treated with UDCA had a complete normalization of GGT during the first year after diagnosis, and this subset had a more favorable 5‐year outcome with 99% survival with their native liver.34 If confirmed in a prospective controlled trial, these findings may suggest that UDCA responders should continue the medication whereas nonresponders could discontinue UDCA therapy for lack of efficacy.
There are several differences between features of PSC in children compared to adults, and there is speculation that pediatric PSC may represent an earlier more immunologically active stage of the disease or even a different disease.11, 35 Overlap of AIH with PSC is present in a much higher percentage of children (up to 50%) compared to adults (6%‐8%). However, there is no universal agreement on how to make the diagnosis of overlap in children, especially based on histologic features, or consensus on optimal treatment.8 It is theoretically possible that the immunomodulatory and anti‐inflammatory properties of UDCA may have a positive impact on the AIH component of overlap disease as well. In the series reported by Miloh et al.,7 children with PSC and positive autoimmune markers without histologic features of AIH responded to UDCA therapy similar to patients with PSC but without autoimmune markers. In the present study, although presence of overlap did not predict response group, 5/8 participants in the flare group had overlap. It is tempting to speculate that UDCA may have a therapeutic impact on AIH/PSC overlap in children and should be studied.
In summary, children with PSC and AIH/PSC overlap on UDCA treatment with biochemically quiescent disease demonstrate a range of biochemical responses to a stepped withdrawal of the drug. Responses range from no change to severe flares with ALT and/or GGT increases to above 10 times ULN, and all appear to be responsive to restarting UDCA therapy. Although normal reference intervals for GGT levels have been defined,36 our study suggests that “normal” GGT levels may not in fact be biologically normal in patients with PSC in light of the significant increase noted in the null group with UDCA withdrawal and reinstitution that did not rise above the normal cutoff (Fig. 2B) as well as significantly higher mean GGT baseline levels noted in the flare group. Factors, such as baseline levels of GGT and albumin as well as IL‐8 and TNF‐α, may be helpful in predicting which patients are most at risk for a significant disease flare off UDCA. More studies are needed to define the long‐term benefit‐to‐risk ratio for the use of UDCA in children with PSC as well as AIH/PSC overlap, which is significantly more prevalent in children than in adults with PSC. As in adults, more reliable biomarkers of disease activity and progression along with well‐designed prospective clinical trials are needed for this progressive disease with no proven effective therapy beyond liver transplantation.
Supporting information
Acknowledgment
The editorial assistance of Dr. Amanda M. Preston, Scientific Editor, Children's Foundation Research Institute, is greatly appreciated.
Supported by the Food and Drug Administration Office of Orphan Product Development (grant R01 FD003709 to D.D.B. and B.L.S.), the Musette and Allen Morgan, Jr. Foundation for the Study of Primary Sclerosing Cholangitis (to D.D.B.), and Le Bonheur Children's Hospital (to D.D.B.).
Registration on ClinicalTrials.gov: NCT01088607.
Potential conflict of interest: Dr. Black consults for Intercept. Dr. Karpen consults for Intercept, Albiero, and Retrophin. Dr. Mack consults for Albireo. Dr. Rosenthal consults for and received grants from Gilead, AbbVie, Roche, and Retrophin; he consults for Intercept, Mirum, Albireo, and Aventis and received grants from Bristol Myers Squibb. Dr. Miloh consults for, is on the speakers’ bureau for, and received grants from Alexion. Dr. Lin received grants from Gilead. The other authors have nothing to report.
References
- 1. Larusso NF, Shneider BL, Black D, Gores GJ, James SP, Doo E, et al. Primary sclerosing cholangitis: summary of a workshop. Hepatology 2006;44:746‐764. [DOI] [PubMed] [Google Scholar]
- 2. Jossen J, Annunziato R, Kim HS, Chu J, Arnon R. Liver Transplantation for children with primary sclerosing cholangitis and autoimmune hepatitis: UNOS database analysis. J Pediatr Gastroenterol Nutr 2017;64:e83‐e87. [DOI] [PubMed] [Google Scholar]
- 3. Deneau MR, El‐Matary W, Valentino PL, Abdou R, Alqoaer K, Amin M, et al. The natural history of primary sclerosing cholangitis in 781 children: a multicenter, international collaboration. Hepatology 2017;66:518‐527. [DOI] [PubMed] [Google Scholar]
- 4. Venkat VL, Ranganathan S, Mazariegos GV, Sun Q, Sindhi R. Recurrence of primary sclerosing cholangitis in pediatric liver transplant recipients. Liver Transpl 2014;20:679‐686. [DOI] [PubMed] [Google Scholar]
- 5. Miloh T, Anand R, Yin W, Vos M, Kerkar N, Alonso E; Studies of Pediatric Liver Transplantation Research Group . Pediatric liver transplantation for primary sclerosing cholangitis. Liver Transpl 2011;17:925‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shneider BL. Diagnostic and therapeutic challenges in pediatric primary sclerosing cholangitis. Liver Transpl 2012;18:277‐281. [DOI] [PubMed] [Google Scholar]
- 7. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A retrospective single‐center review of primary sclerosing cholangitis in children. Clin Gastroenterol Hepatol 2009;7:239‐245. [DOI] [PubMed] [Google Scholar]
- 8. Mieli‐Vergani G, Vergani D, Baumann U, Czubkowski P, Debray D, Dezsofi A, et al. Diagnosis and management of pediatric autoimmune liver disease: ESPGHAN Hepatology Committee position statement. J Pediatr Gastroenterol Nutr 2018;66:345‐360. [DOI] [PubMed] [Google Scholar]
- 9. Gregorio GV, Portmann B, Karani J, Harrison P, Donaldson PT, Vergani D, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16‐year prospective study. Hepatology 2001;33:544‐553. [DOI] [PubMed] [Google Scholar]
- 10. Ibrahim SH, Lindor KD. Current management of primary sclerosing cholangitis in pediatric patients. Paediatr Drugs 2011;13:87‐95. [DOI] [PubMed] [Google Scholar]
- 11. Mehta R, Black DD. Primary sclerosing cholangitis In: Kleinman RE, Goulet OJ, Mieli‐Vergani G, Sanderson IR, Sherman PM, Shneider BL, eds. Walker's Pediatric Gastrointestinal Disease. Volume 2 6th ed Raleigh, NC: People's Medical Publishing House USA Ltd; 2018:1229‐1245. [Google Scholar]
- 12. Davies YK, Cox KM, Abdullah BA, Safta A, Terry AB, Cox KL. Long‐term treatment of primary sclerosing cholangitis in children with oral vancomycin: an immunomodulating antibiotic. J Pediatr Gastroenterol Nutr 2008;47:61‐67. [DOI] [PubMed] [Google Scholar]
- 13. Damman JL, Rodriguez EA, Ali AH, Buness CW, Cox KL, Carey EJ, et al. Review article: the evidence that vancomycin is a therapeutic option for primary sclerosing cholangitis. Aliment Pharmacol Ther 2018;47:886‐895. [DOI] [PubMed] [Google Scholar]
- 14. Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High‐dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50:808‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010;51:660‐678. [DOI] [PubMed] [Google Scholar]
- 16. Gilger MA, Gann ME, Opekun AR, Gleason WA Jr. Efficacy of ursodeoxycholic acid in the treatment of primary sclerosing cholangitis in children. J Pediatr Gastroenterol Nutr 2000;31:136‐141. [DOI] [PubMed] [Google Scholar]
- 17. Wunsch E, Trottier J, Milkiewicz M, Raszeja‐Wyszomirska J, Hirschfield GM, Barbier O, et al. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology 2014;60:931‐940. [DOI] [PubMed] [Google Scholar]
- 18. Turner D, Otley AR, Mack D, Hyams J, de Bruijne J, Uusoue K, et al. Development, validation, and evaluation of a pediatric ulcerative colitis activity index: a prospective multicenter study. Gastroenterology 2007;133:423‐432. [DOI] [PubMed] [Google Scholar]
- 19. Kappelman MD, Crandall WV, Colletti RB, Goudie A, Leibowitz IH, Duffy L, et al. Short pediatric Crohn's disease activity index for quality improvement and observational research. Inflamm Bowel Dis 2011;17:112‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Want EJ, Coen M, Masson P, Keun HC, Pearce JT, Reily MD, et al. Ultra performance liquid chromatography‐mass spectrometry profiling of bile acid metabolites in biofluids: application to experimental toxicology studies. Anal Chem 2010;82:5282‐5289. [DOI] [PubMed] [Google Scholar]
- 21. Li S, Tan HY, Wang N, Hong M, Li L, Cheung F, et al. Substitutes for bear bile for the treatment of liver diseases: research progress and future perspective. Evid Based Complement Alternat Med 2016;2016:4305074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rost D, Rudolph G, Kloeters‐Plachky P, Stiehl A. Effect of high‐dose ursodeoxycholic acid on its biliary enrichment in primary sclerosing cholangitis. Hepatology 2004;40:693‐698. [DOI] [PubMed] [Google Scholar]
- 23. Lazaridis KN, Gores GJ, Lindor KD. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J Hepatol 2001;35:134‐146. [DOI] [PubMed] [Google Scholar]
- 24. Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 2002;36:525‐531. [DOI] [PubMed] [Google Scholar]
- 25. Beuers U. Drug insight: mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat Clin Pract Gastroenterol Hepatol 2006;3:318‐328. [DOI] [PubMed] [Google Scholar]
- 26. Roma MG, Toledo FD, Boaglio AC, Basiglio CL, Crocenzi FA, Sanchez Pozzi EJ. Ursodeoxycholic acid in cholestasis: linking action mechanisms to therapeutic applications. Clin Sci (Lond) 2011;121:523‐544. [DOI] [PubMed] [Google Scholar]
- 27. Vesterhus M, Holm A, Hov JR, Nygard S, Schrumpf E, Melum E, et al. Novel serum and bile protein markers predict primary sclerosing cholangitis disease severity and prognosis. J Hepatol 2017;66:1214‐1222. [DOI] [PubMed] [Google Scholar]
- 28. van Milligen de Wit AW, Kuiper H, Camoglio L, van Bracht J, Jones EA, Tytgat GN, , et al. Does ursodeoxycholic acid mediate immunomodulatory and anti‐inflammatory effects in patients with primary sclerosing cholangitis? Eur J Gastroenterol Hepatol 1999;11:129‐136. [DOI] [PubMed] [Google Scholar]
- 29. Mack CL. Serum cytokines as biomarkers of disease and clues to pathogenesis. Hepatology 2007;46:6‐8. [DOI] [PubMed] [Google Scholar]
- 30. Kaplan GG, Laupland KB, Butzner D, Urbanski SJ, Lee SS. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population‐based analysis. Am J Gastroenterol 2007;102:1042‐1049. [DOI] [PubMed] [Google Scholar]
- 31. Stanich PP, Bjornsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis 2011;43:309‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tabibian JH, Lindor KD. Ursodeoxycholic acid in primary sclerosing cholangitis: if withdrawal is bad, then administration is good (right?). Hepatology 2014;60:785‐788. [DOI] [PubMed] [Google Scholar]
- 33. Cabrera‐Abreu JC, Green A. Gamma‐glutamyltransferase: value of its measurement in paediatrics. Ann Clin Biochem 2002;39:22‐25. [DOI] [PubMed] [Google Scholar]
- 34. Deneau M, Perito E, Ricciuto A, Gupta N, Kamath BM, Palle S, et al. Ursodeoxycholic acid therapy in pediatric primary sclerosing cholangitis: predictors of gamma glutamyltransferase normalization and favorable clinical course. J Pediatr 2019;209:92‐96.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adike A, Carey EJ, Lindor K. Primary sclerosing cholangitis in children versus adults: lessons for the clinic. Expert Rev Gastroenterol Hepatol 2018;12:1025‐1032. [DOI] [PubMed] [Google Scholar]
- 36. Colantonio DA, Kyriakopoulou L, Chan MK, Daly CH, Brinc D, Venner AA, et al. Closing the gaps in pediatric laboratory reference intervals: a CALIPER database of 40 biochemical markers in a healthy and multiethnic population of children. Clin Chem 2012;58:854‐868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials