

Abstract
Each year, more than 25,000 people succumb to liver cancer in the United States, and this neoplasm represents the second cause of cancer‐related death globally. R‐spondins (RSPOs) are secreted regulators of Wnt signaling that function in development and promote tissue stem cell renewal. In cancer, RSPOs 2 and 3 are oncogenes first identified by insertional mutagenesis screens in tumors induced by mouse mammary tumor virus and by transposon mutagenesis in the colonic epithelium of rodents. RSPO2 has been reported to be activated by chromosomal rearrangements in colorectal cancer and overexpressed in a subset of hepatocellular carcinoma. Using human liver tumor gene expression data, we first discovered that a subset of liver cancers were characterized by high levels of RSPO2 in contrast to low levels in adjacent nontumor tissue. To determine if RSPOs are capable of inducing liver tumors, we used an in vivo model from which we found that overexpression of RSPO2 in the liver promoted Wnt signaling, hepatomegaly, and enhanced liver tumor formation when combined with loss of transformation‐related protein 53 (Trp53). Moreover, the Hippo/yes‐associated protein (Yap) pathway has been implicated in many human cancers, influencing cell survival. Histologic and gene expression studies showed activation of Wnt/β‐catenin and Hippo/Yap pathways following RSPO2 overexpression. We demonstrate that knockdown of Yap1 leads to reduced tumor penetrance following RSPO2 overexpression in the context of loss of Trp53. Conclusion: RSPO2 overexpression leads to tumor formation in the mouse liver in a Hippo/Yap‐dependent manner. Overall, our results suggest a role for Yap in the initiation and progression of liver tumors and uncover a novel pathway activated in RSPO2‐induced malignancies. We show that RSPO2 promotes liver tumor formation in vivo and in vitro and that RSPO2's oncogenic activity requires Hippo/Yap activation in hepatocytes. Both RSPO2 and YAP1 are suggested to represent novel druggable targets in Wnt‐driven tumors of the liver.
Abbreviations
- ACTB
actin beta
- APC
adenomatous polyposis coli
- AXN2
axis inhibitor protein 2
- Bcl2l1
B‐cell lymphoma‐2‐like protein 1
- Birc5
baculovirus inhibitor of apoptosis protein repeat containing 5
- Cas9
clustered regularly interspaced short palindromic repeats‐associated protein 9
- CNV
copy number variation
- CRC
colorectal cancer
- CT
threshold cycle
- CTNNB1
cadherin‐associated protein beta 1
- Fah
fumarylacetoacetate hydrolase
- GFP
green fluorescent protein
- GOI
gene of interest
- gRNA
guide RNA
- HCC
hepatocellular carcinoma
- HH
human hepatocyte
- IHC
immunohistochemistry
- LEF1
lymphoid enhancer factor 1
- LGR
leucine‐rich repeat‐containing G‐protein‐coupled receptor
- mRNA
messenger RNA
- NRG
nonobese diabetic/recombination activating gene
- NTBC
2‐(2‐nitro‐4‐trifluoromethylbenzoyl)‐1,3‐cyclohexanedione
- PHI
peptide histidine isoleucine
- qRT‐PCR
quantitative reverse‐transcriptase polymerase chain reaction
- RFP
red fluorescent protein
- RSPO
R‐spondin
- shRNA
short hairpin RNA
- TCF7
T‐cell factor 7
- TCGA
The Cancer Genome Atlas
- Trp53
transformation‐related protein 53
- Yap
yes‐associated protein
Hepatocellular carcinoma (HCC) remains one of the deadliest human carcinomas, with more than 40,000 diagnoses and 25,000 deaths each year in the United States alone.1, 2 At advanced stages of HCC, systemic therapies improve clinical outcomes, although the median overall survival remains a little more than 1 year.2, 3 It is therefore necessary to better understand the molecular pathways involved in HCC development and invasion in order to find novel strategies to treat such tumors. Moreover, chemotherapy, radiation, and surgery have limited effects in the treatment of HCC. More than a third of human HCCs exhibit gene expression profiles consistent with Wnt signaling activation, with cadherin‐associated protein beta 1 (CTNNB1)‐activating mutations occurring in approximately one third of these cases.2, 3 However, multiple strategies targeting β‐catenin in the Wnt pathway have had limited success in clinical trials2 because of toxicity caused by systemic off‐target effects.4, 5 It remains possible that some alterations in Wnt pathway regulators are better drug targets as well as other downstream Wnt effectors.6
R‐spondins (RSPOs) are secreted proteins that bind leucine‐rich repeat‐containing G‐protein‐coupled receptor (LGR) 4/5/6 transmembrane receptors and enhance Wnt signaling by means of Frizzled receptors.7 Sustained Wnt signaling by RSPO/LGR interaction is through inhibition of a negative feedback loop by causing membrane clearance of zinc and ring finger 3 (ZNRF3), resulting in enhanced Wnt signaling.8, 9, 10 Recurrent rearrangements that result in RSPO2 and RSPO3 overexpression were identified in 4% to 10% of human colorectal cancer (CRC) cases, mutually exclusive of adenomatous polyposis coli (APC) mutations.11, 12 Sleeping Beauty transposon‐based insertional mutagenesis screens performed in APC wild‐type mice identified Rspo2 as a common insertion site in gastrointestinal tumors, and Rspo2 and Rspo3 are common insertions sites in mouse mammary tumor virus‐driven breast cancer models.13, 14, 15, 16, 17, 18 Targeting RSPO2 can inhibit cancer by promoting differentiation, which reduces proliferation and stemness.19, 20, 21 These studies support the hypothesis that RSPOs act as oncogenes that activate canonical Wnt signaling. On the other hand, recent studies have shown interactions between the Wnt and Hippo signaling pathways because the yes‐associated protein (YAP) can be a substrate for the APC destruction complex.22 The Hippo/YAP pathway has also been shown to be necessary for liver regeneration and control of organ size and hepatocyte differentiation, leading to HCC regression.23, 24 The Hippo pathway's main effector, the YAP/tafazzin (TAZ) complex, promotes proliferation and inhibits apoptosis when translocated to the nucleus, where it acts as a transcriptional coactivator.22 Because of these molecular interactions and Hippo/Yap physiological functions in the liver, we hypothesized that RSPO2 activates both Wnt/β‐catenin and Hippo/Yap signaling pathways and that YAP is necessary for development of RSPO2‐driven HCC.
In this study, we provide in vitro and in vivo models of RSPO2‐driven HCC/hepatocyte transformation and show strong dual canonical Wnt and Hippo pathway activation. We show that RSPO2 is overexpressed in a subset of human HCC, it activates the Wnt and Hippo pathways, and abrogation of YAP inhibits RSPO2‐driven liver tumor formation. Both RSPO2 and YAP1 represent potential druggable targets against a subset of HCCs, opening a new avenue to treating some cancers characterized by activation of Wnt/β‐catenin signaling.
Materials and Methods
Acquisition of RNA Sequencing and Somatic Mutation Data from the Cancer Genome Atlas
RNA sequencing (RNA‐seq) and somatic mutation data were extracted from The Cancer Genome Atlas (TCGA) data matrix. RNA‐seq normalized counts were obtained for a set of 69 genes of interest related to RSPOs, Wnt signaling, and tissue‐specific markers of differentiation and stemness for 50 normal livers and 200 with HCC. Somatic mutation data were obtained for APC, CTNNB1, RSPO1, RSPO2, RSPO3, and RSPO4 from 200 HCC livers.
Primary Liver Tissues and Microarray Gene Expression Analysis
Gene expression profiling of 319 primary HCC samples, 199 cirrhotic or premalignant samples, and 23 normal liver samples were analyzed for this study. HCC samples were classified according to described subtypes.25 Five gene expression classes were defined by hierarchical clustering for a training set of HCCs. Marker genes were identified that were differentially expressed in each class (CTNNB1, interferon, proliferation, polysomy of chromosome 7, and unannotated). Expression of marker genes was used to classify subsequent HCC samples.
Tissue Culture Reagents and Cell Lines
The human hepatocyte (HH)7 cell line was kindly donated.26 HH7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 5% nonessential amino acids, glutamine, and 1% penicillin/streptomycin. All cells were grown on tissue culture‐treated plates at 37°C and 5% CO2.
In Vitro Gene Knockdown and Overexpression and Proliferation Assays
For overexpression experiments, lentiviral expression vectors were cloned with RSPO2 or Discosoma sp. red fluorescent protein (DsRed) regulated by an AAV Blank Control Vector promoter and followed by an internal ribosome entry site‐red fluorescent protein (RFP) to monitor transduction efficiency. Lentiviral particles were produced in 293T cells by cotransfection with helper plasmids. For both knockdown and overexpression experiments, viral supernatant was collected after 24 hours of virus production, cleared, and applied to transduce experimental cells with 12 μg/mL polybrene overnight. Transduced cells were selected with 1 μg/mL puromycin. Knockdown efficiency and overexpression levels were assayed by quantitative reverse‐transcriptase polymerase chain reaction (qRT‐PCR).
Mouse Strains, Hydrodynamic Injection, and Liver Analysis
All animal work was conducted according to an institutionally approved animal welfare protocol. Mouse strains and hydrodynamic injection protocols were as described.27, 28 Briefly, doubly transgenic mice (fumarylacetoacetate hydrolase [Fah]−/−; Rosa26‐SB11) received 20 μg plasmid DNA per plasmid by hydrodynamic tail vein injection. Plasmid DNA encoded transposon‐based sequences expressing (1) Fah, green fluorescent protein (GFP), luciferase, and either RSPO2, CTNNB1‐S33Y, or GFP control; and (2) transformation‐related protein 53 (Trp53) short hairpin RNA (shRNA), shYap1 shRNA, or no plasmid control. Experimental cohort details are listed in Supporting Table S2. Construct shYap1 was selected based on knockdown efficiency in vitro, as depicted in Supporting Fig. S6. Mice were maintained on 2‐(2‐nitro‐4‐trifluoromethylbenzoyl)‐1,3‐cyclohexanedione (NTBC)‐supplemented drinking water until after injections. At the indicated time points, mice were euthanized, and lungs, hearts, spleens, pancreases, livers, and all abnormal tissues were removed and visually inspected for macroscopic tumors. Macroscopic hyperplastic nodules were counted. Nodules at least 1 mm in size were isolated and snap frozen for RNA extraction. Larger nodules were fixed in 10% formalin, paraffin‐embedded, mounted, and sectioned at 5 µm. Slides were processed and stained with hematoxylin and eosin. Brightfield images of hepatic parenchyma were obtained using an Olympus BX45 microscope equipped with an Olympus DP70 camera and analyzed by two board‐certified pathologists (K.A. and M.L., American Board of Pathology). A reticulin stain was performed on select blocks using a Leica automated stainer. Immunohistochemistry (IHC) for CTNNB1 was performed using β‐catenin (6B3) rabbit monoclonal antibody (mAb) (#9582; Cell Signaling Technology) at 1:100.29 CTNNB1 localization and intensity were scored by two independent blinded reviewers (C.C. and B.T.). YAP staining was performed in the same way (H.H. and G.K.) using YAP (D8H1X) rabbit mAb (#1074; Cell Signaling) at 1:400.
qRT‐PCR
RNA was isolated from cell lines and mouse liver tissues using the PureLink RNA Mini Kit (Ambion). RNA samples were analyzed by gel electrophoresis to assess quality and treated with deoxyribonuclease to remove contaminating genomic DNA (Turbo DNA‐free Kit; Ambion). Complementary DNA (cDNA) was synthesized from 1 μg template RNA per sample using random hexamer primers (SuperScript III First‐Strand Synthesis System; Invitrogen). qRT‐PCR reactions were conducted with FastStart Universal SYBR Green Master Mix (Roche) using 0.5 μL cDNA template per 25 μL reaction. Primer sequences for qRT‐PCR reactions are listed in Supporting Table S3. Data were normalized to actin beta (ACTB) using the following equation: relative expression = 2CT_ACTB/2CT_GOI.
Oncogenic Potential In Vitro Studies of RSPO2 in HH7 Cells
Soft‐agar assays were performed using 0.48% low melting point agarose in sterile water. A total of 7,500 cells were plated per well in a six‐well plate. Pictures were taken in a dissecting microscope at 1.5× and analyzed using ImageJ. Xenograft models were performed in nonobese diabetic/recombination activating gene (NRG) mice, and 3 million cells were injected subcutaneously in media containing Matrigel (1:1). Tumors were harvested 4 months after injection. Cells were lysed with a hypotonic solution, and nuclear fractions were isolated using Nonidet P40 followed by centrifugation. Pellets were resuspended in radio immunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors.
CRISPR/Cas9 Knockout of Yap1
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR‐associated protein 9 (Cas9) HH7‐RSPO2 Yap1 modified cell lines were generated using lentiviral vectors expressing Cas9 and a guide RNA (gRNA) directed against Yap1. Lentiviral vectors were generated by transfecting 293T cells with two viral packaging plasmids and CC9 v2 Cas9/gRNA‐containing plasmid from the Zhang Lab at the Massachusetts Institute of Technology (https://zlab.bio/guide-design-resources). gRNA sequences were cloned into a stuffer region of the plasmid using BsmB1 restriction sites. gRNA sequences were 5′‐GGCGTAGCCCTCGCTCGC‐3′ and 5′‐CGGCGCTGTCCTCGCTCT‐3′ and targeted the translational start site.
Results
RSPO2 is Up‐Regulated in Human Liver Cancer and is Associated with an Activated Wnt Signaling Gene Expression Profile
To characterize the role of RSPOs in HCC, we first analyzed RSPO2/3 messenger RNA (mRNA) levels and gene copy number variation (CNV) in two HCC cohorts (training and validation), including 255 HCC, 31 adjacent cirrhotic tissue, and 10 normal liver samples. Microarray analysis displayed uniformly low RSPO2 mRNA levels in normal and cirrhotic livers but significantly higher RSPO2 levels in the molecular class of HCC characterized by activated Wnt signaling and CTNNB1 mutations30 (P < 0.01; Fig. 1A,B; Supporting Fig. S1C). In addition, chromosomal high‐level amplifications ([cHLAs]; defined as copy number gains >3.8) involving RSPO2 were identified in 8% of the tumor samples but not seen in adjacent cirrhotic/nontumor tissues (P < 0.001; Fig. 1C). RPSO2 cHLAs were significantly enriched in the CTNNB1 class of HCC (P < 0.001; Fig. 1D,E). However, RSPO2 mRNA level and gene copy number were not significantly correlated (Spearman r = 0.10), suggesting that overexpression of RSPO2 is not entirely driven by CNVs.
Figure 1.

RSPO2 is up‐regulated in HCC and associated with Wnt signaling activation. (A) RSPO2 mRNA levels in normal liver, adjacent nontumor/cirrhotic liver, and HCC tumor samples (training cohort; n = 134). Box plots display percentiles 5% to 95%, with 50th percentile lines. (B) RSPO2 mRNA levels in nontumor/Ci liver and HCC grouped by molecular subclass, as defined in a previous study.25 (C) Estimated RSPO2 gene copy number in HCC and adjacent nontumor/Ci liver. (D) Estimated RSPO2 gene copy number in HCC by molecular subclass; *P < 0.05, **P < 0.01, ***P < 0.001. (E) Wnt target gene mRNA levels in nontumor/Ci liver versus HCC with low and high RSPO2 levels in the training cohort. Abbreviations: Ci, cirrhotic; NS, not significant.
Next, we assessed expression of Wnt target genes and enrichment of Wnt signatures in HCC. Tumors that expressed high levels of RSPO2 had significantly elevated expression of Wnt target genes (axis inhibitor protein 2 [AXIN2], lymphoid enhancer factor 1 [LEF1], T‐cell factor 7 [TCF7], and LGR5) compared to normal liver or tumors with low RSPO2 levels (Fig. 1E). Moreover, gene set enrichment analysis showed that Wnt‐related signatures were significantly enriched in samples with increased copy number of RSPO2, similar to samples with CTNNB1 mutation (false discovery rate q value, <25%; Supporting Table S1). Indeed, CTNNB1 mutation was associated with increased RSPO2 mRNA expression and copy number, indicating these events tended to co‐occur (Supporting Fig. S1E).
RSPO3 mRNA levels were decreased in HCC overall and in all subclasses compared to normal, cirrhotic, or premalignant samples (P < 0.001; Supporting Fig. S1A). Low RSPO3 transcript levels were accompanied by RSPO3 gene copy number losses, which were identified in 25% of the tumors but absent in the nontumor tissue (P < 0.001; Fig. 1E; Supporting Fig. S1B).
These findings were confirmed by analysis of gene expression data from an additional 200 liver tumors and 50 normal liver samples (TCGA data set). RSPO2 was overexpressed in HCC overall, with 12% of samples having more than 4‐fold increased RSPO2 mRNA level compared to normal liver (Supporting Fig. S2C). RSPO2‐high status was associated with CTNNB1 mutation (P = 1.72E‐09) and elevated expression of Wnt target genes (AXIN2, TCF7, LEF1, LGR5; Supporting Fig. S2A,B). RSPO3 mRNA expression was suppressed in HCCs overall, and only one sample (0.5%) was identified as RSPO3 high (Supporting Figs. S2A and S6).
Regarding the mechanism of RSPO2 overexpression, deFuse analysis of RSPO‐high liver tumors in the TCGA set did not identify expression of RSPO2 or RSPO3 gene fusions. Further, in a murine model of HCC driven by activating a mutation of β‐catenin (CTNNB1‐S33Y), Rspo2 was not overexpressed (Supporting Fig. S3). These results suggest that, although high RSPO2 expression co‐occurs with CTNNB1 mutation in liver cancer, elevated RSPO2 expression is not merely a consequence of Wnt signaling activated by CTNNB1 but rather an independently selected event.
RSPO2 Promotes Wnt Signaling and Tumorigenesis in the Mouse Liver
To study the role of elevated RSPO2 expression in liver cancer, we next generated mice that somatically expressed RSPO2, alone or with knockdown of Trp53, by hydrodynamic injection and Fah selection. The Fah mouse model lacks the Fah gene product, the last step in the metabolism of tyrosine. These mice accumulate hepatotoxic metabolites. The mice are maintained with NTBC, a hepatoprotective drug that inhibits tyrosine metabolism upstream of Fah. This model takes advantage of this by rescuing Fah, along with our gene of interest, effectively repopulating the mouse liver with humanized hepatocytes. Control mice were injected with a transposon vector expressing Fah, GFP, and luciferase to track liver repopulation. Experimental mice were injected with a transposon vector expressing Fah, GFP, luciferase, and RSPO2, and additional GFP and RSPO2 cohorts were injected with a second transposon vector expressing Trp53 shRNA (Fig. 2A). Necropsies were performed to assess liver mass and tumor formation and analyzed at the molecular level. Hepatic expression of GFP was confirmed at necropsy by visualization with GFP goggles. At early time points (day 15 to 25 after hydrodynamic injection of peptide histidine isoleucine [PHI]), mice injected with RSPO2 had slightly enlarged livers compared to GFP controls (1.2‐fold to 1.5‐fold; P > 0.05). At later time points, there was a prominent phenotype of enlarged liver (hepatomegaly) in mice injected with RSPO2 with or without Trp53 shRNA compared to GFP controls (1.6‐fold to 2.2‐fold enlarged; P < 0.01), whereas mice injected with Trp53 shRNA alone did not develop enlarged livers (Fig. 2B,C). To characterize the enlarged liver phenotype, sections of grossly normal liver were stained with 4ʹ,6‐diamidino‐2‐phenylindole to quantify nuclear density. Mice injected with RSPO2 or RSPO2 and Trp53 shRNA had 30% to 40% increased nuclear density compared to control mice receiving GFP or GFP and Trp53 shRNA, suggesting enlarged livers in these mice arose due to hyperplastic growth (Fig. 2D,E).
Figure 2.
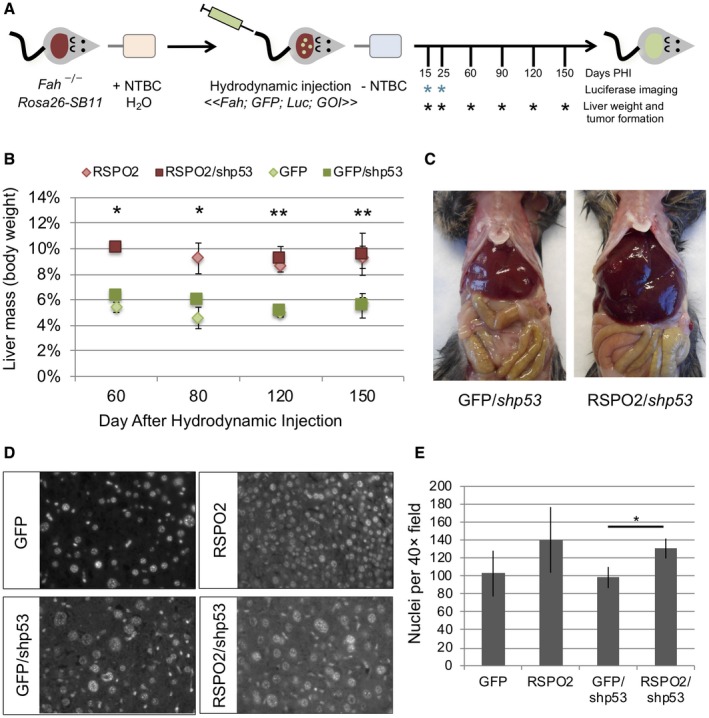
RSPO2 expression drives hyperplastic growth in the mouse liver. (A) Experimental diagram. (B) Average liver mass was significantly increased in mice injected with RSPO2 alone or in combination with Trp53 shRNA (shp53) compared to GFP or GFP/shp53 control mice at all time points 60 to 150 days PHI; *P < 0.05. (C) Representative necropsy images illustrating hepatomegaly of mice injected with RSPO2. (D) Sections of grossly normal liver from mice at 150 days after hydrodynamic injection were stained with DAPI to visualize nuclear density at 40× magnification. (E) Nuclei per 40× field were quantified for six livers per group. Nuclear density increased 30% to 40% with RSPO2 expression, consistent with hyperplastic growth. Bars show mean + SD. *P < 0.05, **P < 0.01. Abbreviations: DAPI, 4ʹ,6‐diamidino‐2‐phenylindole; Luc, luciferase.
At 150 days PHI, mice injected with GFP or GFP and Trp53 shRNA had low tumor penetrance (4%‐5%), consistent with the background level of tumor formation in this model (Fig. 3A). Mice injected with RSPO2 alone had a nonsignificant increase in tumor penetrance (18%). However, expression of RSPO2 combined with Trp53 shRNA dramatically increased tumor penetrance (63% of RSPO2/Trp53 shRNA mice vs. 5.2% of GFP/Trp53 shRNA mice; P = 3.37E‐5; Fig. 3A). RSPO2/Trp53 shRNA‐injected mice formed 1.8 tumors/mouse on average (range, 1‐5). Histologic examination determined the tumors were HCCs with areas of necrosis and lymphocytic infiltration (Fig. 3B,C).
Figure 3.
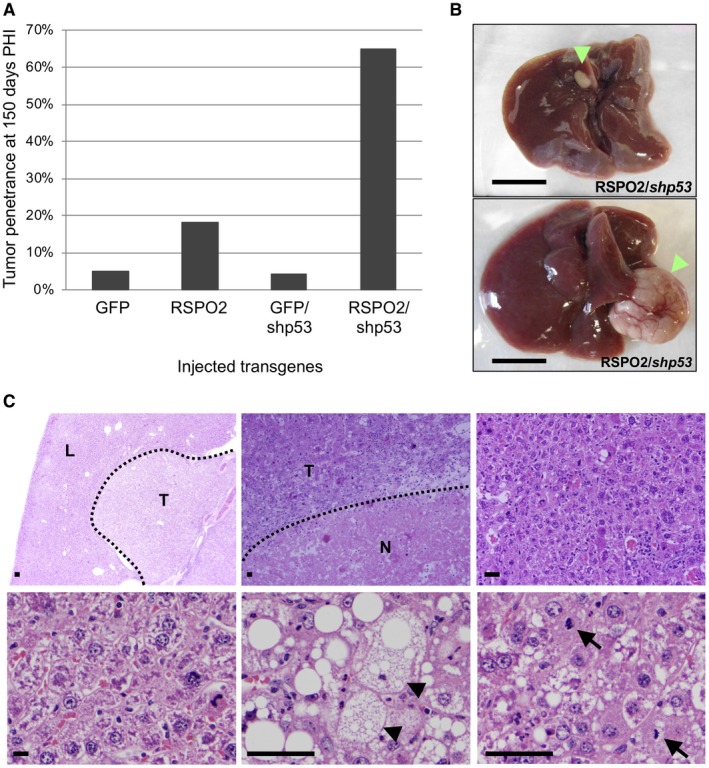
RSPO2 promotes tumor formation in the mouse liver. (A) Tumor penetrance in Fah‐null mice injected with GFP (18 animals), RSPO2 (19 animals), GFP plus shRNA against Trp53 (GFP/shp53) (20 animals), or RSPO2 plus shRNA against Trp53 (RSPO2/shp53) (19 animals) at 150 days PHI (P = 3.37E‐5). (B) Gross images of representative livers from RSPO2/shp53‐injected mice at 150 days PHI. Tumors are indicated by green arrowheads; scale bar, 1 cm. (C) Representative hematoxylin and eosin‐stained sections of RSPO2/shp53‐injected mouse liver and tumor. Upper left, low‐power view of liver nodule; magnification ×2. Upper middle, hepatocellular carcinoma with lymphoid infiltrate and necrosis; magnification ×10. Upper right, dysplastic liver parenchyma; magnification ×10. Lower left, high‐power view of dysplastic liver parenchyma; magnification ×50. Lower middle, hepatocellular carcinoma with clear cell change, indicated by black arrowheads; magnification ×50. Lower right, high‐power view of hepatocellular carcinoma with mitotic figures, indicated by black arrows; magnification ×50. Scale bars, 100 μm. Abbreviations; L, liver; N, necrosis; T, tumor.
Expression of RSPO2 and its effect on Wnt signaling were assessed by qRT‐PCR and IHC. Expression of RSPO2 alone compared to GFP‐injected controls increased expression of Wnt target genes (Axin2, Tcf7, and Lgr5) at early time points (days 15‐25 PHI) (Supporting Fig. S4). At day 150 PHI, expression of Wnt target genes and nuclear localization of CTNNB1 were significantly increased in both livers and tumors from mice injected with RSPO2 and Trp53 compared to GFP/Trp53 shRNA‐injected controls (Fig. 4A‐C). These data confirm that RSPO2 expression enhances Wnt/β‐catenin signaling and promotes HCC formation in the mouse liver.
Figure 4.

RSPO2 increases Wnt signaling in the mouse liver. (A,B) Expression of (A) RSPO2 and (B) endogenous Wnt target genes were assessed by qRT‐PCR in grossly normal liver and tumor samples from mice injected with GFP/Trp53 shRNA or RSPO2/Trp53 shRNA. Wnt target genes were significantly elevated in murine liver and tumors expressing RSPO2. Data points represent gene expression value per animal. Lines represent the mean. (C) β‐catenin protein expression and localization were examined by IHC staining in normal liver and tumor samples from mice injected with GFP/Trp53 shRNA or RSPO2/Trp53 shRNA. Cytoplasmic and nuclear expressions of β‐catenin were elevated in tumors from RSPO2/shp53‐injected mice. Bars represent mean + SD; *P < 0.05.
The Hippo/YAP Pathway is Activated Following RSPO2 Signaling in the Mouse Liver
Recent studies have shown the Hippo pathway interacts with Wnt signaling by Yap binding to the β‐catenin destruction complex and that it is required for CRC cell survival.21, 22 Based on the pronounced phenotype of hepatomegaly in liver overexpressing RSPO2, we tested the effect of RSPO2 overexpression on Yap nuclear localization as a measurement of Hippo activation. We stained liver tissue and scored each section for Yap nuclear and cytoplasmic localization. We assessed Yap staining qualitatively and scored each field of view using 0 (no staining), 1 (low), 2 (medium), and 3 (high) (Fig. 5A). We found that, following RSPO2 overexpression, Yap nuclear localization and overall levels were increased (Fig. 5B). Further, RNA was isolated from normal and tumor liver tissue from the Fah/RSPO2/shp53 mice described above and quantified by qRT‐PCR. We found RSPO2 overexpression in the mouse liver results in overexpression of the Yap target gene B‐cell lymphoma‐2‐like protein 1 (Bcl2l1) in RSPO2‐high and tumor tissue (Fig. 5C). On the other hand, we saw a significant overexpression of the Yap target gene baculovirus inhibitor of apoptosis protein repeat containing 5 (Birc5) in RSPO2 tumor tissue (Fig. 5D).
Figure 5.

RSPO2 overexpression in the mouse liver results in Hippo signaling activation. RPSO2 overexpression in the Fah mouse model results in an increased expression of the Hippo/Yap target genes. (A) Mouse liver sections stained for Yap. Bottom panels show increased levels of expression and nuclear translocation in RSPO2 overexpression livers. Scale bars, 100 μm. (B) Quantification of (A) based on IHC scoring described in the text; each data point represents an individual animal. (C) Bcl2l1. (D) Birc5. Black lines represent SD and *P < 0.05. Abbreviation: NS, not significant.
Knockdown of Yap1 Inhibits RSPO2‐Induced Tumor Formation in Mouse Liver
In order to determine if RSPO2‐driven liver tumors depend on YAP for development and maintenance, we hydrodynamically injected an expression vector encoding an shRNA versus Yap1 in the presence or absence of RSPO2 and/or shTrp53. Control mice were injected with a transposon vector expressing Fah, GFP, and luciferase to track liver repopulation. Experimental mice were injected with a transposon vector expressing Fah, GFP, luciferase, and RSPO2, and additional GFP and RSPO2 cohorts were injected with a second transposon vector expressing Trp53 shRNA, all containing a second and/or third vector expressing an shRNA against Yap1 (Fig. 6A). Necropsies were performed to assess tumor formation 150 days PHI. Yap1 knockdown led to a dramatic decrease in tumor penetrance, suggesting a direct role of Yap1 in RSPO2‐mediated tumorigenesis. Only one of nine (11%) experimental animals hydrodynamically injected with RPSO2, shTrp53, and shYap1 expression vectors formed tumors, whereas RSPO2/shp53‐injected mice again had 68% tumor penetrance (Fig. 6D,E). Yap1 knockdown was confirmed by IHC (Fig. 6B,C), with both cytoplasmic and nuclear stains showing reduced staining for Yap when the shRNA vectors were co‐injected.
Figure 6.

Loss of Yap1 results in decreased RSPO2‐driven tumor formation. (A) Experimental diagram. (B) IHC showing in vivo knockdown of Yap1 in the mouse liver. Scale bars, 100 μm. (C) Quantification of (B). Bar represents the mean of each cohort. (D) Tumor penetrance in Fah‐null mice injected with GFP/shYap1 (10 animals), RSPO2/shp53/shYap1 (9 animals), or RSPO2/shp53 (19 animals) at 150 days PHI (P < 0.05). Bars represent percent of animals that show tumor formation. (E) Two nodules were found in the RSPO2/shp53/shYap1. Only one animal showed tumor penetrance. Abbreviation: Luc, luciferase.
RSPO2 Overexpression in Immortalized HHs Results in Anchorage‐Independent Growth and Xenograft Tumor Formation
Next, we overexpressed RSPO2 in HH7 cells.26 We first evaluated the effects of RSPO2 overexpression in YAP nuclear localization in HHs. We observed in a western blot that levels of total and nuclear YAP protein were increased, accompanied by levels of β‐catenin in RSPO2‐high HH7 cells and posttranslational stabilization of YAP and β‐catenin in both cytoplasmic and whole‐cell fractions (Fig. 7A). We tested the oncogenic effects of RSPO2 in HH7 cells by measuring growth in soft agar and tumor size in xenografts in NRG mice. We found RSPO2 overexpression led to cellular transformation, including both anchorage‐independent growth and tumor formation in contrast to an RFP control (Fig. 7B‐D). These effects were abrogated by a gRNA targeting YAP1.
Figure 7.
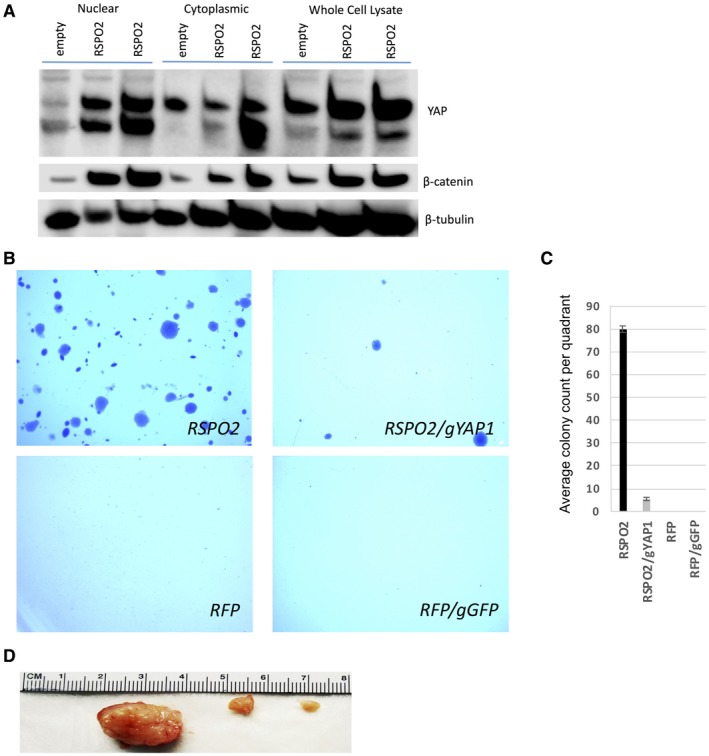
Ectopic expression of RSPO2 in HH7 cells was protumorigenic. (A) Western blots for YAP using nuclear, cytoplasmic, and whole‐cell lysates from control‐HH7 (RFP) cells and two replicates of RSPO2‐HH7 cells. Bands below YAP are nonspecific. (B) RSPO2 overexpression caused anchorage‐independent growth in soft agar. (C) Quantification of (B). Bars show mean + SD. (D) Tumors formed in the NRG xenograft model of RSPO2‐HH7 cells injected subcutaneously.
Discussion
RSPO2 has been described as an oncogene in CRC, and new therapeutic approaches are currently being tested. In fact, clinical trials have been developed using porcupine inhibitors to treat RSPO‐driven metastatic colorectal adenocarcinomas.25 Here, we describe an oncogenic role for RSPO2 in the liver, acting by means of both the Wnt/β‐catenin and Hippo/Yap pathways. Moreover, we show that YAP is necessary for RSPO2‐driven hepatocyte cell transformation in vitro and a downstream effector in vivo. This discovery uncovers an additional downstream mechanism that results in the survival of RSPO2‐high–CTNNB1 subtype HCC and implicates the Hippo/YAP pathway as a potential drug target in these HCCs.
Most human liver cancers maintain a low level of RSPO2, with a small subset having significantly elevated levels. These RSPO2‐high tumors are mainly found in the CTNNB1 subtype of HCC, as defined by Chiang et al.25 Wnt target genes are significantly up‐regulated in these RSPO2‐high liver cancers. The mechanism of RSPO2 overexpression remains an open question. Although RSPO2 overexpression co‐occurred with activating mutation of β‐catenin in human liver cancer, our murine model of HCC initiated by activated β‐catenin suggests these are independently selected events. Further, we were not able to detect gene fusions in RSPO2‐high liver cancers, as previously characterized in CRC.11 Chromosomal amplifications were detected at the RSPO2 locus in 8% of tumors; however, there was not a significant correlation with mRNA levels. Loss of function of a border element, such as by CCCTC‐binding factor (CTCF) mutation or DNA hypermethylation, could also be an epigenetic mechanism that results in the ectopic overexpression of RSPO2. We hypothesize that elevated RSPO2 expression is independent of activated Wnt/CTNNB1 signaling.
Using our in vivo liver cancer mouse model, we were able to directly test the role of RSPO2 in tumorigenesis. We found ectopic overexpression of RSPO2 with loss of Trp53 led to more tumors in mice compared to loss of Trp53 alone. The resulting tumors had elevated levels of RSPO2 and Wnt target genes as well as increased levels of nuclear and cytoplasmic Ctnnb1. These findings further indicate RSPO2 functions as an oncogene in liver cancer.29
Several groups have reported that Wnt and Hippo signaling pathways interact.22, 31 We found RSPO2 overexpression led to accumulation of cytoplasmic and nuclear Yap in the mouse liver. Yap target genes involved in the regulations of apoptosis, Bcl2l1, and Birc5, were up‐regulated in some RSPO2‐driven liver tumors. Hippo/YAP activation mechanisms are varied. As demonstrated by Azollin and colleagues,31 we hypothesize that Hippo signaling occurs by means of a Wnt activation mechanism and that, in the case of activating mutations in CTNNB1, Hippo signaling is enhanced by further inactivation of APC, stabilizing YAP posttranslationally. We also demonstrated YAP activation downstream of RSPO2 in immortalized HHs and found that ectopic RSPO2 expression led to anchorage‐independent growth and tumor formation in NRG mice. Interestingly, these transformation events were abrogated after the loss of YAP1.
Moreover, we have shown that knockdown of Yap1 in the mouse liver results in direct abrogation of RSPO2‐driven tumor development and maintenance. This finding opens new routes for the development of therapies against RSPO2‐driven cancer. The Hippo/Yap pathway is activated following lower density/size in the liver, and its inhibition has been shown to restore hepatocyte differentiations in HCC.23, 24 Activation of the pathway gives rise to cell proliferation and survival. Although Yap1 is necessary for liver regeneration and regulation of organ size,23 we demonstrate that Yap1 is stabilized at the posttranscriptional level following RSPO2 overexpression. Targeting of YAP would be a reasonable approach, versus RSPO2‐driven HCC, to promote apoptosis of tumor cells. Verteporfin and statins are being tested as inhibitors of the Hippo pathway where verteporfin inhibits the Yap‐TEA domain transcription factor 1 (TEAD) interaction, whereas statins act by inhibition of ras homolog family member A (RhoA) signaling.32 Inhibiting YAP may selectively target HCC tumor cells that depend on it for survival. Porcupine inhibitors are candidate drugs to target RSPO‐high tumors.33
Future studies are required to determine whether RSPO2 expression and downstream activation of canonical Wnt and Hippo pathways are required for tumor maintenance in RSPO2‐high cancers. Also unclear is the role of noncanonical Wnt signaling in RSPO2‐high cancers. In summary, we show that RSPO2 overexpression activates both canonical Wnt and Hippo signaling and promotes tumor formation in the liver. This raises the enticing possibility that both RSPOs and/or YAP could be used as biomarkers or therapeutic targets. Indeed, initial studies attempting therapeutic targeting of RSPOs are showing promise.19, 21 We have developed useful in vitro and in vivo model systems for the testing of targeting RSPOs or combination therapies with Wnt and/or Hippo/YAP inhibitors.
Supporting information
Acknowledgment
We thank Susan K. Rathe for helping with the review of the manuscript, the University of Minnesota Supercomputing Institute for providing computing resources, and the University of Minnesota Genomics Center for providing Sanger sequencing and primer synthesis services.
Supported by the National Institutes of Health (grants T32 GM008244 and F30 CA171547 to C.B.C.; T32 GM008244 and T32 HL007741 to G.V.R.; T32 AI083196‐04 to B.R.T.; R01 CA134759, R01 CA113636 to D.A.L.; 5R00CA151672‐03, P30‐CA77598 to T.K.S.; CA150272P3, HEPCAR 667273‐2, SAF 2016‐76390 to J.M.L.), the American Cancer Society (ACS) Research Professor Award (#123939 to D.A.L.); the Mezin‐Koats Colon Cancer Research Fund (ACS PF‐06‐282‐01‐MGO to T.K.S.), and the MN Colorectal Cancer Research Foundation (to T.K.S.).
The results published in this manuscript are in part based on data generated by The Cancer Genome Atlas Research Network (http://cancergenome.nih.gov).
Potential conflict of interest: Dr. Largaespada consults for, advises, and is employed by Biotechne; he consults for, advises, owns stock in, and is employed by Recombinetics; he consults for, advises, and owns stock in Luminary Therapeutics; he owns stock in NeoClone Biotechnologies and ImmuSoft and received grants from Genentech. Dr. Llovet consults for and received grants from Bayer, Blueprint, Bristol‐Myers Squibb, EisaI Inc., and Ipsen; he consults for Eli Lilly, Celsion, Exelixis, Merck, Gloycotest, Navigant, Leerink, Midatech, Fortress Biotech, Spring Bank, Nucleix, and Can Fite and received grants from Incite. The other authors have nothing to report.
References
- 1. Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 2009;27:1485‐1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lachenmayer A, Alsinet C, Savic R, Cabellos L, Toffanin S, Hoshida Y, et al. Wnt‐pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin Cancer Res 2012;18:4997‐5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy‐number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012;44:694‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov 2006;5:997‐1014. [DOI] [PubMed] [Google Scholar]
- 5. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov 2014;13:513‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 2018;15:599‐616. [DOI] [PubMed] [Google Scholar]
- 7. Ugolini F, Charafe‐Jauffret E, Bardou VJ, Geneix J, Adelaide J, Labat‐Moleur F, et al. WNT pathway and mammary carcinogenesis: loss of expression of candidate tumor suppressor gene SFRP1 in most invasive carcinomas except of the medullary type. Oncogene 2001;20:5810‐5817. [DOI] [PubMed] [Google Scholar]
- 8. Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, et al. ZNRF3 promotes Wnt receptor turnover in an R‐spondin‐sensitive manner. Nature 2012;485:195‐200. [DOI] [PubMed] [Google Scholar]
- 9. Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, et al. Tumour suppressor RNF43 is a stem‐cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012;488:665‐669. [DOI] [PubMed] [Google Scholar]
- 10. Wu C, Qiu S, Lu L, Zou J, Li WF, Wang O, et al. RSPO2‐LGR5 signaling has tumour‐suppressive activity in colorectal cancer. Nat Commun 2014;5:3149. [DOI] [PubMed] [Google Scholar]
- 11. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R‐spondin fusions in colon cancer. Nature 2012;488:660‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shinmura K, Kahyo T, Kato H, Igarashi H, Matsuura S, Nakamura S, et al. RSPO fusion transcripts in colorectal cancer in Japanese population. Mol Biol Rep 2014;41:5375‐5384. [DOI] [PubMed] [Google Scholar]
- 13. Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, et al. A transposon‐based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009;323:1747‐1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeda H, Wei Z, Koso H, Rust AG, Yew CC, Mann MB, et al. Transposon mutagenesis identifies genes and evolutionary forces driving gastrointestinal tract tumor progression. Nat Genet 2015;47:142‐150. [DOI] [PubMed] [Google Scholar]
- 15. Lowther W, Wiley K, Smith GH, Callahan R. A new common integration site, Int7, for the mouse mammary tumor virus in mouse mammary tumors identifies a gene whose product has furin‐like and thrombospondin‐like sequences. J Virol 2005;79:10093‐10096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, Jonkers J, et al. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet 2007;39:759‐769. [DOI] [PubMed] [Google Scholar]
- 17. March HN, Rust AG, Wright NA, ten Hoeve J, de Ridder J, Elridge M, et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet 2011;43:1202‐1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Starr TK, Scott PM, Marsh BM, Zhao L, Than BL, O'Sullivan MG, et al. A Sleeping Beauty transposon‐mediated screen identifies murine susceptibility genes for adenomatous polyposis coli (Apc)‐dependent intestinal tumorigenesis. Proc Natl Acad Sci U S A 2011;108:5765‐5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Storm EE, Durinck S, de Sousa e Melo F, Tremayne J, Kljavin N, Tan C, et al. Targeting PTPRK‐RSPO3 colon tumours promotes differentiation and loss of stem‐cell function. Nature 2016;529:97‐100. [DOI] [PubMed] [Google Scholar]
- 20. Fischer MM, Yeung VP, Cattaruzza F, Hussein R, Yen WC, Murriel C, et al. RSPO3 antagonism inhibits growth and tumorigenicity in colorectal tumors harboring common Wnt pathway mutations. Sci Rep 2017;7:15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chartier C, Raval J, Axelrod F, Bond C, Cain J, Dee‐Hoskins C, et al. Therapeutic targeting of tumor‐derived R‐spondin attenuates β‐catenin signaling and tumorigenesis in multiple cancer types. Cancer Res 2016;76:713‐723. [DOI] [PubMed] [Google Scholar]
- 22. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, et al. β‐catenin driven cancers require a YAP1 transcriptional complex for survival and tumorgenesis. Cell 2012;151:1457‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao B, Li L, Lei Q, Guan KL. The Hippo‐YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 2010;24:862‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fitamant J, Kottakis F, Benhamouche S, Tian HS, Chuvin N, Parachoniak CA, et al. YAP inhibition restores hepatocyte differentiation in advanced HCC, leading to tumor regression. Cell Rep 2015;10:1692‐1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 2008;68:6779‐6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clayton RF, Rinaldi A, Kandyba EE, Edward M, Willberg C, Klenerman P, et al. Liver cell lines for the study of hepatocyte function and immunological response. Liver Int 2005;25:389‐402. [DOI] [PubMed] [Google Scholar]
- 27. Bell JB, Podetz‐Pedersen KM, Aronovich EL, Belur LR, McIvor RS, Hackett PB. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Protoc. 2007;2:3153‐3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keng VW, Tschida BR, Bell JB, Largaespada DA. Modeling hepatitis B virus X‐induced hepatocellular carcinoma in mice with the Sleeping Beauty transposon system. Hepatology 2011;53:781‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yin X, Yi H, Wang L, Wu W, Wu X, Yu L. R‐spondin 2 promotes proliferation and migration via the Wnt/β‐catenin pathway in human hepatocellular carcinoma. Oncol Lett 2017;14:1757‐1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin A, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603‐607. Erratum in: Nature 2019;565:E5‐E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, et al. YAP/TAZ incorporation in the β‐catenin destruction complex orchestrates the Wnt response. Cell 2014;158:157‐170. [DOI] [PubMed] [Google Scholar]
- 32. Tanaka K, Osada H, Murakami‐Tonami Y, Horio Y, Hida T, Sekido Y. Statin suppresses Hippo pathway inactivated malignant mesothelioma cells and blocks the YAP/CD44 growth stimulatory axis. Cancer Lett 2017;385:215‐224. [DOI] [PubMed] [Google Scholar]
- 33. Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Targeting Wnt‐driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A 2013;110:20224‐20229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials