

Abstract
Intrathecal (IT) delivery and pharmacology of antisense oligonucleotides (ASOs) for the CNS have been successfully developed to treat spinal muscular atrophy. However, ASO pharmacokinetic (PK) and pharmacodynamic (PD) properties remain poorly understood in the IT compartment. We applied multimodal imaging techniques to elucidate the IT PK and PD of unlabeled, radioactively labeled, or fluorescently labeled ASOs targeting ubiquitously expressed or neuron-specific RNAs. Following lumbar IT bolus injection in rats, all ASOs spread rostrally along the neuraxis, adhered to meninges, and were partially cleared to peripheral lymph nodes and kidneys. Rapid association with the pia and arterial walls preceded passage of ASOs across the glia limitans, along arterial intramural basement membranes, and along white-matter axonal bundles. Several neuronal and glial cell types accumulated ASOs over time, with evidence of probable glial accumulation preceding neuronal uptake. IT doses of anti-GluR1 and anti-Gabra1 ASOs markedly reduced the mRNA and protein levels of their respective neurotransmitter receptor protein targets by 2 weeks and anti-Gabra1 ASOs also reduced binding of the GABAA receptor PET ligand 18F-flumazenil in the brain over 4 weeks. Our multimodal imaging approaches elucidate multiple transport routes underlying the CNS distribution, clearance, and efficacy of IT-dosed ASOs.
Keywords: Neuroscience
Keywords: Neuroimaging, Pharmacology
Multimodal in vivo and ex vivo imaging demonstrates the CNS pharmacology of antisense oligonucleotides administered to the intrathecal space.
Introduction
Antisense oligonucleotides (ASOs) are promising drugs for the treatment of CNS disorders due to their specific molecular targeting and extended pharmacological properties (1). ASOs can elicit their pharmacological effect by several means including RNase H–mediated RNA degradation, or targeted modification of RNA splicing. Currently, delivery of ASOs for the treatment of neurological diseases to their site of action in the CNS requires direct delivery to the central compartment, as these molecules do not readily cross the blood-brain barrier (BBB). The intrathecal (IT) dosing route offers a relatively straightforward solution for bypassing the BBB and delivering drugs directly to the CNS. Use of this dosing route has recently led to the successful development and regulatory approval of the ASO drug Spinraza, the first treatment to slow down neurodegeneration in spinal muscular atrophy (1). In spinal muscular atrophy patients and in nonhuman primates, IT-delivered ASOs were confirmed to distribute rostrally along the neuroaxis throughout the CNS (2–4). Several IT-dosed therapeutics including ASOs, proteins, gene therapy vectors, and stem cells are also currently in clinical development for many CNS disorders (5–7). However, the kinetics of distribution and the PK and PD of IT-dosed therapeutics remains poorly understood due to the unique anatomical and functional properties of the IT space (8, 9).
Although many types of molecules can exchange between cerebrospinal fluid (CSF) and the CNS interstitial fluid (ISF), the exact anatomical routes and physiological processes connecting CSF and ISF are still being elucidated (10–15). Multiple potential routes exist, and each class of molecule likely uses the various pathways to different degrees. Small anesthetic molecules have been shown to move directly across isolated pia mater membranes (16) and human pia mater has also been shown to have fenestrations in certain regions of the neuroaxis (17). While ions have been shown to permeate the glia limitans to submillimeter distances (18), it is unclear whether trans-pial transport or fenestrations allow large molecules to also penetrate the glia limitans and enter the interstitial space to a significant depth. Direct passage of molecules across the pia and glia limitans is also possible through cell-to-cell transfer mechanisms and the shuttling of multiple molecules between astrocytes, neurons, and oligodendrocytes has also been documented via transporters and other transcellular exchange mechanisms (14, 16). Movement of fluid along white-matter axonal tracts offers yet another route for molecular distribution (17–20).
Numerous animal experiments have also demonstrated that molecules in the CSF can readily communicate with ISF bidirectionally along basement membranes located in the walls of penetrating cerebral blood vessels (21–26). This unique conductive aspect of cerebral vessel walls has been variously referred to as the paravascular, perivascular, or intramural periarterial drainage (IPAD) path, and has been shown to convey fluid, molecules, and nanoparticles between the CSF and ISF (12, 21–27). Although still under active exploration, the driving force for molecular exchange along this perivascular path may in part arise from cerebral arterial pulsations (27) and may also depend on the pattern and orientation of aquaporin-4 water channels distributed in the end feet of the astrocytic sheaths surrounding brain capillaries (12, 13). This newer insight for involvement of glial aquaporin-4 channels has led to the emergence of the term “glymphatic system” also being applied to the overall perivascular CSF-ISF exchange process (13). The exact details of mechanisms governing this process remain a matter of debate.
It is not clear how the several known CNS molecular movement processes summarized above impact the CNS delivery of ASO drugs dosed into the IT space. Therapeutic ASOs, which are typically less than 10 kDa in size but display significant protein binding, could potentially be transported in an unbound form into the CNS parenchyma via passage across the pia and glia limitans, along white-matter tracts, via transcellular exchange, in perivascular basement membranes, or by utilizing all of these mechanisms. Competing mechanisms for clearing ASOs from the CSF after IT administration also exist. In addition to clearance from the CSF by uptake into the CNS parenchyma ASOs can also exit to the systemic circulation from the IT compartment. Large molecules have long been demonstrated to be cleared from CSF to the head, neck, and other peripheral lymph nodes via lymphatic vessels located in the meningeal dura mater as well as in cranial and spinal nerve sheaths in several animal species (20, 22–24). This meningeal lymphatic efflux route has recently been reconfirmed by several investigators using newer transgenic and imaging techniques (21, 25, 26). ASOs may be cleared from the CSF to the periphery in larger protein complexes via these meningeal lymphatics. A better understanding of the CNS tissue permeation and CSF clearance processes influencing the behavior and actions of IT-dosed ASOs is important for understanding their drug action and could lead to approaches for improving their efficacy.
Imaging techniques have proven useful in better defining the IT space and the behavior of labeled molecules injected into this compartment (28, 29). CSF-ISF fluid exchange of molecules can be imaged ex vivo and in vivo following IT dosing of small-molecular-weight hydrophilic molecules that are either fluorescent (12) or radioactively tagged, respectively (21). Small-molecule MRI probes such as gadopentate dimeglumine (30) and gadolinium-DTPA (31) have also been used for this purpose. We recently highlighted several parameters and molecular features that affect neuraxial PK and CNS distribution, including the chosen injection site, dose volume, protein binding properties, tissue affinity, and CSF clearance routes of specific molecules using dual isotope imaging of various IT-dosed radiolabeled molecules (29). Here, we extend our previous work by using multimodal in vivo and ex vivo imaging approaches to track the PK of ASOs from the lumbar subarachnoid space to their site of action in brain cells. By combining in vivo single photon emission computed tomography (SPECT) nuclear imaging, IHC, and ex vivo block-face cryofluorescence tomography (CFT), we define the regional 2- and 3-dimensional (2D and 3D) spatiotemporal PK of ASO penetration into CNS structures and demonstrate their interaction with multiple molecular movement paths. Using ASOs targeting neurotransmitter receptors we demonstrate their spatiotemporal PD effect on mRNA and protein knockdown. We also employ GABAA receptor mRNA–targeting (Gabra1-targeting) ASOs and the PET ligand 18F-flumazenil, which binds the benzodiazepine site of the GABAA receptor, for the longitudinal visualization of the kinetics of the ASO PD effects by nuclear imaging in the live rat.
Results
125I-ASO SPECT/CT imaging studies of neuraxial PK.
To determine the kinetics of ASO distribution up the neuraxis following IT delivery, we labeled an ASO targeted to the non–protein-coding RNA Malat1 with 125I and followed it via SPECT/CT live imaging. We previously demonstrated that variation in IT bolus parameters can markedly influence the neuraxial spread of IT-dosed molecules and developed a dosing protocol for rats to spread injected material along the neuraxis and reduce variability of the postbolus intracranial PK (29). Here, this IT injection protocol resulted in rapid delivery of 125I-Malat1-ASO throughout the neuraxial IT space (Figure 1A and Supplemental Video 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.129240DS1). Cranial entry of signal was observed immediately after dosing (Figure 1A). Clearance from the IT space into the periphery was observed much later, with accumulation of the radioactive signal observed in head and neck lymph nodes and kidneys by 4 hours after dosing, with the kidney signal peaking 1 day after dosing. This may be a limitation of the imaging technique, as ASO is most likely present in these structures at earlier time points, but at levels too low to be detected with the imaging modality. Although the overall signal declined with time, the relative whole-body distribution pattern of ASOs remained the same between 1 and 7 days after dosing. SPECT/CT images and whole-body autoradiography identified high retention of signal in the meninges covering the spinal cord and nerve roots, at the base of the brain, and in the region of the tentorium cerebelli and superior sagittal sinus (Figure 1, A and B). Radioactive signal appeared in the nasal turbinates immediately after IT dosing and the appearance of this signal preceded the observed accumulation into the submandibular, deep cervical, paraspinal, and abdominal lymph nodes (Figure 1, A and B, and Supplemental Video 1). The strong meningeal SPECT signal limited the visualization of parenchymal penetration of ASO with live nuclear imaging. Thus, brain penetration of 125I-ASO was visualized by autoradiography (Figure 1B).
Figure 1. SPECT/CT live imaging of IT-dosed ASO.

125I-Malat1 ASO was given as an IT bolus (~180 μg; 230.7 ± 42.6 μCi) in male Sprague-Dawley Rats (n = 5, 270.8 ± 30.6 g). (A) Representative whole-body SPECT/CT and autoradiogram (far right) performed at indicated times after dosing. (B) Head and neck close-up of SPECT/CT images showing egress to nasal turbinates and lymph nodes (white arrows). Head and neck closeup of autoradiogram (far right) shows parenchymal radioactive signal in cerebral cortex and cerebellum (yellow arrows). This is imaging from 1 representative animal from a group of 4 that were imaged in this experiment.
Immunohistochemical tracking of ASO PK and PD in the CNS.
To simultaneously evaluate the kinetics of ASO distribution and target suppression, we designed an ASO to suppress the GluR1 subunit of the rat glutamate AMPA receptor and delivered this to rats via IT lumbar bolus injection. At various times after injection, brains and spinal cords were excised for immunohistochemical localization of ASO (32) and the GluR1 protein product, along with the quantification of GluR1 mRNA target suppression and ASO tissue concentrations. Following IT injection, ASOs were rapidly associated with the meningeal pia mater by 15 minutes after dosing (Figure 2A). Between 1 and 8 hours after IT bolus, diffuse ASO staining permeated the gray and white matter of the spinal cord and the brain. In the spinal cord, lumbar uptake preceded thoracic and cervical uptake. In the brain, a centripetal accumulation pattern was seen, with gray-matter regions closest to the subarachnoid CSF accumulating signal first, beginning as early as 1 hour and progressing to cover most of the gray matter by 8 hours after dose (Figure 2A). ASO accumulated in substructures of the cerebral cortex, hippocampus, and cerebellar cortex by 24 hours, remained until at least 8 weeks, and was cleared to nearly undetectable levels by 16 weeks after dosing.
Figure 2. Ex vivo PK and PD effects of IT-dosed ASOs on brain neurotransmitter receptor mRNA and protein expression.

(A) IHC of GluR1 ASO uptake by brain and spinal cord at various times after dosing (0.7 mg IT bolus) from animals representative of the groups of 4 at each time point. (B) Regional GluR1 ASO PK and PD effect on GluR1 mRNA knockdown via PCR graphed versus time after dosing (in days). Dashed lines in A indicate regions used for analysis. (C) IHC for GluR1 protein at various time after dosing from animals representative of the groups of 4 at each time point. (D) Regional relationship between GluR1 ASO IHC and GluR1 mRNA and protein levels as determined by IHC. (E) AUC analysis of ASO concentrations in frontal cortex, lumbar, thoracic, and cervical spinal cord samples versus time. All data are graphed as mean ± SD with n values of 4 for all groups. Analysis of differences between AUCs of the tissues was by 1-way ANOVA with Bennett’s post hoc test; *P < 0.05.
To directly compare target RNA suppression to the kinetics of ASO distribution, GluR1 mRNA levels were quantified in tissues collected from slides immediately adjacent to the immunohistochemical slides. Dotted lines overlaid on the vehicle control section in Figure 2A indicate the regions collected for GluR1 mRNA quantification. ASO tissue concentrations were quantified by hybridization ELISA (HELISA) in spinal cord and frontal cortex contralateral to the immunohistochemical samples. GluR1 mRNA is inhibited to maximal effect by 3 days in the spinal cord segments and by 7 days in the cortex (Figure 2A and Supplemental Figure 4A with regraphing to visualize early time points). The inhibition of GluR1 mRNA expression then almost completely resolved to untreated values by 16 weeks after dosing. This tracked the ASO accumulation in the various tissues, with RNA suppression resolving as ASO was cleared.
We further extended these findings into smaller brain regions to understand the protein kinetics and to verify the PK/PD properties with additional techniques. In single sections, we performed fluorescence IHC for the ASO and GluR1 protein and compared to mRNA quantified from regions collected from adjacent slides. In treatment-naive animals, diffuse brain staining of GluR1 was seen in gray-matter structures with enrichment in hippocampus, cerebellum, and striatum (Figure 2C). Quantitative image analysis revealed that following ASO treatment, GluR1 mRNA and protein expression both declined over time and the expression levels of both were clearly suppressed by 2 weeks after ASO administration (Figure 2D and Supplemental Figure 4B).
The extent and duration of the GluR1 mRNA and protein suppression largely correlated with the GluR1 ASO concentration kinetics (Figure 2, C and D). The protein expression reductions were delayed with respect to the reduction of the mRNA expression. ASOs lead to the direct degradation of the RNA template, so the reduction in protein expression will be observed only after the already translated proteins are degraded via natural processes. This slight delay in protein level suppression is likely due to the half-life of the protein. Conversely, as mRNA levels return to normal, protein levels follow without delay, as expected (Figure 2D).
Subcellular kinetics of CNS permeation patterns of ASOs.
To determine the cellular and subcellular resolution of ASO kinetics, we performed a higher-resolution analysis of the PK/PD study (Figure 2). Permeation of ASOs from the CSF space into the sub-pial CNS tissue was observed as early as 30 minutes after injection, in the spinal cord, cerebral cortex, and cerebellar cortex tissue (Figure 3). At the earliest time point assessed, 30 minutes after injection, high densities of ASO staining were already seen along axonal bundles in the spinal cord white matter leading to the central gray matter. In adjacent sections, these ASO-laden tracts overlapped with glial fibrillary acidic protein (GFAP), a marker of astrocytic intracellular intermediate filaments. Strong intracellular staining of several spinal cord motor neurons could also be seen at this early time point. In the cerebral cortex (Figure 3), early (30 minutes after dose) ASO association with the pia was also accompanied by diffuse staining of tissue below the adjacent glia limitans, an area rich in GFAP-positive astrocytes.
Figure 3. ASO colocalization with axonal and glial structures.

IHC performed for ASO and GFAP at 30 minutes after lumbar IT dosing of anti-GluR1 ASOs. From left to right, staining is shown associated with lumbar spinal cord axonal bundles, axon- and astrocyte-rich sub-pial tissue in cerebral cortex, and the Bergmann glia-rich layers of the cerebellar cortex. Arrowheads show early ASO signal that was observed in what appear to be Bergman glial cell bodies adjacent to Purkinje cells, as stained by GFAP IHC in adjacent sections Images in the left 3 columns are at ×20 magnification and the right 2 images have additional ×2 digital magnification.
In contrast with the lumbar spinal cord, only sparse intracellular staining in the neurons was observed in the cerebral cortex at 30 minutes. A similar pattern was also seen in the cerebellar cortex, where diffuse extracellular ASO staining was seen in molecular and granule cell layers 30 minutes after dosing. Early accumulation of ASO signal could be clearly seen in the molecular layer of the cerebellum, the same area where what appeared to be Bergman glial processes, as detected by GFAP-positive staining, were observed to be touching the pia and glia limitans in adjacent sections. Early ASO signal was also observed in what appear to be Bergman glial cell bodies adjacent to Purkinje cells, as stained by GFAP IHC in adjacent sections (arrowheads in Figure 3). No accumulation of ASO was visible in Purkinje or granule cell neurons at 30 minutes after dosing.
With longer postinjection intervals, a progressive translocation of ASO signal towards a strong intracellular staining pattern was observed (Figure 4). Uptake into cell bodies located in both the white and gray matter was most rapidly observed in the spinal cord, the neuraxial region most proximal to the injection site. While only some spinal cord motor neuron cell bodies were ASO positive at 30 minutes after dose (Figure 3), between 3 and 24 hours after dose nearly all cell bodies in the white and gray matter accumulated ASOs, with strong intracellular staining of cells being the predominant pattern by 24 hours (Figure 4A). In the brain, ASO signal permeated tissue in a centripetal manner along with a caudal to rostral progression of signal accumulation (Figure 4A). Tissue at the base of the brain or adjacent to anatomical CSF recesses accumulated signal earlier and most prominently. In both the cerebral cortical gray and white matter, there was a translocation of ASO signal from a diffuse pattern to an intracellular pattern between 3 and 24 hours (Figure 4A). The diffuse extracellular staining in the cerebral white matter in the right side of Figure 4A at the 3-hour time point may be indicative of ASO distribution along white-matter tracts.
Figure 4. Cellular uptake patterns of ASOs.

(A) Time-dependent ASO uptake patterns in white and gray matter (WM and GM) of the spinal cord and cerebral cortex. (B) Temporal ASO uptake into layers of the cerebellar cortex (×10 magnification). (C) Early diffuse staining in the cerebellar molecular layer up to 8 hours after dosing transformed to predominately intracellular staining pattern in Purkinje cells by 24 hours that persisted out to 7 days (×20 magnification).
Although the early diffuse staining pattern appeared to be extracellular, the possibility of this ASO signal being located within astrocytic cells was suggested by the progressive staining pattern observed in the cerebellar cortex. Here, the early pattern of the diffuse staining in the molecular and granule cell layers shifted over time to a pattern marked by intracellular accumulation in neurons in all 3 layers (Figure 4B). Prominent intracellular signal appeared in Purkinje cells between 8 and 24 hours and remained for at least 7 days (Figure 4C).
CFT map of IT ASO brain penetration.
To more directly track the regionally heterogeneous brain entry patterns of ASOs in 2 and 3 dimensions, we directly labeled an ASO with the fluorescent tag Cy7 and tracked tissue penetration following IT dosing using fluorescence microscopy and a block-face CFT procedure. For these experiments, we used Cy7-ASOs targeting Gabra1 mRNA, which encodes the GABAA receptor α1 subunit. This target was chosen to allow subsequent correlation of 2D and 3D Cy7-ASO tissue PK and distribution with the PD effect of ASOs, as illustrated by the brain binding pattern of the GABAA receptor ligand 18F-flumazenil assessed via live PET imaging. At various times following lumbar IT injection of the Gabra1 Cy7-ASOs, rat brains were removed and processed for imaging. CFT was performed by using an assembly of lasers and camera fiber optic cables placed over the frozen tissue block and photographing the block face serially as the block was sliced in 25-μm increments. Both white light and fluorescence images were obtained after every slice. Some brain sections were also collected on glass slides for fluorescence microscopic analysis and additional IHC. Following sectioning and photography of the entire brains, the photographic data were digitally processed to create a 3D space, which allowed viewing of the data in any spatial plane of section desired. As shown in Figure 5A, in which the white light and fluorescence images are overlaid, the Gabra1-targeting ASOs entered centripetally from the CSF first into the brain parenchyma adjacent to the subarachnoid space. As with the GluR1 ASO, the Gabra1-targeting Cy7-ASO rapidly associated with the meninges and surface vessels, penetrated along vessel walls, and eventually accumulated in brain cells by 8 hours after dose, with the strongest intracellular ASO signal in what appeared to be predominantly neurons (Figure 5, A–C).
Figure 5. Cryofluorescence tomography (CFT) of ASO permeation into brain tissue.

(A) Cy7-labeled Gabra1 ASOs (700 μg) were dosed IT, brains removed for CFT at various times after dosing, and photographed under white and fluorescent light after each 25-μm block-face section. (B) Trans-pia movement of Gabra1 Cy7-ASOs (×10 magnification). (C) Vascular association and cell uptake of Gabra1 Cy7-ASOs (×20 magnification). (D) Peri-arterial association of Cy7-ASO in 3D reconstruction of CFT data. Arrowhead points to basilar and middle cerebral arteries. Deeper vessels seen include the interhemispheric anterior cerebral artery.
Heterogeneity of ASO penetration into deep brain structures was obvious, with the striatum and deep cerebellar nuclei having slower and lesser ASO accumulation than other deep brain regions (Figure 5A). A close-up view of Cy7-ASO tissue permeation also shows early evidence of trans-pial ASO movement at early time periods, with a more cellular localization pattern by 8 hours (Figure 5B). Clear association of Cy7-ASOs with several brain surface vessels was seen in brain sections based on colocalization of ASO and the staining signal for vascular α-smooth muscle actin (αSMA) or the vascular endothelium marker Reca1 (Figure 5C). When 3D images of the Cy7-ASOs were constructed from the CFT procedure, the widespread ASO signal associated with the pial covering of the brain could be clearly seen as well as the signal strongly outlining cerebral arteries and their fine branches (Figure 5D, white arrowhead).
Further visible on the 2D maps is the centripetal Cy7-ASO brain penetration apparent in sagittal, coronal, and horizontal virtual sections (Figure 6 and Supplemental Video 2). Deeper CNS structures furthest from the subarachnoid space, such as the parts of the thalamus, brainstem, and striatum, were slower to accumulate ASO over the examined postinjection times (Figure 6). The 3D CFT maps of the fluorescence data show early association of Cy7-ASOs with major cerebral arteries and their branches (Figure 6 and Supplemental Video 3). Cy7-ASOs prominently labeled large arteries traversing the surface of the brain as well as those in the circle of Willis and meninges (Figure 6 and Supplemental Video 3). Between 2 and 8 hours after dosing, Cy7-ASO signal penetrated into the brain surface and a progressive loss of signal from the surface arteries occurred as the ASOs moved into the deeper tissue. These relationships can best be seen in the 3D display of the data, as shown in Supplemental Video 3.
Figure 6. 2D and 3D CFT display of IT-dosed Gabra1 Cy7-ASO brain penetration over time.
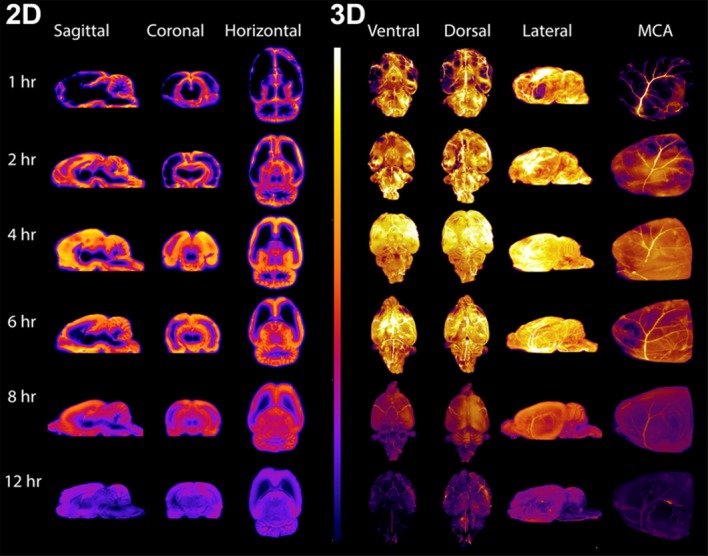
Left side shows sagittal, coronal, and horizontal virtual sections through the 3D-reconstructed CFT fluorescence images of animals euthanized 1, 2, 4, 6, 8, and 12 hours after IT dosing of Cy7-labeled Gabra1 ASOs (700 μg). Right side shows the surface images of the ventral, dorsal, and lateral aspects of the 3D-reconstructed CFT fluorescence images at the same time points. On the far right are images of the forebrain surface of the 3D-reconstructed CFT fluorescence images showing the fluorescently labeled ASO association with the left middle cerebral artery (MCA) at the different time points. For all images the fluorescence intensity was kept constant to evaluate the relative distribution of the ASO in the brain.
The distribution of ASOs within the perivascular compartment in the brain.
A closer evaluation of the immunohistochemical data also revealed the presence of larger punctate ASO signals appearing throughout the brain, which increased over 24 hours and remained prominent out to 16 weeks (Figure 2, A and B, and Figure 7A). The large puncta of ASO signal matched the pattern of brain blood vessels, as confirmed by the immunohistochemical colocalization of the endothelium-specific marker Reca1 (Figure 7B) as well as αSMA and the vascular basement membrane components laminin α2 and perlecan (Figure 7, C and D). By contrast, the more diffuse immunofluorescence signal of smaller puncta was consistent with cell bodies and nuclei of brain cells (Figure 7, B, E, and F).
Figure 7. Perivascular and neuronal association of ASO 24 hours after dosing.

(A) Punctate perivascular staining of ASOs by IHC throughout brain at 24 hours after IT dosing. (B) Colocalization of ASOs and the vascular endothelial marker Reca1 in cerebral cortex (×10 magnification left 2 images, ×40 magnification right image). (C) Colocalization of Cy7-ASO with vascular α-smooth muscle actin (αSMA) and basement membrane component laminin α2 (×20 magnification). (D) Colocalization of ASOs with basement membrane component perlecan (×20 magnification). (E) ASO colocalization with neurons and vessels in cerebral cortex (×10 magnification). (F) ASO colocalization with neuronal cells in hippocampus (×40 magnification).
To explore the relative ASO PK in the perivascular and neuronal compartments we administered increasing IT doses (10 μg to 700 μg) of the Gabra1 ASO and euthanized animals at 14 days after dosing. Serial adjacent brain tissue sections were subsequently double stained for ASO immunoreactivity (Figure 8A) and either the neuronal marker NeuN or the vascular endothelial cell marker RecA1. To track the ASO signal intensity in the respective compartments as a function of dose, marker-specific masks were digitally overlaid onto the ASO immunostaining images (Figure 8B). The Reca1 mask was additionally gated on objects that showed higher than background ASO signal. In addition, the pial signal was also tracked using a mask placed over this layer. A clear dose-dependent increase in the brain-wide ASO immunostaining was observed, with progressive increases in neuronal structures. However, the pial and large punctate vascular signals had maximal staining at the lower doses, and the immunoreactivity did not increase with increasing doses. By contrast, the neuronal signal increased in a dose-dependent manner (Figure 8A). Digital overlay analysis of ASO staining with the neuronal (NeuN) and vascular (Reca1) staining pattern (Figure 8B) confirmed that ASO signal intensity in these compartments behaves differently as a function of dose (Figure 8C). The pial and perivascular compartments showed near maximal signal at the lowest tested ASO dose of 10 μg, while the neuronal staining pattern required a 100-fold higher dose to reach similar intensity in the cortex (Figure 8C). Moreover, regional analysis of the neuronal ASO uptake showed a dose-dependent narrowing of the caudal (retrosplenial cortex) to rostral (orbital cortex) ASO gradient. Striatal neurons accumulated less overall ASO signal than cortical neurons, but a clear dose-dependent delivery of ASO to the striatal neurons was achievable (Figure 8C).
Figure 8. IT dose response of ASO interaction with vascular and neuronal elements.

(A) IHC of GluR1 ASO at 14 days following IT administration at different doses. (B) Neuronal (NeuN) versus vascular (Reca1) gaiting of ASO signal. (C) Differential dose response of ASO interaction with pia, perivascular space, and neuronal cells. Data in C are graphed as mean ± SD, with n values of 4 for each group. All histological images are of 12-μm-thick sections. Cx, cortex.
Live imaging of the PD effect of the Gabra1 ASO.
Having established the dose-dependent anatomical PK of Gabra1 ASOs, we next focused on the PD effect of ASOs in a live brain and used 18F-flumazenil, a high-affinity PET ligand for the benzodiazepine site of the GABAA receptor, to track the effect of Gabra1 ASOs on receptor knockdown in vivo with live imaging. For these experiments, baseline microPET imaging of 2 cohorts of animals (n = 4 each) was collected following intravenous injection with 18F-flumazenil. The cohorts then received a lumbar IT dose (1 mg) of ASOs targeting either the Gabra1 mRNA or a control ASO (ASO targeting ubiquitously expressed long noncoding RNA Malat1). Animals were then serially imaged by PET after intravenous administration of 18F-flumazenil at 1, 2, 3, and 4 weeks after ASO dosing. Figure 9 shows the static and temporal effects of the Malat1 control versus Gabra1-targeted ASOs on brain 18F-flumazenil imaging of the GABAA receptor. As shown for individual brain examples, animals from both groups had a similar baseline level of brain 18F-flumazenil uptake. However, compared with baseline, IT treatment with Gabra1-targeting ASOs markedly reduced brain 18F-flumazenil uptake over time, while treatment with the Malat1 control ASO did not (Figure 9A). The time-course analysis of regional brain 18F-flumazenil uptake in all ASO-treated animals showed a progressive decline in cerebral cortical 18F-flumazenil signal in the Gabra1 ASO–treated animals, but not the Malat1 control ASO–treated animals (Figure 9B). This effect was also seen in the thalamus but was less pronounced and took longer to develop in the striatum. When the AUC values for 18F-flumazenil tissue concentration versus time were calculated, statistically significant differences between Gabra1 ASO and Malat1 control ASO treatment groups were observed at weeks 2, 3, and 4 in the cortex and thalamus and at weeks 3 and 4 in the striatum (Figure 9C).
Figure 9. Live pharmacodynamic PET imaging of Gabra1 ASO–mediated receptor knockdown.

(A) 18F-flumazenil brain uptake in representative Malat1 ASO– or Gabra1 ASO–treated animals. Horizontal and coronal images. (B) 18F-flumazenil tissue concentration curves in Malat1 ASO (black) or Gabra1 ASO (red) groups at baseline or various time points after dosing. (C) AUC of 18F-flumazenil tissue concentration curves at each time point expressed as a percentage of baseline AUC for different brain regions. Malat1 ASO group is shown in red and Gabra1 ASO group is shown in black. Data are graphed as mean ± SD, with n values of 4 for each group. Analysis of differences between AUCs between groups and time points was by 2-way ANOVA with Bonferroni’s post hoc test; *P < 0.05.
Voxel-level maps of 18F-flumazenil AUC were created for each animal by calculating the AUC at each voxel in the brain. 3D group-level brain 18F-flumazenil AUC maps for the Malat1 ASO– versus Gabra1 ASO–treated groups were created by averaging the individual AUC maps of all animals in each group after co-registration. There was a marked overall reduction in brain 18F-flumazenil AUC values in the Gabra1 ASO–treated rats compared with the Malat1 control ASO–treated rats (Figure 10A). By subtracting the 18F-flumazenil AUC maps between the 2 groups from scans at 4 weeks after IT ASO injection, we generated a difference map displaying the brain regions most impacted by the ASO treatment (Figure 10B). This relative PD anatomical map of Gabra1 ASO action on brain 18F-flumazenil uptake matched the pattern of Cy7-labeled Gabra1 ASO distribution observed in individual animal samples at earlier time points in both 3D and 2D reconstructions of the CFT experiment with Cy7-labeled Gabra1 ASO (Figure 10C). These data suggest a tight temporal and regional anatomical relationship between the PK and PD effects of IT-delivered ASOs in the brain.
Figure 10. Nuclear and fluorescence imaging maps of ASO PK and PD.

(A) Group-averaged AUC images of 18F-flumazenil PET data from Malat1 ASO– or Gabra1 ASO–treated animals (n = 6 each) at 4 weeks. (B) Map of 18F-flumazenil PET signal difference between Malat1 ASO– and Gabra1 ASO–treated animals at 4 weeks. (C) 3D and 2D CFT images of Gabra1 Cy7-ASO at 8 and 12 hours, respectively.
Discussion
ASOs exert their pharmacological effects by gaining intracellular access to reach their targeted RNA at the desired anatomical site of action. Although IT-delivered ASOs achieve this quite readily throughout the CNS, a deeper understanding of their PK and PD has been challenging due to the unique anatomy and physiology of the IT space. We used advanced multimodal in vivo imaging techniques combined with ex vivo imaging and traditional histological analysis to track the path of ASOs from their site of delivery via lumbar puncture to cells in various regions of the rat CNS. Following ASOs along this path has highlighted several of the natural processes that govern movement of macromolecules in and out of the CSF.
Detailed PK and PD analysis confirmed that once ASOs access the CNS they have a rapid onset of action, with intracellular uptake leading to target RNA suppression that is maximal between 1 and 3 days after injection. It takes approximately 24 hours for the ASO to traverse through the CSF into the parenchyma and into the neurons, with suppression of target RNA expression then being sustained for multiple weeks after a single dose. This long-lasting target suppression is correlated with ASO presence and reverses as the ASOs are cleared from the tissue. This is consistent with previous reports of other CNS-delivered ASOs of similar chemistry in both rodents and primates (33, 34).
The bulk macro distribution of ASOs throughout the CSF, into the CNS tissues, and ultimately out into the periphery was also as expected based on IT-dosing principles recently revealed through imaging (35, 36). Using radiolabeled ASOs and live SPECT imaging we documented the rapid passage of lumbar IT-dosed ASOs rostrally along the rat neuraxis into the cranium, as well as their egress from the IT compartment across the nasal turbinates and into peripheral lymph nodes. These patterns of ASO molecular movement are consistent with the classical behavior of other large IT-dosed molecules into rodents (37–42). However, egress of molecules from the CNS into nasal passages is less prominent in primates than other mammals with more prominent snouts (43) and the exact mechanism for clearance of molecules from CSF to the periphery in humans remains unknown. Proteins, protein-binding dyes (Evans blue), or particulate material (India ink) injected into the IT compartment have been demonstrated to pass into perispinal, abdominal, deep cervical, and submandibular lymph nodes via lymphatic channels located in the meninges or in the coverings of spinal and cranial nerves (20, 23). Although the molecular weight of therapeutic ASOs is typically less than 10 kDa, their protein binding characteristics (37) are consistent with the prominent association we observed with meninges and nerve sheaths, as well as their subsequent passage to lymph nodes. The eventual accumulation of some signal in the kidneys is also consistent with the known mechanisms of renal clearance of ASOs from the peripheral circulation (38, 39).
The transfer of ASOs from the CSF into and through the brain parenchyma likely utilizes multiple mechanisms. ASOs first associate and track with the surface pia and major arterial branches at early time points, suggesting, at least in part, that ASOs likely gain access to the parenchyma via the pial-glial basement membranes of penetrating cerebral arterioles. Our fluorescence tomography technique confirmed the association of unlabeled and labeled ASOs with major cerebral surface arteries at early time points before permeation into deeper brain tissue. Long-lasting association of ASO signal with vascular elements was illustrated by costaining with the Reca1 and αSMA vascular markers, as well as colocalization with the basement membrane components laminin α2 and perlecan. These observations suggest that intramural perivascular compartments may participate in the movement of ASOs in and out of the CNS parenchyma, as shown previously with other molecules (12, 13, 22–26). The IT dose-response evaluation of ASOs at 14 days after dosing showed that the meningeal and vascular wall ASO IHC signal was high at all doses, while neuronal ASO signal increased dose dependently. These data may suggest that the vascular intramural basement membranes and the surface pia quickly absorb the ASOs and increased doses allow the ASO to penetrate these membranes into the parenchyma of the CNS.
ASO IHC data at early time points also suggested a mass movement of ASO from high CSF concentrations directly into the adjacent cortex and cerebellum by passage across the glia limitans, potentially through astrocytic end-feet gap junctions. We also cannot rule out a role for transcellular exchange across the astrocytic cells of the glia limitans to the parenchyma based on observed ASO staining within astrocytes preceding that seen within neurons. Indeed, in the spinal cord, we observed a rapid centripetal movement of ASOs along axonal tracts of the ventral and dorsal root bundles, preceding intracellular accumulation of ASO in neurons. These axonal bundles contain GFAP-positive astrocytes. Astrocyte-like radial glia are known to bridge the central core of the neuraxis to the pial surface during embryonic development and persist into adult life in some brain regions. Thus, it is conceivable that astrocyte-assisted uptake of ASOs across the pia and glia limitans may be broadly involved in ASO transfer from the CSF to neurons throughout the parenchyma. Astrocyte-to-neuron passage of many intermediary metabolites and larger molecules is well appreciated (14), and further experiments are needed to resolve whether fluid movement into and across astrocytes via aquaporin channels, as espoused by the glymphatic concept, contributes to ASO transcellular movement (13).
Early distribution of the ASO along white-matter tracts was observed as diffuse extracellular staining in the corpus callosum. This diffuse staining changed by 24 hours after dosing to an intracellular pattern in the cells presumably resembling oligodendrocytes and astrocytes. It is possible that, following early access to white-matter tracts, ASOs may travel along the extramyelin water compartments into deeper brain structures as well as into resident oligodendrocytes and astrocytes of the white matter. Centripetal movement along the extramyelin water compartments of the ventral and dorsal root axonal bundles could conceivably explain the very rapid distribution of ASOs into the cells of the gray matter of the spinal cord by 30 minutes after dosing.
Taken together, these data support a model in which following lumbar cistern IT delivery, ASOs distribute rapidly throughout the CSF along the neuraxis and associate with the meningeal layer covering the CNS and penetrating cerebral arteries. ASO movement into and within the parenchyma likely involves perivascular pathways, white-matter tracts, and possibly direct migration through the glial limitans via gap junctions or transcellular exchange mechanisms between the pial cells and adjacent astrocytes. Once distributed to the various regions of the CNS, ASOs are taken up into cells where their action is exerted on the target RNA. Our results also indicate that in rats movement across the nasal turbinates, through meningeal lymphatics and into peripheral lymph nodes, and then to systemic circulation is a route of clearance of ASO from the CNS compartment.
Although the mechanistic underpinning of our largely descriptive work requires further elucidation, our work does provide a proof of concept for utilizing advanced imaging techniques to track the PK and PD of RNA therapeutics both spatially and temporally. This is exemplified by the Gabra1-targeting ASOs, where brain entry and tissue distribution took place over several hours, as demonstrated by the block-face CFT. This was then followed by a protracted PD effect demonstrated by 18F-flumazenil PET imaging. Comparison of the 2D difference maps of the summated 18F-flumazenil PET images between the Gabra1 ASO–treated and Malat1 ASO control groups to the 2D Cy7-labeled Gabra1 ASO images demonstrated that the most prominent regional brain effect of Gabra1 ASOs on reduction of 18F-flumazenil binding correlated with the regions of greatest Cy7-labeled-ASO uptake. These studies confirmed that imaging of ASO PD effects is representative of ASO PK in tissue samples. Such invaluable imaging insights can be used to further optimize ASO delivery, as well as understand the distribution of other protein, RNA, and gene therapy therapeutics.
One limitation of our work is that it focused on IT dosing in rats. Even though this species is largely representative of ASO PK/PD properties (34), species-specific variations in the IT anatomy of primates may yield different observations. The large nasal cavity in the rats may, for example, result in faster clearance of the ASO than what may be seen in primates with smaller nasal cavities (43). ASO distribution along the neuraxis in primates has been shown to be very broad, with similar target reduction in the spinal cord as in the cortex (40). Fortunately, the methods developed here are directly translatable to primates, with the benefit of detailed kinetics possible with fewer animals needed than traditional necropsy studies. Future studies will explore IT dosing optimization in primates.
ASOs have the potential to target underlying disease pathophysiology and deliver dramatic benefits to patients. The success of the IT ASO Spinraza in spinal muscular atrophy (41) and the preclinical work across many neurodegenerative diseases (30, 31, 35, 36, 42, 44) is extremely encouraging. Together, our multimodal imaging approaches have helped overcome the challenges of studying the PK and PD of these molecules in the IT space and have helped provide insights into IT ASO pharmacology. The unique pharmacological properties of ASOs make them promising therapeutics, and the detailed understanding of their IT pharmacology provided here will help to deliver on this promise.
Methods
Animals.
All animals used in the studies described herein were male Sprague-Dawley rats 6 to 8 weeks of age maintained on a 12-hour light/12-hour dark cycle and provided food and water ad libitum. Animals used in the GluR1 ASO time course and dose-response studies were obtained from Envigo. Animals used in the 125I-Malat1 ASO imaging, CFT, and Gabra1 18F-flumazenil imaging studies were obtained from Charles River Laboratories. IT administration of substances was accomplished by bolus injection of the substances in 30 μL followed by 40 μL vehicle flush via lumbar IT catheterization, as described by Mazur et al. (33).
Imaging of ASO distribution using 125I-Malat1 ASO and in vivo SPECT imaging.
Piperidine-modified ASO (Ionis Pharmaceuticals, Inc.) was radiolabeled with 125I using a Bolton-Hunter approach by 3D Imaging. Fifty microliters of a 50 μM sodium phosphate buffer containing 125I was combined with 20 μL Bolton-Hunter reagent and 10 μL Chloramine-T, allowed to react for 30 seconds, and was then quenched with 10 μL sodium metabisulfite. Five microliters of DMF was added to the vessel and the 125I-labeled Bolton-Hunter reagent was extracted using benzene (2 × 250 μL), and then the solvent was removed under reduced pressure and the solute reconstituted in a minimal amount of DMSO. In a separate reaction vessel, 100 μg of the piperidine-modified Malat1 ASO was added to 20 μL 0.1 M sodium borate buffer (pH = 8.5), and then the 125I-Bolton-Hunter reagent was added in DMSO/borate buffer (1:1 ratio), which was allowed to react at room temperature for 2 hours. The reaction was quenched with 50 μL of 2 M NaOH and purified via G-25 Sephadex column. 125I-ASO product was analyzed via HPLC and determined to have a specific activity of 1.14 mCi/mg at a concentration of 6 mg/mL.
125I-Malat1 ASO imaging studies were performed at MPI Research. All animals received 20 mM potassium iodide water at least 72 hours prior to 125I-ASO dosing to block thyroid uptake of radioactive iodine. For imaging, anesthesia was induced with 3%–4% isoflurane and maintained with 1%–2% isoflurane in 0.5–1 L/min oxygen during each scan.
Immediately prior to the start of image acquisition, 230.7 ± 42.6 μCi (180 μg/rat) of 125I-Malat1 ASO was delivered by IT injection. Whole-body SPECT image data were acquired on the NanoSPECT/CT (Mediso Medical Imaging Systems). Semidynamic data were acquired from 0–1 hour (4 × 9-minute frames) after injection. Static data (1 × 30 minute scans) were then acquired at 4 hours, 1 day, 2 days, and 7 days after injection. SPECT acquisition parameters were as follows: energy window of 23.1–32.9 keV, 20 projections per axial field of view, 30 seconds per projection, and 9-pinhole aperture (diameter of 1.4 mm). Each SPECT scan was followed by an 8- to 10-minute CT scan (peak energy 65 kVp, 180 projections per rotation, 0.5 seconds per projection) for anatomical registration and analysis. Reconstructed images from the NanoSPECT/CT were generated in units of radioactivity and the values assigned to the voxels comprising the 3D-reconstructed SPECT images are in units of kBq.
Ex vivo ASO IHC staining time course.
Studies were performed at Ionis Pharmaceuticals, Inc. Thirteen groups of 4 animals were dosed with 0.7 mg of the GluR1 ASO and 1 group of 4 animals was dosed with artificial CSF vehicle. The animals were euthanized by inhalation of CO2 and decapitated at 15, 30, and 60 minutes; 3, 6, 8 and 24 hours; and 3, 7, 14, 28, 56, and 112 days (16 weeks). After euthanasia the brain and spinal cord were harvested from each animal and the right brain hemisphere and portions of the cervical, thoracic, and lumbar spinal cord embedded and frozen in OCT. The orbital cortex from the left hemisphere and portions of the cervical, thoracic, and lumbar spinal cord were frozen for analysis of GluR1 ASO tissue concentrations by HELISA and these concentrations were graphed versus time and the AUC values for these data were calculated using the trapezoidal rule. Twelve-micron-thick sagittal sections of the OCT-embedded tissues were sectioned on a cryostat onto charged slides and kept at –80°C. At the start of the staining protocol, sections were dried at room temperature for 5 minutes. Sections were fixed in 4% paraformaldehyde in 1× PBS. Sections were then washed 3 times 5 minutes in PBS and subsequently blocked in 10% goat serum with 1% BSA for 30 minutes at room temperature. The solution was aspirated off and replaced with 10% goat serum/1% PBS with primary antibodies. Antibodies used were ASO (in-house generated rabbit polyclonal, 1:1000) and NeuN (Millipore chicken anti-NeuN, ABN91, 1:1000). Sections were incubated for 1 hour at room temperature and then washed in 1× PBS for 3 times 5 minutes. Secondary antibodies goat anti-rabbit Alexa Fluor 647 (Thermo Fisher Scientific, A-21244) and goat anti-chicken Alexa Fluor 555 (Thermo Fisher Scientific, A21437) were diluted 1:500 in 10% goat serum/1% BSA and incubated for 30 minutes at room temperature before a final wash of 3 times 5 minutes with 1× PBS and a brief rinse in H2O.
Adjacent sections were stained for GluR1 protein (Millipore mouse anti-GluR1, clone RH95) and counterstained with NeuN (listed above) and secondary antibodies (goat anti-mouse IgG[H+L], Alexa Fluor 555 and goat anti-chicken listed above) were used to visualize the primary antibodies. Slides were dried and scanned in an Agilent G2505B scanner at 5-μm resolution and 50% PMT. Images were processed in ImageJ 1.47v (NIH). To evaluate the GluR1 mRNA expression in the cortex, spinal cord segments, hippocampus, and cerebellum were scraped off adjacent histological sections under a dissecting microscope and used for qRT-PCR analysis.
Diaminobenzidine IHC staining for ASO and GFAP.
Brain hemispheres and portions of the cervical, thoracic, and lumbar spinal cord were harvested and immersion fixed in 10% neutral buffered formalin solution. The tissue was processed on a Sakura Tissue-Tek tissue processor; after embedding, slides were cut at 5 μm, air dried overnight, and then dried at 60°C for 1 hour. Adjacent slides were stained with Ionis Pharmaceuticals rabbit polyclonal anti-ASO and rabbit polyclonal anti-GFAP (Dako, Z033401-2) antibodies on a Ventana Ultra staining system. ASO slides were treated enzymatically with trypsin (Sigma-Aldrich, T8003) and the GFAP slides were treated enzymatically with protease 2 (Ventana, 760-2019). All the slides were then blocked with an Endogenous Biotin Blocking Kit (Ventana, 760-050) and normal goat serum. The primary antibodies were diluted with Discovery Antibody Diluent (Ventana, 760-108) and incubated for 1 hour at 37°C. The antibodies were detected with biotin-labeled goat anti-rabbit secondary antibody (Jackson ImmunoLabs, 111-005-003). The secondary antibody was labeled with a DABMap Kit (Ventana, 760-124). The slides were scanned on a Hamamatsu S360 scanner at ×40 resolution.
Tissue HELISA.
Previously frozen cortex and spinal cord tissue samples were weighed, minced, and homogenized with homogenization buffer (20 mM Tris pH 8, 20 mM EDTA, 0.1 M NaCl, 0.5% NP40) and beads (Matrix Green, Bio 101 no. 6040-801) in a 96-well format plate. A calibration curve and QC samples of known ASO concentrations were prepared in normal homogenized brain and analyzed on the same plate. The ASO was extracted from the tissue, curve, and QC samples via a liquid-liquid extraction using ammonium hydroxide and phenol/chloroform/isoamyl alcohol (25:24:1) (Sigma-Aldrich). The aqueous layer was evaporated to dryness and reconstituted in normal K2EDTA human plasma (Bioreclamation). Reconstituted tissue, curve, and QC samples were further diluted in normal human plasma, as necessary, and analyzed via HELISA, which is a variation on the method reported by Yu et al. (44), on a Molecular Devices Spectramax Gemini XPS. Briefly, the assay involves binding of the analyte ASO drug to the sense DNA probe and subsequent detection of the double-stranded hybridized drug using an antibody that recognizes a 5′-linked digoxigenin. Selection of the double-stranded hybrid is accomplished by elimination of single-stranded (nonhybridized) probe using S1 nuclease digestion. Sample quantitation was calculated from the coextracted calibration curve using SoftMax Pro (Molecular Devices, LLC).
Ex vivo ASO IHC staining dose response.
Studies were performed at Ionis Pharmaceuticals, Inc. Six groups of 4 animals were dosed IT with 10, 30, 100, 300, or 1000 μg of the GluR1 ASO or CSF vehicle. The animals were euthanized by inhalation of CO2 and decapitation at 14 days after injection. After euthanasia the brain was harvested from each animal and the right brain hemisphere embedded and frozen in OCT. Tissues were sectioned at 12-μm thickness, processed, and stained as described above in the Ex vivo ASO IHC staining time course section. Antibodies used were ASO (in-house generated rabbit polyclonal, 1:1000), RECA-1 (Bio-Rad chicken anti-RECA1, MCA970, 1:1000), NeuN (Millipore chicken anti-NeuN, ABN91, 1:1000), goat anti-rabbit Alexa Fluor 647 (Thermo Fisher Scientific, A-21244), and goat anti-chicken Alexa Fluor 555 (Thermo Fisher Scientific, A21437). Slides were dried and scanned in an Agilent G2505B scanner at 5-μm resolution and 50% PMT or imaged on an EVOS microscope (Thermo Fisher Scientific). Images were processed in ImageJ 1.47v.
GABRA Cy7 labeling and CFT.
Gabra1 ASO was labeled with sulfo-Cy7 NHS ester (Lumiprobe). Briefly, 5′-hexylamino–modified ASO dissolved in 0.1 M sodium tetraborate buffer, pH 8.5 (20 mM) was mixed with Cy7 NHS ester (3 eq.) dissolved in DMSO (40 mM). Conjugation was complete after 3 hours. Conjugate was purified using strong anionic-exchange HPLC and desalted using C18 reverse-phase HPLC.
Cy7-ASO was injected IT (33) and allowed to distribute for 1, 2, 4, 6, 8, and 12 hours after dosing when the animals were euthanized by CO2 inhalation and decapitation. Immediately following euthanasia brains were resected, embedded whole in OCT, and frozen on dry ice. Once fully frozen, blocks were loaded into a Leica 3050M cryostat (Leica Camera AG) and sectioned in 25-μm-thick increments in a ventral to dorsal orientation, with the temperature maintained at –17°C. Fluorescence and white-light images were acquired after every section with the FLARE surgical fluorescence imaging system (Curadel LLC) using the NIR-2 channel at 770 nm. The fluorescence images were processed using a Lucy-Richardson deconvolution (45, 43) with measured point-spread function resulting in a 3D fluorescence distribution recovery.
The series of white-light images was self-registered using a mutual information alignment method with a dynamically updated reference image formed by a proximity-weighted linear combination of adjacent slice images. The fluorescence and white-light images were natively registered due to the construction of the imaging system, allowing the same registration transforms to be applied to the fluorescence images. Individual slices were stacked to form 3D white-light and fluorescence images (46). The resulting voxel size of the image stack was 28 μm × 28 μm × 25 μm. The 3D images were resampled to isotropic 25-μm voxels for the purposes of qualitative analysis and rendering (47).
Costaining ASO and vascular markers by IHC.
Frozen sections collected during CFT imaging were rehydrated with wash buffer for 10 minutes and fixed with 4% paraformaldehyde for 15 minutes. Following a 30-minute permeabilization step with 0.05% Triton X-100, sections were blocked with Background Sniper (Biocare Medical, BS966) for 15 minutes followed by 5% normal donkey serum in PBS/0.03% Triton X-100 for 45 minutes at room temperature. Primary antibodies were incubated overnight at 4°C and included rabbit polyclonal anti-GFAP (1:8000, Novus Biological, NB300-141), rabbit polyclonal anti-αSMA (1:800, Novus Biological, NB600-531), mouse monoclonal anti-NeuN (1:800, EMD Millipore, MAB377), and mouse monoclonal anti-RecA1 (1:800, GeneTex, GTX6652). A secondary donkey anti-mouse or anti-rabbit conjugated to Alexa Fluor 555 was incubated for 1 hour at room temperature (Abcam, ab150074 or ab150106). Sections were mounted with Prolong Gold (Thermo Fisher Scientific, P36966) and coverslipped.
In another series of ASO and vascular marker costaining experiments, animals injected IT with either the GluR1 or the Cy7-Gabra1 ASO at 0.7 mg were euthanized and the brains and spinal cords frozen. Brain and spinal cord tissues were sectioned on a cryostat at 12-μm thickness and stored frozen at –80°C until shipped to the University of Southampton, United Kingdom, where they were stained for ASO and the vascular markers by IHC. The sections were stained for ASO using the Ionis Pharmaceuticals rabbit polyclonal anti-ASO antibody at 1:3000 dilution. The rabbit antibody was detected with a secondary goat anti-rabbit conjugated to Alexa Fluor 594 at 1:400 dilution. Sections were counterstained for the basement membrane marker perlecan, the glia limitans basement membrane marker laminin α2, and αSMA. Perlecan was stained using a rat anti-perlecan monoclonal antibody at 1:100 dilution (Sigma-Aldrich; clone A716) and visualized with a secondary goat anti-rat antibody conjugated with Alexa Fluor 488 at 1:400 dilution. Adjacent sections were stained for the glia limitans basement membrane marker laminin α2 using a rat anti–laminin α2 antibody at 1:200 dilution (Abcam; clone 4H8-2). This antibody was visualized with a goat anti-rat antibody conjugated to Alexa Fluor 488 at 1:400 dilution. αSMA was stained using an anti-αSMA antibody conjugated to FITC at 1:200 dilution (Sigma-Aldrich; clone 1A4). Sections with immunofluorescence were analyzed using an SP8 confocal laser-scanning microscope (Milton Keys) and exported to ImageJ software. Montages were created with Adobe Photoshop CS6.
18F-flumazenil PET imaging experiment: ASO treatment.
Malat1 and Gabra1 ASO dosing and 18F-flumazenil imaging experiments were performed at Invicro, LLC. Animals were dosed IT with 1 mg of either the Gabra1 or the Malat1 ASO.
18F-flumazenil PET imaging experiment: 18F-flumazenil production/radiochemistry.
No-carrier-added 18F produced via 18O(p,n) 18F reaction via cyclotron was purchased from PETNET. Flumazenil precursor nitromazenil (4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid, 5,6-dihydro-5-methyl-8-nitro-6-oxo-, ethyl ester; CAS no. 84377-97-9) was purchased from Advanced Biochemical Compounds. Flumazenil reference standard (4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid 8-fluoro-5,6-dihydro-5-methyl-6-oxo-, ethyl ester; CAS no. 78755-81-4) was purchased from Advanced Biochemical Compounds. 18F-flumazenil was prepared according to the method previously described by Ryzhikov et al. (48). Radiochemical purity ranged from 98% to 99% and specific activity ranged from 37 to 740 GBq/μmol at the end of synthesis (n = 6).
18F-flumazenil PET imaging experiment: PET imaging procedure.
18F-flumazenil PET data were acquired from each animal at 5 different time points to measure GABAA receptor availability and change in GABAA receptor availability due to ASO treatment over time. Baseline 18F-flumazenil scans were performed on all animals 1 day prior to ASO administration (day –1). After ASO administration, 18F-flumazenil imaging data were acquired weekly for 4 weeks.
Single intravenous doses of 18F-flumazenil were administered over 5 seconds on the PET scanner bed via a pre-placed catheter line beginning 10 seconds after the start of the scan. The line was flushed with saline, removed, and residual activity was recorded. For each synthesis of 18F-flumazenil, 4 pairs of rats were injected and imaged, except week 1, where 2 syntheses were performed due to low tracer yield. The injected radioactivity dose for each group at each time point is displayed in Table 1.
Table 1. Doses of 18F-flumazenil prepared and injected into the groups of rats treated with either the Malat1 ASO or Gabra1 ASO at each time point in the PET imaging study.

Focused-head PET data were acquired on the microPET Focus 220 (Siemens Medical Solutions USA, Inc.). Two animals were imaged during each scan. Animals were positioned side by side within the scanner on a specially designed, 3D-printed rat bed. Dynamic data were acquired in 3D listmode for 60 minutes. PET data were reconstructed with image resolution of 1.5 mm FWHM and corrections for decay, random coincidences, dead time, nonuniform radial sampling, and normalization by an OSEM 3D fast MAP (2 OSEM iterations, 16 MAP) algorithm. Reconstructed images were binned into frames of 3 × 10 seconds, 1 × 30 seconds, 4 × 60 seconds, 5 × 300 seconds, 3 × 600 seconds and produced in units of radioactivity. Namely, the values assigned to the voxels (volume elements) comprising the 3D-reconstructed PET images were in units of nCi/cm3.
PET image quantification and registration/time-activity curves.
18F-flumazenil PET time-activity curves of decay-corrected percentage injected dose per gram (%ID/g) were generated from the dynamic PET reconstructions by registering each animal’s PET scan (summed 0–60 minutes image) to a rat brain atlas. Volumes of interest included cerebral cortex, thalamus, and striatum. An area under the curve (AUC) of each region time-activity curve for each animal was calculated at each imaging time point. Group-averaged voxel-wise AUC PET images were generated for Malat1 ASO and Gabra1 ASO treatment groups.
Approvals.
All study procedures using animals performed at MPI Research were approved by the Institutional Animal Care and Use Committee (IACUC) of MPI Research under IACUC protocol 1881-085. All study procedures using animals performed at Invicro, LLC were approved by the IACUC of Invicro LLC and performed under IACUC protocol 010-2014. All study procedures using animals performed at Ionis Pharmaceuticals were approved by the Ionis Pharmaceuticals IACUC committee and performed under IACUC protocol P-0282. MPI Research, Invicro, LLC, and Ionis Pharmaceuticals, Inc. are all AALAC accredited.
Statistics.
All statistical analyses were performed with GraphPad Prism version 8.1.2. In all cases where error bars are presented they represent standard deviation around the mean value. The AUC for the GluR1 concentrations versus time and the 18F-flumazenil signal versus time were calculated using the trapezoidal rule. GluR1 concentration AUC values were compared between tissues using 1-way ANOVA with Bennett’s post hoc analysis. Differences in 18F-flumazenil PET activity between Gabra1 ASO– and Malat1 ASO–treated animals were evaluated with 2-way repeated-measures ANOVA with Bonferroni’s post hoc test to compare between treatments at each time point. For all statistical analyses P < 0.05 was considered statistically significant.
Author contributions
CM assisted in designing the experiments, conducted experiments, collected and analyzed data, and is primary author of the manuscript. BP and FK conducted experiments, collected and analyzed data, and assisted in writing the manuscript. MO worked out the chemistry of labeling ASOs with fluorescent and radioactive conjugates and provided reagents. AV, RC, JMS, KZ, HD, and JH assisted in designing the experiments, analyzed data, and assisted in writing the manuscript. MS, VG, NJA, and LK conducted experiments and collected and analyzed data. RWH and MQ analyzed data. IP conducted experiments and collected data. ROC and MB assisted in designing the experiments, conducted experiments, and collected and analyzed data. JM conducted experiments, collected and analyzed data. CMG collected, analyzed, and managed data. HK assisted in writing the manuscript. EES assisted in designing the experiments.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Tracy Reigle for her graphics artistry in preparing the figures for this manuscript. The authors would like to thank the laboratory, project management, and analysis teams of Invicro and MPI Research for their support and expert execution of the nuclear and fluorescence imaging studies. The authors would also like to thank Dan Norris for his input in the writing of this manuscript. The research described in this publication was wholly funded by Biogen and Ionis Pharmaceuticals, Inc.
Version 1. 10/17/2019
Electronic publication
Footnotes
AV’s present address is: Codiak Biosciences, Cambridge, Massachusetts, USA.
Conflict of interest: CM, BP, JM, FK, MO, HK, and EES are employees of and stock holders in Ionis Pharmaceuticals, Inc. JH is a cofounder and managing partner of Invicro, LLC as well as an equity shareholder. KZ, HD, MS, JMS, RWH, MQ, IP, RC, VG, and CMG were employees of Invicro, LLC when the studies were performed. JMS is currently an employee of Biogen Inc. NJA, LK, and ROC are employees of the University of Southampton. MB is an employee of 3D Imaging, and AV was an employee of Biogen Inc. at the time that this work was performed.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(20):e129240.https://doi.org/10.1172/jci.insight.129240.
Contributor Information
Curt Mazur, Email: CMazur@ionisph.com.
Berit Powers, Email: BPowers@ionisph.com.
Kenneth Zasadny, Email: kzasadny@invicro.com.
Hemi Dimant, Email: hdimant@invicro.com.
Fredrik Kamme, Email: fkamme@ionisph.com.
Jacob Hesterman, Email: hesterman@invicro.com.
John Matson, Email: jmatson@ionisph.com.
Marc Seaman, Email: marcseaman84@gmail.com.
Ildiko Polyak, Email: ipolyak@invicro.com.
Richard Coelho, Email: rcoelho@invicro.com.
Vijay Gottumukkala, Email: vgottumukkala@invicro.com.
Nazira J. Albargothy, Email: nazira.albargothy@liu.se.
Louise Kelly, Email: l.kelly@soton.ac.uk.
Roxana O. Carare, Email: rcn@soton.ac.uk.
Jack Hoppin, Email: hoppin@invicro.com.
Holly Kordasiewicz, Email: hkordasiewicz@ionisph.com.
Ajay Verma, Email: ajay.verma@precisionmedicinegrp.com.
References
- 1.Wurster CD, et al. Intrathecal administration of nusinersen in adolescent and adult SMA type 2 and 3 patients. J Neurol. 2019;266(1):183–194. doi: 10.1007/s00415-018-9124-0. [DOI] [PubMed] [Google Scholar]
- 2.Finkel RS, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–3026. doi: 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 3.Southwell AL, Skotte NH, Bennett CF, Hayden MR. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med. 2012;18(11):634–643. doi: 10.1016/j.molmed.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 4.DeVos SL, Miller TM. Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics. 2013;10(3):486–497. doi: 10.1007/s13311-013-0194-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao J, et al. Treatment of Epstein-Barr virus associated central nervous system diseases after allogeneic hematopoietic stem cell transplantation with intrathecal donor lymphocyte infusion. Bone Marrow Transplant. 2019;54(6):821–827. doi: 10.1038/s41409-018-0409-9. [DOI] [PubMed] [Google Scholar]
- 6.Giugliani R, et al. Neurological manifestations of lysosomal disorders and emerging therapies targeting the CNS. Lancet Child Adolesc Health. 2018;2(1):56–68. doi: 10.1016/S2352-4642(17)30087-1. [DOI] [PubMed] [Google Scholar]
- 7.Hardcastle N, Boulis NM, Federici T. AAV gene delivery to the spinal cord: serotypes, methods, candidate diseases, and clinical trials. Expert Opin Biol Ther. 2018;18(3):293–307. doi: 10.1080/14712598.2018.1416089. [DOI] [PubMed] [Google Scholar]
- 8.Logan MR, McClure JH, Wildsmith JA. Plain bupivacaine: an unpredictable spinal anaesthetic agent. Br J Anaesth. 1986;58(3):292–296. doi: 10.1093/bja/58.3.292. [DOI] [PubMed] [Google Scholar]
- 9.Sallerin-Caute B, Lazorthes Y, Monsarrat B, Cros J, Bastide R. CSF baclofen levels after intrathecal administration in severe spasticity. Eur J Clin Pharmacol. 1991;40(4):363–365. doi: 10.1007/BF00265844. [DOI] [PubMed] [Google Scholar]
- 10.Hocking G, Wildsmith JA. Intrathecal drug spread. Br J Anaesth. 2004;93(4):568–578. doi: 10.1093/bja/aeh204. [DOI] [PubMed] [Google Scholar]
- 11.Abbott NJ, Pizzo ME, Preston JE, Janigro D, Thorne RG. The role of brain barriers in fluid movement in the CNS: is there a ‘glymphatic’ system? Acta Neuropathol. 2018;135(3):387–407. doi: 10.1007/s00401-018-1812-4. [DOI] [PubMed] [Google Scholar]
- 12.Albargothy NJ, et al. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018;136(1):139–152. doi: 10.1007/s00401-018-1862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iliff JJ, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4(147):147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tarasoff-Conway JM, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457–470. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev. 2018;98(1):239–389. doi: 10.1152/physrev.00042.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magistretti PJ. Neuron-glia metabolic coupling and plasticity. Exp Physiol. 2011;96(4):407–410. doi: 10.1113/expphysiol.2010.053157. [DOI] [PubMed] [Google Scholar]
- 17.Geer CP, Grossman SA. Interstitial fluid flow along white matter tracts: a potentially important mechanism for the dissemination of primary brain tumors. J Neurooncol. 1997;32(3):193–201. doi: 10.1023/A:1005761031077. [DOI] [PubMed] [Google Scholar]
- 18.Budnik V, Ruiz-Cañada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci. 2016;17(3):160–172. doi: 10.1038/nrn.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carare RO, Hawkes CA, Jeffrey M, Kalaria RN, Weller RO. Review: cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination failure angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol Appl Neurobiol. 2013;39(6):593–611. doi: 10.1111/nan.12042. [DOI] [PubMed] [Google Scholar]
- 20.Harling-Berg C, Knopf PM, Merriam J, Cserr HF. Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J Neuroimmunol. 1989;25(2-3):185–193. doi: 10.1016/0165-5728(89)90136-7. [DOI] [PubMed] [Google Scholar]
- 21.Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun. 2017;8(1):1434. doi: 10.1038/s41467-017-01484-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zenker W, Bankoul S, Braun JS. Morphological indications for considerable diffuse reabsorption of cerebrospinal fluid in spinal meninges particularly in the areas of meningeal funnels. An electronmicroscopical study including tracing experiments in rats. Anat Embryol. 1994;189(3):243–258. doi: 10.1007/BF00239012. [DOI] [PubMed] [Google Scholar]
- 23.Andres KH, von Düring M, Muszynski K, Schmidt RF. Nerve fibres and their terminals of the dura mater encephali of the rat. Anat Embryol. 1987;175(3):289–301. doi: 10.1007/BF00309843. [DOI] [PubMed] [Google Scholar]
- 24.Miura M, Kato S, von Lüdinghausen M. Lymphatic drainage of the cerebrospinal fluid from monkey spinal meninges with special reference to the distribution of the epidural lymphatics. Arch Histol Cytol. 1998;61(3):277–286. doi: 10.1679/aohc.61.277. [DOI] [PubMed] [Google Scholar]
- 25.Aspelund A, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212(7):991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Louveau A, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aldea R, Weller RO, Wilcock DM, Carare RO, Richardson G. Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front Aging Neurosci. 2019;11:1. doi: 10.3389/fnagi.2019.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papisov MI, et al. Delivery of proteins to CNS as seen and measured by positron emission tomography. Drug Deliv Transl Res. 2012;2(3):201–209. doi: 10.1007/s13346-012-0073-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf DA, et al. Dynamic dual-isotope molecular imaging elucidates principles for optimizing intrathecal drug delivery. JCI Insight. 2016;1(2):e85311. doi: 10.1172/jci.insight.85311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Southwell AL, et al. Huntingtin suppression restores cognitive function in a mouse model of Huntington’s disease. Sci Transl Med. 2018;10(461):eaar3959. doi: 10.1126/scitranslmed.aar3959. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen AD, et al. Murine knockin model for progranulin-deficient frontotemporal dementia with nonsense-mediated mRNA decay. Proc Natl Acad Sci USA. 2018;115(12):E2849–E2858. doi: 10.1073/pnas.1722344115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagemann TL, et al. Antisense suppression of glial fibrillary acidic protein as a treatment for Alexander disease. Ann Neurol. 2018;83(1):27–39. doi: 10.1002/ana.25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazur C, Fitzsimmons B, Kamme F, Nichols B, Powers B, Wancewicz E. Development of a simple, rapid, and robust intrathecal catheterization method in the rat. J Neurosci Methods. 2017;280:36–46. doi: 10.1016/j.jneumeth.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 34.Hung G, et al. Characterization of target mRNA reduction through in situ RNA hybridization in multiple organ systems following systemic antisense treatment in animals. Nucleic Acid Ther. 2013;23(6):369–378. doi: 10.1089/nat.2013.0443. [DOI] [PubMed] [Google Scholar]
- 35.Kordasiewicz HB, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCampbell A, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest. 2018;128(8):3558–3567. doi: 10.1172/JCI99081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaus HJ, Gupta R, Chappell AE, Østergaard ME, Swayze EE, Seth PP. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2019;47(3):1110–1122. doi: 10.1093/nar/gky1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geary RS, et al. Pharmacokinetic properties of 2′-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J Pharmacol Exp Ther. 2001;296(3):890–897. [PubMed] [Google Scholar]
- 39. Mathison B, Ushiro-Watanabe T, Savides MC, Henry SP, Levin AA. Second generation antisense oligonucleotide pharmacokinetics and mass balance following intravenous administration in rats. In: Society of Toxicology. San Francisco, California, USA: Oxford University Press; 2001:343. [Google Scholar]
- 40.Rigo F, et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther. 2014;350(1):46–55. doi: 10.1124/jpet.113.212407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finkel RS, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–3026. doi: 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 42.Niu C, et al. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci Transl Med. 2018;10(465):eaap8677. doi: 10.1126/scitranslmed.aap8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmes TJ, Liu YH. Richardson-Lucy/maximum likelihood image restoration algorithm for fluorescence microscopy: further testing. Appl Opt. 1989;28(22):4930–4938. doi: 10.1364/AO.28.004930. [DOI] [PubMed] [Google Scholar]
- 44.Yu RZ, Baker B, Chappell A, Geary RS, Cheung E, Levin AA. Development of an ultrasensitive noncompetitive hybridization-ligation enzyme-linked immunosorbent assay for the determination of phosphorothioate oligodeoxynucleotide in plasma. Anal Biochem. 2002;304(1):19–25. doi: 10.1006/abio.2002.5576. [DOI] [PubMed] [Google Scholar]
- 45.Holmes TJ, Liu YH. Richardson-Lucy/maximum likelihood image restoration algorithm for fluorescence microscopy: further testing. Appl Opt. 1989;28(22):4930–4938. doi: 10.1364/AO.28.004930. [DOI] [PubMed] [Google Scholar]
- 46.Roy D, Steyer GJ, Gargesha M, Stone ME, Wilson DL. 3D cryo-imaging: a very high-resolution view of the whole mouse. Anat Rec (Hoboken) 2009;292(3):342–351. doi: 10.1002/ar.20849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steyer GJ, Roy D, Salvado O, Stone ME, Wilson DL. Removal of out-of-plane fluorescence for single cell visualization and quantification in cryo-imaging. Ann Biomed Eng. 2009;37(8):1613–1628. doi: 10.1007/s10439-009-9726-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ryzhikov NN, et al. Preparation of highly specific radioactivity [18F]flumazenil and its evaluation in cynomolgus monkey by positron emission tomography. Nucl Med Biol. 2005;32(2):109–116. doi: 10.1016/j.nucmedbio.2004.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.