

In this Review, Rashkovan et al. discuss the role of cancer metabolic circuitries feeding anabolism and redox potential in leukemia development and recent progress in translating these important findings to the clinic.
Keywords: glycolysis, leukemia, metabolism, mitochondria, targeted therapy, Warburg effect
Abstract
Leukemia cell proliferation requires up-regulation and rewiring of metabolic pathways to feed anabolic cell growth. Oncogenic drivers directly and indirectly regulate metabolic pathways, and aberrant metabolism is central not only for leukemia proliferation and survival, but also mediates oncogene addiction with significant implications for the development of targeted therapies. This review explores leukemia metabolic circuitries feeding anabolism, redox potential, and energy required for tumor propagation with an emphasis on emerging therapeutic opportunities.
Tumor cell proliferation is tightly coupled with anabolic cell growth, as each cycle of cell duplication implies a 100% increase in cellular biomass for cell size to be maintained. As a result, activation of anabolic pathways together with the energy and redox potential to feed them are essential components of oncogenic transformation. Moreover, cancer cells do not simply highjack anabolic mechanisms active in highly proliferating normal tissues, but they reorganize these, bypassing cell growth control checkpoints, securing maximum anabolic output, and gaining enhanced protection from oxidative stress. This aberrant nature of cancer metabolic circuitries was first recognized by Otto Warburg when he postulated that tumor tissues have an increased rate of glucose uptake compared with normal tissues and rely primarily on glucose to produce ATP (Warburg et al. 1924; Warburg 1956). While our understanding of cancer metabolism is still developing, altered metabolism is already recognized as a cornerstone mechanism of tumorigenesis, a hallmark of cancer (Hanahan and Weinberg 2011). In addition, tumor metabolism represents a readily druggable molecular space, bringing much interest in targeting metabolic vulnerabilities in cancer. Importantly, major oncogenic drivers broadly active in human leukemia such as MYC and RAS are prime drivers of metabolic rewiring in leukemia. Increased MYC expression is a hallmark of Burkitt's lymphoma and B-cell and T-cell acute lymphoblastic leukemias (ALL) harboring chromosomal translocations placing the MYC locus in the vicinity of the immunoglobulin and T cell receptor loci, respectively (Dalla-Favera et al. 1983; Erikson et al. 1986). In addition, enhancer-driven MYC expression downstream from NOTCH1 is a driver of cell proliferation, growth, and survival in over 60% of T-ALLs with activating mutations in NOTCH1 (Palomero et al. 2006; Herranz et al. 2014) and as regulator of cell proliferation and negative regulator of cell differentiation in acute myeloid leukemia (Zuber et al. 2011). Furthermore, oncogenic MYC expression promotes glycolysis, glutaminolysis, mitochondrial biogenesis, lipid synthesis, and nucleotide biosynthesis (Shim et al. 1997; Osthus et al. 2000; Li et al. 2005; Liu et al. 2008; Wise et al. 2008; Gao et al. 2009; Morrish et al. 2010). MYC expression and activity are modulated by HIF1A and mTORC1 signaling, which are controlled by oxygen and metabolic sensors, thus integrating the anabolic and pro-proliferative effects of MYC with metabolic determinants of cell growth (Zhang et al. 2007; Pourdehnad et al. 2013). Activating mutations in the RAS-MAPK pathway are characteristic of juvenile myelomonocytic leukemia (Stieglitz et al. 2015), but can also be found in ALL and acute myeloid leukemia (AML) (Roberts and Mullighan 2015; Belver and Ferrando 2016; Bullinger et al. 2017). As in the case of MYC, RAS mutations promote glutaminolysis, enhance glucose uptake, and promote a shift to anabolic metabolism by shunting glycolytic intermediates to anabolic pathways and by regulating the activity of rate-limiting enzymes in the nonoxidative arm of the pentose phosphate pathway (PPP) and in hexosamine biosynthesis (Ying et al. 2012; Lyssiotis et al. 2013; Son et al. 2013). Furthermore, RAS-driven cancers scavenge nutrients via increased micropinocytosis and autophagy, which promote tumor growth and proliferation (Bar-Sagi and Feramisco 1986; Guo et al. 2011; Lock et al. 2011; Yang et al. 2011; Commisso et al. 2013). Finally, PTEN, a negative regulator of the PI3K-AKT signaling pathway is frequently mutated and deleted in T-ALL (Palomero et al. 2007), and loss of PTEN results in increased glycolysis, lipid biogenesis, protein translation, and mitochondrial metabolism (Stiles et al. 2004). These oncogene-driven metabolic circuitries and interactions substantiate that metabolic alterations in cancer cells are closely intertwined with the genetic, transcriptional, and epigenetic changes associated with malignant transformation and serve as critical downstream effectors of oncogenic pathways mediating tumor growth. As corollary, oncogene-driven metabolic circuitries constitute important mediators of oncogene addiction, and as such, represent true cancer-specific vulnerabilities, further substantiating their relevance as therapeutic targets. In this review, we discuss the role of cancer metabolic pathways in leukemia development and recent progress in translating these important findings to the clinic.
Glucose as source of energy and anabolic building blocks
Glycolysis converts glucose to pyruvate and yields energy in the form of two molecules of ATP. Ultimately, pyruvate is converted to lactate by aerobic glycolysis or introduced into the tricarboxylic acid (TCA) cycle, which increases energy production by fueling mitochondrial oxidative phosphorylation (Fig. 1). However, and paradoxically, tumor cells frequently prefer the use of pyruvate for aerobic glycolysis over oxidative phosphorylation, despite the availability of oxygen, which results in lower energy yields. B precursor ALL lymphoblasts show up-regulation of genes involved in glycolysis and concomitant down-regulation of TCA cycle genes compared with normal CD34+ progenitor cells (Boag et al. 2006). In addition, 2-deoxy-D-glucose (2-DG), a glucose analog and inhibitor of glycolysis, induces apoptosis, supporting a dependence on glycolysis for cell survival (Boag et al. 2006). In agreement, 3-bromo-2-oxopropionate-1-propyl ester (3-BrOP), an inhibitor of hexokinase, the first enzyme in the glycolytic pathway, can deplete the ATP pool in AML and B cell lymphoma cell lines and synergizes with inhibition of mTOR to induce apoptosis (Xu et al. 2005). Furthermore, deletion of Solute Carrier Family 2 Member 1 (SLC2A1), a major glucose transporter in lymphoid cells, induces metabolic reprogramming in favor of catabolism over anabolic metabolism, and blocks cell proliferation, in B-ALL lymphoblasts harboring the BCR-ABL1 tyrosine kinase fusion oncogene (Liu et al. 2014). Moreover, treatment of BCR-ABL1 B-ALL cells with 2-DG increases apoptosis and enhances the antitumor activity of dasatinib, a tyrosine kinase inhibitor, in vivo. Similarly, AML cell lines and primary patient leukemia samples with activating mutations in the FLT3 kinase oncogene are also dependent on glycolysis, and treatment of these cells with 3-BrOP or 2-DG increases the efficacy of tyrosine kinase inhibition with sorafenib (Huang et al. 2016; Ju et al. 2017). However, it should be noted that 2-DG also works as an inhibitor of protein glycosylation and as a result it can impair the transport of active mutant FLT3 and KIT tyrosine receptors to the cell surface, which may contribute to its antileukemic effects (Larrue et al. 2015).
Figure 1.
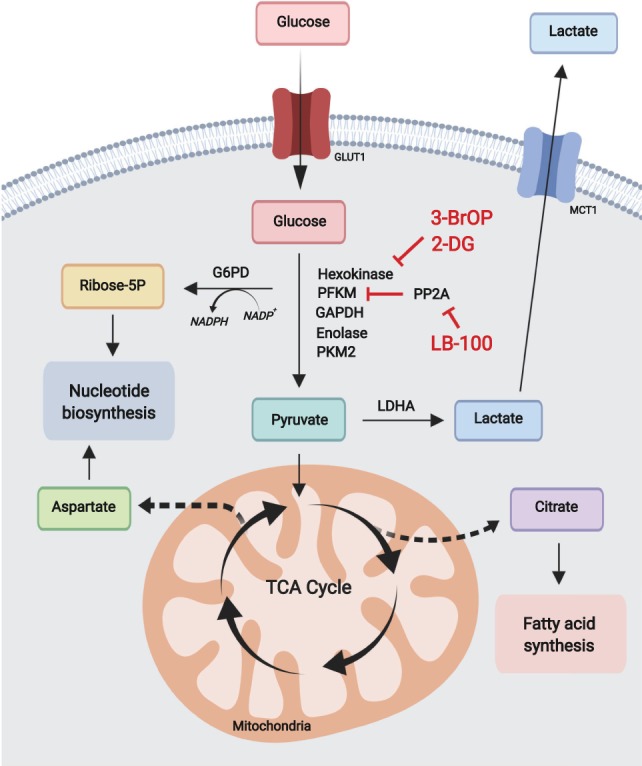
Glycolysis and pentose phosphate pathway. Catabolically, glycolysis converts glucose to pyruvate ultimately producing lactate, or acetyl-CoA in the TCA cycle for mitochondrial oxidative phosphorylation. Anabolically, glycolysis generates metabolic precursors for serine biosynthesis and supports redox potential and nucleotide biosynthesis via the pentose phosphate pathway. Most leukemias are highly dependent on glycolysis and can be targeted using glycolytic inhibitors.
The preferred use of glycoIysis over oxidative phosphorylation in tumor cells is linked with the role of glycolytic intermediates as important precursors for multiple biosynthetic metabolic routes including serine biosynthesis and the pentose phosphate pathway. In AML, activation of mTORC1 supports protein translation and cell growth and promotes glycolysis and high-glucose flux through the PPP, contributing to glucose addiction (Xu et al. 2003; Tamburini et al. 2009). In addition, marked dependence on oxidative PPP for cell proliferation and survival in AML makes glucose 6 phosphate dehydrogenase (G6PD), the first and rate-limiting enzyme in the PPP, a potentially relevant therapeutic target in this disease (Xu et al. 2016; Poulain et al. 2017). A key function of the PPP is the generation of redox potential in the cell via conversion of nicotinamide adenine dinucleotide phosphate (NADP+) to its reduced form (NADPH) for biosynthesis and protection from oxidative stress. The serine/threonine-protein phosphatase 2A (PP2A) functions as a tumor suppressor and it is down-regulated in multiple types of cancer. However, in B-ALL PP2A functions to redirect glucose from glycolysis to the PPP as a guard against oxidative stress (Xiao et al. 2018). This metabolic shunt compensates for the activity of the B-cell transcription factors PAX5 and IKZF1, which normally down-regulate the expression of G6PD and other key PPP enzymes, resulting in low PPP activity in B cells (Xiao et al. 2018). As a result, inhibition of PP2A with LB-100 abrogates the shunt of glucose carbon to the PPP and results in marked antitumor effects (Xiao et al. 2018). The relevance of the metabolic wiring in normal hematopoietic cells for leukemia tumor initiation and maintenance is further supported by studies on the role of glycolysis in hematopoietic stem cell (HSC) self-renewal and transformation. HSCs normally rely heavily on glycolysis and loss of the glycolytic enzymes pyruvate kinase M2 (Pkm2) or lactate dehydrogenase A (Ldha), which forces a shift from glycolysis to mitochondrial respiration, results in impaired long-term repopulating capacity upon transplantation (Wang et al. 2014). Notably, genetic inactivation of Pkm2 and Ldha in HSCs also delays the development of myeloid leukemia following oncogene expression in support of active glycolysis as an important requirement for leukemia initiation (Wang et al. 2014). Beyond its role in tumor initiation and maintenance, increased glycolytic activity has also been linked with leukemia resistance to therapy. In ALL, tumors with primary resistance to prednisolone, a glucocorticoid central in the treatment of this disease, show up-regulation of genes involved in glycolysis and carbohydrate metabolism (Holleman et al. 2004). Moreover, 2-DG synergizes with prednisolone in these glucocorticoid-resistant cells and induces apoptosis, supporting a role for the inhibition of glycolysis in rendering ALL cells more sensitive to glucocorticoid treatment (Hulleman et al. 2009). Finally, systemic regulation of glucose levels in the context of AML has been proposed as a mechanism favoring glucose uptake by myeloblasts (Ye et al. 2018). Mechanistically, AML can increase serum levels of Insulin-like growth factor binding protein 1 (IGFBP1), which results in desensitization of normal tissues to insulin, decreasing their glucose uptake. In this way, leukemic cells, which do not require insulin for glucose uptake, would have a competitive advantage in the acquisition and utilization of systemic glucose.
Taking aim at the mitochondria
Despite the growth advantage conferred by the use of aerobic glycolysis as a source of anabolic precursors and redox potential, many hematopoietic malignancies utilize both glycolysis and oxidative phosphorylation for ATP production, and some tumors still favor mitochondrial oxidation as a source of energy (Fig. 2; Weinberg et al. 2010; Guo et al. 2011; Marin-Valencia et al. 2012; Birsoy et al. 2015; Boroughs and DeBerardinis 2015; Hensley et al. 2016). The control of nutrient utilization is regulated in part by prolyl hydroxylase domain proteins, which hydroxylate proline residues in substrate proteins involved in fuel switching. Among these, PHD3 can suppress fatty acid oxidation in response to nutrient abundance via acetyl-coA carboxylase 2 (ACC2) hydroxylation, and low levels of PHD3 expression in AML make these tumors rely on lipid catabolism regardless of external nutrient availability (German et al. 2016). Metformin, an inhibitor of mitochondrial complex I, in clinical use for the treatment of type 2 diabetes, has shown antitumor effects via disruption of oxidative phosphorylation in tumor cells (Bailey and Turner 1996) and is in clinical trials in combination with standard chemotherapy for the treatment of relapsed ALL and chronic lymphocytic leukemia (CLL) (NCT01750567, NCT01324180). In CLL lymphocytes, tyrosine kinase inhibition with dasatinib can increase glucose consumption while reducing lactate production, suggesting a shift in glucose use from aerobic glycolysis to the TCA cycle and oxidative phosphorylation (Martinez Marignac et al. 2013). In agreement with this, dasatinib sensitive CLL cells are also selectively sensitive to metformin (Martinez Marignac et al. 2013). Moreover, in T-ALL, mitochondrial complex I inhibition with metformin has been reported to synergize with daunorubicin, vincristine, L-asparaginase, and etoposide, enhancing the antileukemic effects of these drugs (Pan et al. 2012; Rosilio et al. 2013; Yi et al. 2017). In addition, in chronic myeloid leukemia (CML) and AML, metformin suppresses leukemia cell proliferation, clonogenic activity, and survival, consistent with higher mitochondrial biogenesis and higher dependency on mitochondrial oxidation in leukemia myeloblasts compared with normal hematopoietic precursor cells (Green et al. 2010; Vakana et al. 2011). Similarly, inhibition of mitochondrial function with tigecycline, a third generation tetracycline antibiotic that interferes with translation by blocking the interaction of aminoacyl-tRNA with the A site of the ribosome in bacteria and mitochondria, has been proposed as a potential therapy in AML (Škrtić et al. 2011). Furthermore, inhibition of ClpP, a mitochondrial protease frequently overexpressed in AML, can induce leukemia cell death while sparing normal hematopoietic cells (Cole et al. 2015). Importantly, AML cells resistant to cytarabine chemotherapy show higher levels of oxidative phosphorylation and are highly sensitive to mitochondrial targeting with tigecycline or inhibitors of the electron transport chain in vitro and in vivo (Farge et al. 2017).
Figure 2.

Mitochondrial metabolic therapeutic targets. The mitochondria also serves as a major energy factory in leukemia cells in support of targeting oxidative phosphorylation with biguanide drugs (metformin and phenformin) and mitochondrial protein biosynthesis with tigecycline. Leukemia cells, which are highly dependent on arginine–creatine metabolism, are sensitive to cyclocreatine-mediated inhibition of creatine kinase. Activation of the intrinsic apoptosis pathway with BH3 mimetic drugs such as ABT199 activate the BAK–BAX complex to introduce pores in the mitochondrial membrane, enhancing the activity of mitochondrial metabolism-targeting drugs.
An important consideration is that, even within the same tumor, leukemia populations may not be metabolically homogeneous and that chemoresistant leukemia-initiating cells responsible for tumor progression and therapeutic failure may show different metabolic characteristics and dependencies from bulk blast progenitors. In this setting, it has been proposed that leukemia stem cells are characterized by relatively low levels of reactive oxygen species and overexpression of the BCL2 antiapoptotic factor, and that these features can be therapeutically targeted by reducing oxidative phosphorylation with ABT-199, a small molecule BCL2 inhibitor (Lagadinou et al. 2013). In addition, and most notably, ABT-199 plus azacitidine, a DNA demethylating agent, can further reduce oxidative phosphorylation in AML cells and effectively induces durable clinical responses in older, treatment-naïve patients (Pollyea et al. 2018). In accord, CML leukemia-initiating cells show increased oxidative metabolism compared with normal hematopoietic stem cells, which can be targeted with tigecycline (Kuntz et al. 2017). Of note, the combination of tyrosine kinase inhibition with imatinib plus mitochondrial disruption with tigecycline can effectively eliminate most leukemia stem cells in xenograft models (Kuntz et al. 2017).
Decreased energy levels in cells activate AMP-activated protein kinase (AMPK), a crucial energy sensor, which promotes ATP production by increasing the activity and expression of proteins involved in catabolism including glycolysis and fatty acid oxidation, while conserving ATP by switching off biosynthetic pathways such as fatty acid, glycogen, and protein synthesis (Merrill et al. 1997; Marsin et al. 2000, 2002; Li et al. 2011). Oncogenic NOTCH1 promotes glycolysis in T-ALL, but also induces metabolic stress and AMPK activation, which in turn restrains aerobic glycolysis and favors oxidative phosphorylation rendering T-ALL lymphoblasts sensitive to phenformin, a metformin-related biguanide complex I inhibitor (Kishton et al. 2016). Consistently, treatment of T-ALL cells with metformin stimulates AMPK to trigger autophagy and apoptosis (Grimaldi et al. 2012). However, and importantly, AMPK also regulates metabolic energy balance at the whole-body level. Dietary restriction in mice harboring MLL-AF9-induced AML leads to activation of AMPK, which is essential to support leukemia development (Saito et al. 2015). Thus, AMPK deletion significantly delays leukemogenesis and depletes leukemia-initiating cells by reducing glucose uptake, and increasing oxidative stress and DNA damage (Saito et al. 2015). Notably, AML leukemia-initiating cells are particularly dependent on AMPK to suppress oxidative stress in the hypoglycemic bone marrow environment and AMPK inhibition synergizes with dietary restriction to suppress leukemogenesis (Saito et al. 2015). In addition, it should be considered that diet may affect the activity and antileukemic effects of metabolic drugs and targeted therapies as a result of the replenishment of metabolic pathways (Polet et al. 2016) and signaling feedback mechanisms (Hopkins et al. 2018), respectively. The effects of diet in therapy response is exemplified by the impact of ketogenic diet on the effect of PI3K inhibitor in AML (Hopkins et al. 2018). Ketogenic diet in animal leukemia models accelerates the onset of MLL-AF9-induced AML, however it also enhances the response to PI3K inhibitors, by precluding the development of tumor-enhancing hyperinsulinemia.
The ecotropic virus integration site 1 (EVI1) gene encodes a transcription factor with chromatin remodeling activity (Hinai and Valk 2016). Aberrant expression of EVI1 is an oncogenic driver in AML bearing chromosomal rearrangements inv(3)(q21q26·2) or t(3;3)(q21;q26·2) involving the EVI1 locus, and also an important independent adverse prognostic marker in 6%–11% cases with aberrant EVI1 expression (Hinai and Valk 2016). Among other oncogenic effects, EVI1 expression remodels the metabolic landscape of hematopoietic stem cells altering arginine metabolism and de novo nucleotide synthesis (Fenouille et al. 2017). In addition, EVI1 overexpressing AMLs show high expression of mitochondrial creatine kinase CKMT1, the enzyme responsible for the transfer of a high-energy phosphate from mitochondria to the cytosolic carrier, creatine (Fenouille et al. 2017). Notably, CKMT1 inhibition in these cells compromises mitochondrial respiration and ATP production by impairing the arginine–creatine pathway (Fenouille et al. 2017). As a result, EVI1-expressing leukemia cells are dependent on arginine–creatine metabolism and become sensitive to creatine kinase inhibition with cyclocreatine (Fenouille et al. 2017).
Targeting nucleotide biosynthesis and degradation pathways
Nucleotide biosynthesis is an essential component of cell metabolism and a major vulnerability in actively proliferating cells. The synthesis of purine and pyrimidine nucleotides is centrally connected to anabolic pathways as it utilizes energy and building blocks from multiple metabolic routes engaging amino acid metabolism, the pentose phosphate pathway, the one-carbon-unit cycle and the tricarboxylic acid cycle (Fig. 3). In leukemia cells, nucleotide synthesis critically supports DNA replication and ribosome biogenesis as essential processes for cell proliferation and growth. Proof of principle for the targeting of nucleotide metabolism in the clinic emerged early on with the seminal identification of the antileukemic effects of aminopterin, a folate antagonist inhibitor of nucleotide biosynthesis, in acute lymphoblastic leukemia (Farber and Diamond 1948). Today, antimetabolites targeting nucleotide biosynthesis are a stalwart component of the treatment of hematologic malignancies. Commonly used antimetabolites in the treatment of leukemia include folic acid antagonists, such as methotrexate, purine antimetabolites, such as 6-mercaptopurine, fludarabine phosphate, pentostatin, clofarabine, and cladribine, and pyrimidine antimetabolites such as cytarabine. New antimetabolite drugs are actively being incorporated in the antileukemic armamentarium targeting these pathways. Recently, dihydroorotate dehydrogenase (DHODH), an enzyme mediating the fourth step in de novo pyrimidine biosynthesis (Löffler et al. 1997), has been proposed as a target in AML, where depletion of the pyrimidine pool not only abrogates proliferation, but also induces a HOXA9-dependent differentiation block, triggering myeloid differentiation and suppressing leukemia-initiating cell activity (Sykes et al. 2016; Christian et al. 2019). In addition, nucleotide degradation pathways have started to gain attention with the realization of important dependencies mediated by the clearance of endogenous and drug-generated toxic nucleotide metabolic byproducts. The purine nucleotide phosphorylase (PNP) enzyme mediates nucleotide degradation by catalyzing the cleavage of inosine, deoxyinosine, guanosine, and deoxyguanosine (dGuo) to their corresponding base and sugar 1-phosphate. In the absence of PNP, nucleosides accumulate, active deoxycytidine kinase metabolizes the excess deoxyguanosine to produce dGTP, which in turn inhibits ribonucleotide diphosphate reductase, blocking the synthesis of dCDP and dUDP and DNA synthesis, particularly in dividing T-cells (Schramm 2002). Consistently, forodesine, a transition state analog inhibitor of PNP has shown antitumor effects in T-ALL (Homminga et al. 2011) and clinical activity in peripheral T-cell lymphomas (Makita et al. 2018).
Figure 3.

Nucleotide metabolism. Purine and pyrimidine biosynthesis are central to DNA replication and can be inhibited with antimetabolite drugs. In addition, the purine degradation activity of NT5C2, which contributes to resistance to 6-MP, sensitizes leukemia cells to inhibition of purine biosynthesis with mizoribine.
6-mercaptopurine (6-MP), a purine base analog commonly used in the treatment of ALL, is incorporated in salvage pathway of purine biosynthesis and induces antitumor effects via inhibition of de novo purine biosynthesis and by inducing apoptosis upon incorporation into DNA (De Abreu 1994). The cytosolic 5′-Nucleotidase II (NT5C2), an enzyme responsible for the intracellular 5′-dephosphorylation of the purine biosynthesis precursors inosine monophosphate (IMP), guanosine monophosphate (GMP), and xanthine monophosphate (XMP), can antagonize the activity of 6-MP by facilitating the dephosphorylation and cellular export of the thiopurine-derived metabolites thio-IMP, thio-XMP, and thio-GMP (Dieck and Ferrando 2019). Notably, somatic activating mutations in NT5C2 are present in 3%–10% of relapsed B-ALL and 20% of relapsed T-ALL cases (Meyer et al. 2013; Tzoneva et al. 2013; Oshima et al. 2016) and can also be found in acute promyelocytic leukemia (APL) relapse samples from patients who have been treated with 6-MP (Lehmann-Che et al. 2018). The direct mechanistic link between increased NT5C2 activity and resistance to 6-MP supports a role of NT5C2 inhibition in the abrogation of 6-MP resistant clones. Moreover, excess NT5C2 activity and consequent increased export of purine nucleosides renders NT5C2 mutant ALL cells more sensitive to mizoribine, a purine biosynthesis inhibitor blocking inosine monophosphate dehydrogenase (IMPDH) (Tzoneva et al. 2018).
Amino acids for protein biosynthesis and beyond
Amino acids are the building blocks of protein biosynthesis, but they also serve as a source of carbon and nitrogen in nucleotide synthesis and other biosynthetic pathways. In addition, amino acid degradation can also constitute an important source of precursors for energy production. Ketogenic amino acids leucine and lysine generate acetyl-CoA, while glycogenic amino acids generate pyruvate and TCA cycle intermediates. Among these, glutamine plays a prominent role as a carbon and nitrogen source both in normal cells and in tumor metabolism. Glutamine conversion to glutamate and then α-ketoglutarate feeds carbon into the TCA cycle and glutamine metabolism contributes to the synthesis of lipids, nucleotides, and nonessential amino acids (DeBerardinis et al. 2007). AML cells are particularly dependent on glutamine, as glutamine controls oxidative phosphorylation in these cells, and removal of glutamine leads to apoptosis (Willems et al. 2013; Jacque et al. 2015). Consistently, glutaminase (GLS), the enzyme that catalyzes the conversion of glutamine to glutamate, has been proposed as a therapeutic target in this disease (Matre et al. 2016; Gallipoli et al. 2018; Gregory et al. 2018). Moreover, glutamine deprivation in AML cells results in the up-regulation of 3-phosphoglycerate dehydrogenase (PHGDH) and phosphoglycerate aminotransferase (PSAT), two essential enzymes in the serine biosynthesis pathway, and serine removal enhances the effects of GLS inhibition therapy (Polet et al. 2016). In addition, glutamine use is also prominently active in T-ALL and glutaminolysis critically contributes to leukemia cell growth and survival in the context of suppression of oncogenic NOTCH1 signaling, making glutaminase inhibitors highly synergistic with anti-NOTCH1 therapies in this disease (Herranz et al. 2015).
Asparagine is a nonessential amino acid in most tissues as it is synthesized via a transamination reaction catalyzed by asparagine synthetase (ASNS), which converts aspartate and glutamine into asparagine and glutamate. However, acute lymphoblastic leukemia cells are auxotropic for asparagine as they express low levels of ASNS. As a result, these tumors are highly vulnerable to asparaginase treatment, which induces systemic degradation of asparagine and depletes extracellular sources of this amino acid required for leukemia cell growth (Kidd 1953a,b; Broome 1963a,b; Miller et al. 1969; Ohnuma et al. 1970; Capizzi et al. 1971). Mechanistically, asparaginase can deplete not only the asparagine but also the glutamine intracellular pool. Moreover, metabolomic profiling following asparaginase treatment reveals global metabolic shifts with decreased translation and inhibited glycolysis, activation of autophagy and increased fatty-acid oxidation and cellular respiration (Hermanova et al. 2016). Altogether, these perturbations induce profound antileukemic effects and, as a result, asparaginase treatment contributes significantly to the success of multiagent chemotherapy regimens for ALL (Sallan et al. 1983; Clavell et al. 1986). Moreover, activation of the Wnt pathway via GSK3-dependent inhibition, could sensitize leukemia cells to asparaginase (Hinze et al. 2019).
Arginine, a semi-essential amino acid, can be synthesized from citrulline, but it must also be taken up from the diet in order to meet the cellular demands (Wheatley 2005). AML blast cells frequently show defects in the arginine-recycling pathway enzymes argininosuccinate synthase and ornithine transcarbamylase, which makes them arginine auxotrophic and dependent on arginine uptake by the CAT-1 and CAT-2B arginine transporters. Consequently, depletion of extracellular arginine with BCT-100, a pegylated human recombinant arginase, leads to a rapid decrease of the intracellular arginine pool and induces proliferation arrest, apoptosis, and decreased AML engraftment in vivo (Mussai et al. 2015), a therapeutic strategy that may also apply for the treatment of T-ALL (Hernandez et al. 2010; Morrow et al. 2013). However, altered arginine availability can also be induced by AML cells, which express and release arginase II into the plasma, as a mechanism to suppress immunosurveillance by interfering with T-cell proliferation and inducing monocyte polarization towards a suppressive M2-like phenotype (Mussai et al. 2013).
In CML blast crisis cells, the synthesis of essential branched-chain amino acids leucine, valine, and isoleucine is enhanced by up-regulation of branched chain amino acid transaminase 1 (BCAT1), an enzyme catalyzing the amination of branched-chain keto acids. In this setting, BCAT1 inhibition induces cellular differentiation and impairs CML blast crisis propagation in vitro and in vivo in support of an essential role for branched amino acid production in this disease (Hattori et al. 2017).
The multiple roles of amino acid metabolism in leukemia growth and survival beyond protein biosynthesis is further substantiated by the use of cysteine as a precursor for the biosynthesis of glutathione (GSH) required for the maintaining of intracellular redox potential (Orlowski and Meister 1971; Wu et al. 2004a). Tumor B cells in CLL have high levels of intracellular reactive oxygen species and are highly dependent on SLC7A11-mediated cysteine import from the microenvironment to support GSH production and to maintain their intracellular redox balance (Zhang et al. 2012). Consequently, treatment of CLL cells with a cysteinase enzyme alone or in combination with fludarabine led to significant increases in apoptosis both in the presence and in the absence of stromal cells (Cramer et al. 2017).
Lipids also fuel leukemia growth
Cholesterol, phospholipids, and triglycerides serve as structural components of membranes, and even though most tissues can satisfy their lipid requirements through the uptake of free fatty acids and lipoproteins from the blood, cancer cell growth requires active biosynthesis to meet the high demand for the biogenesis of membrane structures. Fatty acid biosynthesis is initiated by the carboxylation of acetyl-CoA to produce malonyl-CoA, which is subsequently condensed by fatty acid synthase (FASN) to generate palmitate (Maier et al. 2008). Resulting fatty acids, along with those taken up from the extracellular environment, serve as building blocks for the synthesis of triacylglycerides, glycerophospholipids, cardiolipids, sphingolipids, and eicosanoids.
In addition, degradation of fatty acids by fatty acid oxidation in the mitochondria is a prominent source of acetyl-CoA for oxidative phosphorylation and energy production in cancer cells. Transfer of fatty acids to the mitochondria requires first their coupling with to coenzyme A, and then the transfer of the acyl group to carnitine, a reaction catalyzed by the carnitine palmitoyltransferase 1a (CPT1A) enzyme. Targeting fatty acid transport and β-oxidation with the CPT1a inhibitor ST1326 induces cell growth arrest, mitochondrial damage, and apoptosis in AML cells (Ricciardi et al. 2015). Of note, cell growth and survival in AML monocytic cells seems to be supported by bone marrow adipocytes, which promote fatty acid β-oxidation (Tabe et al. 2017). Consistently, the lipolysis inhibitor orlistat, which blocks the generation of free fatty acids from triglycerides and phospholipids, and pharmacologic inhibition of fatty acid oxidation with etomoxir or ranolazine, inhibit AML cell proliferation, and sensitize human leukemia blasts to apoptosis induced by ABT-737, a small molecule inhibitor of the BCL2 and BCLXL antiapoptotic factors (Samudio et al. 2010). In an analogous way, CLL cells, which express high levels of CPT1 and CPT2, are sensitive to suppression of fatty acid transport into the mitochondria with perhexiline, a CPT inhibitor, leading to depletion of cardiolipin, a key component of mitochondrial membranes, and consequent loss of mitochondrial integrity and induction of programmed cell death (Liu et al. 2016).
Lipid steroids play important roles in membrane structure and fluidity (cholesterol), as components of the electron transport chain (ubiquinone), in protein glycosylation (dolichol), and in protein isoprenylation (farnesyl pyrophosphate and geranylgeranyl pyrophosphate). Synthesis of steroids from acetyl-CoA in the mevalonate pathway can be blocked with statins, which prevent the conversion of HMG-CoA to mevalonate by inhibiting HMG-CoA reductase (HMGCR) (Goldstein and Brown 1990). An antitumor effect of statin drugs was recognized early on (Newman et al. 1994) and in vitro experiments have shown that statins can induce apoptosis in hematologic malignancies (Dimitroulakos et al. 2000; Xia et al. 2001; Wong et al. 2002; Wu et al. 2004b). Mechanistically, statins can induce apoptosis in leukemia cells at least in part by interfering with the prenylation of the Rho and Ras GTPases (Mangues et al. 1998; Cafforio et al. 2005). Notably, statins are broadly used as hypolipidemic agents for reduction of cardiovascular risk, and a recent retrospective population-based study has shown both an association between CLL and dislipidemia and improved CLL survival in patients receiving hypolipidemic therapy (Mozessohn et al. 2017). These results are in agreement with a role for lipid metabolism in CLL and the observation of increased cell proliferation in early-stage indolent CLL blasts in response to increased levels of blood low-density lipoproteins (Chow et al. 2016). In addition, statins have been evaluated clinically for the treatment of AML in combination with chemotherapy. These studies support the findings that statin treatment may enhance complete remission rates in favorable-risk AML groups, but have a less clear role in high-risk AML (Kornblau et al. 2007; Advani et al. 2018). In addition, treatment of AML and CLL cells with statins increases their sensitivity to BCL2 inhibition with ABT-199 as a result of inhibition of protein geranylgeranylation with consequent up-regulation of the protein p53 up-regulated modulator of apoptosis (PUMA). Moreover, and in support of clinical relevance, retrospective analyses of three CLL clinical studies showed that statin use was associated with enhanced response to ABT-199 therapy (Lee et al. 2018).
Sphingolipids are lipid components of cell membranes with important roles in control of cell proliferation, cell–cell interactions, signaling, and cell survival. De novo sphingolipid biosynthesis is initiated through the action of serine palmitoyltransferase (SPT), which joins palmitoyl-CoA to serine to produce sphingosine, the backbone of all sphingolipids. Conjugation of sphingosine to a fatty acid generates ceramide, and addition of a polar head group to ceramide leads to the production of more complex sphingolipids, including sphingomyelin (after the addition of phosphocholine) and glycosphingolipids (after the addition of a sugar). In this group, ceramides are proapoptotic and promote cell cycle arrest and senescence (Obeid et al. 1993). Ceramide synthase (CerS), the enzyme that catalyzes the formation of ceramide from sphingosine, is increased in CLL upon treatment with the BCL2-family inhibitor ABT-263, resulting in enhanced apoptosis (Beverly et al. 2013). In addition, in AML leukemia cells, FLT3 signaling is associated with lower CerS1 expression and CerS1 up-regulation as result of FLT3 inhibition leads to mitophagy-dependent cell death (Dany et al. 2016). On the other hand, sphingosine-1-phosphate (S1P), a secreted sphingolipid, promotes cell migration and plays important roles in tissue homing of lymphoid and myeloid cells. Sphingosine kinase (SPHK) generates S1P via phosphorylation of sphingosine and inhibition of this enzyme is synergistic with vincristine chemotherapy in T-ALL (Evangelisti et al. 2014). Similarly, inhibition of SPHK1 in combination with cytarabine enhanced leukemia cell death in AML (Powell et al. 2017). Moreover, genetic deletion of Sphk1 in BCR-ABL1-induced mouse models of ALL delayed leukemia development and was synergistic with tyrosine kinase inhibitor treatment against BCR-ABL1-positive cell lines, further supporting a relevant role for S1P signaling in promoting leukemia development (Wallington-Beddoe et al. 2019).
Beyond metabolism
Cancer metabolic networks control not only energy balance and growth, but are also increasingly recognized as important modulators of epigenetic mechanisms of gene expression via modulation of epigenetic enzymes implicated in DNA methylation and as writers and erasers of nucleosome epigenetic marks in histone tails. In fact, acetyl-coA is required for histone acetylation, and α-ketoglutarate, a metabolic intermediate in the TCA cycle, serves as a substrate for the TET family of DNA demethylases and the Jumonji family of histone demethylases (Fig. 4; Tsukada et al. 2006; Tahiliani et al. 2009).
Figure 4.

Metabolic targeting of epigenetic mechanisms in leukemia. TET enzymes, which mediate DNA demethylation, use α-ketoglutarate as a substrate and ascorbate as a cofactor and are inhibited in AML and myeloproliferative disorders as a result of loss-of-function mutations in TET2 or because of 2HG produced by neomorphic mutations in the IDH1 and IDH2 genes. Small molecule inhibitors of mutant IDH1 and IDH2, and vitamin C supplementation can abrogate the production of 2HG or enhance TET2 activity, respectively, restoring DNA methylation patterns and myeloid cell differentiation.
The isocitrate dehydrogenase enzymes IDH1 and IDH2, which catalyze the conversion of isocitrate to α-ketoglutarate, are mutated in 20% of AML cases (Schnittger et al. 2010; Green et al. 2011). Recurrent alterations in the IDH1 and IDH2 genes in AML are characteristically heterozygous missense mutations involving arginine residues implicated in isocitrate binding (Dang et al. 2009; Mardis et al. 2009; Yan et al. 2009). These gain-of-function mutations change substrate specificity and result in the conversion of α-ketoglutarate into 2-hydroxyglutarate (2-HG) (Dang et al. 2009; Kranendijk et al. 2010; Ward et al. 2010), an oncometabolite that functions as an inhibitor of DNA and histone demethylases, leading to consequent DNA hypermethylation and deregulated gene expression (Figueroa et al. 2010; Xu et al. 2011). Notably, mutant IDH enzymes can be targeted with small molecule inhibitors AG-120 (Ivosidenib) for IDH1 and AG-221 (Enasidenib) for mutant IDH2 (Stein et al. 2017; DiNardo et al. 2018). In a phase I clinical trial, Ivosidenib induced differentiation and maturation of mutant IDH1 AML cells, decreased 2-HG levels in serum and resulted in durable remissions in patients (DiNardo et al. 2018). Similarly, Enasidenib induced differentiation and maturation of malignant myeloblasts with IDH2 mutations in phase I and induced clinically relevant responses in patients who had previously failed conventional AML therapy (Stein et al. 2017). Conversely, heterozygous loss-of-function mutations in TET2, which impair the ability of this enzyme to mediate DNA demethylation via oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), are present in about 30% of AML tumors (Abdel-Wahab et al. 2009; Delhommeau et al. 2009). Notably, TET proteins use vitamin C as a cofactor (Fritz et al. 2014) and vitamin C supplementation can overcome the effects of partial Tet2 deficiency in AML, leading to myeloid differentiation (Cimmino et al. 2017). Finally, overexpression of BCAT1 in AML leukemic stem cells decreases α-ketoglutarate levels by coupling this metabolite to α-amino groups from branched chain amino acids. In turn, depletion of α-ketoglutarate results in altered TET activity and DNA hypermethylation, reminiscent of that induced by IDH mutations, in support of the branched chain amino acid-BCAT1-α-ketoglutarate axis as an oncogenic mechanism and a potential therapeutic target in this disease (Raffel et al. 2017).
Future perspectives
While the studies highlighted here have significantly enhanced our understanding of the metabolic wiring of leukemia cells, the metabolic programs regulating leukemia initiation and progression remain to be fully understood. Deep exploration of leukemia metabolomics using improved analytical techniques suitable for the analysis of small cell numbers and for in vivo analyses is sure to bring new and improved perspectives on the metabolic circuitries operating in leukemia populations and the interaction between leukemia cells and their microenvironment. Moreover, while targeting metabolic pathways deregulated in leukemia, and most relevantly in leukemia-initiating cell populations offers much prospect to improve the treatment of these hematologic malignancies, realizing this opportunity will require deep exploration of metabolic vulnerabilities across different genetic and biological leukemia groups as well as dedicated efforts to investigate these in primary leukemia samples, relevant mouse leukemia models, and patient-derived xenografts in vivo. However, it should be noted that while many of the targets and drugs covered in this review display great promise in the laboratory setting, many are not yet ready for deployment in the clinic.
Acknowledgments
This work was supported by a Translational Research Grant 6531-18 from the Leukemia & Lymphoma Society (A.F.), the National Institutes of Health grants CA206501 (A.F.) and CA210065 (A.F.), and an Innovative Research Award from the Alex Lemonade Stand Foundation (A.F.). M.R. is a Damon Runyon-Sohn Pediatric Cancer Fellow supported by the Damon Runyon Cancer Research Foundation (DRSG 20-17).
Author contributions: M.R. and A.F. wrote the manuscript
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.326470.119.
Freely available online through the Genes & Development Open Access option.
References
- Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, et al. 2009. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 114: 144–147. 10.1182/blood-2009-03-210039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Advani AS, Li H, Michaelis LC, Medeiros BC, Liedtke M, List AF, O'Dwyer K, Othus M, Erba HP, Appelbaum FR. 2018. Report of the relapsed/refractory cohort of SWOG S0919: a phase 2 study of idarubicin and cytarabine in combination with pravastatin for acute myelogenous leukemia (AML). Leuk Res 67: 17–20. 10.1016/j.leukres.2018.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CJ, Turner RC. 1996. Metformin. N Engl J Med 334: 574–579. 10.1056/NEJM199602293340906 [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D, Feramisco JR. 1986. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233: 1061–1068. 10.1126/science.3090687 [DOI] [PubMed] [Google Scholar]
- Belver L, Ferrando A. 2016. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 16: 494–507. 10.1038/nrc.2016.63 [DOI] [PubMed] [Google Scholar]
- Beverly LJ, Howell LA, Hernandez-Corbacho M, Casson L, Chipuk JE, Siskind LJ. 2013. BAK activation is necessary and sufficient to drive ceramide synthase-dependent ceramide accumulation following inhibition of BCL2-like proteins. Biochem J 452: 111–119. 10.1042/BJ20130147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. 2015. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162: 540–551. 10.1016/j.cell.2015.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boag JM, Beesley AH, Firth MJ, Freitas JR, Ford J, Hoffmann K, Cummings AJ, de Klerk NH, Kees UR. 2006. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia 20: 1731–1737. 10.1038/sj.leu.2404365 [DOI] [PubMed] [Google Scholar]
- Boroughs LK, DeBerardinis RJ. 2015. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17: 351–359. 10.1038/ncb3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome JD. 1963a. Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. I. Properties of the L-asparaginase of guinea pig serum in relation to those of the antilymphoma substance. J Exp Med 118: 99–120. 10.1084/jem.118.1.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome JD. 1963b. Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. II. Lymphoma 6C3HED cells cultured in a medium devoid of L-asparagine lose their susceptibility to the effects of guinea pig serum in vivo. J Exp Med 118: 121–148. 10.1084/jem.118.1.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullinger L, Döhner K, Döhner H. 2017. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol 35: 934–946. 10.1200/JCO.2016.71.2208 [DOI] [PubMed] [Google Scholar]
- Cafforio P, Dammacco F, Gernone A, Silvestris F. 2005. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis 26: 883–891. 10.1093/carcin/bgi036 [DOI] [PubMed] [Google Scholar]
- Capizzi RL, Bertino JR, Skeel RT, Creasey WA, Zanes R, Olayon C, Peterson RG, Handschumacher RE. 1971. L-asparaginase: clinical, biochemical, pharmacological, and immunological studies. Ann Intern Med 74: 893–901. 10.7326/0003-4819-74-6-893 [DOI] [PubMed] [Google Scholar]
- Chow S, Buckstein R, Spaner DE. 2016. A link between hypercholesterolemia and chronic lymphocytic leukemia. Leuk Lymphoma 57: 797–802. 10.3109/10428194.2015.1088651 [DOI] [PubMed] [Google Scholar]
- Christian S, Merz C, Evans L, Gradl S, Seidel H, Friberg A, Eheim A, Lejeune P, Brzezinka K, Zimmermann K, et al. 2019. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia. 10.1038/s41375-019-0461-5 [DOI] [PubMed] [Google Scholar]
- Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, et al. 2017. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170: 1079–1095.e20. 10.1016/j.cell.2017.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavell LA, Gelber RD, Cohen HJ, Hitchcock-Bryan S, Cassady JR, Tarbell NJ, Blattner SR, Tantravahi R, Leavitt P, Sallan SE. 1986. Four-agent induction and intensive asparaginase therapy for treatment of childhood acute lymphoblastic leukemia. N Engl J Med 315: 657–663. 10.1056/NEJM198609113151101 [DOI] [PubMed] [Google Scholar]
- Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, Mattson R, Hurren R, Babovic S, Maclean N, et al. 2015. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 27: 864–876. 10.1016/j.ccell.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. 2013. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497: 633–637. 10.1038/nature12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, Triplett K, Lamb C, Alters SE, Rowlinson S, et al. 2017. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med 23: 120–127. 10.1038/nm.4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla-Favera R, Martinotti S, Gallo RC, Erikson J, Croce CM. 1983. Translocation and rearrangements of the c-myc oncogene locus in human undifferentiated B-cell lymphomas. Science 219: 963–967. 10.1126/science.6401867 [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462: 739–744. 10.1038/nature08617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dany M, Gencer S, Nganga R, Thomas RJ, Oleinik N, Baron KD, Szulc ZM, Ruvolo P, Kornblau S, Andreeff M, et al. 2016. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 128: 1944–1958. 10.1182/blood-2016-04-708750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Abreu RA. 1994. Nucleotide metabolism: mode of action of thiopurines in leukemia. Adv Exp Med Biol 370: 195–200. 10.1007/978-1-4615-2584-4_42 [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. 2007. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci 104: 19345–19350. 10.1073/pnas.0709747104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, et al. 2009. Mutation in TET2 in myeloid cancers. N Engl J Med 360: 2289–2301. 10.1056/NEJMoa0810069 [DOI] [PubMed] [Google Scholar]
- Dieck CL, Ferrando A. 2019. Genetics and mechanisms of NT5C2-driven chemotherapy resistance in relapsed ALL. Blood 133: 2263–2268. 10.1182/blood-2019-01-852392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitroulakos J, Thai S, Wasfy GH, Hedley DW, Minden MD, Penn LZ. 2000. Lovastatin induces a pronounced differentiation response in acute myeloid leukemias. Leuk Lymphoma 40: 167–178. 10.3109/10428190009054894 [DOI] [PubMed] [Google Scholar]
- DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, Swords R, Collins RH, Mannis GN, Pollyea DA, et al. 2018. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 378: 2386–2398. 10.1056/NEJMoa1716984 [DOI] [PubMed] [Google Scholar]
- Erikson J, Finger L, Sun L, ar-Rushdi A, Nishikura K, Minowada J, Finan J, Emanuel BS, Nowell PC, Croce CM. 1986. Deregulation of c-myc by translocation of the alpha-locus of the T-cell receptor in T-cell leukemias. Science 232: 884–886. 10.1126/science.3486470 [DOI] [PubMed] [Google Scholar]
- Evangelisti C, Evangelisti C, Teti G, Chiarini F, Falconi M, Melchionda F, Pession A, Bertaina A, Locatelli F, McCubrey JA, et al. 2014. Assessment of the effect of sphingosine kinase inhibitors on apoptosis, unfolded protein response and autophagy of T-cell acute lymphoblastic leukemia cells; indications for novel therapeutics. Oncotarget 5: 7886–7901. 10.18632/oncotarget.2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber S, Diamond LK, Mercer RD, Sylvester RF Jr, Wolff JA. 1948. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid (Aminopterin). N Engl J Med 238: 787–793. 10.1056/NEJM194806032382301 [DOI] [PubMed] [Google Scholar]
- Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M, et al. 2017. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov 7: 716–735. 10.1158/2159-8290.CD-16-0441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenouille N, Bassil CF, Ben-Sahra I, Benajiba L, Alexe G, Ramos A, Pikman Y, Conway AS, Burgess MR, Li Q, et al. 2017. The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat Med 23: 301–313. 10.1038/nm.4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. 2010. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18: 553–567. 10.1016/j.ccr.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz H, Flower G, Weeks L, Cooley K, Callachan M, McGowan J, Skidmore B, Kirchner L, Seely D. 2014. Intravenous vitamin C and cancer: a systematic review. Integr Cancer Ther 13: 280–300. 10.1177/1534735414534463 [DOI] [PubMed] [Google Scholar]
- Gallipoli P, Giotopoulos G, Tzelepis K, Costa ASH, Vohra S, Medina-Perez P, Basheer F, Marando L, Di Lisio L, Dias JML, et al. 2018. Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 131: 1639–1653. 10.1182/blood-2017-12-820035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. 2009. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458: 762–765. 10.1038/nature07823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- German NJ, Yoon H, Yusuf RZ, Murphy JP, Finley LW, Laurent G, Haas W, Satterstrom FK, Guarnerio J, Zaganjor E, et al. 2016. PHD3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2. Mol Cell 63: 1006–1020. 10.1016/j.molcel.2016.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. 1990. Regulation of the mevalonate pathway. Nature 343: 425–430. 10.1038/343425a0 [DOI] [PubMed] [Google Scholar]
- Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C, Boyer O, Bardet V, Park S, Foretz M, et al. 2010. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood 116: 4262–4273. 10.1182/blood-2010-02-269837 [DOI] [PubMed] [Google Scholar]
- Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC, Gale RE. 2011. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood 118: 409–412. 10.1182/blood-2010-12-322479 [DOI] [PubMed] [Google Scholar]
- Gregory MA, Nemkov T, Reisz JA, Zaberezhnyy V, Hansen KC, D'Alessandro A, DeGregori J. 2018. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp Hematol 58: 52–58. 10.1016/j.exphem.2017.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi C, Chiarini F, Tabellini G, Ricci F, Tazzari PL, Battistelli M, Falcieri E, Bortul R, Melchionda F, Iacobucci I, et al. 2012. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: therapeutic implications. Leukemia 26: 91–100. 10.1038/leu.2011.269 [DOI] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25: 460–470. 10.1101/gad.2016311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, Tayyari F, McSkimming D, Kannan N, Tojo A, et al. 2017. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 545: 500–504. 10.1038/nature22314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. 2016. Metabolic heterogeneity in human lung tumors. Cell 164: 681–694. 10.1016/j.cell.2015.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanova I, Arruabarrena-Aristorena A, Valis K, Nuskova H, Alberich-Jorda M, Fiser K, Fernandez-Ruiz S, Kavan D, Pecinova A, Niso-Santano M, et al. 2016. Pharmacological inhibition of fatty-acid oxidation synergistically enhances the effect of l-asparaginase in childhood ALL cells. Leukemia 30: 209–218. 10.1038/leu.2015.213 [DOI] [PubMed] [Google Scholar]
- Hernandez CP, Morrow K, Lopez-Barcons LA, Zabaleta J, Sierra R, Velasco C, Cole J, Rodriguez PC. 2010. Pegylated arginase I: a potential therapeutic approach in T-ALL. Blood 115: 5214–5221. 10.1182/blood-2009-12-258822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navas D, Cordon-Cardo C, et al. 2014. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 20: 1130–1137. 10.1038/nm.3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Sudderth J, Sánchez-Martín M, Belver L, Tosello V, Xu L, Wendorff AA, Castillo M, Haydu JE, et al. 2015. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med 21: 1182–1189. 10.1038/nm.3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinai AA, Valk PJ. 2016. Review: Aberrant EVI1 expression in acute myeloid leukaemia. Br J Haematol 172: 870–878. 10.1111/bjh.13898 [DOI] [PubMed] [Google Scholar]
- Hinze L, Pfirrmann M, Karim S, Degar J, McGuckin C, Vinjamur D, Sacher J, Stevenson KE, Neuberg DS, Orellana E, et al. 2019. Synthetic lethality of Wnt pathway activation and asparaginase in drug-resistant acute leukemias. Cancer Cell 35: 664–676.e7. 10.1016/j.ccell.2019.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleman A, Cheok MH, den Boer ML, Yang W, Veerman AJ, Kazemier KM, Pei D, Cheng C, Pui CH, Relling MV, et al. 2004. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med 351: 533–542. 10.1056/NEJMoa033513 [DOI] [PubMed] [Google Scholar]
- Homminga I, Zwaan CM, Manz CY, Parker C, Bantia S, Smits WK, Higginbotham F, Pieters R, Meijerink JP. 2011. In vitro efficacy of forodesine and nelarabine (ara-G) in pediatric leukemia. Blood 118: 2184–2190. 10.1182/blood-2011-02-337840 [DOI] [PubMed] [Google Scholar]
- Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR, et al. 2018. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560: 499–503. 10.1038/s41586-018-0343-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Ju HQ, Liu K, Zhan G, Liu D, Wen S, Garcia-Manero G, Huang P, Hu Y. 2016. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer Lett 377: 149–157. 10.1016/j.canlet.2016.04.040 [DOI] [PubMed] [Google Scholar]
- Hulleman E, Kazemier KM, Holleman A, VanderWeele DJ, Rudin CM, Broekhuis MJ, Evans WE, Pieters R, Den Boer ML. 2009. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood 113: 2014–2021. 10.1182/blood-2008-05-157842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M, et al. 2015. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 126: 1346–1356. 10.1182/blood-2015-01-621870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju HQ, Zhan G, Huang A, Sun Y, Wen S, Yang J, Lu WH, Xu RH, Li J, Li Y, et al. 2017. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia 31: 2143–2150. 10.1038/leu.2017.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JG. 1953a. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. I. Course of transplanted cancers of various kinds in mice and rats given guinea pig serum, horse serum, or rabbit serum. J Exp Med 98: 565–582. 10.1084/jem.98.6.565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JG. 1953b. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. II. Studies on the nature of the active serum constituent: histological mechanism of the regression: tests for effects of guinea pig serum on lymphoma cells in vitro: discussion. J Exp Med 98: 583–606. 10.1084/jem.98.6.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, Macintyre AN, Goraksha-Hicks P, de Cubas AA, Liu T, et al. 2016. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab 23: 649–662. 10.1016/j.cmet.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblau SM, Banker DE, Stirewalt D, Shen D, Lemker E, Verstovsek S, Estrov Z, Faderl S, Cortes J, Beran M, et al. 2007. Blockade of adaptive defensive changes in cholesterol uptake and synthesis in AML by the addition of pravastatin to idarubicin + high-dose Ara-C: a phase 1 study. Blood 109: 2999–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranendijk M, Struys EA, van Schaftingen E, Gibson KM, Kanhai WA, van der Knaap MS, Amiel J, Buist NR, Das AM, de Klerk JB, et al. 2010. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science 330: 336 10.1126/science.1192632 [DOI] [PubMed] [Google Scholar]
- Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, Helgason GV, Gottlieb E. 2017. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med 23: 1234–1240. 10.1038/nm.4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O'Dwyer KM, et al. 2013. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12: 329–341. 10.1016/j.stem.2012.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrue C, Saland E, Vergez F, Serhan N, Delabesse E, Mansat-De Mas V, Hospital MA, Tamburini J, Manenti S, Sarry JE, et al. 2015. Antileukemic activity of 2-deoxy-d-glucose through inhibition of N-linked glycosylation in acute myeloid leukemia with FLT3-ITD or c-KIT mutations. Mol Cancer Ther 14: 2364–2373. 10.1158/1535-7163.MCT-15-0163 [DOI] [PubMed] [Google Scholar]
- Lee JS, Roberts A, Juarez D, Vo TT, Bhatt S, Herzog LO, Mallya S, Bellin RJ, Agarwal SK, Salem AH, et al. 2018. Statins enhance efficacy of venetoclax in blood cancers. Sci Transl Med 10: eaaq1240. 10.1126/scitranslmed.aaq1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann-Che J, Bally C, Letouzé E, Berthier C, Yuan H, Jollivet F, Ades L, Cassinat B, Hirsch P, Pigneux A, et al. 2018. Dual origin of relapses in retinoic-acid resistant acute promyelocytic leukemia. Nat Commun 9: 2047 10.1038/s41467-018-04384-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O'Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV. 2005. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol 25: 6225–6234. 10.1128/MCB.25.14.6225-6234.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al. 2011. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 13: 376–388. 10.1016/j.cmet.2011.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI, Dang CV. 2008. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 3: e2722 10.1371/journal.pone.0002722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Kishton RJ, Macintyre AN, Gerriets VA, Xiang H, Liu X, Abel ED, Rizzieri D, Locasale JW, Rathmell JC. 2014. Glucose transporter 1-mediated glucose uptake is limiting for B-cell acute lymphoblastic leukemia anabolic metabolism and resistance to apoptosis. Cell Death Dis 5: e1470 10.1038/cddis.2014.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PP, Liu J, Jiang WQ, Carew JS, Ogasawara MA, Pelicano H, Croce CM, Estrov Z, Xu RH, Keating MJ, et al. 2016. Elimination of chronic lymphocytic leukemia cells in stromal microenvironment by targeting CPT with an antiangina drug perhexiline. Oncogene 35: 5663–5673. 10.1038/onc.2016.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. 2011. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 22: 165–178. 10.1091/mbc.e10-06-0500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löffler M, Jöckel J, Schuster G, Becker C. 1997. Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides. Mol Cell Biochem 174: 125–129. 10.1023/A:1006859115450 [DOI] [PubMed] [Google Scholar]
- Lyssiotis CA, Son J, Cantley LC, Kimmelman AC. 2013. Pancreatic cancers rely on a novel glutamine metabolism pathway to maintain redox balance. Cell Cycle 12: 1987–1988. 10.4161/cc.25307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Leibundgut M, Ban N. 2008. The crystal structure of a mammalian fatty acid synthase. Science 321: 1315–1322. 10.1126/science.1161269 [DOI] [PubMed] [Google Scholar]
- Makita S, Maeshima AM, Maruyama D, Izutsu K, Tobinai K. 2018. Forodesine in the treatment of relapsed/refractory peripheral T-cell lymphoma: an evidence-based review. Onco Targets Ther 11: 2287–2293. 10.2147/OTT.S140756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangues R, Corral T, Kohl NE, Symmans WF, Lu S, Malumbres M, Gibbs JB, Oliff A, Pellicer A. 1998. Antitumor effect of a farnesyl protein transferase inhibitor in mammary and lymphoid tumors overexpressing N-ras in transgenic mice. Cancer Res 58: 1253–1259. [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. 2009. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361: 1058–1066. 10.1056/NEJMoa0903840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, et al. 2012. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab 15: 827–837. 10.1016/j.cmet.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. 2000. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol 10: 1247–1255. 10.1016/S0960-9822(00)00742-9 [DOI] [PubMed] [Google Scholar]
- Marsin AS, Bouzin C, Bertrand L, Hue L. 2002. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J Biol Chem 277: 30778–30783. 10.1074/jbc.M205213200 [DOI] [PubMed] [Google Scholar]
- Martinez Marignac VL, Smith S, Toban N, Bazile M, Aloyz R. 2013. Resistance to Dasatinib in primary chronic lymphocytic leukemia lymphocytes involves AMPK-mediated energetic re-programming. Oncotarget 4: 2550–2566. 10.18632/oncotarget.1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matre P, Velez J, Jacamo R, Qi Y, Su X, Cai T, Chan SM, Lodi A, Sweeney SR, Ma H, et al. 2016. Inhibiting glutaminase in acute myeloid leukemia: metabolic dependency of selected AML subtypes. Oncotarget 7: 79722–79735. 10.18632/oncotarget.12944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. 1997. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 273: E1107–E1112. [DOI] [PubMed] [Google Scholar]
- Meyer JA, Wang J, Hogan LE, Yang JJ, Dandekar S, Patel JP, Tang Z, Zumbo P, Li S, Zavadil J, et al. 2013. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45: 290–294. 10.1038/ng.2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller HK, Salser JS, Balis ME. 1969. Amino acid levels following L-asparagine amidohydrolase (EC.3.5.1.1) therapy. Cancer Res 29: 183–187. [PubMed] [Google Scholar]
- Morrish F, Noonan J, Perez-Olsen C, Gafken PR, Fitzgibbon M, Kelleher J, VanGilst M, Hockenbery D. 2010. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J Biol Chem 285: 36267–36274. 10.1074/jbc.M110.141606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow K, Hernandez CP, Raber P, Del Valle L, Wilk AM, Majumdar S, Wyczechowska D, Reiss K, Rodriguez PC. 2013. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia 27: 569–577. 10.1038/leu.2012.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozessohn L, Earle C, Spaner D, Cheng SY, Kumar M, Buckstein R. 2017. The association of dyslipidemia with chronic lymphocytic leukemia: a population-based study. J Natl Cancer Inst 109: djw226 10.1093/jnci/djw226 [DOI] [PubMed] [Google Scholar]
- Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, Qureshi A, Dazzi F, Vyas P, Cerundolo V. 2013. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 122: 749–758. 10.1182/blood-2013-01-480129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussai F, Egan S, Higginbotham-Jones J, Perry T, Beggs A, Odintsova E, Loke J, Pratt G, U KP, Lo A, et al. 2015. Arginine dependence of acute myeloid leukemia blast proliferation: a novel therapeutic target. Blood 125: 2386–2396. 10.1182/blood-2014-09-600643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A, Clutterbuck RD, Powles RL, Millar JL. 1994. Selective inhibition of primary acute myeloid leukaemia cell growth by simvastatin. Leukemia 8: 2023–2029. [PubMed] [Google Scholar]
- Obeid LM, Linardic CM, Karolak LA, Hannun YA. 1993. Programmed cell death induced by ceramide. Science 259: 1769–1771. 10.1126/science.8456305 [DOI] [PubMed] [Google Scholar]
- Ohnuma T, Holland JF, Freeman A, Sinks LF. 1970. Biochemical and pharmacological studies with asparaginase in man. Cancer Res 30: 2297–2305. [PubMed] [Google Scholar]
- Orlowski M, Meister A. 1971. Partial reactions catalyzed by γ-glutamylcysteine synthetase and evidence for an activated glutamate intermediate. J Biol Chem 246: 7095–7105. [PubMed] [Google Scholar]
- Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, Sanchez-Martin M, Carpenter Z, Penson A, Perez-Garcia A, et al. 2016. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci 113: 11306–11311. 10.1073/pnas.1608420113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. 2000. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 275: 21797–21800. 10.1074/jbc.C000023200 [DOI] [PubMed] [Google Scholar]
- Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O'Neil J, Neuberg D, Weng AP, et al. 2006. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci 103: 18261–18266. 10.1073/pnas.0606108103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, et al. 2007. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 13: 1203–1210. 10.1038/nm1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Chen C, Jin Y, Fuentes-Mattei E, Velazquez-Tores G, Benito JM, Konopleva M, Andreeff M, Lee MH, Yeung SC. 2012. Differential impact of structurally different anti-diabetic drugs on proliferation and chemosensitivity of acute lymphoblastic leukemia cells. Cell Cycle 11: 2314–2326. 10.4161/cc.20770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polet F, Corbet C, Pinto A, Rubio LI, Martherus R, Bol V, Drozak X, Gregoire V, Riant O, Feron O. 2016. Reducing the serine availability complements the inhibition of the glutamine metabolism to block leukemia cell growth. Oncotarget 7: 1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, D'Alessandro A, Culp-Hill R, Riemondy KA, Gillen AE, et al. 2018. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 24: 1859–1866. 10.1038/s41591-018-0233-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulain L, Sujobert P, Zylbersztejn F, Barreau S, Stuani L, Lambert M, Palama TL, Chesnais V, Birsen R, Vergez F, et al. 2017. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 31: 2326–2335. 10.1038/leu.2017.81 [DOI] [PubMed] [Google Scholar]
- Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. 2013. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci 110: 11988–11993. 10.1073/pnas.1310230110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JA, Lewis AC, Zhu W, Toubia J, Pitman MR, Wallington-Beddoe CT, Moretti PAB, Iarossi D, Samaraweera SE, Cummings N, et al. 2017. Targeting sphingosine kinase 1 induces MCL1-dependent cell death in acute myeloid leukemia. Blood 129: 771–782. 10.1182/blood-2016-06-720433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, Bullinger L, Poschet G, Nonnenmacher Y, Barnert A, et al. 2017. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551: 384–388. 10.1038/nature24294 [DOI] [PubMed] [Google Scholar]
- Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, Foa R, Nicolai R, Peluso G, Tafuri A. 2015. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood 126: 1925–1929. 10.1182/blood-2014-12-617498 [DOI] [PubMed] [Google Scholar]
- Roberts KG, Mullighan CG. 2015. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol 12: 344–357. 10.1038/nrclinonc.2015.38 [DOI] [PubMed] [Google Scholar]
- Rosilio C, Lounnas N, Nebout M, Imbert V, Hagenbeek T, Spits H, Asnafi V, Pontier-Bres R, Reverso J, Michiels JF, et al. 2013. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett 336: 114–126. 10.1016/j.canlet.2013.04.015 [DOI] [PubMed] [Google Scholar]
- Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. 2015. AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell 17: 585–596. 10.1016/j.stem.2015.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallan SE, Hitchcock-Bryan S, Gelber R, Cassady JR, Frei E III, Nathan DG. 1983. Influence of intensive asparaginase in the treatment of childhood non-T-cell acute lymphoblastic leukemia. Cancer Res 43: 5601–5607. [PubMed] [Google Scholar]
- Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, et al. 2010. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 120: 142–156. 10.1172/JCI38942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittger S, Haferlach C, Ulke M, Alpermann T, Kern W, Haferlach T. 2010. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood 116: 5486–5496. 10.1182/blood-2010-02-267955 [DOI] [PubMed] [Google Scholar]
- Schramm VL. 2002. Development of transition state analogues of purine nucleoside phosphorylase as anti-T-cell agents. Biochim Biophys Acta 1587: 107–117. 10.1016/S0925-4439(02)00073-X [DOI] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. 1997. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci 94: 6658–6663. 10.1073/pnas.94.13.6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Škrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, et al. 2011. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20: 674–688. 10.1016/j.ccr.2011.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. 2013. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496: 101–105. 10.1038/nature12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, et al. 2017. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130: 722–731. 10.1182/blood-2017-04-779405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieglitz E, Taylor-Weiner AN, Chang TY, Gelston LC, Wang YD, Mazor T, Esquivel E, Yu A, Seepo S, Olsen S, et al. 2015. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47: 1326–1333. 10.1038/ng.3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, Sherwin R, Devaskar S, Lesche R, Magnuson MA, et al. 2004. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity. Proc Natl Acad Sci 101: 2082–2087. 10.1073/pnas.0308617100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, Lewis TA, Schajnovitz A, Jain E, Lee D, et al. 2016. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell 167: 171–186.e15. 10.1016/j.cell.2016.08.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, Mogushi K, Shikami M, Ruvolo V, Ishizawa J, et al. 2017. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res 77: 1453–1464. 10.1158/0008-5472.CAN-16-1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935. 10.1126/science.1170116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburini J, Green AS, Bardet V, Chapuis N, Park S, Willems L, Uzunov M, Ifrah N, Dreyfus F, Lacombe C, et al. 2009. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 114: 1618–1627. 10.1182/blood-2008-10-184515 [DOI] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. 2006. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439: 811–816. 10.1038/nature04433 [DOI] [PubMed] [Google Scholar]
- Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, Paietta E, Racevskis J, Rowe JM, Tallman MS, et al. 2013. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med 19: 368–371. 10.1038/nm.3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzoneva G, Dieck CL, Oshima K, Ambesi-Impiombato A, Sánchez-Martín M, Madubata CJ, Khiabanian H, Yu J, Waanders E, Iacobucci I, et al. 2018. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 553: 511–514. 10.1038/nature25186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakana E, Altman JK, Glaser H, Donato NJ, Platanias LC. 2011. Antileukemic effects of AMPK activators on BCR-ABL-expressing cells. Blood 118: 6399–6402. 10.1182/blood-2011-01-332783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallington-Beddoe CT, Xie V, Tong D, Powell JA, Lewis AC, Davies L, Pitson SM, Bradstock KF, Bendall LJ. 2019. Identification of sphingosine kinase 1 as a therapeutic target in B-lineage acute lymphoblastic leukaemia. Br J Haematol 184: 443–447. 10.1111/bjh.15097 [DOI] [PubMed] [Google Scholar]
- Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG, Scadden DT. 2014. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158: 1309–1323. 10.1016/j.cell.2014.07.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. 1956. On respiratory impairment in cancer cells. Science 124: 269–270. [PubMed] [Google Scholar]
- Warburg O, Posener K, Negelein E. 1924. On the metabolism of carcinoma cells. Biochem Z 152: 309–344. [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. 2010. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17: 225–234. 10.1016/j.ccr.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. 2010. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci 107: 8788–8793. 10.1073/pnas.1003428107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley DN. 2005. Arginine deprivation and metabolomics: important aspects of intermediary metabolism in relation to the differential sensitivity of normal and tumour cells. Semin Cancer Biol 15: 247–253. 10.1016/j.semcancer.2005.04.002 [DOI] [PubMed] [Google Scholar]
- Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, Schmitt A, Poulain L, Green AS, Uzunov M, et al. 2013. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 122: 3521–3532. 10.1182/blood-2013-03-493163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. 2008. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci 105: 18782–18787. 10.1073/pnas.0810199105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WWL, Dimitroulakos J, Minden MD, Penn LZ. 2002. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 16: 508–519. 10.1038/sj.leu.2402476 [DOI] [PubMed] [Google Scholar]
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. 2004a. Glutathione metabolism and its implications for health. J Nutr 134: 489–492. 10.1093/jn/134.3.489 [DOI] [PubMed] [Google Scholar]
- Wu J, Wong WW, Khosravi F, Minden MD, Penn LZ. 2004b. Blocking the Raf/MEK/ERK pathway sensitizes acute myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer Res 64: 6461–6468. 10.1158/0008-5472.CAN-04-0866 [DOI] [PubMed] [Google Scholar]
- Xia Z, Tan MM, Wong WW, Dimitroulakos J, Minden MD, Penn LZ. 2001. Blocking protein geranylgeranylation is essential for lovastatin-induced apoptosis of human acute myeloid leukemia cells. Leukemia 15: 1398–1407. 10.1038/sj.leu.2402196 [DOI] [PubMed] [Google Scholar]
- Xiao G, Chan LN, Klemm L, Braas D, Chen Z, Geng H, Zhang QC, Aghajanirefah A, Cosgun KN, Sadras T, et al. 2018. B-cell-specific diversion of glucose carbon utilization reveals a unique vulnerability in B cell malignancies. Cell 173: 470–484.e18. 10.1016/j.cell.2018.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. 2003. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 102: 972–980. 10.1182/blood-2002-11-3429 [DOI] [PubMed] [Google Scholar]
- Xu RH, Pelicano H, Zhang H, Giles FJ, Keating MJ, Huang P. 2005. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia 19: 2153–2158. 10.1038/sj.leu.2403968 [DOI] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. 2011. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19: 17–30. 10.1016/j.ccr.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SN, Wang TS, Li X, Wang YP. 2016. SIRT2 activates G6PD to enhance NADPH production and promote leukaemia cell proliferation. Sci Rep 6: 32734 10.1038/srep32734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. 2009. IDH1 and IDH2 mutations in gliomas. N Engl J Med 360: 765–773. 10.1056/NEJMoa0808710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev 25: 717–729. 10.1101/gad.2016111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Adane B, Khan N, Alexeev E, Nusbacher N, Minhajuddin M, Stevens BM, Winters AC, Lin X, Ashton JM, et al. 2018. Subversion of systemic glucose metabolism as a mechanism to support the growth of leukemia cells. Cancer Cell 34: 659–673.e6. 10.1016/j.ccell.2018.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Y, Gao L, Wu M, Ao J, Zhang C, Wang X, Lin M, Bergholz J, Zhang Y, Xiao ZJ. 2017. Metformin sensitizes leukemia cells to vincristine via activation of AMP-activated protein kinase. J Cancer 8: 2636–2642. 10.7150/jca.19873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. 2012. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149: 656–670. 10.1016/j.cell.2012.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. 2007. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11: 407–420. 10.1016/j.ccr.2007.04.001 [DOI] [PubMed] [Google Scholar]
- Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W, et al. 2012. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol 14: 276–286. 10.1038/ncb2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. 2011. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478: 524–528. 10.1038/nature10334 [DOI] [PMC free article] [PubMed] [Google Scholar]