

Abstract
Mutations in the liver glycogen phosphorylase (Pygl) gene are associated with the diagnosis of glycogen storage disease type VI (GSD‐VI). To understand the pathogenesis of GSD‐VI, we generated a mouse model with Pygl deficiency (Pygl −/−). Pygl −/− mice exhibit hepatomegaly, excessive hepatic glycogen accumulation, and low hepatic free glucose along with lower fasting blood glucose levels and elevated blood ketone bodies. Hepatic glycogen accumulation in Pygl −/− mice increases with age. Masson's trichrome and picrosirius red staining revealed minimal to mild collagen deposition in periportal, subcapsular, and/or perisinusoidal areas in the livers of old Pygl −/− mice (>40 weeks). Consistently, immunohistochemical analysis showed the number of cells positive for alpha smooth muscle actin (α‐SMA), a marker of activated hepatic stellate cells, was increased in the livers of old Pygl −/− mice compared with those of age‐matched wild‐type (WT) mice. Furthermore, old Pygl −/− mice had inflammatory infiltrates associated with hepatic vessels in their livers along with up‐regulated hepatic messenger RNA levels of C‐C chemokine ligand 5 (Ccl5/Rantes) and monocyte chemoattractant protein 1 (Mcp‐1), indicating inflammation, while age‐matched WT mice did not. Serum levels of aspartate aminotransferase and alanine aminotransferase were elevated in old Pygl −/− mice, indicating liver damage. Conclusion: Pygl deficiency results in progressive accumulation of hepatic glycogen with age and liver damage, inflammation, and collagen deposition, which can increase the risk of liver fibrosis. Collectively, the Pygl‐deficient mouse recapitulates clinical features in patients with GSD‐VI and provides a model to elucidate the mechanisms underlying hepatic complications associated with defective glycogen metabolism.
Abbreviations
- α‐SMA
alpha smooth muscle actin
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CCL5/Rantes
C‐C chemokine ligand 5
- Col1a1
collagen type I alpha 1
- Col1a2
collagen type I alpha 2
- Col4a2
collagen type IV alpha 2
- Ctgf
connective tissue growth factor
- Fn1
fibronectin
- FRT
flippase recognition target
- G1P
glucose 1‐phosphate
- GNG
gluconeogenesis
- GSD
glycogen storage disease
- H&E
hematoxylin & eosin
- HSC
hepatic stellate cell
- KOMP
Knockout Mouse Project
- lacZ
beta‐galactosidase
- loxP
locus of X‐over P1
- Mcp‐1
monocyte chemoattractant protein 1
- mRNA
messenger RNA
- PCR
polymerase chain reaction
- Pygl
liver glycogen phosphorylase
- Tgf‐β
transforming growth factor beta
- Tnf‐a
tumor necrosis factor alpha
- WT
wild type
Glycogen storage disease type VI (GSD‐VI; MIM#232700), or Hers disease, is caused by a deficiency or mutation in the human glycogen phosphorylase (PYGL) gene, which codes for the metabolic enzyme liver glycogen phosphorylase. GSD‐VI is an autosomal recessive disease that affects approximately 1 in 65,000 to 85,000 live births.1, 2 An integral enzyme in glucose metabolism, PYGL catalyzes the rate‐limiting step of glycogenolysis, converting glycogen into glucose 1‐phosphate (G1P). Breakdown of glycogen in the liver requires the step‐wise activation of several cytosolic liver enzymes. Phosphorylase kinase, which phosphorylates liver PYGL, triggers a conformation switch from phosphorylase b (inactive form) to phosphorylase a (active form), which catalyzes the breakdown of glycogen into chains of G1P monomers. Through the addition of the glycogen debranching enzyme, which assists to further cleave α‐1,6 glycosidic linkages, free G1P monomers are converted to glucose 6‐phosphate, which can be released from the liver or shunted into alternative pathways.3 Additionally, glycogen is processed through the autophagy–lysosomal pathway, mediated by the enzyme acid α‐glucosidase, to produce low quantities of free glucose.4
People with GSD‐VI typically present with growth retardation and hepatomegaly early in life. Unlike the more severe form of GSD‐I, patients with GSD‐VI experience mild hypoglycemia.2 In fact, several clinical reports detail asymptomatic patients with GSD‐VI presenting with isolated hepatomegaly and no prior history of hypoglycemia.5 While hypoglycemia can be mild in GSD‐VI, blood analyses often demonstrate hyperlipidemia, hepatic transaminase elevation, and hyperketosis.2, 5, 6, 7, 8, 9, 10 Protein deficiency can occur from over dependence on protein as an alternative fuel and a precursor for gluconeogenesis. As a result of the tendency for ketosis and a low protein concentration, most patients with GSD‐VI benefit from therapy even if severe hypoglycemia is not occurring.
Based on the nonspecific and variable nature of the disease, GSD‐VI is almost certainly underdiagnosed in the general population. There is a paucity of literature on the condition, but severe complications can occur, including liver fibrosis, liver tumors, and cirrhosis.2, 7, 8 In order to better understand the pathophysiology of the condition and develop new therapies, we generated this novel mouse model with a deficiency in the Pygl gene that exhibits the metabolic abnormalities and clinical manifestations associated with GSD‐VI.
Materials and Methods
Generation of Pygl‐Deficient Mice
A Pygl knockout allele mouse (Pygl targeted mutation 1a [tm1a/+] or Pygl +/–, C57BL/6N background) line was created in collaboration with the Knockout Mouse Project (KOMP) Repository (#CSD23397) at the University of California, Davis, using the “knockout first (promoter driven)” strategy.11 To generate the knockout allele, a cassette containing flippase recognition target (FRT), locus of X(cross)‐over in P1 (loxP) sequences, engrailed 2 splice acceptor (En2 SA), beta‐galactosidase (lacZ), neomycin, and FRT and loxP sites was inserted in the intron between exon 2 and exon 3 of the Pygl gene. The allele of Pygltm1a was initially in a nonexpressive form with conditional potential. Embryonic stem cells containing the targeted allele (JMN8.4 subline, C57BL/6N background, KOMP repository, Pygl H02) were introduced into donor blastocysts (BALB/c). Chimerism of 50% or greater was identified by coat color, and chimeric male mice were bred with C57BL/6N female mice. Desired heterozygous mice from the bred cross (Pygl tm1a allele) were genotyped to confirm germline transmission. For all experiments, heterozygous mating (Pygl+/–) pairs were used to generate Pygl‐deficient mice (Pygl −/−). Human and mouse PYGL proteins share 94% of sequence similarity (the Ensembl genome browser www.ensembl.org).
Animal Studies
All animal studies were performed in accordance with the guidelines and approval of the Institutional Animal Care and Use Committee of the University of Connecticut Health Center. All mice were maintained in a pathogen‐free animal facility at 22°C‐24°C under a 12‐hour:12‐hour light–dark cycle in individually ventilated caging systems. Standard rodent chow (Envigo, Madison, WI) and water were provided ad libitum. Animals were weaned and separated according to sex at postnatal day 21 and housed in groups of four to five mice. Littermate (Pygl+/+) animals were used as wild‐type (WT) control mice. Phenotypes evaluated in this study were indistinguishable between male and female mice; therefore, both male and female mice were used in all studies without preference. The mice were classified by age group as young (4‐20 weeks) and old (>40 weeks). Total number of mice used were young WT (n = 27) and Pygl −/− (n = 36) and old WT (n = 27) and Pygl −/− (n = 13) mice. At the end of the study, mice were fasted for 24 hours before being killed. Liver and kidney weights were expressed as percentage of body weight. The numbers of mice analyzed were young WT (n = 13) and Pygl −/− (n = 24) mice for liver weight and young WT (n = 7) and Pygl −/− (n = 17) mice for kidney weight.
Genotyping
Tail biopsies were lysed in tail lysis solution (DirectPCR tail lysis; 20 mg/mL proteinase K) at 55°C overnight and then at 85°C for 1 hour. Genotyping was performed using Accupower polymerase chain reaction (PCR) premix (Bioneer, Oakland, CA) tubes. The introduced cassette containing lacZ in intron 2‐3 was identified using the following primer pair: lacZ forward (5′‐TAATCACGACGCGCTGTACT‐3′) and lacZ reverse (5′‐CGGATAAACGGAACTGGAAA‐3′); this was expected to amplify a fragment of 500 base pairs (bp) in the inserted lacZ cassette. The WT allele was identified using the following primer pair: WT forward (5′‐TGCTGAAACACATCAGCACA‐3′) and WT reverse (5′‐ATGTCCAATCCA AGCTGAGG‐3′); this was expected to amplify a fragment of 800 bp in the WT allele. However, if the introduced cassette exists in the DNA, it will fail to amplify the fragment.
Fasting Glucose and Ketone Test
Fasting glucose tests and ketone tests were performed at 0, 2, 4, 6, and 24 hours. Blood glucose levels were measured with a blood glucose meter and cuvettes (HemoCue Glucose 201 System; HemoCue, Brea, CA), and blood ketone (β‐hydroxybutyrate) concentration was determined using a blood ketone meter and ketone strips (Precision Xtra; Abbott Laboratories, Abbott Park, IL). Young WT (n = 12) and Pygl −/− (n = 17) mice were used for fasting glucose tests, and young WT (n = 18) and Pygl −/− (n = 18) mice were used in ketone studies.
Hepatic Glycogen and Free Glucose Content Determination
To determine hepatic glycogen and free glucose content, liver tissues from 24‐hour fasted young WT (n = 5) and Pygl −/− (n = 6) mice and old WT (n = 5) and Pygl −/− (n = 6) mice were homogenized. Hepatic glycogen and free glucose content were measured according to the manufacturers' instructions using a glycogen colorimetric/fluorometric assay kit (BioVision, Milpitas, CA) and a D‐glucose assay kit (Megazyme, Chicago, IL), respectively, and normalized to hepatic protein concentration measured by the bicinchoninic acid (BCA) assay kit from Thermo Fisher Scientific (Louisville, CO).
Hepatic Hydroxyproline Determination
To quantify hepatic collagen content, hepatic hydroxyproline was measured using a colorimetric assay kit from BioVision. Briefly, livers from both young and old WT (n = 11) and Pygl −/− (n = 13) mice were homogenized in 100 μL of distilled water. We added 100 μL of 10 N NaOH to the homogenate, incubated this at 120°C for 1 hour, and neutralized the mixture with 100 μL of 10 N HCl. After centrifugation at 10,000g for 5 minutes, the hydroxyproline content in the resulting hydrolysate was measured with the SpectraMax i3x (Molecular Devices, Sunnyvale, CA) according to the manufacturer's instructions and normalized to the hepatic protein concentration in the homogenate as measured by the BCA assay kit.
Serum Biochemistry
To determine serum metabolites, serum was collected from 24‐hour fasted young WT (n = 6) and Pygl −/− (n = 7) mice and analyzed for triglyceride, cholesterol, lactic acid, and uric acid. The colorimetric kits used for serum biochemistry were the cholesterol and uric acid kits from Thermo Fisher Scientific, the triglyceride kit from Sigma‐Aldrich (St. Louis, MO), and the lactic acid kit from Pointe Scientific (Canton, MI). To analyze liver function, serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and bilirubin were determined using the respective colorimetric kits from BioVision.
Histopathology of GSD‐VI
Liver samples from young WT (n = 6) and Pygl −/− (n = 9) and old WT (n = 7) and Pygl −/− (n = 13) mice were fixed in 10% neutral‐buffered formalin and embedded in paraffin using standard methods (Histoserv, Inc., Germantown, MD). Adjacent 4‐5 µm sections stained with hematoxylin and eosin (H&E) and Masson's trichrome were evaluated by a certified veterinary pathologist (J.A.S.). Liver sections were also stained by picrosirius red and periodic acid–Schiff for evaluation of collagen and glycogen, respectively.
Immunohistochemical Analysis
The formalin‐fixed paraffin‐embedded liver sections of young WT (n = 5) and Pygl −/− (n = 6) mice and those of old WT (n = 6) and Pygl −/− (n = 13) mice were sectioned and deparaffinized by xylene. The resulting sections were then incubated in citrate antigen unmasking buffer (Cell Signaling Technology, Danvers, MA) for 10 minutes at 100°C. Endogenous peroxidase activity in tissue sections was quenched with 3% hydrogen peroxide solution. Tissue sections were blocked with normal goat serum, incubated with polyclonal antibody to alpha smooth muscle actin (α‐SMA; Cell Signaling Technology), and followed with the biotinylated secondary antibody in the VECTASTAIN Elite ABC kit (Vector Laboratories). The resulting complexes in sections were visualized with the ImmPACT EQV DAB kit (Vector Laboratories). Tissue sections were also counterstained with hematoxylin (Vector Laboratories). All stained sections were imaged with Laxco's LMC 4000 series compound microscope (Laxco, Inc., Mill Creek, WA) using a SeBaCam5c digital camera and SeBaView software (Laxco, Inc.).
Gene Expression Analysis
Total RNA was isolated from liver samples from young WT (n = 9) and Pygl −/− mice (n = 24) and old WT (n = 16) and Pygl −/− (n = 13) mice using TRIzol Reagent (Invitrogen, Carlsbad, CA) and the RNeasy Protect Mini Kit (QIAGEN) according to the manufacturers' instruction. We synthesized complementary DNA (cDNA) using the iScript genomic DNA Clear cDNA Synthesis Kit (Bio‐Rad Laboratories, Hercules, CA). Messenger RNA (mRNA) expression was quantified by the CFX96 One Touch Real‐Time PCR Detection System (Bio‐Rad Laboratories). Data were analyzed using CFX Maestro software (Bio‐Rad Laboratories) and normalized to mouse ribosomal protein L19 mRNA expression. The PrimePCR quantitative PCR assay primers (Bio‐Rad Laboratories) used are summarized in Supporting Table S1.
Statistical Analysis
Statistical analyses of ketone bodies, serum metabolites, gene expression, hepatic metabolites, and hepatic hydroxyproline were performed using Wilcoxon rank sum tests with SAS, version 9.4 (SAS, Inc., Cary, NC). Box‐and‐whisker plots were generated using GraphPad Prism, version 7 (GraphPad Software Inc., San Diego, CA). Other data were analyzed by unpaired t tests using GraphPad Prism, version 7. All tests were two sided.
Results
Generation of Pygl‐Deficient Mice and Presentation of GSD‐VI
PCR analysis confirmed integration of the internal ribosome entry site:LacZ cassette and floxed promoter‐driven neomycin cassette between exon 2‐3 of the Pygl gene (Fig. 1A,B). Hepatic Pygl deficiency was also confirmed by real‐time PCR in young (4‐20 weeks) and old (>40 weeks) mice (Fig. 1C). Pygl −/− mice exhibited no significant difference in body weight or physical appearance compared with age‐matched WT mice, although we observed a slight reduction in body weight in old Pygl −/− mice compared to age‐matched WT mice (Supporting Fig. S1). Pygl −/− mice exhibited hepatomegaly with an elevated ratio of liver weight to body weight compared with WT mice, while no significant difference in kidney size and appearance was observed between WT and Pygl −/− mice (Fig. 1D,E).
Figure 1.
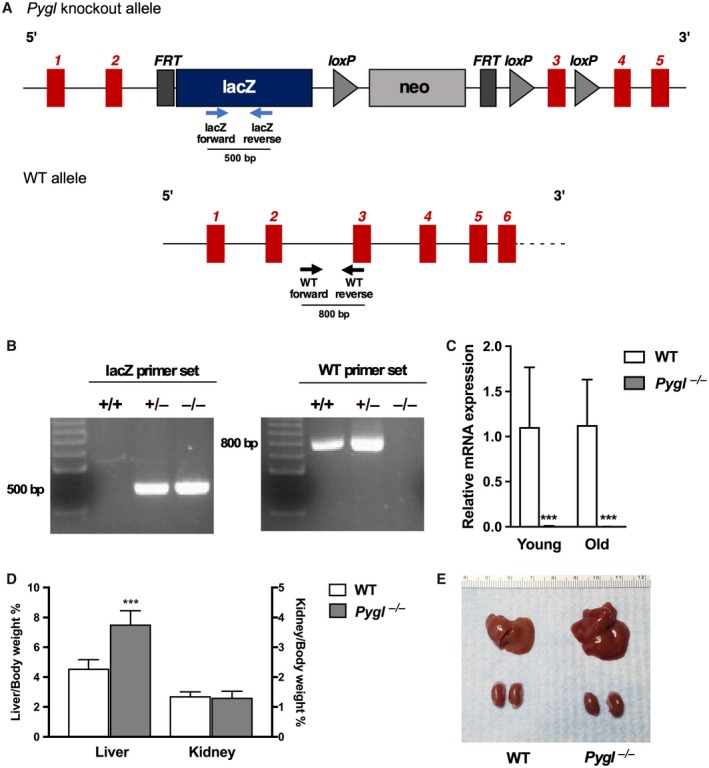
Generation of Pygl‐deficient mice. (A) Schematic representation of the Pygl‐knockout and WT alleles. Insertion of the FRT/loxP cassette in the intron between exon 2‐3 disrupts Pygl mRNA expression. Arrows indicate WT or lacZ primers used for genotyping. (B) PCR genotype analysis of WT (+/+), heterozygous (+/–), and Pygl −/− mice. The lacZ primer set and the WT primer set are expected to amplify a fragment of 500 bp and 800 bp, respectively. (C) Quantification of hepatic mRNA for Pygl in young WT (n = 9) and Pygl −/− (n = 24) mice and old WT (n = 16) and Pygl −/− (n = 13) mice. (D) Liver and kidney weights expressed as percentage of body weight. Numbers of mice analyzed were young WT (n = 13) and Pygl −/− (n = 24) mice for liver weight and young WT (n = 7) and Pygl −/− (n = 17) mice for kidney weight. (E) Representative image of liver and kidney in WT and Pygl −/− mouse. Scale shows length in centimeters. Data in (C,D) represent mean ± SD. ***P < 0.0001.
GSD‐VI Mice Exhibit Excessive Hepatic Glycogen Accumulation and Ketotic Hypoglycemia
Periodic acid–Schiff staining revealed glycogen accumulation in Pygl −/− mice compared with WT mice (Supporting Fig. S2). Quantification of hepatic glycogen revealed significantly increased glycogen accumulation in young and old Pygl −/− mice compared with age‐matched WT mice (Fig. 2A). Hepatic glycogen levels in old Pygl −/− mice were about 3‐fold higher than those in young Pygl −/− mice, suggesting hepatic glycogen accumulation with age. Consistent with impaired glycogen breakdown reflected by excessive glycogen accrual, hepatic free glucose levels were significantly decreased in Pygl −/− mice compared with age‐matched WT mice (Fig. 2B).
Figure 2.
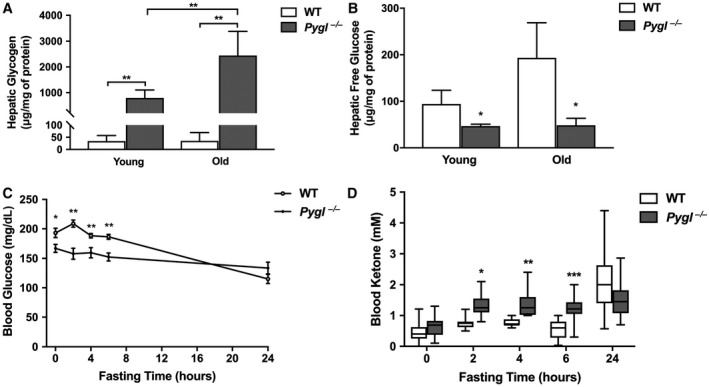
Pygl −/− mice exhibit excessive hepatic glycogen accumulation along with fasting ketotic hypoglycemia. (A,B) Hepatic glycogen and free glucose levels in 24‐hour fasted young and old WT (n = 5) and Pygl −/− (n = 6) mice. (C) Fasting glucose test in young WT (n = 12) and Pygl −/− (n = 17) mice. (D) Box‐and‐whisker plots of fasting blood ketone levels in young WT (n = 18) and Pygl −/− (n = 18) mice. Statistical analyses of box‐and‐whisker plots were performed using the Mann‐Whitney test and show the interquartile range (box), median (horizontal line), and maximum and minimum observations (whiskers). Hepatic glycogen and free glucose data (A,B) present the mean ± SD. Fasting glucose data (C) are expressed as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Patients with GSD‐VI display mild hypoglycemia with elevated blood ketone bodies during fasting.2, 12 Consistently, compared with WT mice, Pygl −/− mice displayed lower blood glucose levels at all tested fasting time points except 24 hours of fasting (Fig. 2C) and displayed elevated blood ketones levels at 2, 4, and 6 hours of fasting (Fig. 2D), representing ketotic hypoglycemia observed in patients with GSD‐VI. However, we did not witness any hypoglycemic seizures in Pygl −/− mice, a common complication observed during fasting in GSD‐Ia mice.13 Additionally, analysis of serum biochemistry revealed 24‐hour fasted Pygl −/− mice expressed significantly lower serum levels of triglyceride, cholesterol, and lactic acid compared with WT mice (Supporting Fig. S3).
Histologic Findings in GSD‐VI Mouse Liver
Examination of H&E stained sections revealed that both young and old Pygl −/− mice had histologic evidence of glycogen accumulation in hepatocytes, with associated moderate enlargement to ballooning of hepatocytes that resulted in inconspicuous hepatic sinusoids (Fig. 3; Supporting Fig. S4). Eight of 13 old Pygl −/− mice had small to large (>200 cells) aggregates of mononuclear cells associated with blood vessels (Fig. 3), while none of the WT mice did. Picrosirius red staining and Masson's trichrome staining revealed that in 13 old Pygl −/− mice, eight mice had minimal but occasionally mild collagen deposition in the subcapsular, sinusoidal, and/or periportal area of their livers (Fig. 3D1‐D4,E1‐E4,F1‐F4) compared to old WT mice (Fig. 3C1‐C4), and one mouse had regionally severe central to central fibrosis with a collapse of the intervening lobular structure (Fig. 3G1‐G4).
Figure 3.
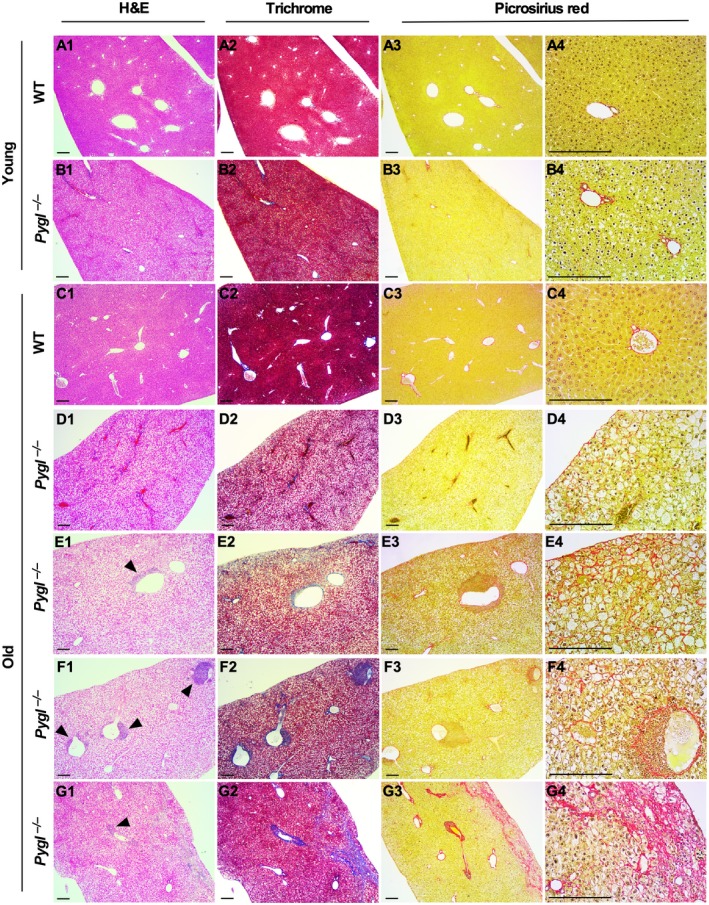
Histologic analysis of livers from WT and Pygl −/− mice. Livers from young WT (n = 6) and Pygl −/− (n = 9) mice and old WT (n = 7) and Pygl −/− (n = 13) mice were evaluated histologically by a veterinary pathologist. Representative images of liver sections stained with H&E, Masson's trichrome, and picrosirius red in young WT mice (A1‐A4), young Pygl −/− mice (B1‐B4), old WT mice (C1‐C4), and four individual old Pygl −/− mice (D1‐D4, E1‐E4, F1‐F4, G1‐G4). With picrosirius red staining, individual old Pygl −/− mouse exhibited distinct pathological patterns, including minimal collagen deposition in subcapsular areas (D3,D4); subcapsular and sinusoidal mild collagen deposition (E3,E4); minimal collagen deposition in perisinusoidal and periportal areas (F3,F4); and regionally severe fibrosis with central to central bridging and collapse of the intervening lobular structure (G3,G4). In picrosirius red staining, the images in the fourth column (A4‐G4) present higher magnification views of the images in the third column (A3‐F3). Scale bars represent 200 µm. Arrow heads indicate immune cell infiltrations.
Old GSD‐VI Mice Exhibit Elevated Serum Transaminases and Activated Hepatic Stellate Cells
Quantitative analysis of hepatic hydroxyproline level, indicative of collagen content, revealed no significant difference between WT and Pygl −/− mice at both young and old age, although there were some individual variations between old Pygl −/− mice (Fig. 4A). Elevated serum transaminases has been reported in patients with GSD‐VI.2 Consistently, serum levels of AST and ALT were elevated in old Pygl −/− mice, indicating liver injury, while there was no significant difference in serum levels of bilirubin and ALP between WT and Pygl −/− mice at both young and old age (Fig. 4B). Activated hepatic stellate cells (HSCs) play a critical role in liver fibrosis.14 When HSCs are activated by liver injury, they transdifferentiate into myofibroblasts, which produce and secrete various extracellular matrix proteins, leading to liver fibrosis.14 To investigate HSC activation in liver, we performed immunohistochemistry of α‐SMA, a marker of activated HSCs. Notably, old Pygl −/− mice displayed increased but variable numbers of α‐SMA‐positive cells in periportal and perisinusoidal areas in their livers compared with old WT mice, while there was no difference between young WT and Pygl −/− mice (Fig. 4C). Collectively, these results suggest that liver injury in old Pygl −/− mice may activate HSCs, which are responsible for liver fibrosis.
Figure 4.
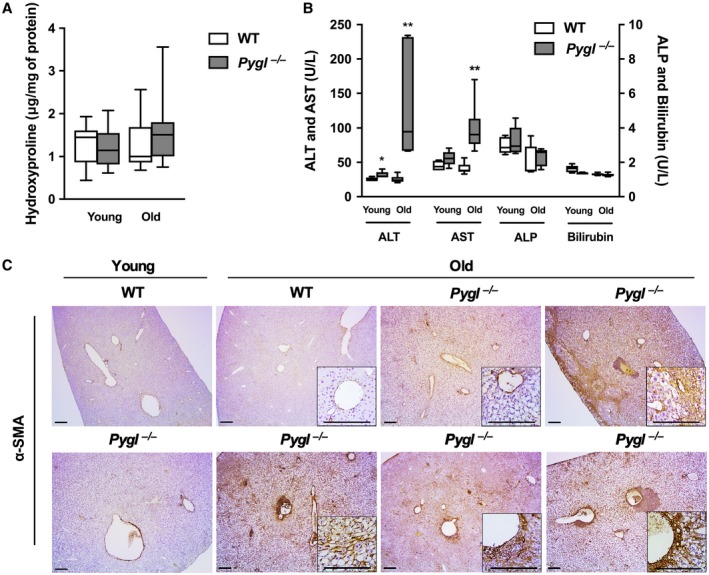
Elevated serum transaminases and activated HSCs in old Pygl −/− mice. (A) Box‐and‐whisker plots for quantification of hepatic hydroxyproline in both young and old WT (n = 11) and Pygl −/− (n = 13) mice. (B) Box‐and‐whisker plots showing serum levels of ALT, AST, ALP, and bilirubin in young WT (n = 5) and Pygl −/− (n = 5) mice and old WT (n = 6) and Pygl −/− (n = 6) mice. Data in (A,B) show the interquartile range (box), median (horizontal line), and maximum and minimum observations (whiskers). Statistical analyses were performed using the Mann‐Whitney test. *P < 0.05, **P < 0.01. (C) Immunohistochemical analysis of α‐SMA. Old Pygl −/− mice exhibited elevated but variable numbers of α‐SMA‐positive cells in periportal, perisinusoidal, and/or lobular areas in their liver sections. The insets show higher magnification views. Scale bars represent 200 µm.
Expression Profile of Fibrotic and Inflammatory Genes in GSD‐VI Mice
To understand the underlying pathological mechanism in Pygl‐deficient mice, hepatic mRNA expressions in young and old WT and Pygl −/− mice were evaluated by quantitative real‐time PCR analysis. Target mRNA markers analyzed were connective tissue growth factor (Ctgf) and transforming growth factor β (Tgf‐β) related to liver fibrosis; interleukin‐6 (Il‐6) and tumor necrosis factor alpha (Tnf‐α) related to inflammation; collagen I a1/a2 (Col1a1/a2), collagen IV a2 (Col4a2), and fibronectin (Fn1) related to extracellular matrix; and monocyte chemoattractant protein 1 (Mcp‐1), C‐C chemokine ligand 5 (Ccl5/Rantes), C‐X‐C chemokine ligand 1 (Cxcl1/KC), and macrophage inflammatory protein 2α (Mip‐2α/Cxcl2) related to immune cell recruitment. Hepatic mRNA levels of Tgf‐β (P < 0.001), Tnf‐α (P < 0.01), and Ccl5/Rantes (P < 0.05) along with Col1a1 (P < 0.001), Col1a2 (P < 0.05), Col4a2 (P < 0.001), and Fn1 (P < 0.01) were elevated in young Pygl −/− mice compared with WT mice (Fig. 5A). However, examination of H&E, Masson's trichrome, and picrosirius red staining (Fig. 3) showed that increased mRNA expression of these genes in young Pygl −/− mice did not correlate with liver fibrosis. Consistent with pathological phenotypes observed in old Pygl −/− mice (Fig. 3), we also observed up‐regulated hepatic mRNA levels of Ctgf mRNA (P < 0.0001) and Mcp‐1 (P < 0.001) and Ccl5/Rantes (P < 0.05), which play important roles in fibrosis and immune cell recruitment (Fig. 5B).
Figure 5.
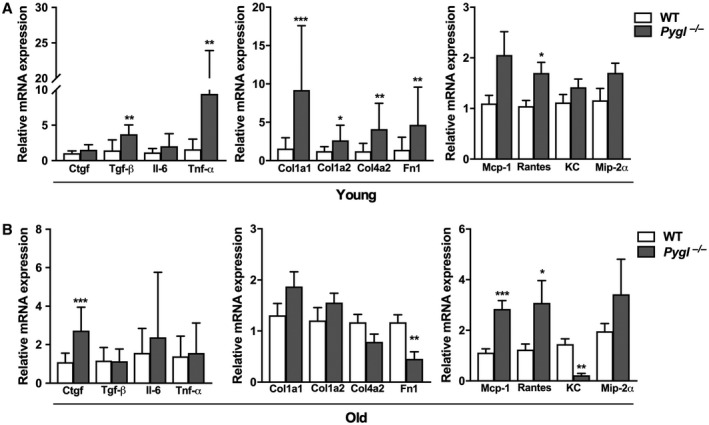
Expression profile of fibrotic and inflammatory genes in WT and Pygl −/− mice. Quantification of hepatic mRNA for fibrogenic genes (Ctgf, Tgf‐β), inflammatory genes (Il‐6, Tnf‐a), extracellular matrix (Col1a1, Col1a2, Col4a2, Fn 1), and chemokines (Mcp‐1, Ccl5/Rantes, Cxcl1/KC, Mip‐2α/Cxcl2) in (A) young WT (n = 9) and Pygl −/− (n = 24) mice and (B) old WT (n = 16) and Pygl −/− (n = 13) mice. Data represent the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: Fn1, fibronectin; Il‐6, interleukin‐6; KC, C‐X‐C chemokine ligand 1; Mip‐2α, macrophage inflammatory protein 2α.
Discussion
GSD‐VI is an inheritable metabolic disorder characterized by hepatomegaly, mild fasting hypoglycemia, and hyperketosis during fasting.2, 5, 6, 7, 8, 9, 10 GSD‐VI is frequently underdiagnosed because of its mild phenotype. However, severe complications, including liver fibrosis, liver tumors, and cirrhosis, have been reported in patients with GSD‐VI.2, 7, 8 Currently, there are limited resources available to study this disease besides clinical case reports. Thus, we developed a mouse model with a genetic deficiency of Pygl that recapitulates the phenotype reported in human GSD‐VI. Indeed, Pygl‐deficient mice exhibit hepatomegaly, excessive hepatic glycogen accumulation, mild fasting hypoglycemia, and elevated blood ketone bodies during prolonged fasting. Furthermore, we show PYGL deficiency leads to progressive accumulation of hepatic glycogen with age and increases the risk of liver damage and inflammation along with collagen deposition in old Pygl −/− mice (Fig. 6).
Figure 6.
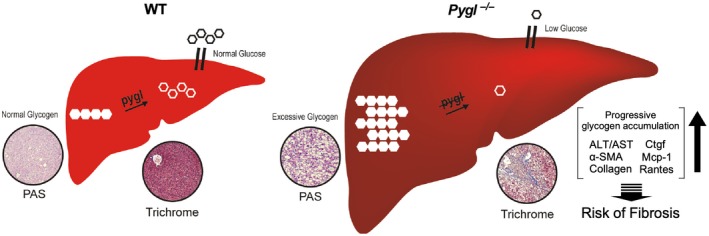
Pathophysiology of Pygl‐deficient liver. In a fasting period, WT mice maintain blood glucose by breakdown of the hepatic glycogen store to glucose through glycogenolysis. However, in Pygl‐deficient mice, hepatic glycogen accumulates instead of being released as glucose into the blood stream. As a result, Pygl‐deficient mice exhibit hepatomegaly and fasting hypoglycemia. Moreover, hepatic glycogen accumulation increases with age in Pygl‐deficient mice. Thus, prolonged Pygl deficiency leads to excessive buildup of hepatic glycogen, liver damage, inflammation, and collagen deposition, all of which may increase the risk of liver fibrosis.
During fasting periods, the liver plays a critical role in endogenous glucose production through glycogenolysis and gluconeogenesis (GNG).15 Short‐term fasting activates hepatic glycogenolysis, including liver glycogen phosphorylase‐mediated degradation of glycogen into G1P, which is further metabolized into glucose. Once hepatic glycogen storage is depleted by prolonged fasting, the liver induces GNG, which uses amino acids, lactate, pyruvate, glycerol, and fatty acids to produce glucose. This study shows that Pygl‐deficient mice following a fasting challenge display mild ketotic hypoglycemia along with reduced levels of serum metabolites, including triglyceride, cholesterol, and lactic acid, which can be substrates for GNG. Intriguingly, it has been suggested that elevated ketone bodies in ketotic GSDs reflect increased mitochondria fatty acid oxidation, which is elicited by GNG.16 Therefore, elevated blood ketone bodies and reduced levels of serum metabolites in fasted Pygl −/− mice raise the possibility that Pygl deficiency may induce GNG instead of defective glycogenolysis to compensate low glucose production in fasted periods, as suggested.17
Distinct hepatic complications have been reported in each type of GSD. Untreated patients with GSD‐I and animal models frequently develop hepatocellular adenoma (HCA) or carcinoma (HCC), while liver fibrosis or cirrhosis was not observed in GSD‐I.13, 15 In contrast, liver fibrosis and cirrhosis as well as hepatic tumors have been reported in patients with GSD‐III.18, 19 However, it is still unclear which hepatic complications are prevalent in GSD‐VI, although there are a few case reports showing that patients with GSD‐VI develop hepatic tumor, fibrosis, and cirrhosis.2, 7, 8 In this study, we have shown that old Pygl −/− mice are at risk of liver fibrosis while there was no evidence of HCA and HCC in spite of excessive glycogen accumulation.
Hepatic fibrosis is triggered by chronic liver injury caused by viral infection, drugs, toxins, or metabolic disorders.14 Once HSCs are activated by cytokines and chemokines released by immune cells and damaged hepatocytes, they undergo transdifferentiation into myofibroblasts, which are mainly responsible for collagen deposition in liver fibrosis.14 In this study, several lines of evidence show that prolonged Pygl deficiency leads to the pathological features associated with liver fibrosis. First, Pygl −/− mice displayed age‐dependent excessive hepatic glycogen accumulation, which can damage hepatocytes as well as the liver architecture. It is noteworthy that average hepatic glycogen levels in old Pygl −/− mice are at least 60‐fold higher than those in WT mice. Moreover, we observed liver injury reflected by elevated serum ALT and AST in old Pygl −/− mice. Second, α‐SMA‐positive cells, indicative of activated HSCs, were increased in old Pygl −/− mice compared with age‐matched WT mice. Third, collagen deposition and inflammatory infiltrates associated with hepatic vessels were variably but overall elevated in the livers of old Pygl −/− mice along with up‐regulated hepatic mRNA levels of fibrogenic gene (Ctgf) and chemokine genes (Ccl5/Rantes and Mcp‐1). Based on these results, we suggest that prolonged hepatic Pygl deficiency leads to an excessive buildup of hepatic glycogen that can increase the risk of liver damage, inflammation, and fibrosis (Fig. 6). Additional mechanistic studies are needed to understand the complete metabolic impact of defective glycogen metabolism on hepatic pathogenesis in GSD‐VI.
While the Pygl‐deficient mice appear to be an outstanding model for studying GSD‐VI in humans, it is important to note that several differences were identified. Growth retardation, presumably from chronic ketosis, is common in humans, but body weight differences were only identified in aged mice in this study. The severity of hypoglycemia is also worse in humans compared with this mouse model. In humans with GSD‐VI, hyperlipidemia is common, but this is not found in this model. Finally, results from initial fasting studies were similar to those seen in humans, but humans are much more prone to severe ketosis with prolonged fasting. One explanation for the discrepancy may result from the broad spectrum of residual liver glycogen phosphorylase activities in human GSD‐VI patients.2, 5, 9 in contrast to the complete absence of the enzyme activity in the Pygl −/− mouse. A second possibility is a differential dependency on alternative metabolic pathways to compensate low blood glucose. Indeed, GSD‐VI mice exhibit decreased serum levels of lactic acid, triglyceride, and cholesterol that can be used for gluconeogenesis in a prolonged fasting state. Additionally, the difference shown in serum metabolites might result from the varying fasting conditions to collect blood in clinical cases1, 2, 7 compared to the current study.
In summary, our newly generated GSD‐VI murine model faithfully recapitulates the features of human GSD‐VI (Fig. 6), including hepatic glycogen accumulation, ketotic hypoglycemia, and hepatic collagen deposition, and provides a model to elucidate the mechanisms underlying hepatic complications associated with defective glycogen metabolism.
Supporting information
Acknowledgment
We thank the KOMP Repository at the University of California, Davis, for collaboration and production efforts to generate the Pygl‐knockout mice. All funding support was managed by the foundations at the University of Connecticut Health Center and the University of Florida.
Supported by the Global Center for Glycogen Storage Disease (GSD) (to D.A.W.), the Ralph & Alice Brown Type VI GSD Research Fund (to D.A.W.), and the Crozet Crusader's Fund for GSD Research (to D.A.W.).
Potential conflict of interest: Dr. Weinstein received grants from Nestle/Vitaflo, Ultragenyx, and Moderna. The other authors have nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Burwinkel B, Bakker HD, Herschkovitz E, Moses SW, Shin YS, Kilimann MW. Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI. Am J Hum Genet 1998;62:785‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roscher A, Patel J, Hewson S, Nagy L, Feigenbaum A, Kronick J, et al. The natural history of glycogen storage disease types VI and IX: long‐term outcome from the largest metabolic center in Canada. Mol Genet Metab 2014;113:171‐176. [DOI] [PubMed] [Google Scholar]
- 3. Sutherland EW Jr, Wosilait WD. Inactivation and activation of liver phosphorylase. Nature 1955;175:169‐170. [DOI] [PubMed] [Google Scholar]
- 4. Roach PJ, Depaoli‐Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem J 2012;441:763‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang NL, Hui J, Young E, Worthington V, To KF, Cheung KL, et al. A novel mutation (G233D) in the glycogen phosphorylase gene in a patient with hepatic glycogen storage disease and residual enzyme activity. Mol Genet Metab 2003;79:142‐145. [DOI] [PubMed] [Google Scholar]
- 6. Chang S, Rosenberg MJ, Morton H, Francomano CA, Biesecker LG. Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI. Hum Mol Genet 1998;7:865‐870. [DOI] [PubMed] [Google Scholar]
- 7. Ogawa A, Ogawa E, Yamamoto S, Fukuda T, Sugie H, Kohno Y. Case of glycogen storage disease type VI (phosphorylase deficiency) complicated by focal nodular hyperplasia. Pediatr Int 2010;52:e150‐e153. [DOI] [PubMed] [Google Scholar]
- 8. Manzia TM, Angelico R, Toti L, Cillis A, Ciano P, Orlando G, et al. Glycogen storage disease type Ia and VI associated with hepatocellular carcinoma: two case reports. Transplant Proc 2011;43:1181‐1183. [DOI] [PubMed] [Google Scholar]
- 9. Davit‐Spraul A, Piraud M, Dobbelaere D, Valayannopoulos V, Labrune P, Habes D, et al. Liver glycogen storage diseases due to phosphorylase system deficiencies: diagnosis thanks to non invasive blood enzymatic and molecular studies. Mol Genet Metab 2011;104:137‐143. [DOI] [PubMed] [Google Scholar]
- 10. Beauchamp NJ, Taybert J, Champion MP, Layet V, Heinz‐Erian P, Dalton A, et al. High frequency of missense mutations in glycogen storage disease type VI. J Inherit Metab Dis 2007;30:722‐734. [DOI] [PubMed] [Google Scholar]
- 11. Testa G, Schaft J, van der Hoeven F, Glaser S, Anastassiadis K, Zhang Y, et al. A reliable lacZ expression reporter cassette for multipurpose, knockout‐first alleles. Genesis 2004;38:151‐158. [DOI] [PubMed] [Google Scholar]
- 12. Dagli AI, Weinstein DA. Glycogen storage disease type VI In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., eds. GeneReviews. Seattle, WA: University of Seattle; 2009, updated 2011. [Google Scholar]
- 13. Chou JY, Jun HS, Mansfield BC. Glycogen storage disease type I and G6Pase‐beta deficiency: etiology and therapy. Nat Rev Endocrinol 2010;6:676‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut 2015;64:830‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gjorgjieva M, Mithieux G, Rajas F. Hepatic stress associated with pathologies characterized by disturbed glucose production. Cell Stress 2019;3:86‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Derks TG, van Rijn M. Lipids in hepatic glycogen storage diseases: pathophysiology, monitoring of dietary management and future directions. J Inherit Metab Dis 2015;38:537‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernandes J, Pikaar NA. Ketosis in hepatic glycogenosis. Arch Dis Child 1972;47:41‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu KM, Wu JY, Chen YT. Mouse model of glycogen storage disease type III. Mol Genet Metab 2014;111:467‐476. [DOI] [PubMed] [Google Scholar]
- 19. Haagsma EB, Smit GP, Niezen‐Koning KE, Gouw AS, Meerman L, Slooff MJ. Type IIIb glycogen storage disease associated with end‐stage cirrhosis and hepatocellular carcinoma. The Liver Transplant Group. Hepatology 1997;25:537‐540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials