

Abstract
We report ion mobility spectrometry and mass spectrometry studies of the non-enzymatic step-by-step degradation of substance P (subP), an eleven-residue neuropeptide, with the sequence Arg1-Pro2-Lys3-Pro4-Gln5-Gln6-Phe7-Phe8-Gly9-Leu10-Met11-NH2, in ethanol. At elevated solution temperatures (55 to 75 °C) several reactions are observed, including: a protonation event, i.e., [subP+2H]2+ + H+ → [subP+3H]3+, that appears to be regulated by a configurational change; and, two sequential bond cleavages (the Pro2-Lys3 peptide bond is cleaved to form the smaller nonapeptide Lys3-Met11-NH2 [subP(3–11)], and subsequently, subP(3–11) is cleaved at the Pro4-Gln5 peptide bond to yield the heptapeptide Gln5-Met11-NH2 [subP(5–11)]). Each of the product peptides [subP(3–11) and subP(5–11)] is accompanied by a complementary diketopiperazine (DKP): cyclo-Arg1-Pro2 (cRP) for the first cleavage, and cyclo-Lys3-Pro4 (cKP) for the second. Insight about the mechanism of degradation is obtained by comparing kinetics calculations of trial model mechanisms with experimental data. The best model of our experimental data indicates that the initial cleavage of subP is regulated by a conformational change, likely a trans→cis isomerization of the Arg1-Pro2 peptide bond. The subP(3–11) product has a long lifetime (t½ ~ 30 hrs at 55 °C) and appears to transition through several structural intermediates prior to dissociation, suggesting that subP(3–11) is initially formed with a Lys3-trans-Pro4 peptide bond configuration and that slow trans→cis isomerization regulates the second bond cleavage event as well. From these data and our model mechanisms, we obtain transition state thermochemistry ranging from ΔH‡ = 41 to 85 kJ·mol−1 and ΔS‡ = −43 to −157 J·mol−1·K−1 for each step in the reaction.
Keywords: ion mobility spectrometry-mass spectrometry, peptide conformation, dissociation kinetics, proline isomerization, penultimate proline
Graphical Abstract
Introduction
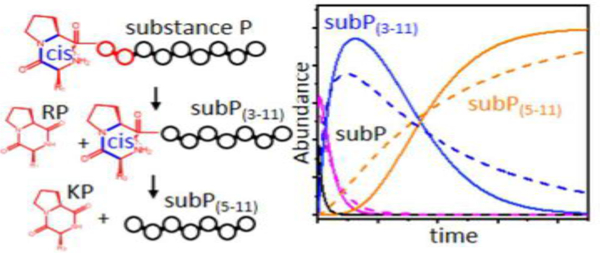
Substance P (subP), an undecapeptide with the sequence Arg1-Pro2-Lys3-Pro4-Gln5-Gln6-Phe7-Phe8-Gly9-Leu10-Met11-NH2, was discovered in 1931 by Euler & Gaddum[1] and was among the first known neurotransmitters. This neuropeptide is found throughout the nervous system as well as in the smooth-muscle tissue of the gastrointestinal tract.[2] It is a member of the tachykinin[3] family of molecules that are associated with many functions, including: stimulation of smooth-muscle tissue, inflammation, pain, and regulation of blood pressure. Early studies of subP noted its instability, even when stored at low temperatures.[4, 5] A number of processes may be responsible for the loss of function. At mM concentrations subP can form oligomers that are associated through hydrophobic residues near the C terminus.[6, 7] SubP is susceptible to methionine oxidation, which decreases biological activity.[8] Additionally, when stored for long times, subP degrades to form cyclic diketopiperazine (DKP) forms of the dipeptides Arg1-Pro2 and Lys3-Pro4.[9]
Here, we use a combination of ion mobility spectrometry (IMS) with mass spectrometry (MS) to examine key steps associated with the spontaneous dissociation of subP in heated ethanol solutions. Recently, this approach was used to study the degradation of the nonapeptide bradykinin.[10] When heated, in the absence of enzymes, bradykinin undergoes a spontaneous and highly-specific bond cleavage to produce the cyclo-Arg1-Pro2 DKP product. Upon monitoring the abundances of species that are resolved in the IMS-MS spectrum over time, we found evidence for at least five resolvable steps for this reaction, including a slow configurationally-coupled protonation. A similar protonation reaction was observed previously in the isomerization of polyproline-7.[11] In 1-propanol, polyproline-7 favors a singly-protonated polyproline-I helix, in which all of the peptide bonds are in the cis configuration. Upon dissolution into water the polyproline-I structure undergoes a cooperative cis→trans isomerization of all of the Pro-Pro peptide bonds, forming a doubly-protonated polyproline-II helix.[11] Although dissociation was not observed, this study reveals how the conformations of proline peptide bonds regulate protonation.
Below, we show that subP undergoes sequential bond cleavages, producing both cyclo-Arg1-Pro2 (cRP) and cyclo-Lys3-Pro4 (cKP) in the form of DKPs. Similar to bradykinin,[10] the first cleavage is preceded by a configurationally-coupled protonation event. While the second cleavage involves only one charge state, a long induction period prior to cleavage is observed. Comparisons of kinetics profiles for possible trial models with experimentally measured kinetics suggest the presence of several intermediates prior to cleavage to form cKP. Thermochemistry is derived for barriers associated with specific transition states. The overall free energy barriers for different steps associated with configurational changes and dissociation are similar; however, there is substantial variation associated with enthalpic and entropic contributions for different steps.
Spontaneous formation of DKP is not limited to subP and bradykinin.[10] Recombinant human growth hormone undergoes a similar reaction in the absence of enzymes.[12] DKP is observed as a side product in laboratory peptide synthesis and necessitates that certain precautions are taken to minimize its formation.[13–16] Studies on dipeptides and tripeptides have noted the tendency for these short sequences to cyclize to form DKPs, and often these transformations lead to sequence inversions.[17–19] Capasso et al. investigated DKP formation from the Ala1-Pro2-NH2 dipeptide and proposed a mechanism that involves an attack by the nucleophilic N-terminal amine on the electrophilic carbonyl carbon of the second residue, ultimately forming the DKP product.[20] This mechanism requires a cis-configured peptide bond between the first and second residues (e.g., Xxx1-cis-Xxx2, where Xxx is any amino acid) – the same configuration that we assigned to a critical intermediate for cleavage of the peptide bond of bradykinin.[10] Capasso also found that cyclization of an Ala1-Pro2-NH2 dipeptide has a significant pH dependence under more basic conditions, but was pH-independent in acidic conditions.[20] These results appear to be sequence dependent, as analysis of His1-Pro2-NH2 cyclization produces a more bell-shaped pH dependence profile.[21] In loss of cRP from bradykinin, there was no evidence that the concentration of acetic acid influenced the rate of dissociation.[10]
Each of the aforementioned peptides shares a common sequence motif – a penultimate proline. Because of the unique nature of the pyrrolidine ring of proline, a cis-configured peptide bond is uniquely accessible for proline residues, compared with any other naturally-occurring amino acids, which favor trans-configurations.[22–25] The presence of this amino acid residue in a polypeptide chain often introduces interesting structural features,[26–31] and interconversion of cis- and trans-configurations is a rate-limiting step in folding.[32–34] In the work presented below, the presence of multiple prolines in subP makes it possible to explore the step-by-step configurational changes that are associated with peptide processing – in this case providing a rare glimpse into the sequence of motions prior to bond cleavage.
Experimental
Peptide synthesis and sample preparation.
The peptide subP (≥95% purity) was obtained from Sigma Aldrich (St. Louis, MO, U.S.A.). The truncated subP(3–11) peptide, and several subP analogues involving a range of Pro→Ala substitutions, were synthesized in house, using standard FMOC solid-phase peptide synthesis carried out on an Applied Biosystems 433A Peptide Synthesizer (Applied Biosystems, Foster City, CA).[35] Each purified peptide was dissolved into pure ethanol solutions to concentrations of 500 µM and stored at −22 °C. To initiate the degradation process, these samples were diluted to 50 µM into solutions of ethanol and 0.5% acetic acid (by volume) and immediately incubated at specified temperatures (from 55 to 75 °C) using a water bath.
IMS-MS measurements.
The theory of IMS-MS[36–39] and instrument operation[40–42] have been described. Aliquots of the incubating samples were taken at regular intervals, cooled to room temperature, and quickly analyzed using a home-built IMS-MS instrument.[43] Ions were produced using a Nanomate (Advion, Ithica, NY) autosampler and nanospray ionization source that produces reproducible ion signals for IMS-MS analysis. Once formed, ions are accumulated in an ion funnel before being released as narrow packets into a 2 m drift tube filled with ~3.0 Torr He buffer gas. Ions traverse the drift tube region under the influence of a uniform electric field (~10 V·cm−1) where species of different sizes and shapes are separated by collisions with the He buffer gas. These mobility-separated ions exit the drift tube into a time-of-flight (TOF) mass spectrometer for mass-to-charge analysis.[44]
Determination of experimental collision cross sections.
Drift times are related to an ion’s average collision cross section (Ω) by the following equation:[36]
| (1) |
Equation 1 includes terms for ion charge (z), elementary charge (e), the masses of the ion (mI) and buffer gas (mB), Boltzmann’s constant (kb), temperature in kelvin (T), and the neutral number density of the buffer gas (N) at standard temperature and pressure. The experimentally controlled parameters are the electric field (E), the length of drift tube (L), and the buffer gas pressure (P).
Normalization of MS peak areas in order to determine relative solution abundances.
The experiments described below were carried out over a period of two years using several instruments. During this time, we noticed that the ionization efficiency and detector responses for different ions can vary from instrument to instrument as well as between types of molecules (e.g., DKP compared with the larger peptides). To account for differences in peak intensities for species having the same solution concentration but different response factors we normalized the MS intensities (from kinetics studies) using values measured for a solution that was prepared such that every species observed in these experiments was at the same initial concentration. These studies revealed that efficiency of ionizing and detecting cRP and cKP was low, relative to the larger peptides, as can be observed in the mass spectra presented below. For the kinetics plots shown below, in order to estimate the solution abundances of the different species that are present, we multiply the [subP(3–11)+2H]2+, [cRP+H]+, and [cKP+H]+ ion signals by measured response factors of 1.4, 12, and 140, respectively.
Methods for investigating proline configurations.
Pierson et al.[45] assigned differences in peptide conformations arising from different cis- and trans-configurations of the three prolines found in the nonapeptide bradykinin on the basis that an alanine residue substituted for a proline restricts the peptide bond (on the N-terminal side of the residue) to the trans form.[22, 24, 25] While any change in amino acid sequence could introduce other changes in conformation, creation of sequence analogues for testing proline configurations appears in most cases to be a valuable approach, and we use this method here. Below, we examine three substituted sequences: RAKPQQFFGLM-NH2 [subP(P2A)], RPKAQQFFGLM-NH2 [subP(P4A)], and RAKAQQFFGLM-NH2 [subP(P2,4A)]. After accounting for differences in the intrinsic sizes of the proline and alanine,[46] the cross section distributions of these peptides are compared to unmodified subP to assign the proline configurations. In addition to comparison of cross sections between the natural sequence and the Ala-substituted analogues, we incubated each analogue (at 75 °C for 24 hr.) to examine their reactivity. This provides an additional test of how the backbone configuration influences bond cleavage.
Effects of acetic acid and peptide concentration.
Acetic acid (0.5% by volume) was added to ethanol because it aids ionization - increasing our sensitivity to various reactants and products while suppressing adduction of salts. Unlike similar systems, some amount of acetic acid appears to be necessary for the reaction to proceed, though we found that at 0.5% and higher concentrations (by volume) the rate is not affected.[10, 11] Similarly, studies were carried out over a range of subP precursor concentrations. Measurements over the 10 to 50 µM range of concentrations showed no discernable differences in the final distribution of products.
Methods for obtaining insight about reaction mechanisms.
Insight about pathways associated with structural changes and bond cleavages can be obtained by comparing experimental data with calculated kinetics for differential rate equations corresponding to models of trial reaction pathways.[10, 47–49] The differential rate expressions establish the relationship of each step of the mechanism and the rate constants derived from optimized fits are a measure of the reaction kinetics for each step. While this approach does not unambiguously establish a single pathway, it is valuable for ruling out many mechanisms and in most cases, those trial models that accurately represent the pathway tend to be closely related. To compare the goodness of fit between the kinetics calculated for each modeled pathway and the experimental data, the sum of residual sum of squares (ΣRSS) associated with the differences between the calculated and experimental data for each pathway are calculated, with the lowest ΣRSS representing the best-fit reaction mechanism.
An interesting feature of some experimental datasets is the observation of an induction period prior to product formation, giving rise to a kinetics profile that deviates from expected first-order reaction kinetics for unimolecular transitions.[50] The presence of such an induction period is an indication of unresolved (or unobservable) intermediate states along the dissociation pathway. We have previously discussed that it is possible to develop models that constrain the number of possible unobserved intermediate states.[49] The modeled abundances of these intermediates are summed at each point in time to produce the observed abundance profile, and are critical for capturing the shape of an induction period in the experimental data. One expects that there may be many different solutions (different combinations of intermediate states, mechanisms, and rate constants) that would capture the shapes associated with induction periods observed in the experimental kinetics data. However, we have investigated how much one can vary such parameters and find that in general the best fits appear to be those in which rate constants associated with transitions between multiple intermediate states are equal to each other.10,49 Since we lack direct information about the states that give rise to the induction periods observed here, this is also the simplest interpretation of these data.
Methods for determining transition state thermochemistry.
The rate constants determined from the optimized kinetics fits to data obtained over a range of temperatures are used to derive thermochemistry for each transition state that is described in our best fit model of the data. The present studies were performed at five temperatures [55, 60, 65, 70, and 75 (all ± 1 °C)] and replicated in triplicate measurements carried out on different days with samples that were incubated independently. Plotting the natural log of each average rate constant (k) as a function of temperature (T) allows for construction of an Arrhenius plot in the form of Equation 2,
| (2) |
that can be used to obtain an activation energy (Ea) and pre-exponential factor (A) for each step. From Ea and A we derive enthalpy of activation (ΔH‡, Equation 3), entropy of activation (ΔS‡, Equation 4), and Gibbs free energy of activation (ΔG‡, Equation 5):
| (3) |
| (4) |
| (5) |
where R is the gas constant, T is the temperature in solution, kb is Boltzmann’s constant, and h is Planck’s constant.
Results and discussion
Mass spectra for subP incubated in ethanol at 55 °C.
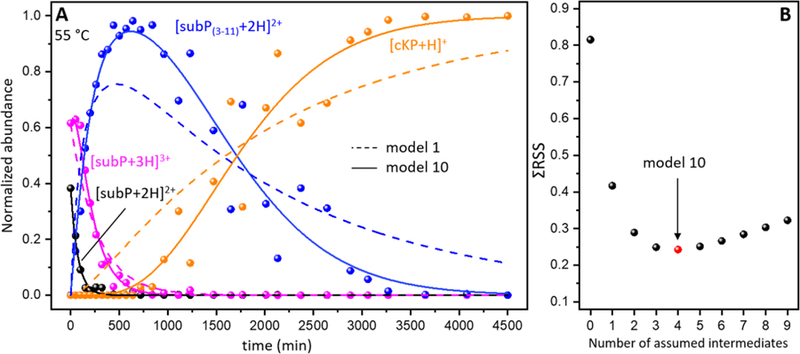
Figure 1 shows representative mass spectra obtained upon incubating subP in a solution of ethanol for 0, 150, 1110, 2880 and 4500 min. at 55 °C. At short incubation times two peaks, at m/z = 674 and 450, corresponding to [subP+2H]2+ and [subP+3H]3+, respectively, are observed. The intensities of these peaks are interesting. Initially, [subP+H]2+ comprises ~40% of the ion signal. At longer times the [subP+2H]2+ peak decreases, becoming insignificant by ~150 min. The [subP+3H]3+ intensity over this same time period changes only slightly. In our replicate experiments this peak sometimes increases or decreases slightly, or stays the same. From our experience with these types of systems, we interpret the combined behaviors of [subP+2H]2+ and [subP+3H]3+ as an indication of a slow protonation reaction, i.e., [subP+2H]2+ + H+ → [subP+3H]3+, as has been reported for polyproline-7 as well as bradykinin.[10, 11] Below, we show that this interpretation is consistent with the proposed reaction pathway. That addition of the lightest and fastest chemical moiety (a proton) to the peptide occurs so slowly, indicates that this reaction is regulated by a motion in this system that is much slower. In the case of bradykinin,[10] we ascribed this regulation to a trans→cis configurational change associated with the penultimate proline residue; it is likely that an analogous conformational change also regulates the protonation reaction in subP. At longer incubation times (e.g., 150 min in Figure 1), two new peaks in the mass spectra can be observed at m/z = 548 and 254. These peaks can be assigned to products associated with cleavage of the Pro2-Lys3 peptide bond (i.e., [subP(3–11)+2H]2+ and [cRP+H]+), analogous to the solution bond cleavage reported for bradykinin.[10] At still longer incubation times, (e.g., ~1110 min.), the original subP precursor peptide has completely disappeared (within our detection limits) and only the [subP(3–11)+2H]2+ and [cRP+H]+ species are observed in the mass spectrum.
Figure 1.
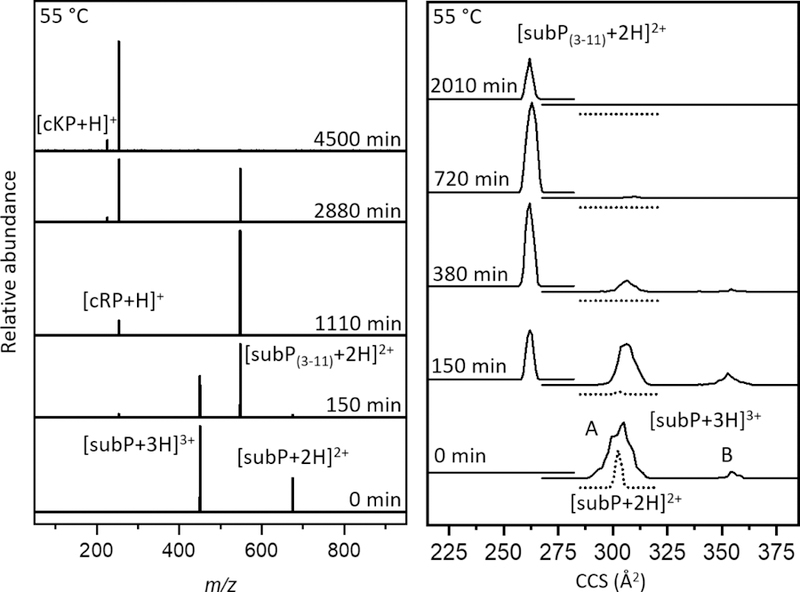
(Left) Representative mass spectra of solutions containing subP at various incubation times in ethanol and 0.5% (by volume) acetic acid at 55 °C. An apparently sequential dissociation is observed involving degradation of subP into subP(3–11) and cRP, followed by dissociation of subP(3–11) into subP(5–11) and cKP. (Right) Cross section distributions of [subP+3H]3+, [subP+2H]2+, and [subP(3–11)+2H]2+ species at different time points throughout incubation of the solution at 55 °C. For [subP+3H]3+, two peaks are observed, as defined in the text: conformer A at Ω = 305 Å2 conformer B at Ω = 354 Å2. As the abundance of [subP+3H]3+ ions decrease relative to the other species, the relative abundance of A and B is nearly constant. The cross section distribution of [subP+2H]2+ ions (dotted line) contains one peak at Ω = 302 Å2. A peak at Ω = 261 Å2 is observed in the [subP(3–11)+2H]2+ distribution. No changes in cross section are observed for [subP+2H]2+ or [subP(3–11)+2H]2+ ions, throughout the incubation.
The subP(3–11) product also contains a penultimate proline residue. As the system continues to incubate, (e.g., 2880 min.) a new peak is observed at m/z = 226. This peak corresponds to [cKP+H]+ - the DKP product that forms by dissociation of subP(3–11) at the Pro4- Gln5 peptide bond. The anticipated complementary fragment ion of this dissociation corresponding to Gln5-Met11-NH2 [subP(5–11)] is absent from all of the mass spectra. This is somewhat unsatisfying. It could be that once formed, subP(5–11) rapidly disappears (due to aggregation or rapid decomposition); or, perhaps more likely is that the loss of the basic N-terminal amino acid Arg1 and Lys3 residues leads to a peptide with no highly basic groups and thus ionizes poorly. For the remainder of our analysis, we assume that the subP(5–11) species is present in solution but not ionized efficiently. By 4500 minutes, no evidence of subP(3–11) remains, indicating that the cleavage reaction has reached completion. At this (and longer times) cRP and cKP are the only observable products.
We note that the delayed appearance of [cKP+H]+, and absence of any Arg1-Pro2-Lys3-Pro4 product, cyclic or otherwise, requires that the cleavages of the antecedent peptides to produce cRP and cKP, occur sequentially. That is, the Pro4-Gln5 bond is not cleaved until after the loss of the N-terminal Arg1-Pro2 residues. This conclusion is consistent with the fact that we observe no ions corresponding to an Arg-Pro-Lys-Pro sequence, which would be observed if the tetrapeptide was formed directly. Finally, these bond cleavages are remarkably specific. Each of our proline containing peptides react to completion, with no evidence for any competing side products. This indicates that the mechanism for bond cleavage is not accessible to other peptide bonds along the polypeptide chain. As discussed below, data for several Ala-substituted subP analogues also show this specificity.
Ion mobility cross section distributions for subP and products.
Figure 1 also shows cross section distributions recorded for subP over the course of incubation. While the overall populations of different ions change (in accordance with the mass spectra shown above), in all cases, the relative populations of the peaks in each cross section distribution are largely unchanged over the course of the experiment – as can be observed from inspection of Figure 1. For example, in the [subP+3H]3+ distribution the peaks labeled A and B disappear at roughly the same rate.
Normalized solution abundances as a function of incubation time at 55 °C.
A summary of the solution abundances (normalized by the response factor, as described above) for each observed species as a function of incubation time is shown in Figure 2. In the first 100 min. the abundance of [subP+2H]2+ rapidly disappears while [subP+3H]3+ species is largely unchanged. The abundances of the first bond cleavage products, subP(3–11) and cRP begin to increase soon after experiments are initiated, reaching a maximum abundance of ~60 to 70% of the total distribution at ~600 minutes. While we anticipate that these two products should track one another exactly, even after normalization the cRP abundance is somewhat lower than that for subP(3–11). This is a reflection of the uncertainty in these abundance measurements. At ~1000 min, the cKP product is observed and the abundance of cKP continues to increase until all of the subP(3–11) has dissociated. As mentioned above, presumably the Gln5-Met11-NH2 product (which was not detected) has a solution concentration that is comparable to that for cKP. A visual comparison of the kinetics profiles associated with degradation of both [subP+2H]2+ and [subP+3H]3+ shows that these species are degraded more rapidly than subP(3–11). The difference in these cleavage rates are likely associated with variations in the residues adjacent to the relevant proline, i.e., Arg1-Pro2-Lys3 for subP and Lys3-Pro4-Gln5 for subP(3–11).
Figure 2.
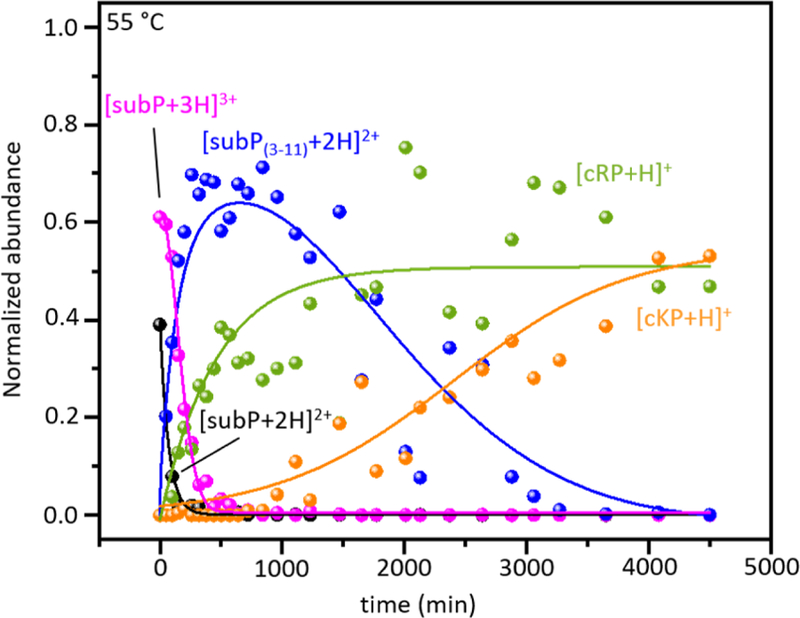
Normalized solution abundance profiles (obtained from measured ion intensities as discussed in the text) of reactants, intermediates and products that appear throughout incubation of subP in ethanol at 55 °C. The lines are shown only as visual aids.
Analysis of Pro→Ala analogues of subP to determine peptide bond configurations.
A summary of the cross section distributions recorded by ion mobility measurements for the subP(P2A), subP(P4A), and subP(P2,4A) analogues is shown in Figure 3. The [subP+3H]3+ ion shows two peaks that have been characterized previously. The relatively broad peak at Ω = 305 Å2 corresponds to a set of structures that emerge as the final water molecules evaporate from the peptide.[51] These structures are collectively referred to as solution structures - conformer A, as assigned previously. A very small peak at Ω = 354 Å2 is also observed. This feature is formed upon activation of the conformer A structures in the gas phase; thus, it has been assigned as a gas-phase structure – conformer B.[51]
Figure 3.
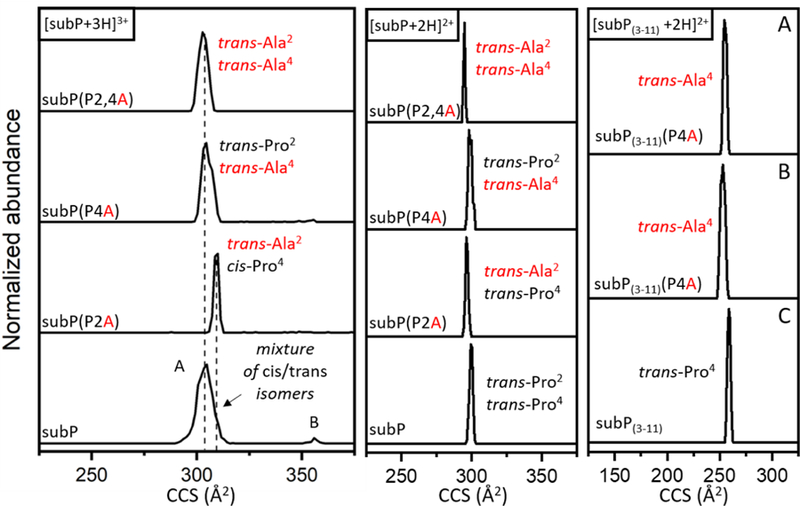
Cross section distributions of [subP+3H]3+ (left), [subP+2H]2+ (middle), and [subP(3- 11)+2H]2+ (right) and corresponding ions of Pro→Ala analogues, for the purpose of assigning Xxx-Pro peptide bonds as cis or trans. For [subP(3–11)+2H]2+, the top spectrum (A) shows the cross section distribution of [subP(3–11)(P4A)+2H]2+ ions corresponding to subP(3–11)(P4A) produced from incubation of subP(P4A), the middle spectrum (B) shows the cross section distribution of [subP(3–11)(P4A)+2H]2+ from the synthesized subP(3–11)(P4A) peptide, and the bottom spectrum (C) shows the mobility profile of [subP(3–11)+2H]2+ produced during incubation of subP in ethanol at 55 °C after 150 min.
As we begin this analysis, we note that the differences in the cross sections associated with the different substitutions discussed below are very small. This suggests that most of the population is likely due to trans-configured peptide bonds. With this said, it is worthwhile to go through this analysis in detail. We begin by assigning configurations associated with the [subP+3H]3+ ion. Consider the cross section distribution for the doubly-substituted [subP(P2,4A)+3H]3+ species, which has only trans-configured peptide bonds. The cross section for this ion falls near the center of the [subP+3H]3+ distribution. Thus, the population of states near the center of the [subP+3H]3+ distribution is consistent with a population of trans-Pro2,trans-Pro4 bonds. Further insight is obtained by examining the distribution associated with the singly-substituted [subP(P2A)+3H]3+ cross section distribution. This ion shows a peak at a slightly higher cross sections, near the unresolved shoulder on the right side of the [subP+3H]3+ conformer A peak. This slight shift suggests that some peptides having trans-Pro2, cis-Pro4 configurations may also be present in the conformer A peak. We note that left side of the peak associated with conformer A could correspond to cis-Pro2,trans-Pro4 or cis-Pro2,cis-Pro4 configured proline peptide bonds.
Each of the cross section distributions for [subP+2H]2+ and [subP(3–11)+2H]2+, and their associated analogues in Figure 3 show only a single very sharp peak. The cross sections for the Ala-substituted analogues show that the trans-configured Ala-substituted peptides are nearly identical (within 1% relative difference) of the [subP+2H]2+ cross section. Thus, it appears that both prolines in [subP+2H]2+ are trans-configured. The product ion formed from bond cleavage, [subP(3–11)+2H]2+, is also appears to be trans-configured. In this case, we generated [subP(3- 11)(P4A)-2H]2+ in two ways: by peptide synthesis, giving rise to the distribution in Figure 3B; and, by bond cleavage of the antecedent P4A-substituted subP precursor. These identical species have identical cross sections that are consistent with a trans-Pro4 configuration in the [subP(3- 11)+2H]2+ ion. We note that the P2A-substituted subP precursor does not undergo bond cleavage. This is an important clue in these systems – suggesting that the trans-configured proline must isomerize to the cis configuration in order to initiate bond cleavage.
Proposed mechanism for cRP and cKP formation.
Scheme 1 depicts a proposed dissociation mechanism for subP that results in DKP formation. This mechanism is similar to one proposed previously[20] where nucleophilic attack of the N-terminal amine on the carbonyl carbon of the penultimate Pro residue results in DKP formation. In order for this interaction to occur, the Arg1-Pro2 peptide bond must have a cis configuration, as the nucleophile cannot readily access the carbonyl carbon if the peptide bond is trans-configured. This configurational requirement is in agreement with the finding that incubated Pro→Ala analogues containing penultimate alanine were prohibited from dissociation, and provides an explanation of the slow rate of this process as well as the protonation reaction; that is, trans→cis isomerization often involves a substantial barrier that is rate limiting in structural transitions of biological systems.[32–34] Once cRP has been formed, the subP(3–11) peptide is expected to follow a similar mechanism in order to cleave the Pro4-Gln5 peptide bond, forming subP(5–11) and cKP. In the subP precursor, the high specificity, associated with the N-terminal attack towards Pro2, but not Pro4, suggests that the stability of the six-membered ring formed by the DKP helps to favor this product relative to elimination of a larger ring involving the tetrapeptide. Additionally, it is likely that entropic effects arising from the increased interaction distance associated with the N-terminus and the Pro4 residue also disfavor tetrapeptide elimination.
Scheme 1.
Proposed dissociation pathway of subP. Initially, cleavage occurs at the Pro2-Lys3 bond by attack of the N-terminal amine on the carbonyl carbon of Pro2. This reaction results in a cyclo-Arg1-Pro2 diketopiperazine and the complementary subP(3–11) product. The newly formed subP(3–11) spontaneously cleaves the Pro4-Gln5 bond by the same mechanism to form cyclo-Lys3-Pro4 and subP(5–11).
Characterizing favored reaction pathways.
While subP is a relatively simple peptide, the data described above provide the opportunity to understand the dynamics associated with structural changes involved in non-enzymatic processing of the Pro2-Lys3 and Pro4-Gln5 peptide bonds. To do this, we have calculated kinetics for a number of assumed (candidate) pathways and compared these calculated kinetics with our experimental data, a method successfully utilized previously[10, 47–49] and described briefly in the experimental section. We begin by modeling the most straight-forward pathway - the direct dissociation of both [subP+2H]2+ and [subP+3H]3+ to form cRP and its complementary product, subP(3–11); once formed the subP(3–11) product dissociates to form cKP and subP(5–11), an overall process that is described by Equation 6.
| (6) |
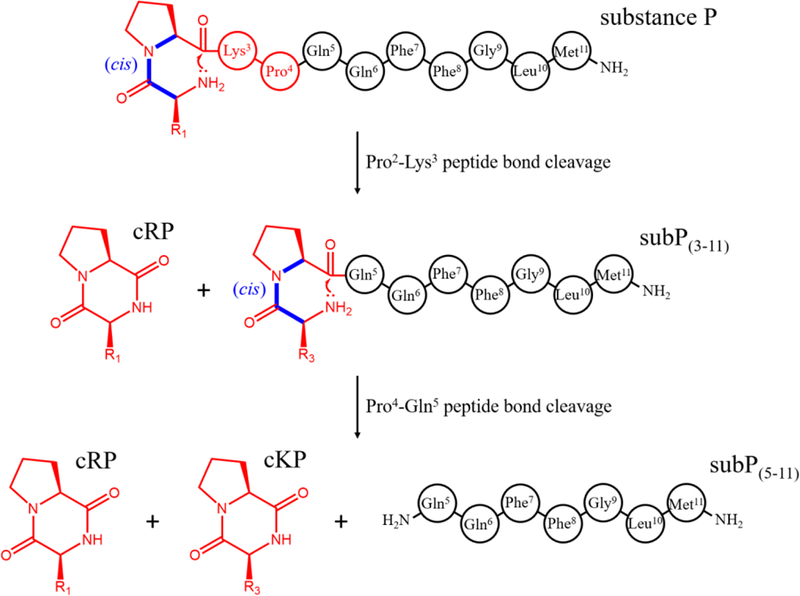
We refer to this process as model 1 as shown in Table 1. Figure 4A shows a best fit comparison of this model with the measured experimental kinetics data. The model captures the abundance profile of the [subP+2H]2+ ions nicely; but it fails to capture the shape of the [subP+3H]3+ curve as well as the profile associated with dissociation (i.e., [subP(3–11)+2H]2+ and [cKP+H]+). To better understand our pathway, we calculated the kinetics of 15 candidate pathways, listed in Table 1 and compared these with our experimental data. Assessment of each model’s ‘goodness of fit’ was accomplished by a residual sum of squares analysis (shown in Figure 4B). The outcome of this analysis shows that the abundance profile of subP(3–11), is captured best upon inclusion of ~3 to 5 intermediate states, as described by Equation 7 (for model 10 having four intermediates).
Table 1.
Modeled reaction mechanisms compared with experimental kinetics data of the subP dissociation reaction.
 |
List of assumed reaction pathways leading to sequential dissociation of subP. For each mechanism we solve a system of differential rate equations and compare the calculated kinetics for each model with the experimentally measured kinetics. The abundances of modeled intermediates (in) are summed with the species’ indicated by red brackets to produce the calculated kinetics profiles.
The quality of each fit is assessed from analysis of the residuals (i.e., by comparing each model’s calculated kinetics with the kinetics measured experimentally). See text for details.
Figure 4.
The left panel (A) depicts normalized abundances of subP charge states, subP(3–11), and cKP as the incubation progresses at 55 °C. cRP was not incorporated in the normalization and fitting of the kinetic data as it shares the same channel as subP(3–11). Kinetic fits from two model pathways are shown, including model 1 shown as dashed lines, and model 10 as solid lines (see Table 1 for details). The right panel (B) shows a list of summed residuals (ΣRSS) from models involving a number of subP(3–11) intermediates prior to dissociation. Model 10, possessing the lowest calculated ΣRSS, is indicated in (B).
| (7) |
Model 10 involves three key steps, as follows: protonation of [subP+2H]2+ to [subP+3H]3+ (regulated by a trans→cis isomerization of the Pro2 peptide bond); dissociation of [subP+3H]3+ to form cRP and subP(3–11); and finally, transition of subP(3–11) through four structural intermediates culminating in dissociation of subP(3–11) to form cKP and subP(5–11). We note that although model 10 had the lowest ΣRSS value, models 9 and 11 (Table 1) represent the experimental kinetics nearly as well. Although the intermediates prior to dissociation of subP(3–11) were not observed directly it is likely that they are associated with trans→cis isomerization of the Pro4 peptide bond, as this must occur in order for bond cleavage to occur. These intermediates effectively explain very different rates of dissociations of subP and subP(3–11) (t½ ~100 min. for dissociation of subP and ~1700 min. for dissociation of subP(3–11) at 55 °C). Moreover, these models indicate a highly sequential, step-by-step process for product creation.
Transition state thermochemistry.
Figure 5 shows a summary of kinetics data sets acquired at 55, 60, 65, 70, and 75 °C. By fitting all of these data with the pathway given by equation 7 (i.e., the model that best fits our experimental data), it is possible to determine rate constants at each temperature and use the Arrhenius equation to derive transition state thermochemistry. The Arrhenius plots are shown in Figure 6. For the first step in our pathway, (configurationally-coupled protonation, i.e., [subP+2H]2+ + H+ → [subP+3H]3+) we find a relatively low transition state energy barrier of ΔH‡ = 41 ± 5 kJ·mol−1. However, this transition state is tight, i.e., there is an entropic barrier, ΔS‡ = −157 ± 12 J·mol−1·K−1. Combination of these values yields the Gibbs free energy, ΔG‡ = 88 ± 6 kJ·mol−1. This is followed by cleavage of the Pro2-Lys3 peptide bond. The transition state for bond cleavage requires more energy, ΔH‡ = 55 ± 9 kJ·mol−1 and is also entropically disfavored (ΔS‡ = −130 ± 22 J·mol−1·K−1) as might be expected as nucleophilic attack (see Scheme 2) leading to peptide bond cleavage and ring formation requires a well-defined configuration. The higher Gibbs free energy, ΔG‡ = 94 ± 11 kJ·mol−1, slows the system down as the peptide bond is cleaved and subP(3–11) and the cRP DKP products are formed. The initial subP(3–11) (with a trans-configured penultimate proline) goes through a complex set of five unresolved transition states with the final step leading to bond cleavage and formation of the subP(5–11) peptide and the cKP DKP product. Our analysis treats these states identically and yields a larger energy barrier (ΔH‡ = 85 ± 5 kJ·mol−1) for each step and while each transition state is still entropically disfavored (ΔS‡ = −43 ± 2 J·mol−1·K−1) this analysis suggests that this set of barriers is far more accessible than the preceding transition states. These barriers have the largest overall Gibbs free energy barriers - ΔG‡ = 98 ± 5 kJ·mol−1.
Figure 5.
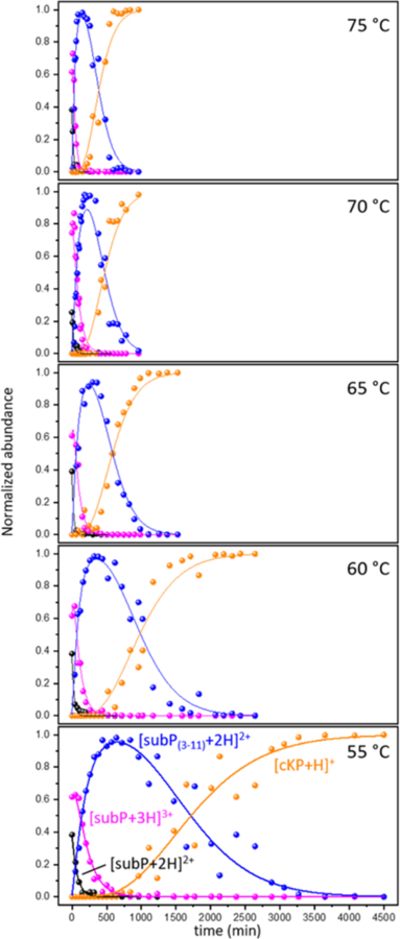
Normalized abundances of each species involved in fitting the kinetics plots ([subP+2H]2+, [subP+3H]3+, [subP(3–11)+2H]2+, and [cKP+H]+) as a function of time as solutions are incubated at 55, 60, 65, 70, 75 °C. Solid lines show the optimized calculated kinetics from model 10 fit to each species.
Figure 6.
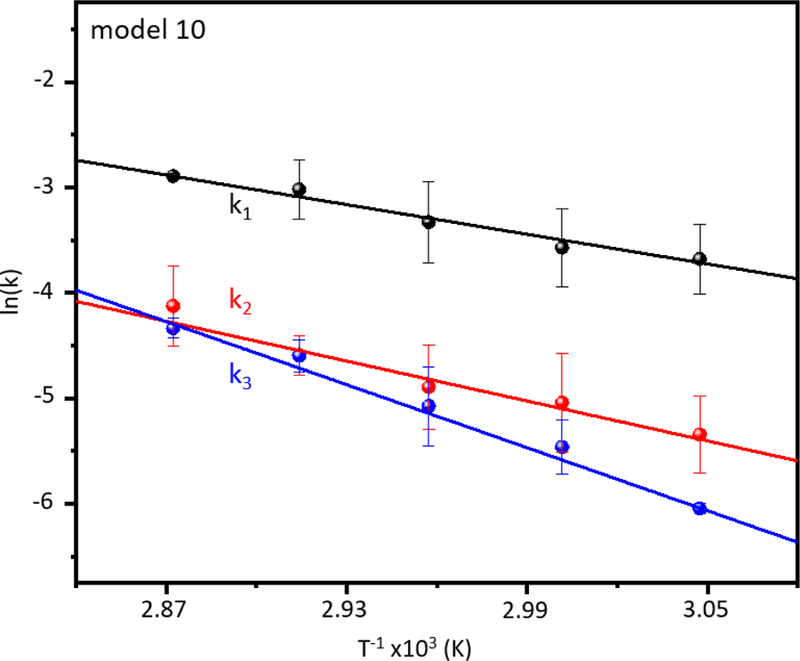
Arrhenius plot of rate constants derived from fitting the kinetics of model #10 to the experimental data. Three rate constants correspond to the three differentiated steps of the reaction: k1 for [subP+2H]2+ + H+ → [subP+3H]3+, k2 for [subP+3H]3+ → [cRP+H]+ + [subP(3- 11)+2H]2+, and k3 for [subP(3–11)+2H]2+ → i1 → i2 → i3 → i4 → [subP(5–11)+H]+ + [cKP+H]+, where each step involving an intermediate, in, is assumed to have the same rate constant, k3. The dissociation was performed at five different temperatures in triplicate, and the averaged rate constants derived from each temperature are points color-coded to their respective transition step. Each color-coded line is a weighted linear fit for that rate constant.
Scheme 2.
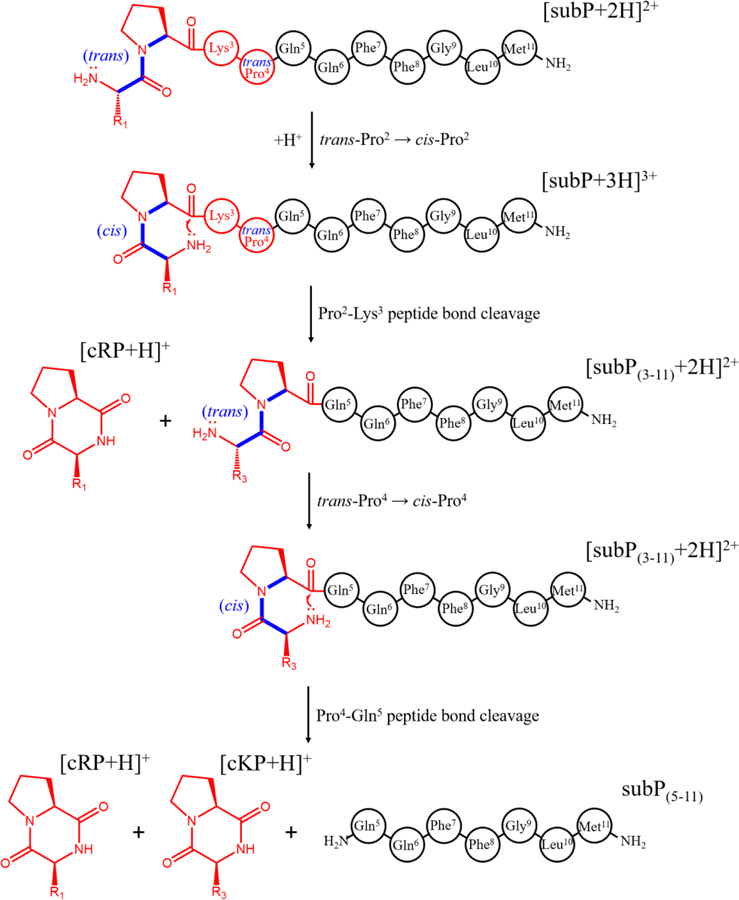
Dissociation pathway of subP based on the best-fit model determined from Figure 5. The first step involves a trans→cis isomerization of the Arg1-Pro2 peptide bond, followed by dissociation of cRP. The complementary product of this cleavage, subP(3–11), undergoes a similar trans→cis isomerization of the Lys3-Pro4 peptide bond (as one intermediate out of three to five, predicted), before dissociating to form cKP and subP(5–11).
Comparison of non-enzymatic and enzymatic cleavage mechanisms.
Several enzymes are known to degrade subP at specific sites,[52–55] and the resulting peptide fragments exhibit varying degrees of bioactivity.[56] Dipeptidyl peptidase IV (DPP IV) will sequentially cleave dipeptides from the N-terminal side of subP, first at the Pro2-Lys3 bond, and subsequently at the Pro4-Gln5 bond.[53, 56] Importantly, subP(5–11) shows an increased bioactivity and a high efficiency for cellular uptake relative to subP.[57–59] Thus, there are biological consequences for formation of subP(5–11) by enzyme cleavage or from spontaneous cleavage of subP and subP(3–11) in the absence of enzymes. A notable difference is that DPP IV cleavage results in linear dipeptides rather than cyclical DKP products.[56] Additionally, enzymatic DPP IV is reported to cleave only trans-configured peptide bonds, rather than cis-configured bonds.[60] Thus, if subP were to undergo a spontaneous trans→cis isomerization prior to enzymatic cleavage, the resulting peptide would be trapped along a pathway that leads to DKP formation (rather than formation of the linear dipeptide). From this we might imagine that one role of the DPP IV enzyme may be in processing the peptides into intact dipeptides, thus avoiding formation of cDKP products, which frequently possess some bioactivity themselves.[61] Finally, some diseases are thought to arise upon degradation of unstable sequences;[62–67] thus, spontaneous degradation of systems containing penultimate prolines [10, 12] may have deleterious consequences.
Conclusions
IMS-MS techniques have been used to investigate the spontaneous degradation of subP over time in solutions of ethanol. From comparisons of calculated kinetics for assumed pathways with experimental kinetics we find evidence for a complex, step-by-step pathway, that leads to two bond cleavage events. There is experimental evidence for seven transition states (four that appear to be hidden, but necessary to fit the data). Overall, bond cleavage hinges upon the configuration of peptide bonds associated with penultimate-proline residues. Once the pathway is established, kinetics measurements at different temperatures are used to derive thermochemistry for each of the transition state. The observation of intermediates, delineation of a preferred pathway, and characterization of transition-state thermochemistry provides a rare glimpse into the detailed, step-by-step motions associated with spontaneous peptide processing.
Acknowledgements
This work was supported in part by grants from the National Institute of Health, R01 GM121751-03 (DEC). CRC and DRF were supported by fellowships from the Robert and Marjorie Mann Chair (DEC) and TJE was supported by a fellowship from the Indiana University College of Arts and Sciences. The work at TAMU (DHR) was funded by NSF (CHE-1707675) and NIH (P41GM121751-01A1).
References
- 1.Euler UV, Gaddum J: An unidentified depressor substance in certain tissue extracts. The Journal of physiology 72, 74–87 (1931) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hökfelt T, Pernow B, Wahren J: Substance P: a pioneer amongst neuropeptides. Journal of internal medicine 249, 27–40 (2001) [DOI] [PubMed] [Google Scholar]
- 3.Severini C, Improta G, Falconieri-Erspamer G, Salvadori S, Erspamer V: The tachykinin peptide family. Pharmacological reviews 54, 285–322 (2002) [DOI] [PubMed] [Google Scholar]
- 4.Bienert M, Klauschenz E, Ehrlich A, Katzwinkel S, Niedrich H, Tót G, Teplán I: Tritium‐labelling in two phenylalanine residues of norleucine11‐substance P. Journal of Labelled Compounds and Radiopharmaceuticals 16, 673–679 (1979) [Google Scholar]
- 5.Higa T, Desiderio DM: Chemical degradation of 3H-labeled substance P in Tris buffer solution. Analytical biochemistry 173, 463–468 (1988) [DOI] [PubMed] [Google Scholar]
- 6.Mehlis B, Rueger M, Becker M, Bienert M, Niedrich H, Oehme P: Circular Dichroism Studies of Substance P and its C-terminal Sequences. Chemical Biology & Drug Design 15, 20–28 (1980) [PubMed] [Google Scholar]
- 7.Rueger M, Bienert M, Mehlis B, Gast K, Zirwer D, Behlke J: Self‐association of the neuroregulatory peptide substance P and its C‐terminal sequences. Biopolymers 23, 747–758 (1984) [DOI] [PubMed] [Google Scholar]
- 8.Floor E, Leeman SE: Substance P sulfoxide: Separation from substance P by high-pressure liquid chromatography, biological and immunological activities, and chemical reduction. Analytical biochemistry 101, 498–503 (1980) [DOI] [PubMed] [Google Scholar]
- 9.Kertscher U, Bienert M, Krause E, Sepetov NF, Mehlis B: Spontaneous chemical degradation of substance P in the solid phase and in solution. Chemical Biology & Drug Design 41, 207–211 (1993) [DOI] [PubMed] [Google Scholar]
- 10.Fuller DR, Conant CR, El-Baba TJ, Brown CJ, Woodall DW, Russell DH, Clemmer DE: Conformationally regulated peptide bond cleavage in bradykinin. Journal of the American Chemical Society 140, 9357–9360 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi L, Holliday AE, Khanal N, Russell DH, Clemmer DE: Configurationally-coupled protonation of polyproline-7. Journal of the American Chemical Society 137, 8680–8683 (2015) [DOI] [PubMed] [Google Scholar]
- 12.Battersby J, Hancock W, Canova‐Davis E, Oeswein J, O’ONNOR B: Diketopiperazine formation and N‐terminal degradation in recombinant human growth hormone. Chemical Biology & Drug Design 44, 215–222 (1994) [DOI] [PubMed] [Google Scholar]
- 13.Gisin BF, Merrifield R: Carboxyl-catalyzed intramolecular aminolysis. Side reaction in solid-phase peptide synthesis. Journal of the American Chemical Society 94, 3102–3106 (1972) [DOI] [PubMed] [Google Scholar]
- 14.Khosla M, Smeby R, Bumpus F: Failure sequence in solid-phase peptide synthesis due to the presence of an N-alkylamino acid. Journal of the American Chemical Society 94, 4721–4724 (1972) [DOI] [PubMed] [Google Scholar]
- 15.Giralt E, Eritja R, Pedroso E: Diketopiperazine formation in acetamido-and nitrobenzamido-bridgedpolymeric supports. Tetrahedron Letters 22, 3779–3782 (1981) [Google Scholar]
- 16.Pedroso E, Grandas A, de las Heras X, Eritja R, Giralt E: Diketopiperazine formation in solid phase peptide synthesis using p-alkoxybenzyl ester resins and Fmoc-amino acids. Tetrahedron letters 27, 743–746 (1986) [Google Scholar]
- 17.Steinberg S, Bada JL: Diketopiperazine formation during investigations of amino acid racemization in dipeptides. Science 213, 544–545 (1981) [DOI] [PubMed] [Google Scholar]
- 18.Steinberg SM, Bada JL: Peptide decomposition in the neutral pH region via the formation of diketopiperazines. The Journal of Organic Chemistry 48, 2295–2298 (1983) [Google Scholar]
- 19.Sepetov N, Krymsky M, Ovchinnikov M, Bespalova Z, Isakova O, Soucek M, Lebl M: Rearrangement, racemization and decomposition of peptides in aqueous solution. Pept Res 4, 308–313 (1991) [PubMed] [Google Scholar]
- 20.Capasso S, Vergara A, Mazzarella L: Mechanism of 2, 5-dioxopiperazine formation. Journal of the American Chemical Society 120, 1990–1995 (1998) [Google Scholar]
- 21.Møss J, Bundgaard H: Kinetics and mechanism of the facile cyclization of histidyl‐ prolineamide to cyclo (His‐ Pro) in aqueous solution and the competitive influence of human plasma. Journal of Pharmacy and Pharmacology 42, 7–12 (1990) [DOI] [PubMed] [Google Scholar]
- 22.Ramachandran GT, Sasisekharan V: Conformation of polypeptides and proteins Elsevier, (1968) [DOI] [PubMed] [Google Scholar]
- 23.Zimmerman SS, Scheraga HA: Stability of cis, trans, and nonplanar peptide groups. Macromolecules 9, 408–416 (1976) [DOI] [PubMed] [Google Scholar]
- 24.Jorgensen WL, Gao J: Cis-trans energy difference for the peptide bond in the gas phase and in aqueous solution. Journal of the American Chemical Society 110, 4212–4216 (1988) [Google Scholar]
- 25.Stewart DE, Sarkar A, Wampler JE: Occurrence and role of cis peptide bonds in protein structures. Journal of molecular biology 214, 253–260 (1990) [DOI] [PubMed] [Google Scholar]
- 26.MacArthur MW, Thornton JM: Influence of proline residues on protein conformation. Journal of molecular biology 218, 397–412 (1991) [DOI] [PubMed] [Google Scholar]
- 27.Williamson MP: The structure and function of proline-rich regions in proteins. Biochemical journal 297, 249 (1994) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kay BK, Williamson MP, Sudol M: The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. The FASEB journal 14, 231–241 (2000) [PubMed] [Google Scholar]
- 29.Counterman AE, Clemmer DE: Cis− Trans Signatures of Proline-Containing Tryptic Peptides in the Gas Phase. Analytical chemistry 74, 1946–1951 (2002) [DOI] [PubMed] [Google Scholar]
- 30.Glover MS, Bellinger EP, Radivojac P, Clemmer DE: Penultimate proline in neuropeptides. Analytical chemistry 87, 8466–8472 (2015) [DOI] [PubMed] [Google Scholar]
- 31.Glover MS, Shi L, Fuller DR, Arnold RJ, Radivojac P, Clemmer DE: On the split personality of penultimate proline. Journal of the American Society for Mass Spectrometry 26, 444–452 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brandts JF, Halvorson HR, Brennan M: Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 14, 4953–4963 (1975) [DOI] [PubMed] [Google Scholar]
- 33.Brandts JF, Brennan M, Lin L-N: Unfolding and refolding occur much faster for a proline-free proteins than for most proline-containing proteins. Proceedings of the National Academy of Sciences 74, 4178–4181 (1977) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin L-N, Brandts JF: Further evidence suggesting that the slow phase in protein unfolding and refolding is due to proline isomerization: a kinetic study of carp parvalbumins. Biochemistry 17, 4102–4110 (1978) [DOI] [PubMed] [Google Scholar]
- 35.Coin I, Beyermann M, Bienert M: Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences. Nature protocols 2, 3247–3256 (2007) [DOI] [PubMed] [Google Scholar]
- 36.Mason EA, McDaniel EW Wiley Online Library, (1988)
- 37.Mesleh M, Hunter J, Shvartsburg A, Schatz GC, Jarrold M: Structural information from ion mobility measurements: effects of the long-range potential. The Journal of Physical Chemistry 100, 16082–16086 (1996) [Google Scholar]
- 38.Shvartsburg AA, Jarrold MF: An exact hard-spheres scattering model for the mobilities of polyatomic ions. Chemical physics letters 261, 86–91 (1996) [Google Scholar]
- 39.Wyttenbach T, von Helden G, Batka JJ, Carlat D, Bowers MT: Effect of the long-range potential on ion mobility measurements. Journal of the American society for mass spectrometry 8, 275–282 (1997) [Google Scholar]
- 40.Merenbloom SI, Koeniger SL, Valentine SJ, Plasencia MD, Clemmer DE: IMS− IMS and IMS− IMS− IMS/MS for separating peptide and protein fragment ions. Analytical Chemistry 78, 2802–2809 (2006) [DOI] [PubMed] [Google Scholar]
- 41.Bohrer BC, Merenbloom SI, Koeniger SL, Hilderbrand AE, Clemmer DE: Biomolecule analysis by ion mobility spectrometry. Annu. Rev. Anal. Chem 1, 293–327 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Valentine SJ, Counterman AE, Hoaglund CS, Clemmer DE: Peer Reviewed: Injected-Ion Mobility Analysis of Biomolecules. Analytical Chemistry 69, 728A–735A (1997) [Google Scholar]
- 43.Koeniger SL, Merenbloom SI, Valentine SJ, Jarrold MF, Udseth HR, Smith RD, Clemmer DE: An IMS− IMS Analogue of MS− MS. Analytical chemistry 78, 4161–4174 (2006) [DOI] [PubMed] [Google Scholar]
- 44.Hoaglund CS, Valentine SJ, Sporleder CR, Reilly JP, Clemmer DE: Three-dimensional ion mobility/TOFMS analysis of electrosprayed biomolecules. Analytical chemistry 70, 2236–2242 (1998) [DOI] [PubMed] [Google Scholar]
- 45.Pierson NA, Chen L, Russell DH, Clemmer DE: Cis–trans isomerizations of proline residues are key to bradykinin conformations. Journal of the American Chemical Society 135, 3186–3192 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srebalus Barnes CA, Clemmer DE: Assessing intrinsic side chain interactions between i and i+ 4 residues in solvent-free peptides: a combinatorial gas-phase approach. The Journal of Physical Chemistry A 107, 10566–10579 (2003) [Google Scholar]
- 47.Shi L, Holliday AE, Shi H, Zhu F, Ewing MA, Russell DH, Clemmer DE: Characterizing intermediates along the transition from polyproline I to polyproline II using ion mobility spectrometry-mass spectrometry. Journal of the American Chemical Society 136, 12702–12711 (2014) [DOI] [PubMed] [Google Scholar]
- 48.Shi L, Holliday AE, Glover MS, Ewing MA, Russell DH, Clemmer DE: Ion mobility-mass spectrometry reveals the energetics of intermediates that guide polyproline folding. Journal of The American Society for Mass Spectrometry 27, 22–30 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El-Baba TJ, Kim D, Rogers DB, Khan FA, Hales DA, Russell DH, Clemmer DE: Long-Lived Intermediates in a Cooperative Two-State Folding Transition. The Journal of Physical Chemistry B 120, 12040–12046 (2016) [DOI] [PubMed] [Google Scholar]
- 50.Marcus RA: Interaction of Theory and Experiment in Reaction Kinetics (1999)
- 51.Silveira JA, Fort KL, Kim D, Servage KA, Pierson NA, Clemmer DE, Russell DH: From solution to the gas phase: stepwise dehydration and kinetic trapping of Substance P reveals the origin of peptide conformations. Journal of the American Chemical Society 135, 19147–19153 (2013) [DOI] [PubMed] [Google Scholar]
- 52.Pernow B: Inactivation of Substance P by Proteolysis Enzymes. Acta Physiologica 34, 295–302 (1955) [DOI] [PubMed] [Google Scholar]
- 53.Kato T, Nagatsu T, Fukasawa K, Harada M, Nagatsu I, Sakakibara S: Successive cleavage of N-terminal Arg1-Pro2 and Lys3-Pro4 from substance P but no release of Arg1-Pro2 from bradykinin, by X-Pro dipeptidyl-aminopeptidase. Biochimica et Biophysica Acta (BBA)-Enzymology 525, 417–422 (1978) [DOI] [PubMed] [Google Scholar]
- 54.Blumberg S, Teichberg V, Charli J, Hersh L, McKelvy J: Cleavage of substance P to an N-terminal tetrapeptide and a C-terminal heptapeptide by a post-proline cleaving enzyme from bovine brain. Brain research 192, 477–486 (1980) [DOI] [PubMed] [Google Scholar]
- 55.Matsas R, Kenny AJ, Turner AJ: The metabolism of neuropeptides. The hydrolysis of peptides, including enkephalins, tachykinins and their analogues, by endopeptidase-24.11. Biochemical Journal 223, 433 (1984) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang L, Ahmad S, Benter IF, Chow A, Mizutani S, Ward PE: Differential processing of substance P and neurokinin A by plasma dipeptidyl (amino) peptidase IV, aminopeptidase M and angiotensin converting enzyme. Peptides 12, 1357–1364 (1991) [DOI] [PubMed] [Google Scholar]
- 57.Otsuka M, Konishi S: Substance P and excitatory transmitter of primary sensory neurons. Cold Spring Harbor symposia on quantitative biology 40, 135–143 (1976) [DOI] [PubMed] [Google Scholar]
- 58.Yajima H, Kitagawa K, Segawa T: Studies on peptides. XXXVIII. Structure-activity correlations in substance P. Chemical and Pharmaceutical Bulletin 21, 2500–2506 (1973) [Google Scholar]
- 59.Nakata Y, Kusaka Y, Yajima H, Segawa T: Active Uptake of Substance P Carboxy‐ Terminal Heptapeptide (5–11) into Rat Brain and Rabbit Spinal Cord Slices. Journal of neurochemistry 37, 1529–1534 (1981) [DOI] [PubMed] [Google Scholar]
- 60.Fischer G, Heins J, Barth A: The conformation around the peptide bond between the P1-and P2-positions is important for catalytic activity of some proline-specific proteases. Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology 742, 452–462 (1983) [DOI] [PubMed] [Google Scholar]
- 61.Borthwick AD: 2, 5-Diketopiperazines: synthesis, reactions, medicinal chemistry, and bioactive natural products. Chemical reviews 112, 3641–3716 (2012) [DOI] [PubMed] [Google Scholar]
- 62.Brownlee M, Vlassara H, Cerami A: Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Annals of internal medicine 101, 527–537 (1984) [DOI] [PubMed] [Google Scholar]
- 63.Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S: Dominant and differential deposition of distinct β-amyloid peptide species, Aβ N3 (pE), in senile plaques. Neuron 14, 457–466 (1995) [DOI] [PubMed] [Google Scholar]
- 64.Shimizu T, Watanabe A, Ogawara M, Mori H, Shirasawa T: Isoaspartate formation and neurodegeneration in Alzheimer’s disease. Archives of Biochemistry and Biophysics 381, 225–234 (2000) [DOI] [PubMed] [Google Scholar]
- 65.Truscott RJ, Schey KL, Friedrich MG: Old proteins in man: a field in its infancy. Trends in biochemical sciences 41, 654–664 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A: Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proceedings of the National Academy of Sciences 91, 4766–4770 (1994) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miyata T, de Strihou C.v.Y., Kurokawa K, Baynes JW: Alterations in nonenzymatic biochemistry in uremia: origin and significance of “carbonyl stress” in long-term uremic complications. Kidney international 55, 389–399 (1999) [DOI] [PubMed] [Google Scholar]