

Abstract
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a natural substrate for the N-terminal active site of angiotensin-converting enzyme (ACE). We previously reported that Ac-SDKP prevented cardiac fibrosis in rats with renovascular or aldosterone-salt hypertension. However, it is not clear whether Ac-SDKP reverses cardiac fibrosis in hypertension, nor the mechanism(s) involved. In the present study, we tested the hypothesis that Ac-SDKP reversal of hypertension-induced cardiac fibrosis involves a decrease in transforming growth factor-β (TGF-β) and/or connective tissue growth factor (CTGF). In 2-kidney, 1-clip (2K-1C) hypertensive rats, Ac-SDKP at 400 or 800 μg/kg per day SC was started 8 weeks after hypertension and cardiac fibrosis were established and was continued for 8 weeks. Left ventricular (LV) collagen in rats with 2K-1C plus vehicle at 8 and 16 weeks after clipping was similar but higher than in the sham group (P<0.05). Ac-SDKP at 400 and 800 μg/kg per day, which increased plasma Ac-SDKP 2- and 5-fold, respectively, reversed the increase in LV collagen in a dose-dependent manner. The mechanism by which Ac-SDKP reverses LV fibrosis does not appear to depend on ACE inhibition by Ac-SDKP, since we found that Ac-SDKP at various doses did not affect blood pressure responses to exogenous angiotensin I or bradykinin. However, Ac-SDKP reversed the increase in LV TGF-β and CTGF compared with rats with 2K-1C plus vehicle (P<0.005). We concluded that in hypertension, Ac-SDKP reverses cardiac fibrosis, perhaps due in part to a decrease in TGF-β and CTGF in the heart.
Keywords: hypertension, renovascular; collagen; heart; fibrosis; transforming growth factors
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a natural inhibitor of pluripotent hematopoietic stem cell proliferation1,2 that is normally present in human plasma and circulating mononuclear cells.3 It is cleaved to an inactive form by the NH2-terminal catalytic domain of angiotensin-converting enzyme (ACE).4
ACE inhibitors (ACEi) reversed cardiac fibrosis in patients with hypertensive heart disease,5 spontaneously hypertensive rats (SHR),6 and rats with deoxycorticosterone acetate (DOCA)-salt hypertension, and this effect was independent of changes in blood pressure (BP).7 ACEi reversal of cardiac fibrosis could be partially due to Ac-SDKP, since ACE inhibition increased plasma Ac-SDKP.4,8 We previously found that Ac-SDKP not only inhibited cardiac fibroblast proliferation and collagen synthesis in vitro but also prevented enhanced collagen deposition in the left ventricle (LV) in both 2-kidney, 1-clip (2K-1C) and aldosterone-salt hyper-tensive rats.9–11 However, once cardiac fibrosis is established, it is not clear whether it can be reversed by Ac-SDKP, nor the mechanisms involved.
Transforming growth factor-β (TGF-β) is a key profibrotic cytokine whose effect may be mediated by another cytokine, connective tissue growth factor (CTGF), a downstream component of the TGF-β signaling pathway.12–16 These two cytokines play a central role in the development of cardiac fibrosis.16 Thus, we hypothesized that Ac-SDKP reverses hypertension-induced cardiac fibrosis by inhibiting expression of TGF-β and/or CTGF. Boulanger et al17 reported that Ac-SDKP at very high doses impairs angiotensin I (Ang I)–induced contractions of the rat aorta in vitro, suggesting that this effect is mediated by ACE inhibition. Therefore, we tested whether Ac-SDKP at the doses we used in vivo inhibits ACE activity. For this, we examined the pressor and depressor response to Ang I and bradykinin (BK), respectively, in an additional group of rats treated with various doses of Ac-SDKP. Since we previously found that cardiac fibrosis is fully established 8 weeks after induction of 2K-1C hypertension,10 in the present study Ac-SDKP was started at the end of week 8 and continued for 8 weeks.
Methods
Animals and Experimental Design
Male Sprague-Dawley rats (Charles River, 175 to 185 g) were anesthetized with methohexital sodium (Brevital; 50 mg/kg IP). Hypertension was induced by clipping the left renal artery as described previously.10 Ac-SDKP (obtained from Dr Domenico Regoli, University of Sherbrooke, Canada) at 400 or 800 μg/kg per day was started 8 weeks after clipping and continued for 8 weeks. Ac-SDKP was infused subcutaneously through an osmotic minipump (Alzet). Animals were divided into 6 groups: (1) 8-week sham-clipped (sham-8W, n=5), (2) 8-week 2K-1C plus vehicle (2K-1C/vehicle-8W, n=5), (3) 16-week sham-clipped (sham-16W, n=9), (4) 16-week 2K-1C plus vehicle (2K-1C/vehicle-16W, n=10), (5) 2K-1C plus Ac-SDKP 400 μg/kg per day (2K-1C/Ac-SDKP 400, n=9), and (6) 2K-1C plus Ac-SDKP 800 μg/kg per day (2K-1C/Ac-SDKP 800, n=13). To test whether Ac-SDKP at the doses we used inhibits ACE, we studied the effect of Ac-SDKP at 400 (n=5), 800 (n=4), and 1600 μg/kg per day (n=5) for 3 weeks (subcutaneously, through osmotic minipump) on the BP response to Ang I, Ang II, and BK (Bachem). Captopril (Sigma; 50 mg/kg per day) given in drinking water was used as a positive control for blocking ACE (n=5) and was compared with placebo (n=5). Drugs were given through a catheter placed in the carotid artery, and mean arterial pressure (MAP) was measured via the femoral artery.18 These studies were approved by the Henry Ford Hospital Institutional Animal Care and Use Committee.
Measurement of Systolic Blood Pressure and Sample Collection
Systolic blood pressure (SBP) was measured by tail cuff once per week. Tissue was collected as described previously.11
Measurement of Plasma Ac-SDKP
Blood was withdrawn through the vena cava in a heparinized syringe containing lisinopril (Merck; final concentration, 10 μmol/L). Plasma Ac-SDKP was measured with an enzyme immunoassay kit (SPI-BIO).
Hydroxyproline Assay
Collagen content of the LV was determined by hydroxyproline assay, as described previously.11,19,20
Histochemical Analysis of Interstitial Collagen Fraction in the Left Ventricle
Interstitial collagen fraction (ICF) was measured in a double-blind manner, as described previously.11,21,22
Immunohistochemical Staining for TGF-β and CTGF in the Left Ventricle
A monoclonal antibody against bovine TGF-β1,2,3 (1:1000; R&D Systems) and an affinity-purified rabbit polyclonal antibody against a human CTGF peptide (residues 81 to 94, 1 ng/mL)23 were used as the primary antibody for TGF-β and CTGF detection, respectively. Staining was assayed with a Vectastain ABC kit (Vector Laboratories), as described previously.11 Areas that stained positive for TGF-β (Bioquant NOVA Image Analysis System) were expressed as a percentage of total myocardial area, as suggested by Lim et al.24 For each sample, 16 randomly selected fields in the LV were examined. CTGF-positive cells in half of the LV sections were counted and expressed as positive cells per millimeter squared. Immunohistochemical measurements were carried out in a blinded manner.
Statistical Analysis
ANOVA with repeated measures was used to analyze the SBP data. The Student t test was used to compare treatment groups with regard to the ratio of organ weight to body weight (BW), TGF-β and CTGF, collagen content, and ICF. Because of the lack of normally distributed samples, nonparametric Wilcoxon 2-sample exact tests were used to compare plasma Ac-SDKP measurements. The Hochberg step-up method was applied to each group of probability values within each parameter group to adjust for multiple testing.25 Family-wise α levels were preset at α=0.05 within each group. Values are expressed as mean±SEM.
Results
Systolic Blood Pressure, Plasma Ac-SDKP Concentration, and Ratio of Organ Weight to Body Weight
Two weeks after surgery, SBP in the 2K-1C/vehicle group was significantly increased compared with sham, reaching a maximum by week 3 and remaining elevated for up to 16 weeks. Ac-SDKP at 400 or 800 μg/kg per day for 8 weeks had no effect on hypertension (Table). After 8 weeks of Ac-SDKP treatment, plasma Ac-SDKP concentration was increased in a dose-dependent manner compared with the untreated groups (Table). The ratio of LV weight (LVW) to BW in 2K-1C rats was significantly and similarly increased at 8 and 16 weeks after clipping, and treatment with Ac-SDKP for 8 weeks did not reverse this increase. The ratio of atrial weight (AW) or RV weight (RVW) to BW did not differ significantly among groups (Table).
Table.
Measurement of Systolic Blood Pressure, Organ Weight, and Plasma Ac-SDKP Concentration
| 2K-1C |
||||||
|---|---|---|---|---|---|---|
| Parameters | Sham-8W | Sham-16W | Vehicle-8W | Vehicle-16W | Ac-SDKP (400 μg · kg−1 · d−1) |
Ac-SDKP (800 μg · kg−1 · d−1) |
| SBP, mm Hg | 123±7 | 112±3 | 208±7* | 157±3† | 149±3† | 157±3† |
| LVW/BW, mg/100 g | 179±4 | 168±4 | 263±23* | 210±9† | 211±8† | 231±7† |
| RVW/BW, mg/100 g | 51.4±2.2 | 45.2±1.6 | 56.3±3.6 | 46.9±1.5 | 50.2±2.1 | 48.8±3.5 |
| AW/BW, mg/10 g | 1.3±0.1 | 1.5±0.1 | 1.5±0.1 | 1.3±0.1 | 1.6±0.1 | 1.5±0.1 |
| Plasma Ac-SDKP, nmol/L | 3.93±0.30 | 3.55±1.12 | 3.92±0.54 | 2.17±0.20 | 5.77±1.39‡ | 13.19±3.69‡ |
SBP indicates systolic blood pressure; LVW, left ventricular weight; RVW, right ventricular weight; AW, atrial weight; BW, body wt.
P<0.05 vs sham-8W;
P<0.05 vs sham-16W;
P<0.05 vs 2K-1C/vehicle-16W.
Effect of Ac-SDKP on Pressor Response to Ang I and Ang II or Depressor Response to BK
As shown in Figure 1, BP response to Ang I, Ang II, or BK was not affected by Ac-SDKP. However, BP response to Ang I was attenuated by captopril, whereas BP response to BK was enhanced, consistent with ACE inhibition; conversely, BP response to Ang II was not affected by captopril, indicating that Ac-SDKP at the doses used did not inhibit ACE.
Figure 1.

Changes in MAP in response to exogenous Ang I, BK, and Ang II in rats treated with various doses of Ac-SDKP or captopril for 3 weeks. Baseline MAP in each group was control, 120±11 mm Hg; Ac-SDKP 400 μg/kg per day, 112±13 mm Hg; Ac-SDKP 800 μg/kg per day, 126±2 mm Hg; Ac-SDKP 1600 μg/kg per day, 123±9 mm Hg, and captopril, 125±6 mm Hg. Ac-SDKP did not affect the MAP response to Ang I, BK, or Ang II. As expected, captopril attenuated the pressor effect of Ang I and potentiated the depressor effect of BK but did not affect the pressor effect of Ang II. This suggests that Ac-SDKP at the doses used does not affect ACE activity in vivo. *P<0.05 vs control.
Collagen Content of the Left Ventricle
LV collagen content, as estimated by hydroxyproline assay, was increased in the 2K-1C/vehicle group 8 weeks after clipping compared with sham (12.5±0.9 versus 9.7±0.3 μg/mg dry LV; P=0.03), and this increase was maintained for up to 16 weeks (Figure 2). LV collagen was reversed in a dose-dependent manner in rats with 2K-1C plus Ac-SDKP at 16 weeks after clipping, reaching maximum regression at a dose of 800 μg/kg per day (Figure 2).
Figure 2.

LV collagen content (top) and interstitial collagen fraction (ICF; bottom) in 2K-1C hypertensive rats treated with Ac-SDKP for 8 weeks. Collagen content (assessed by hydroxyproline assay) was significantly increased at 8 or 16 weeks after clipping. Ac-SDKP reversed this increase in a dose-dependent manner compared with 2K-1C/vehicle-8W, reaching significance only at a high dose (P<0.001). ICF showed a similar pattern, confirming the collagen data. *P<0.05 vs sham-8W, †P<0.001 vs sham-16W, ‡P<0.05, §P<0.001 vs 2K-1C/vehicle-16W.
Interstitial Collagen Fraction in the Left Ventricle
Histochemical analysis showed that ICF in the LV was significantly increased in the 8-week 2K-1C/vehicle group compared with 8-week sham (P<0.001). There was no difference in ICF between 8-week and 16-week 2K-1C/ vehicle groups. Ac-SDKP at 400 and 800 μg/kg per day reversed the increase in ICF in a dose-dependent manner (Figures 2 and 3), confirming the data for collagen content.
Figure 3.
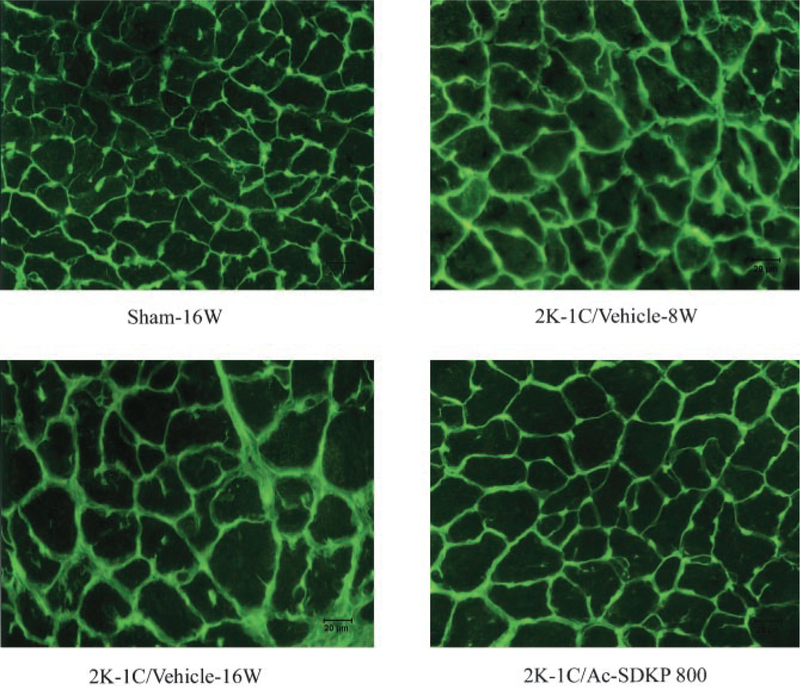
Myocyte cross-sectional area (MCSA) and ICF in LV of sham-16W rats and 2K-1C-8W and 2K-1C-16W rats treated with vehicle or Ac-SDKP at 800 μg/kg per day. Green staining represents collagen. Eight weeks after clipping, LV collagen deposition and myocyte cross-sectional area were increased. LV collagen in 2K-1C/vehicle-8W was similar to 2K-1C/vehicle-16W. Ac-SDKP reversed the increase in LV collagen but not the increase in MCSA, suggesting that Ac-SDKP can reverse cardiac fibrosis but not hypertrophy in 2K-1C hypertensive rats.
TGF-β and CTGF Expression in the Left Ventricle
TGF-β–positive staining was found in the LV interstitial space and perivascular area in both sham-clipped rats and rats with 2K-1C hypertension (Figure 4). It was increased 6-fold in the LV in the 2K-1C/vehicle group 8 weeks after clipping compared with 8-week sham (0.43±0.04% versus 0.07±0.01%; P=0.001) and exhibited a similar increase at 16 weeks, whereas there was no significant difference in TGF-β expression between 8-week and 16-week sham groups (Figures 4 and 5). Ac-SDKP treatment for 8 weeks significantly reversed increased TGF-β in the LV (Figure 5). CTGF-positive staining was seldom observed in vascular endothelial, smooth muscle, or LV interstitial cells in the sham-clipped groups; however, in rats with 2K-1C hypertension, CTGF-positive staining was distributed diffusely throughout the vascular endothelium, interstitial, and perivascular spaces (Figure 6). At either 8 or 16 weeks after clipping, more CTGF-positive cells were seen in the 2K-1C/vehicle group than sham. Both doses of Ac-SDKP also significantly lowered the number of CTGF-positive cells in the LV (Figures 5 and 6).
Figure 4.
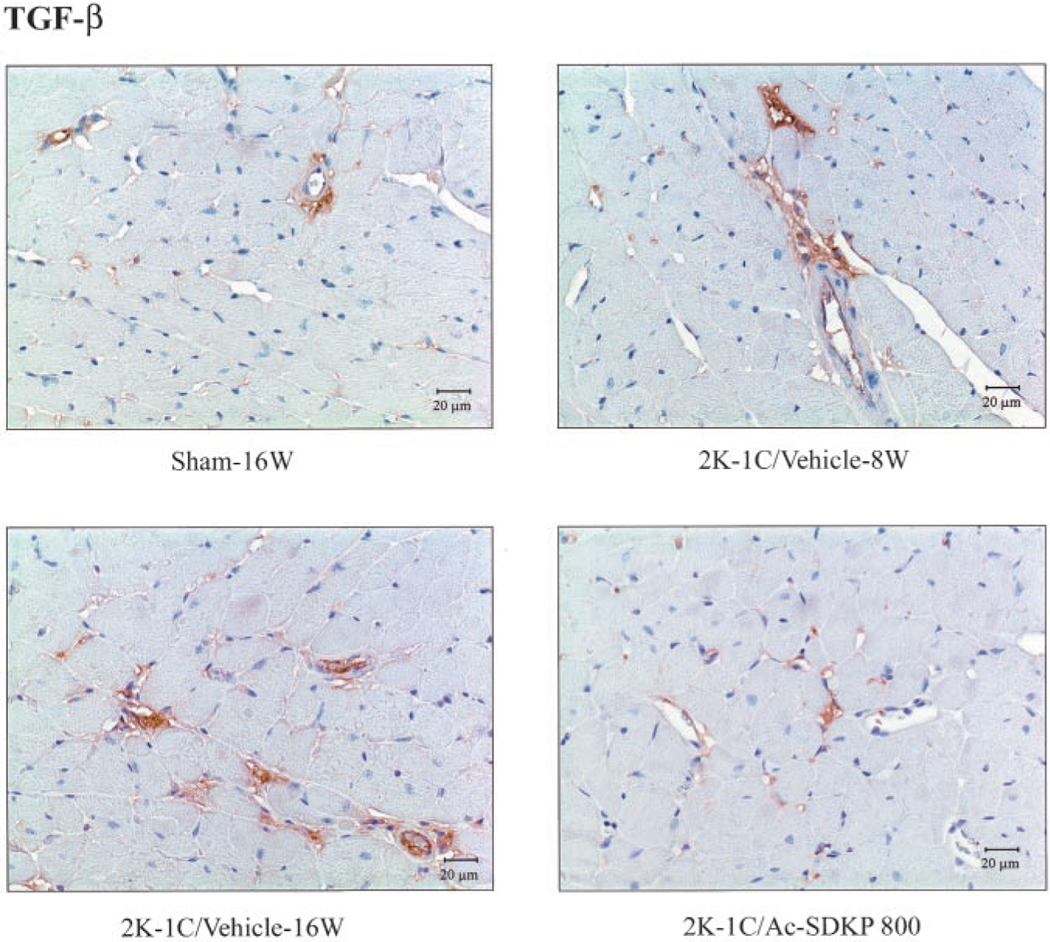
Immunohistochemical staining for TGF-β (with hematoxylin counter-staining) in LV of 2K-1C rats treated with Ac-SDKP. Brown color represents positive staining. TGF-β–positive staining was found in the LV interstitial space, primarily in the perivascular area. TGF-β expression was increased 8 and 16 weeks after clipping, and Ac-SDKP treatment for 8 weeks reversed this increase.
Figure 5.

TGF-β (top) and CTGF (bottom) in LV of 2K-1C rats treated with Ac-SDKP for 8 weeks. Percentage of TGF-β–positive area was significantly increased at 8 and 16 weeks after clipping compared with sham. Ac-SDKP treatment for 8 weeks significantly lowered TGF-β production. CTGF-positive cells showed a similar pattern. This suggests that Ac-SDKP reverses cytokine production in 2K-1C hypertensive rats. *P<0.005 vs sham-8W, †P<0.001 vs sham-16W, ‡P<0.005 vs 2K-1C/vehicle-16W.
Figure 6.
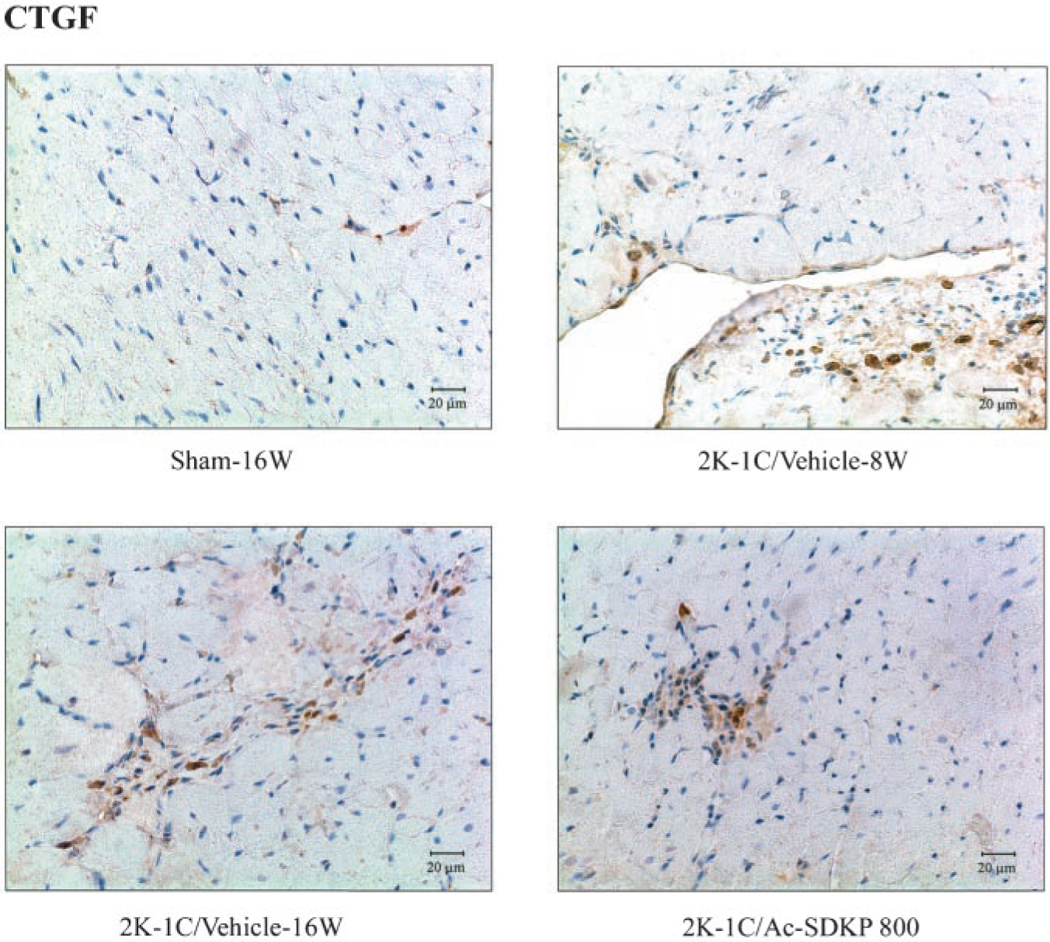
Immunohistochemical staining for CTGF (with hematoxylin counterstaining) in LV of 2K-1C rats treated with Ac-SDKP. Brown color represents positive staining. CTGF-positive cells were observed in vascular endothelial cells and interstitial cells and were increased 8 and 16 weeks after clipping, whereas Ac-SDKP reversed this increase.
Discussion
We previously reported that Ac-SDKP prevents cardiac fibrosis without affecting hypertrophy or BP.10,11 In the present study, we tested whether Ac-SDKP can reverse cardiac fibrosis in 2K-1C hypertensive rats. We found that LV collagen deposition was significantly increased at 8 weeks after clipping and remained elevated at 16 weeks compared with sham-clipped rats, as assessed by hydroxyproline assay and histochemical analysis. Ac-SDKP was started 8 weeks after clipping and continued for 8 weeks. It reversed cardiac fibrosis in a dose-dependent manner, yet had no effect on hypertension or LV hypertrophy. It is interesting to note that RV hypertrophy did not occur in this model of high renin and Ang II hypertension, similar to the study by Brilla et al,26 who reported finding RV fibrosis but no hypertrophy in a slightly different model (abdominal aortic banding plus right renal artery clipping). The present study was a follow-up of our previous work10 and was designed mainly to study whether Ac-SDKP reverses established LV fibrosis. Others have also reported reversing cardiac fibrosis in SHR without altering LV hypertrophy and hypertension with the use of an ACEi.6 The density of collagen is low, which together with its small volume fraction (0.9%) accounts for its lack of any major impact on myocardial tissue weight.
One mechanism by which Ac-SDKP reverses cardiac fibrosis could be inhibition of ACE. Indeed, Boulanger et al17 reported that Ac-SDKP at 10 to 100 μmol/L impaired Ang I–induced contractions of the rat aorta in vitro, suggesting that this effect is mediated by ACE inhibition. However, our present findings indicate that at the doses of Ac-SDKP we used to reverse cardiac fibrosis, or even at a higher dose, ACE activity remained unchanged, since Ac-SDKP did not affect changes in BP in response to exogenous Ang I, bradykinin, or Ang II, suggesting that the antifibrotic effects of Ac-SDKP that we observed were not mediated by ACE inhibition. Although the reason for the differences between Boulanger’s findings and ours is not clear, they could be related to the amount of Ac-SDKP that we used (plasma concentration, 4 to 15 nmol/L), which was at least one order of magnitude lower than in Boulanger’s study.
TGF-β is a cytokine that may play an important role in the development and maintenance of fibrosis, particularly in the presence of an activated renin-angiotensin system (as in 2K-1C hypertension), since Ang II stimulates synthesis of TGF-β.27,28 TGF-β has been shown to stimulate gene expression of extra-cellular matrix components such as fibronectin, laminin, and collagen.16 We confirmed that TGF-β expression was similarly increased in the LV in another model of high Ang II hypertension, transgenic rats characterized by increased cardiac Ang II concentrations [TGR(mRen2)27], as reported by others.28,29 Ac-SDKP treatment for 8 weeks significantly reversed increased TGF-β; however, it is not clear how Ac-SDKP reduced TGF-β production. Several types of cells are able to express TGF-β, including fibroblasts, myocytes, and endothelial and vascular smooth muscle cells.30 In addition, an increased number of inflammatory cells (eg, macrophages) in the hypertensive heart could increase TGF-β, leading to cardiac fibrosis.31–33 We previously reported that Ac-SDKP inhibits infiltration by inflammatory cells (monocytes and macrophages) within the LV interstitial space of rats with 2K-1C hypertension,10 suggesting that Ac-SDKP may inhibit TGF-β expression through its anti-inflammatory effect.
CTGF is a 38-kDa protein belonging to the insulin-like growth factor family and is a mitogenic and chemotactic factor in cultured fibroblasts.34,35 It has been shown to promote proliferation and production of extracellular matrix in the heart and is a downstream component of the TGF-β signaling path-way.16 CTGF is a ubiquitously expressed cytokine that is secreted by fibroblasts and endothelial cells. We found that CTGF was markedly increased in the LV of 2K-1C rats at both 8 and 16 weeks after clipping and that Ac-SDKP reversed overexpression of CTGF in the heart. Therefore, reversal of cardiac fibrosis was associated with reversal of increased LV TGF-β and CTGF. Ac-SDKP could reverse the increase in CTGF by inhibiting TGF-β production. It is possible that inhibition of CTGF by Ac-SDKP is due to suppression of both TGF-β expression and TGF-β–stimulated phosphorylation of Smad2 or protein kinase C/ras/mitogen-activated protein kinase (MAK) pathways, since there is evidence that Ac-SDKP inhibits the effect of TGF-β on Smad signaling in vitro.36,37 TGF-β could also directly activate transcription of the CTGF gene, which contains a specific TGF-β response element; thus, Ac-SDKP may inhibit CTGF expression by decreasing the available amount of TGF-β.38
The antifibrotic effect of Ac-SDKP appears to be compatible with its inhibitory effect on TGF/CTGF expression; still, the changes in collagen did not completely match those of TGF/CTGF, suggesting that other mechanisms could be involved in the antifibrotic effect of Ac-SDKP, including inhibition of fibroblast proliferation or inhibition of collagen synthesis, as we have shown previously.9–11
Reversal of cardiac fibrosis by Ac-SDKP could also be due to enhanced metalloproteinase (MMP) activity and decreased tissue inhibitors of MMPs, but we know of no evidence that Ac-SDKP has any effect on these two parameters. The contribution of collagen degradation to reversal of fibrosis is rather complex and merits a separate study, investigating the effect of Ac-SDKP on temporal changes in the activity of matrix MMPs and their respective endogenous inhibitors.
In summary, our study showed that 2K-1C hypertension increased TGF-β and CTGF in the LV, accompanied by increased LV collagen deposition. Ac-SDKP significantly reversed increased LV fibrosis without affecting BP or cardiac hypertrophy, acting independently of ACE inhibition and most likely through inhibition of TGF-β/CTGF expression.
Perspectives
We found that Ac-SDKP can ameliorate cardiac fibrosis by reversing increased LV TGF-β/CTGF. In a separate study, we compared the antifibrotic effect of ACEi with that of Ac-SDKP in Ang II–hypertensive rats and found that ACEi not only mimicked the antifibrotic effect of Ac-SDKP but also raised plasma Ac-SDKP to levels similar to exogenous Ac-SDKP.39 Since ACEi can increase plasma Ac-SDKP concentration4,8 and reverse cardiac fibrosis,40 we speculate that the beneficial effects of ACEi could be partially mediated by Ac-SDKP and that the antifibrotic effects of ACEi and Ac-SDKP on the heart could be due in part to decreased TGF-β/CTGF expression. More extensive studies are needed to clarify the mechanisms of Ac-SDKP in reducing LV fibrosis. In the future, an Ac-SDKP antagonist or an inhibitor of Ac-SDKP production will be instrumental in fully assessing the role of Ac-SDKP in the antifibrotic effects of ACEi. Reversal of cardiac fibrosis has important clinical relevance, and producing an Ac-SDKP agonist that is resistant to ACE, or an antagonist of the TGF-β/CTGF signaling pathway, might both prevent and reverse cardiac fibrosis.
Acknowledgments
This study was supported by American Heart Association grant 0130128N and National Institutes of Health grants HL71806 – 01 (N.-E.R.) and HL-28982–21 (O.A.C.).
References
- 1.Lenfant M, Wdzieczak-Bakala J, Guittet E, Prome JC, Sotty D, Frindel E. Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci U S A. 1989;86: 779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonnet D, Lemoine FM, Pontvert-Delucq S, Baillou C, Najman A, Guigon M. Direct and reversible inhibitory effect of the tetrapeptide acetyl-N-Ser-Asp-Lys-Pro (Seraspenide) on the growth of human CD34+ subpopulations in response to growth factors. Blood. 1993;82:3307–3314. [PubMed] [Google Scholar]
- 3.Pradelles P, Frobert Y, Créminon C, Liozon E, Massé A, Frindel E. Negative regulator of pluripotent hematopoietic stem cell proliferation in human white blood cells and plasma as analysed by enzyme immunoassay. Biochem Biophys Res Commun 1990;170:986–993. [DOI] [PubMed] [Google Scholar]
- 4.Azizi M, Rousseau A, Ezan E, Guyene T-T, Michelet S, Grognet J-M, Lenfant M, Corvol P, Ménard J. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J Clin Invest 1996;97:839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102: 1388–1393. [DOI] [PubMed] [Google Scholar]
- 6.Brilla CG, Janicki JS, Weber KT. Impaired diastolic function and coronary reserve in genetic hypertension: role of interstitial fibrosis and medial thickening of intramyocardial coronary arteries. Circ Res 1991;69:107–115. [DOI] [PubMed] [Google Scholar]
- 7.Brown L, Duce B, Miric G, Sernia C. Reversal of cardiac fibrosis in deoxy-corticosterone acetate-salt hypertensive rats by inhibition of the renin-angiotensin system. J Am Soc Nephrol 1999;10:S143–S148. [PubMed] [Google Scholar]
- 8.Azizi M, Ezan E, Nicolet L, Grognet JM, Menard J. High plasma level of N-acetyl-seryl-aspartyl-lysyl-proline: a new marker of chronic angiotensin-converting enzyme inhibition. Hypertension. 1997;30:1015–1019. [DOI] [PubMed] [Google Scholar]
- 9.Rhaleb N-E, Peng H, Harding P, Tayeh M, LaPointe MC, Carretero OA. Effect of N-acetyl-seryl-aspartyl-lysyl-proline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension. 2001;37:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rhaleb N-E, Peng H, Yang X-P, Liu Y-H, Mehta D, Ezan E, Carretero OA. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation. 2001;103:3136–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng H, Carretero OA, Raij L, Yang F, Kapke A, Rhaleb N-E. Antifibrotic effects of N-acetyl-seryl-aspartyl-lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension. 2001;37:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-β by anti-TGF-β antibody attenuates kidney hypertrophy and the enhanced extra-cellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–530. [DOI] [PubMed] [Google Scholar]
- 13.Bitzer M, Sterzel RB, Bottinger EP. Transforming growth factor-beta in renal disease. Kidney Blood Press Res 1998;21:1–12. [DOI] [PubMed] [Google Scholar]
- 14.Goumenos DS, Tsamandas AC, Oldroyd S, Sotsiou F, Tsakas S, Petropoulou C, Bonikos D, el Nahas AM, Vlachojannis JG. Transforming growth factor-beta(1) and myofibroblasts: a potential pathway towards renal scarring in human glomerular disease. Nephron 2001;87:240–248. [DOI] [PubMed] [Google Scholar]
- 15.Villarreal FJ, Dillmann WH. Cardiac hypertrophy-induced changes in mRNA levels for TGF-β1, fibronectin, and collagen. Am J Physiol 1992; 262:H1861–H1866. [DOI] [PubMed] [Google Scholar]
- 16.Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF-β in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol 2000;32:1805–1819. [DOI] [PubMed] [Google Scholar]
- 17.Boulanger CM, Ezan E, Massé F, Mathieu E, Lévy BI, Azizi M. The hemoregulatory peptide N-acetyl-ser-asp-lys-pro impairs angiotensin I-induced contractions in rat aorta. Eur J Pharmacol 1998;363:153–156. [DOI] [PubMed] [Google Scholar]
- 18.Rhaleb N-E, Yang X-P, Scicli AG, Carretero OA. Role of kinins and nitric oxide in the antihypertrophic effect of ramipril. Hypertension. 1994;23: 865–868. [DOI] [PubMed] [Google Scholar]
- 19.Chiariello M, Ambrosio G, Cappelli-Bigazzi M, Perrone-Filardi P, Brigante F, Sifola C. A biochemical method for the quantitation of myocardial scarring after experimental coronary artery occlusion. J Mol Cell Cardiol 1986;18: 283–290. [DOI] [PubMed] [Google Scholar]
- 20.Cleutjens JP, Verluyten MJ, Smits JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen-Smith FM, Watson L, Lu DY, Goldstein I. Griffonia simplicifolia I: fluorescent tracer for microcirculatory vessels in nonperfused thin muscles and sectioned muscle. Microvasc Res 1988;36:199–215. [DOI] [PubMed] [Google Scholar]
- 22.Laitinen L. Griffonia simplicifolia lectins bind specifically to endothelial cells and some epithelial cells in mouse tissues. Histochem J. 1987;19:225–234. [DOI] [PubMed] [Google Scholar]
- 23.Brigstock DR, Steffen CL, Kim GY, Vegunta RK, Diehl JR, Harding PA. Purification and characterization of novel heparin-binding growth factors in uterine secretory fluids: identification as heparin-regulated Mr 10,000 forms of connective tissue growth factor. J Biol Chem 1997;272:20275–20282. [DOI] [PubMed] [Google Scholar]
- 24.Lim D-S, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M, Roberts R, Marian AJ. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westfall PH, Tobias RD, Rom D, Wolfinger RD, Hochberg Y. Multiple Comparisons and Multiple Tests Using the SAS System. Cary, NC: SAS Institute; 1999. [Google Scholar]
- 26.Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990;67: 1355–1364. [DOI] [PubMed] [Google Scholar]
- 27.Crawford DC, Chobanian AV, Brecher P. Angiotensin II induces fibronectin expression associated with cardiac fibrosis in the rat. Circ Res 1994;74: 727–739. [DOI] [PubMed] [Google Scholar]
- 28.Pinto YM, Pinto-Sietsma S-J, Philipp T, Engler S, Kossmehl P, Hocher B, Marquardt H, Sethmann S, Lauster R, Merker H-J, Paul M. Reduction in left ventricular messenger RNA for transforming growth factor β1 attenuates left ventricular fibrosis and improves survival without lowering blood pressure in the hypertensive TGR(mRen2)27 rat. Hypertension. 2000;36:747–754. [DOI] [PubMed] [Google Scholar]
- 29.Villarreal FJ, MacKenna DA, Omens JH, Dillmann WH. Myocardial remodeling in hypertensive Ren-2 transgenic rats. Hypertension. 1995;25: 98–104. [DOI] [PubMed] [Google Scholar]
- 30.Kawano H, Do YS, Kawano Y, Starnes V, Barr M, Law RE, Hsueh WA. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation. 2000;101:1130–1137. [DOI] [PubMed] [Google Scholar]
- 31.Porreca E, Di Febbo C, Mincione G, Reale M, Baccante G, Guglielmi MD, Cuccurullo F, Colletta G. Increased transforming growth factor-β production and gene expression by peripheral blood monocytes of hypertensive patients. Hypertension. 1997;30:134–139. [DOI] [PubMed] [Google Scholar]
- 32.Hinglais N, Heudes D, Nicoletti A, Mandet C, Laurent M, Bariéty J, Michel JB. Colocalization of myocardial fibrosis and inflammatory cells in rats. Lab Invest 1994;70:286–294. [PubMed] [Google Scholar]
- 33.Nicoletti A, Heudes D, Mandet C, Hinglais N, Bariety J, Michel JB. Inflammatory cells and myocardial fibrosis: spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc Res 1996;32:1096–1107. [DOI] [PubMed] [Google Scholar]
- 34.Moussad EEA, Brigstock DR. Connective tissue growth factor: what’s in a name? Mol Genet Metab 2000;71:276–292. [DOI] [PubMed] [Google Scholar]
- 35.Steffen CL, Ball-Mirth DK, Harding PA, Bhattacharyya N, Pillai S, Brigstock DR. Characterization of cell-associated and soluble forms of connective tissue growth factor (CTGF) produced by fibroblast cells in vitro. Growth Factors. 1998;15:199–213. [DOI] [PubMed] [Google Scholar]
- 36.Pokharel S, Rasoul S, Roks AJM, van Leeuwen REW, van Luyn MJA, Deelman LE, Smits JF, Carretero O, van Gilst WH, Pinto YM. N-acetyl-Ser-Asp-Lys-Pro inhibits phosphorylation of Smad2 in cardiac fibroblasts. Hypertension. 2002;40:155–161. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Blom IE, Sa S, Goldschmeding R, Abraham DJ, Leask A. CTGF expression in mesangial cells: involvement of SMADs, MAP kinase, and PKC. Kidney Int 2002;62:1149–1159. [DOI] [PubMed] [Google Scholar]
- 38.Grotendorst GR, Okochi H, Hayashi N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ 1996;7:469–480. [PubMed] [Google Scholar]
- 39.Rasoul S, Carretero OA, Peng H, Cavasin MA, Zhou J, Rhaleb N-E. Angiotensin-converting enzyme inhibition and acetyl-seryl-aspartyl-lysyl-proline lower cardiac inflammation and fibrosis in hypertension. Hypertension. 2002; 40:397 Abstract. [Google Scholar]
- 40.Brilla CG, Matsubara L, Weber KT. Advanced hypertensive heart disease in spontaneously hypertensive rats. Lisinopril-mediated regression of myocardial fibrosis. Hypertension. 1996;28:269–275. [DOI] [PubMed] [Google Scholar]