

Abstract
Background—
Angiotensin-converting enzyme (ACE) inhibitors are valuable agents for the treatment of hypertension, heart failure, and other cardiovascular and renal diseases. The cardioprotective effects of ACE inhibitors are mediated by blockade of both conversion of angiotensin (Ang) I to Ang II and kinin hydrolysis. Here, we report a novel mechanism that may explain the cardiac antifibrotic effect of ACE inhibition, involving blockade of the hydrolysis of N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP).
Methods and Results—
To study the role of Ac-SDKP in the therapeutic effects of the ACE inhibitor captopril, we used a model of Ang II–induced hypertension in rats treated with the ACE inhibitor either alone or combined with a blocking monoclonal antibody (mAb) to Ac-SDKP. These hypertensive rats had left ventricular hypertrophy (LVH) as well as increases in cardiac fibrosis, cell proliferation, transforming growth factor-β(TGF-β) expression, and phosphorylation of Smad2 (P-Smad2), a signaling mediator of the effects of TGF-β. The ACE inhibitor did not decrease either blood pressure or LVH; however, it significantly decreased LV collagen from 13.3±0.9 to 9.6±0.6 μg/mg dry wt (P<0.006), and this effect was blocked by the mAb (12.1±0.6; P<0.034, ACE inhibitor versus ACE inhibitor+mAb). In addition, analysis of interstitial collagen volume fraction and perivascular collagen (picrosirius red staining) showed a very similar tendency. Likewise, the ACE inhibitor significantly decreased LV monocyte/macrophage infiltration, cell proliferation, and TGF-β expression, and these effects were blocked by the mAb. Ang II increased Smad2 phosphorylation 3.2±0.9-fold; the ACE inhibitor lowered this to 0.6±0.1-fold (P<0.001), and the mAb blocked this decrease to 2.1±0.3 (P<0.001, ACE inhibitor versus ACE inhibitormAb). Similar findings were seen when the ACE inhibitor was replaced by Ac-SDKP.
Conclusions—
We concluded that in Ang II–induced hypertension, the cardiac antifibrotic effect of ACE inhibitors is a result of the inhibition of Ac-SDKP hydrolysis, resulting in a decrease in cardiac cell proliferation (probably fibroblasts), inflammatory cell infiltration, TGF-β expression, Smad2 activation, and collagen deposition.
Keywords: angiotensin, inhibitors, hypertension, growth substances, collagen
Angiotensin-converting enzyme (ACE) inhibitors are important agents for the treatment of hypertension, heart failure, and other cardiovascular and renal diseases. There is evidence that they act by inhibiting both conversion of angiotensin (Ang) I to Ang II and kinin hydrolysis.1 Here, we report a novel mechanism that mediates the cardiac antiproliferative and antifibrotic effects of ACE inhibition, involving an increase in plasma and tissue concentrations of N-acetylseryl-aspartyl-lysyl-proline (Ac-SDKP), which is a natural inhibitor of pluripotent hematopoietic stem cell proliferation.2,3 ACE inhibitors prevented degradation of endogenous Ac-SDKP and raised its circulating concentrations approximately 5-fold.4,5 We previously reported that Ac-SDKP inhibited rat cardiac fibroblast proliferation and collagen synthesis in vitro6,7 and also prevented left ventricular (LV) fibrosis in hypertensive rats in vivo.8,9 Ang II–induced hypertension has been associated with not only cardiac fibroblast proliferation and interstitial/perivascular fibrosis but also expression of transforming growth factor-β (TGF),10 which is central to stimulation of collagen synthesis.11 Moreover, TGF-β blockade with an anti–TGF-β neutralizing antibody prevented myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats.12 It is well documented that TGF-β signaling occurs via ligand-induced formation of a heteromeric complex of type I and type II serine/threonine kinase receptors.13 When the TGF-β receptor is activated, R-Smads (Smad2 and Smad3) are phosphorylated and form a dimer with a common mediator, Smad4.14 The Smad2/3-Smad4 complex translocates to the nucleus, where the dimer can regulate specific gene transcription either directly or indirectly via interactions with other transcription factors.14
We tested the hypothesis that the antiproliferative and antifibrotic effects of ACE inhibitors are at least partly mediated by Ac-SDKP. For this, we examined whether (1) an ACE inhibitor (captopril) or exogenous Ac-SDKP blunts LV monocyte/macrophage infiltration, cell proliferation, and collagen deposition; (2) a blocking monoclonal antibody (mAb) to Ac-SDKP antagonizes the effects of ACE inhibitors and Ac-SDKP; and (3) the mechanism by which ACE inhibitors and Ac-SDKP suppress cardiac collagen is associated with a decrease in both TGF-β expression and phosphorylation of Smad2, a downstream mediator of the effects of TGF-β. Because the antifibrotic effect of ACE inhibitors in Ang II–induced hypertension is not associated with hemodynamic changes,15,16 we selected this model to test our hypothesis.
Methods
This study was approved by the Henry Ford Hospital Institutional Animal Care and Use Committee.
Animals and Experimental Design
Male Sprague-Dawley rats weighing 200 to 255 g (Charles River, Wilmington, Del) were anesthetized with sodium pentobarbital (50 mg/kg IP). A small incision was made between the shoulder blades and a pocket created subcutaneously, just large enough to hold an osmotic minipump (Alzet 2 ML4). The pump was implanted to deliver Ang II (750 μg · kg−1 · d−1) and/or Ac-SDKP (synthesized by Drs Domenico Regoli and Witold Neugebauer, University of Sherbrooke, Canada) or saline plus 0.01N acetic acid. Captopril was given in drinking water, and immunoglobulin G (IgG, 400 μg/kg) or anti–AcSDKP mAb (400 μg/kg) was given intraperitoneally every other day. Treatment with Ac-SDKP and captopril was begun simultaneously with Ang II and continued for 4 weeks. Rats were divided into 6 groups: (1) sham+IgG, (2) Ang II+IgG, (3) Ang II+captopril (100 mg · kg−1 · d−1)+IgG, (4) Ang II+captopril+mAb, (5) Ang II+Ac-SDKP (800 μg · kg−1 · d−1)+IgG, and (6) Ang II+Ac-SDKP+mAb.
Before the study, we tested whether the mAb (1:1000) blocked the effect of Ac-SDKP on collagen synthesis when adult rat cardiac fibroblasts in culture were stimulated with endothelin-1 (ET-1).6 We found that collagen in control cells was 5.7±0.8 μg/mg protein; it was 14.3±0.5 with ET-1 (10−8 mol/L), 5.3±0.7 with ET-1AcSDKP (10−9 mol/L), and 14.2±1.5 with ET-1Ac-SDKPmAb. In a separate study, we tested whether chronic blockade of endogenous Ac-SDKP with mAb would increase LV collagen deposition compared with sham rats given either intraperitoneal IgG (n=5) or mAb (n=5) for 4 weeks.
Systolic blood pressure (SBP) was measured by tail cuff once a week for 4 weeks. At the end of the experiment, animals were anesthetized with 50 mg/kg pentobarbital sodium. The heart was stopped at diastole with an intraventricular injection of 15% KCl and rapidly excised. The LV (including the septum) was weighed and sectioned transversely from apex to base.
Hydroxyproline Assay
The collagen content of myocardial and renal tissue was determined by hydroxyproline assay as described previously.9,17 Briefly, tissue was dried, homogenized, and hydrolyzed with 6N HCl for 16 hours at 110°C. A standard curve of 0 to 5 μg hydroxyproline was used. Data were expressed as μg collagen/mg dry wt, assuming that collagen contains an average of 13.5% hydroxyproline.18
Collagen Morphometry
Sections measuring 6 μm were deparaffinized, rehydrated, and stained with picrosirius red using a modification of Sweat and Puchtler’s method.19 Briefly, sections were postfixed in Bouin’s fluid, followed by iron hematoxylin stain to show the nuclei. They were then stained with 0.1% picrosirius red for 1 hour and washed twice with 0.5% acetic acid. Images were obtained with a computerized digital camera (Spot Diagnostic) and collagen morphometry examined under a microscope (Nikon E600) using normal and polarized light.20
Determination of Cell Proliferation (Ki-67) and LV Monocyte/Macrophage Count (ED1)
Sections measuring 6 μm were deparaffinized and rehydrated. To reveal antigen, sections were boiled in 10 mmol/L sodium citrate buffer (pH 6.0) for 10 minutes in a microwave oven. They were washed in dH2O and incubated in 3% H2O2 for 10 minutes at room temperature, then preincubated in blocking solution for 30 minutes at room temperature, and finally incubated with a mouse mAb against rat macrophages/monocytes (ED1, 1:1000 dilution; Chemicon) at room temperature for 30 minutes16 or with a monoclonal mouse anti-rat Ki-67 antigen antibody (1:50; Dako) overnight at 4°C.21,22 ED1 and Ki-67 antigen were assayed with a Vectastain ABC kit (Vector Laboratories). Sections were developed with diaminobenzidine substrate (Vector) and counterstained with hematoxylin. Ki-67–positive cells in half of the LV were examined. Proliferating cells with dark brown nuclei were counted and expressed as cells/mm2.
Western Blot for Detection of Tissue TGF-β and Smads
Protein (60 μg) from the LV extracts was subjected to 12% SDS-PAGE under nonreducing conditions for TGF-β, or 10% SDS-PAGE under reducing conditions for Smads. Proteins were transferred to a nitrocellulose membrane. The primary antibodies were mouse mAb against TGF-β1,2,3 (1:1000; R & D Systems), goat polyclonal antibody against Smad2/3 or Smad7 (1:300; Santa Cruz Biotechnology), or a rabbit polyclonal antibody against p-Smad2 (1:1000, Cell Signaling Technology). A goat polyclonal antibody against actin (1:500; Santa Cruz) was added along with the above primary antibodies as a housekeeping protein. Recombinant human TGF-β1 (R & D Systems) and extracts from TGF-β–treated HepG2 cells (cell signaling) were used as a positive control for TGF-β and p-Smad2, respectively. Results were quantified with a bioscanner and expressed as fold increase compared with sham. The molecular weights of positive bands were as follows: TGF-β, 25 kDa; Smad 2/3, 65 kDa; Smad7, 51 kDa; and p-Smad2, 58 kDa.
Ac-SDKP Urine Levels
Urine was collected for 24 hours in vials containing lisinopril (final concentration, 10 μmol/L) and centrifuged at 2000g for 15 minutes. Urine Ac-SDKP was quantified using a competitive enzyme immunoassay kit (SPI-BIO) and expressed as micrograms per 24 hours.23
Statistical Analysis
SBP was examined using ANOVA with repeated measures and a covariate (baseline SBP). All other variables were analyzed by 1-way ANOVA. Logarithmic transformations of variables were used to stabilize the variances. There were 6 groups: (1) sham+IgG, (2) Ang II+IgG, (3) Ang IIcaptopril+IgG, (4) Ang IIcaptoprilmAb, (5) Ang IIAc-SDKP+IgG, and (6) Ang II+Ac-SDKP+mAb. Seven comparisons were prespecified (1 versus 2, 2 versus 3, 2 versus 4, 2 versus 5, 2 versus 6, 5 versus 6, and 3 versus 4). Significance was judged using Hochberg’s method for post hoc comparisons. Values are expressed as mean±SEM. All probability values of P<0.05 are reported.
Results
SBP and LV Hypertrophy
SBP in the Ang II+vehicle group increased significantly (P<0.005) 1 week after Ang II infusion was started and remained elevated for 3 weeks. Neither captopril nor AcSDKP alone or combined with the mAb for 4 weeks had any effect on hypertension (Figure 1). LV weight was significantly increased in the Ang II+vehicle group (P<0.001), and neither captopril nor Ac-SDKP (with or without the mAb) decreased LV hypertrophy (Figure 1). Treatment of sham rats with mAb did not alter the ratio of LV weight to body weight.
Figure 1.

SBP (n=10 to 13, top) and ratio of LV weight to body weight (LVW/ BW) (n=5 to 6, bottom) of rats with Ang II–induced hypertension after 4 weeks of ACE inhibitor or Ac-SDKP. Blood pressure and LV hypertrophy were not affected by any treatment. *P<0.05 vs Ang II+vehicle.
Ac-SDKP Urine Levels
Urine Ac-SDKP was not significantly affected by chronic Ang II infusion (Figure 2) but was increased 5-fold in rats given the ACE inhibitor (P=0.0008) and Ac-SDKP (P=0.0001). Unexpectedly, mAb only blunted the effect of chronic Ac-SDKP infusion, going from 3.81±0.56 to 1.83±0.47 μg/24 hours (P=0.0055), but did not influence the ACE inhibitor (5.37±0.61 μg/24 hours) (Figure 2).
Figure 2.

Twenty-four-hour urinary Ac-SDKP in rats with Ang II–induced hypertension after 4 weeks of ACE inhibitor or Ac-SDKP in the absence or presence of a mAb against Ac-SDKP (n=4 to 7).
LV Collagen Content
LV collagen was significantly increased in the Ang II+vehicle group (13.3±0.9 μg/mg dry LV wt) compared with control (8.9±0.7 μg/mg dry LV wt; P<0.002), and this increase was significantly blunted by either captopril (9.6±0.6 μg/mg dry LV wt; P<0.006) or Ac-SDKP (9.7±0.4 μg/mg dry LV wt; P<0.015) (Figure 3). The mAb significantly blunted the effect of captopril (P<0.039) and Ac-SDKP (P<0.015) (Figure 3). Treatment of the sham group with mAb did not alter LV collagen content in normotensive rats, with 8.9±0.4 μg/mg dry LV wt in sham given IgG and 8.8±0.3 μg/mg dry LV wt in sham given mAb (P=NS). Subsequent protocols did not include comparisons with sham ± mAb.
Figure 3.

Collagen deposition in the LV of rats with Ang II–induced hypertension after 4 weeks of ACE inhibitor or Ac-SDKP. Both prevented cardiac fibrosis, and this effect was blunted by an anti–Ac-SDKP mAb (n=9 to 12).
LV Interstitial and Perivascular Collagen
LV interstitial collagen fraction was significantly increased in the Ang II+vehicle group (8.8±0.3%) compared with control (5.10.3%; P<0.001), and this increase was significantly blunted by either captopril (5.6±0.2%; P<0.001) or AcSDKP (6.0±0.1%; P<0.001) (Figure 4). The mAb significantly blunted the effect of captopril (P<0.001) and AcSDKP (P<0.001) (Figure 4). The ratio of perivascular collagen area to luminal area of the coronary arteries was 4.0±0.9 in the sham group and increased significantly to 9.9±1.4 in the Ang II hypertensive rats (P<0.001). The ACE inhibitor and Ac-SDKP significantly attenuated the increase in perivascular collagen, with the ratio of perivascular collagen area to luminal area falling to 3.6±0.6 (P<0.001) and 3.8±0.7 (P<0.001), respectively (Figure 5). The mAb significantly blunted the effect of captopril (P<0.001) and Ac-SDKP (P<0.001) (Figure 5).
Figure 4.

LV interstitial collagen deposition (red staining; top) in rats with Ang II–induced hypertension after 4 weeks of ACE inhibitor or Ac-SDKP alone or combined with an anti-Ac-SDKP mAb (picrosirius red, ×400); bottom, quantitative analysis of LV interstitial collagen. Both the ACE inhibitor and Ac-SDKP blunted cardiac fibrosis, and this effect was diminished by an anti-Ac-SDKP mAb (n=5).
Figure 5.

Representative images showing LV perivascular collagen deposition (red staining; top) in rats with Ang II–induced hypertension after 4 weeks of ACE inhibitor or Ac-SDKP alone or combined with an anti–Ac-SDKP mAb (picrosirius red, ×400); bottom, quantitative analysis of LV perivascular collagen. Both the ACE inhibitor and Ac-SDKP blunted cardiac fibrosis, and this effect was diminished by an anti–Ac-SDKP mAb (n=5).
Cell Proliferation and Monocytes/Macrophages in the LV
Ki-67–positive cells (a marker for cell proliferation) were localized to the vasculature and the interstitial space of the LV (brown-stained cells; Figure 6). There were more Ki-67– positive cells in the LV of rats given Ang II+vehicle (826 cells/mm2) than sham (512 cells/mm2; P<0.001) (Figure 7A). Treatment with ACE inhibitors or Ac-SDKP normalized the number of proliferating cells in Ang II–infused rats, and the mAb blocked the antiproliferative effect of ACE inhibitors or Ac-SDKP (Figure 7A). We confirmed that ED1positive cells were significantly increased in the Ang II+vehicle group compared with control (P<0.001). Treatment with captopril and Ac-SDKP significantly reduced the number of ED1-positive cells in the LV (P<0.001) (Figure 7B), and the mAb blocked the antiinflammatory effect of ACE inhibitors or Ac-SDKP.
Figure 6.
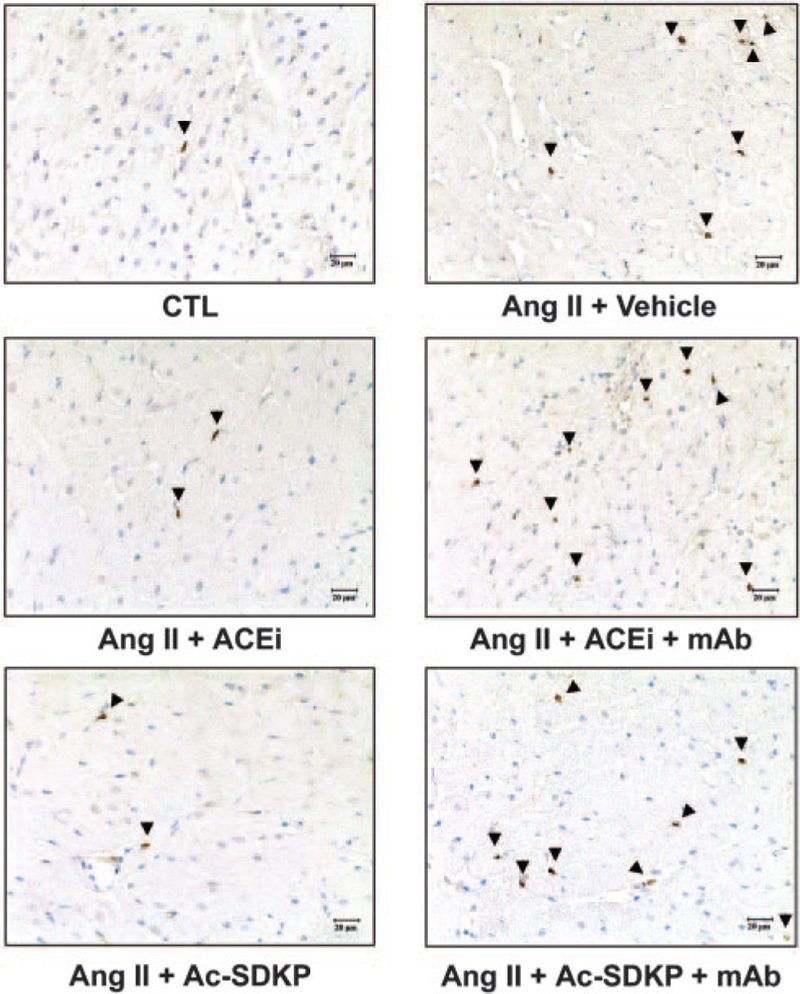
Representative LV immunohistochemical staining for Ki-67–positive cells (indicator for cell proliferation) in rats with Ang II–induced hypertension after 4 weeks of ACE inhibitor or Ac-SDKP treatment in the presence or absence of an anti–Ac–SDKP mAb.
Figure 7.

A, Ki-67–positive cells and B, ED1-positive cells (monocytes/macrophages) in rats with Ang II–induced hypertension after 4 weeks of ACE inhibitor or Ac-SDKP treatment in the presence or absence of an anti–Ac-SDKP mAb. LV inflammatory cell infiltration and cell proliferation were increased in rats given Ang II+vehicle compared with sham. These effects were blunted by the ACE inhibitor or Ac-SDKP, and the anti–Ac-SDKP mAb blocked the effect of both the ACE inhibitor and Ac-SDKP (n=5 to 6).
TGF-β expression in the LV (Western Blot)
TGF-β was highly expressed in the LV in the Ang II+vehicle group. The Western blot for TGF-β showed a band of 25 kDa. This band was almost absent in the sham group and was barely seen in the Ang II–induced hypertension groups treated with captopril or Ac-SDKP, but it was present in the mAb group (Figure 8, top). Quantitative analysis indicated that TGF-β expression was increased 257-fold compared with control (P<0.001) and was significantly reduced by the ACE inhibitor (5±2-fold; P<0.001) or Ac-SDKP (2.1±0.5-fold; P<0.001). The mAb blocked the effect of the ACE inhibitor or Ac-SDKP on TGF-β expression (Figure 8, bottom).
Figure 8.

TGF-β expression in the LV as detected by Western blot (WB), showing bands of the typical homodimer (25 kDa) (top). Quantitative analysis showed that TGF-β was highly expressed in the LV in rats with Ang II–induced hypertension (bottom). The ACE inhibitor or Ac-SDKP lowered TGF-β expression, and an anti–Ac-SDKP mAb blocked the effect of both the ACE inhibitor and Ac-SDKP. P, positive control for TGF-β (n=4 to 7).
p-Smad2, Smad2/3, and Smad7 Expression in the LV
P-Smad2 was highly expressed in the LV of rats with Ang II–induced hypertension (3.2±0.9-fold increase; P<0.017) (Figure 9, top). The ACE inhibitor (0.6±0.1-fold; P<0.001) or Ac-SDKP (1.4±0.3-fold; P<0.034) lowered Smad2 phosphorylation expression, and the mAb blocked the effect of the ACE inhibitor or Ac-SDKP on P-Smad2 (Figure 9, top). Smad2/3 protein content in the LV did not change in Ang II–induced hypertension and was not affected by either the ACE inhibitor or Ac-SDKP (Figure 9, bottom). Expression of Smad7 (51 kDa) did not change significantly in any group (Figure 10).
Figure 9.

A, Western blot showing phosphorylated Smad2 (p-Smad2) expression in the LV as a band of 58 kDa (top). p-Smad2 was highly expressed in the LV of rats with Ang II–induced hypertension (bottom). The ACE inhibitor or Ac-SDKP lowered P-Smad2 expression, and an anti-Ac-SDKP mAb blocked the effect of both the ACE inhibitor and Ac-SDKP. B, Smad2/3 expression (58 kDa) was equally apparent in all groups. β-Actin (40 kDa) showed uniform gel loading. P, positive control for p-Smad2 (n=4 to 7).
Figure 10.

Western blot (top) and quantitative analysis (bottom) of Smad7 expression (51 kDa) in the LV. There was no significant difference among all 6 groups. β-Actin expression was equally apparent in all groups (n=4 to 7).
Discussion
ACE inhibitors are used to treat hypertension and congestive heart failure, to avert remodeling and reinfarction after myocardial infarction, and to ameliorate diabetic nephropathy and other renal diseases. They have also been shown to decrease or slow the development of diabetes, cancer, and aging.24–26 Thus, understanding the mechanism behind the therapeutic effects of ACE could yield new approaches to treating these and other diseases. Although ACE is known to hydrolyze a number of peptides, the therapeutic effects of ACE inhibitors are attributed primarily to inhibition of both conversion of Ang I to Ang II and kinin hydrolysis. Here we show that another peptide, Ac-SDKP, mediates some of the effects of ACE inhibitors.
We previously reported that chronic administration of an ACE inhibitor or exogenous Ac-SDKP at doses that increased plasma Ac-SDKP concentration to similar levels had comparable antiinflammatory and antifibrotic effects.16 We concluded that Ac-SDKP may be an important mediator of the antiinflammatory and antifibrotic effects of ACE inhibitors; however, direct evidence was lacking. In the present study, we hypothesized that the antifibrotic effect of ACE inhibition is mediated by Ac-SDKP. To test the role of Ac-SDKP in the effect of ACE inhibitors, we used a more direct approach, blocking the effect of endogenous Ac-SDKP with a specific mAb. Chronic treatment of normotensive rats with mAb did not alter LV collagen content, indicating that endogenous low levels of Ac-SDKP do not participate in the maintenance of normal LV collagen. In this model of hypertension, both the ACE inhibitor and Ac-SDKP prevented (1) LV collagen deposition (interstitial and perivascular), (2) LV interstitial cell proliferation, (3) LV monocyte/macrophage infiltration, (4) increased TGF-β expression, and (5) activation of Smad2; moreover, all of these effects occurred in the absence of changes in blood pressure or cardiac hypertrophy. The effects of both the ACE inhibitor and Ac-SDKP were significantly blocked by a mAb against Ac-SDKP. Thus, we concluded that in Ang II–induced hypertension, the antifibrotic and antiproliferative effects of ACE inhibition are mediated primarily by Ac-SDKP. This is a novel mechanism whereby ACE inhibition can reduce cardiac fibrosis independent of its antihypertensive effect.
Urinary Ac-SDKP increased 5-fold in rats treated with either the ACE inhibitor or Ac-SDKP; this effect was blocked by mAb only in rats given Ac-SDKP but not the ACE inhibitor, for reasons that are not clear. Further studies focusing on the kidneys and their role in the production and degradation of Ac-SDKP are needed to clarify this. It is known that in humans, Ac-SDKP is found in plasma and circulating mononuclear cells.23 In mice, it is distributed in several tissues, including the lung, kidney, and heart.27 Thymosin-β4 is the most likely precursor of the peptide, because its N-terminus contains Ac-SDKP. We have reported that the enzyme responsible for the release of Ac-SDKP is prolyl oligopeptidase.28 Ac-SDKP is cleaved to an inactive form by the NH2-terminal catalytic domain of ACE.4 It has a 4.5-minute half-life in the circulation and is probably released continuously.5 Previously, we have shown that in vitro Ac-SDKP inhibits fibroblast proliferation and collagen synthesis.6 We have also shown that this tetrapeptide has an antifibrotic effect in rats with aldosterone-salt9 or 2-kidney, 1-clip hypertension (2K-1C)8 and in rat hearts after myocardial infarction.29 Ac-SDKP prevented macrophage/monocyte infiltration and decreased the number of cells that expressed TGF-β and connective tissue growth factor.29 These studies clearly indicate that exogenous administration of Ac-SDKP in vitro and in vivo has an antifibrotic effect. In the present study, we found that the antifibrotic effect of ACE inhibition was mediated by endogenous Ac-SDKP. This is further supported by recent studies by Pokharel et al,30 who reported that transgenic rats with cardioselective overexpression of ACE exhibited marked cardiac fibrosis, but cardiac Ang II concentrations were normal. TGF-β and Smad-R phosphorylation were directly related to the number of ACE genes expressed, whereas Ac-SDKP was inversely related. They suggested that the development of fibrosis in this model involves a decrease in Ac-SDKP.
In the present study, we also tested the mechanism by which ACE inhibition via Ac-SDKP may decrease fibrosis, concentrating on its effects on interstitial cell proliferation, TGF-β expression, and signaling of TGF-β (R-Smads and Smad7). We have shown previously that in vitro Ac-SDKP inhibits fibroblast proliferation,6 and in vivo, it inhibits cell proliferation in the heart, probably fibroblasts.8,9 Here, we demonstrate that suppression of cell proliferation in the heart by ACE inhibition is a result of Ac-SDKP, because the mAb blocked the effect of the ACE inhibitor. Furthermore, we have shown that administration of Ac-SDKP in various models of hypertension decreased the number of cells expressing TGF-β in the heart. The present study clearly shows that both the ACE inhibitor and Ac-SDKP decreased cardiac TGF-β, and these effects were blocked by the mAb. TGF-β is an important profibrotic cytokine that simultaneously stimulates cells to increase synthesis of most matrix proteins, decrease production of matrix-degrading proteases, and increase production of protease inhibitors.12,31,32 TGF-β could be increased in the hypertensive heart either by accelerated infiltration of inflammatory cells (macrophages) or by Ang II acting on cardiac fibroblasts and myofibroblasts.10 Lee et al33 reported that Ang II stimulates autocrine production of TGF-β in adult rat cardiac fibroblasts and suggested that its effect on the adult myocardium may be mediated in part by autocrine/paracrine mechanisms, including production and release of TGF-β by cardiac fibroblasts. TGF-β acts by signaling through 2 different receptor serine/threonine kinases,14 TGF-β receptor type-I (T-βR-I) and T-βR-II, which together form a tetrameric complex once T-βR-I binds to T-βR-II. T-βR-II activates T-βR-I after phosphorylation of the serine residue, which in turn leads to phosphorylation (activation) of R-Smads (Smad2 and Smad3). Activated R-Smads form a heteroligomer with co-Smad4, and these complexes are translocated to the nucleus, where they bind to the promoter region that initiates specific gene transcription either directly or indirectly via interaction with other transcription factors.14 Thus, phosphorylation of Smad2 and translocation to the nucleus are critical steps in the modulation of cell signaling.
Increased TGF-β and Smad2, 3, and 4 protein expression has been found to correlate positively with increased collagen type I expression in the chronic phase of myocardial infarct scar healing.34 In the present study, we showed that ACE inhibitors, acting via endogenous Ac-SDKP, inhibited LV Smad2 phosphorylation, indicating interference with R-Smad activation. This could be because of inhibition of either synthesis or activation and nuclear translocation of Smad protein; but, because neither the ACE inhibitor nor Ac-SDKP significantly affected total protein content of Smad 2/3, it seems more likely that either Ac-SDKP or the ACE inhibitor (acting via endogenous Ac-SDKP) inhibits phosphorylation (activation) of Smad2 through a mechanism that could be associated with decreased TGF-β expression. However, other unknown mechanisms may also be involved, because AcSDKP inhibits the effect of exogenous TGF-β on Smad activation in cardiac fibroblasts and mesangial cells.7,35
Smad7 belongs to the class of inhibitory Smads that prevent R-Smad activation, thereby downregulating TGF-β signaling.36,37 Decreased Smad7 expression has been shown to play a role in the pathogenesis of cardiac fibrosis in the heart after myocardial infarction,38 and transient gene transfer and expression of Smad7 prevent bleomycin-induced Smad2 phosphorylation and lung fibrosis in mice.39 We questioned whether ACE inhibitors or Ac-SDKP might induce Smad7 expression but were unable to detect any differences in the signal for Smad7 by Western blot. This might indicate that (1) Smad7 expression is very low and is not robustly induced by Ac-SDKP7 or (2) the inhibitory effect of Smad7 is not related to its increased expression and quantity in the cell but rather to its ability to translocate from the nucleus to the cytoplasm and bind competitively to T-βR-I, as shown by Kanasaki et al35 in human mesangial cells. They demonstrated that the inhibitory Smad7 was exported from the nucleus to the cytoplasm by treatment with Ac-SDKP, which we were unable to examine in our LV samples. Thus, inhibition of TGF-β expression and Smad2 phosphorylation by Ac-SDKP and ACE inhibitors (acting via endogenous Ac-SDKP) in the LV of Ang II–infused hypertensive rats may also be an important factor in mediating the antifibrotic effect of ACE inhibitors.
In summary, an ACE inhibitor increased plasma AcSDKP, which in turn inhibited LV inflammatory cell infiltration, interstitial cell proliferation (most likely fibroblasts), TGF-β, Smad2 activation, and collagen deposition. This was confirmed by the fact that neutralizing Ac-SDKP in vivo with an anti–Ac-SDKP mAb significantly blunted the actions of the ACE inhibitor without causing any hemodynamic effects. These findings suggest that Ac-SDKP prevents cardiac fibrosis by blocking collagen production in Ang II-hypertensive rats, most likely via inhibition of TGF-β and its subcellular signal transduction (p-Smad2). ACE inhibitors increase plasma5 and tissue Ac-SDKP40 and decrease cardiac and renal fibrosis.1,41–44 We conclude that Ac-SDKP may be an important new mediator of the antiinflammatory and antifibrotic effects of ACE inhibitors in hypertension.
CLINICAL PERSPECTIVE.
ACE inhibitors are used to treat hypertension and congestive heart failure, avert remodeling and reinfarction after myocardial infarction, and ameliorate diabetic nephropathy and other renal diseases. They have also been shown to decrease or slow the development of diabetes, cancer, and aging. Thus, understanding the mechanism(s) behind the therapeutic effects of ACE could yield new approaches to treating these and other diseases. Although ACE is known to hydrolyze a number of peptides, the therapeutic effects of ACE inhibitors are attributed primarily to inhibition of both conversion of Ang I to II and kinin hydrolysis. Here we show that another peptide, Ac-SDKP, which inhibits pluripotent hematopoietic stem cell proliferation, may account for some of the effects of ACE inhibitors. We found that the ACE inhibitor increased plasma Ac-SDKP, which in turn inhibited LV inflammatory cell infiltration, interstitial cell proliferation (most likely fibroblasts), TGF-β, Smad2 activation, and collagen deposition. Neutralizing Ac-SDKP in vivo with an anti–Ac-SDKP mAb significantly blunted the actions of the ACE inhibitor without hemodynamic effects, confirming the importance of Ac-SDKP in the in vivo effects of ACE inhibitors. These findings suggest that Ac-SDKP prevents cardiac fibrosis by blocking collagen production in Ang II-hypertensive rats, most likely via inhibition of TGF-β and its subcellular signal transduction (p-Smad2). Thus, Ac-SDKP may be an important mediator of the antiinflammatory and antifibrotic effects of ACE inhibitors in hypertension.
Acknowledgments
This study was supported by American Heart Association grant 0130128N and NIH grants HL-71806-01 (Dr Rhaleb) and HL-28982 (Dr Carretero).
Disclosure
Dr Carretero is the recipient of a grant from Pfizer (not related to the present research) and has served as a consultant to Novartis. Dr Rhaleb has received research support from the HFH institutional fund A10163.
References
- 1.Brooks WW, Bing OH, Robinson KG, Slawsky MT, Chaletsky DM, Conrad CH. Effect of angiotensin-converting enzyme inhibition on myocardial fibrosis and function in hypertrophied and failing myocardium from the spontaneously hypertensive rat. Circulation. 1997;96: 4002–4010. [DOI] [PubMed] [Google Scholar]
- 2.Lenfant M, Wdzieczak-Bakala J, Guittet E, Prome JC, Sotty D, Frindel E.Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci U S A. 1989;86: 779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonnet D, Lemoine FM, Pontvert-Delucq S, Baillou C, Najman A,Guigon M. Direct and reversible inhibitory effect of the tetrapeptide acetyl-N-Ser-Asp-Lys-Pro (Seraspenide) on the growth of human CD34+subpopulations in response to growth factors. Blood. 1993;82:3307–3314. [PubMed] [Google Scholar]
- 4.Azizi M, Rousseau A, Ezan E, Guyene T-T, Michelet S, Grognet J-M,Lenfant M, Corvol P, Ménard J. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J Clin Invest. 1996;97:839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azizi M, Ezan E, Nicolet L, Grognet JM, Menard J. High plasma level of N-acetyl-seryl-aspartyl-lysyl-proline: a new marker of chronic angiotensin-converting enzyme inhibition. Hypertension. 1997;30:1015–1019. [DOI] [PubMed] [Google Scholar]
- 6.Rhaleb N-E, Peng H, Harding P, Tayeh M, LaPointe MC, Carretero OA.Effect of N-acetyl-seryl-aspartyl-lysyl-proline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension. 2001;37:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pokharel S, Rasoul S, Roks AJM, van Leeuwen REW, van Luyn MJA,Deelman LE, Smits JF, Carretero O, van Gilst WH, Pinto YM. N-Acetyl-Ser-Asp-Lys-Pro inhibits phosphorylation of Smad2 in cardiac fibroblasts. Hypertension. 2002;40:155–161. [DOI] [PubMed] [Google Scholar]
- 8.Rhaleb N-E, Peng H, Yang X-P, Liu Y-H, Mehta D, Ezan E, Carretero OA. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation. 2001;103:3136–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng H, Carretero OA, Raij L, Yang F, Kapke A, Rhaleb N-E. Antifibrotic effects of N-acetyl-seryl-aspartyl-lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension. 2001;37: 794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell SE, Katwa LC. Angiotensin II stimulated expression of transforming growth factor-β1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol. 1997;29:1947–1958. [DOI] [PubMed] [Google Scholar]
- 11.Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGFexpression is induced by TGF-β in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32: 1805–1819. [DOI] [PubMed] [Google Scholar]
- 12.Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K,Imaizumi T. Transforming growth factor-β function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–135. [DOI] [PubMed] [Google Scholar]
- 13.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-β receptor. Nature. 1994;370:341–347. [DOI] [PubMed] [Google Scholar]
- 14.Massague J. TGF-β signal transduction. Annu Rev Biochem. 1998;67: 753–791. [DOI] [PubMed] [Google Scholar]
- 15.Crawford DC, Chobanian AV, Brecher P. Angiotensin II induces fibronectin expression associated with cardiac fibrosis in the rat. Circ Res. 1994;74:727–739. [DOI] [PubMed] [Google Scholar]
- 16.Rasoul S, Carretero OA, Peng H, Cavasin MA, Zhuo J, Sanchez-Mendoza A, Brigstock DR, Rhaleb N-E. Antifibrotic effect of Ac-SDKP and angiotensin-converting enzyme inhibition in hypertension. J Hypertens. 2004;22:593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 18.Chiariello M, Ambrosio G, Cappelli-Bigazzi M, Perrone-Filardi P,Brigante F, Sifola C. A biochemical method for the quantitation of myocardial scarring after experimental coronary artery occlusion. J Mol Cell Cardiol. 1986;18:283–290. [DOI] [PubMed] [Google Scholar]
- 19.Sweat F, Puchtler H, Rosenthal SI. Sirius red F3BA as a stain for connective tissue. Arch Pathol. 1964;78:69–72. [PubMed] [Google Scholar]
- 20.Whittaker P, Boughner DR, Kloner RA. Analysis of healing after myocardial infarction using polarized light microscopy. Am J Pathol. 1989; 134:879–893. [PMC free article] [PubMed] [Google Scholar]
- 21.Schlüter C, Duchrow M, Wohlenberg C, Becker MHG, Key G, Flad H-D, Gerdes J. The cell proliferation-associated antigen of antibody Ki-67: a very large, ubiquitous nuclear protein with numerous repeated elements, representing a new kind of cell cycle-maintaining proteins. J Cell Biol. 1993;123:513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gore SD, Weng L-J, Burke PJ. Validation of flow-cytometric determination of Ki67 expression as a measure of growth factor response in acute myelogenous leukemia. Exp Hematol. 1993;21:1702–1708. [PubMed] [Google Scholar]
- 23.Pradelles P, Frobert Y, Créminon C, Liozon E, Massé A, Frindel E. Negative regulator of pluripotent hematopoietic stem cell proliferation in human white blood cells and plasma as analysed by enzyme immunoassay. Biochem Biophys Res Commun. 1990;170:986–993. [DOI] [PubMed] [Google Scholar]
- 24.Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, MacKinnon PL, Meredith PA, Murray LS, Reid JL, Robertson JWK. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet. 1998;352:179–184. [DOI] [PubMed] [Google Scholar]
- 25.The Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000;342:145–153. [DOI] [PubMed] [Google Scholar]
- 26.de Cavanagh EMV, Piotrkowski B, Basso N, Stella I, Inserra F, Ferder L, Fraga CG. Enalapril and losartan attenuate mitochondrial dysfunction in aged rats. FASEB J. 2003;17:1096–1098. [DOI] [PubMed] [Google Scholar]
- 27.Pradelles P, Frobert Y, Créminon C, Ivonine H, Frindel E. Distribution of a negative regulator of haematopoietic stem cell proliferation (AcSDKP) and thymosin beta 4 in mouse tissues. FEBS Lett. 1991;289:171–175. [DOI] [PubMed] [Google Scholar]
- 28.Cavasin MA, Rhaleb N-E, Yang X-P, Carretero OA. Prolyl oligopeptidase is involved in release of the antifibrotic peptide Ac-SDKP. Hypertension. 2004;43:1140–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang F, Yang X-P, Liu Y-H, Xu J, Cingolani O, Rhaleb N-E, Carretero OA. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension. 2004;43:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pokharel S, van Geel PP, Sharma UC, Cleutjens JPM, Bohnemeier H, Tian X-L, Schunkert H, Crijns HJGM, Paul M, Pinto YM. Increased myocardial collagen content in transgenic rats overexpressing cardiac angiotensin-converting enzyme is related to enhanced breakdown of N-acetyl-Ser-Asp-Lys-Pro and increased phosphorylation of Smad2/3. Circulation. 2004;110:3129–3135. [DOI] [PubMed] [Google Scholar]
- 31.Border WA, Noble NA. Transforming growth factor β in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. [DOI] [PubMed] [Google Scholar]
- 32.Petrov VV, Fagard RH, Lijnen PJ. Stimulation of collagen production bytrans forming growth factor-β1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. 2002;39:258–263. [DOI] [PubMed] [Google Scholar]
- 33.Lee AA, Dillmann WH, McCulloch AD, Villarreal FJ. Angiotensin IIstimulates the autocrine production of transforming growth factor-β1 in adult rat cardiac fibroblasts. J Mol Cell Cardiol. 1995;27:2347–2357. [DOI] [PubMed] [Google Scholar]
- 34.Hao J, Ju H, Zhao S, Junaid A, Scammell-La Fleur T, Dixon IMC.Elevation of expression of Smads 2, 3, and 4, decorin and TGF-β in the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol. 1999;31:667–678. [DOI] [PubMed] [Google Scholar]
- 35.Kanasaki K, Koya D, Sugimoto T, Isono M, Kashiwagi A, Haneda M.N-Acetyl-seryl-aspartyl-lysyl-proline inhibits TGF-β-mediated plasminogen activator inhibitor-1 expression via inhibition of Smad pathway in human mesangial cells. J Am Soc Nephrol. 2003;14:863–872. [DOI] [PubMed] [Google Scholar]
- 36.Massagué J, Chen Y-G. Controlling TGF-β signaling. Genes Dev. 2000; 14:627–644. [PubMed] [Google Scholar]
- 37.Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M, Deng C, Kucherlapati R, Böttinger EP, Roberts AB. Functional characterization of transforming growth factor signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945–19953. [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Hao J, Jones SC, Yee M-S, Roth JC, Dixon IMC. Decreased Smad 7 expression contributes to cardiac fibrosis in the infarcted rat heart. Am J Physiol. 2002;282:H1685–H1696. [DOI] [PubMed] [Google Scholar]
- 39.Nakao A, Fujii M, Matsumura R, Kumano K, Saito Y, Miyazono K, Iwamoto I. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest. 1999;104:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junot C, Nicolet L, Ezan E, Gonzales M-F, Menard J, Azizi M. Effects of angiotensin-converting enzyme inhibition on plasma, urine, and tissue concentrations of hemoregulatory peptide acetyl-Ser-Asp-Lys-Pro in rats. J Cardiovasc Pharmacol. 1999;291:982–987. [PubMed] [Google Scholar]
- 41.Liu Y-H, Yang X-P, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure: role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99: 1926–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun Y, Ratajska A, Zhou G, Weber KT. Angiotensin-converting enzyme and myocardial fibrosis in the rat receiving angiotensin II or aldosterone. J Lab Clin Med. 1993;122:395–403. [PubMed] [Google Scholar]
- 43.Kim S, Ohta K, Hamaguchi A, Omura T, Yukimura T, Miura K, Inada Y,Wada T, Ishimura Y, Chatani F, Iwao H. Role of angiotensin II in renal injury of deoxycorticosterone acetate-salt hypertensive rats. Hypertension. 1994;24:195–204. [DOI] [PubMed] [Google Scholar]
- 44.Kaneto H, Morrissey J, McCracken R, Reyes A, Klahr S. Enalapril reduces collagen type IV synthesis and expansion of the interstitium in the obstructed kidney of the rat. Kidney Int. 1994;45:1637–1647. [DOI] [PubMed] [Google Scholar]