

In this study, Toroney et al. set out to identify the mechanism of Prp43p action in splicing. The authors use biochemical approaches to demonstrate that the 3' end of U6 acts as the key substrate by which Prp43p promotes disassembly and intron release, thereby terminating splicing.
Keywords: pre-mRNA splicing, spliceosome, intron, disassembly, fidelity, U6 snRNA, Prp43p, DEAH box, ATPase, helicase
Abstract
The termination of pre-mRNA splicing functions to discard suboptimal substrates, thereby enhancing fidelity, and to release excised introns in a manner coupled to spliceosome disassembly, thereby allowing recycling. The mechanism of termination, including the RNA target of the DEAH-box ATPase Prp43p, remains ambiguous. We discovered a critical role for nucleotides at the 3′ end of the catalytic U6 small nuclear RNA in splicing termination. Although conserved sequence at the 3′ end is not required, 2′ hydroxyls are, paralleling requirements for Prp43p biochemical activities. Although the 3′ end of U6 is not required for recruiting Prp43p to the spliceosome, the 3′ end cross-links directly to Prp43p in an RNA-dependent manner. Our data indicate a mechanism of splicing termination in which Prp43p translocates along U6 from the 3′ end to disassemble the spliceosome and thereby release suboptimal substrates or excised introns. This mechanism reveals that the spliceosome becomes primed for termination at the same stage it becomes activated for catalysis, implying a requirement for stringent control of spliceosome activity within the cell.
Eukaryotic gene expression requires pre-mRNA splicing to excise introns, a process frequently targeted for regulation and perturbed in disease (Fu and Ares 2014; Scotti and Swanson 2016). An intron is excised from pre-mRNA in two steps: first, an intronic branch-site adenosine attacks the 5′ splice site, yielding a free 5′ exon and a lariat intermediate; second, the free 5′ exon attacks the 3′ splice site, excising the lariat intron and ligating the exons. Conserved intronic sequences define these three reactive sites of the substrate and recruit the catalyst of splicing—the spliceosome—which is conserved from budding yeast to humans and composed of five small nuclear RNAs (snRNAs) and more than 80 proteins, as recently defined structurally by cryoelectron microscopy (Will and Lührmann 2011; Fica and Nagai 2017; Yan et al. 2019). Assembly of the spliceosome is coupled to intron recognition, resulting in a highly dynamic ribonucleoprotein machine (Staley and Guthrie 1998). Once splicing is complete, this coupling necessitates that splicing termination proceeds through disassembly of the spliceosome to release the excised intron product and also to recycle spliceosome components. Disassembly also plays a central role in the fidelity of splicing. Nevertheless, the mechanism of spliceosome disassembly remains under investigation.
Spliceosome assembly begins with the binding of the U1 small nuclear ribonucleoprotein (snRNP) complex to the 5′ splice site and the U2 snRNP to the branch site. Recruitment of the U4/U6.U5 tri-snRNP follows (Will and Lührmann 2011). Then, U1 is replaced by U6 snRNA at the 5′ splice site, and U6 is liberated from base-pairing with U4 snRNA (Raghunathan and Guthrie 1998; Staley and Guthrie 1999), enabling U6 to form catalytic structures including base-paired U2/U6 helix I, which promotes substrate juxtaposition, and the intramolecular stem loop (ISL) of U6, which positions catalytic metals (Madhani and Guthrie 1992; Sun and Manley 1995; Hilliker and Staley 2004; Hang et al. 2015); U2 and U6 are also held together by a nearby helix—U2/U6 helix II (Fig. 1A; Madhani and Guthrie 1994a; Hang et al. 2015). Next, the nineteen complex (NTC) and other factors bind, enabling splicing catalysis (Fabrizio et al. 2009; Hang et al. 2015; Galej et al. 2016; Wan et al. 2016; Bai et al. 2017; Bertram et al. 2017; Fica et al. 2017; Liu et al. 2017; Wilkinson et al. 2017; Yan et al. 2017; Wan et al. 2019). Finally, the mRNA is released from the spliceosome, and then the excised intron is released for degradation in a manner coupled to spliceosome disassembly (Company et al. 1991; Arenas and Abelson 1997; Schwer and Gross 1998; Wagner et al. 1998; Martin et al. 2002).
Figure 1.

The 3′ end of U6 is required for turnover of excised, lariat intron. (A) Secondary structure of U6 and U2/U6 base-pairing interactions at the catalytic core of the spliceosome. Antisense oligos used for RNase H cleavage are depicted by bars above their target U6 sequences; light gray indicates DNA nucleotides in the anti-sense oligo, whereas dark gray indicates 2′-O-methyl nucleotides. (B) Truncation of the 3′ end of U6 by ∼10 nt impedes turnover of the excised intron. The panel shows denaturing PAGE analysis of radiolabeled ACT1 pre-mRNA following in vitro splicing in yeast extracts (yJPS860) that were first subjected to RNase H cleavage to truncate the 3′ end of U6 snRNA, directed by oligonucleotides depicted in A. Quantitation of intron turnover for each reaction was calculated as the molar ratio of excised intron to mRNA and is shown below the gel and is represented as the mean ± one standard deviation for three independent replicates. Cleavage of U6 was monitored by northern blot (bottom panel) with a radioactive probe directed to nucleotides 28–54 of U6. (C,D) Deletions of 5–8 nt in the 3′ end of U6 impede turnover of the excised lariat intron in vivo. Deleted residues are indicated in C. Viable deletions were analyzed by northern in D for pre-U3A snoRNA, the excised lariat intron, mature U3A snoRNA, and U6. Quantitation of excised intron levels, relative to mature U3A, is shown below the northern, after normalization to the levels in wild type; the average and values are shown for at least two biological replicates. See also Supplemental Figure S1.
Intron release and spliceosome disassembly are driven by Prp43p (Arenas and Abelson 1997; Martin et al. 2002; Wan et al. 2017; Zhang et al. 2019), a DExH box member of the SF2 superfamily of nucleic acid–dependent ATPases (Fairman-Williams et al. 2010; Cordin and Beggs 2013; Ozgur et al. 2015). The DExH box members bind ssRNA and are thought to function by translocating 3′ to 5′ along ssRNA in an ATP-dependent manner. In this way, DExH box members are thought to either plow directly through an RNA duplex or RNP complex (Pyle 2008) or pull on ssRNA to disrupt an RNA duplex or RNP complex at a distance (Semlow et al. 2016).
Prp43p also plays a role in the fidelity of splicing, discarding rejected, suboptimal substrates (Semlow and Staley 2012). For example, two distinct DExH box ATPases, Prp16p and Prp22p, reject suboptimal sites on a substrate by antagonizing their usage during branching and exon ligation, respectively (Burgess and Guthrie 1993; Mayas et al. 2006; Koodathingal et al. 2010). These activities allow for the selection of alternative, optimal sites (Semlow et al. 2016), but in the absence of such sites, the substrate is discarded from the spliceosome by Prp43p (Pandit et al. 2006; Koodathingal et al. 2010; Mayas et al. 2010; Chen et al. 2013). Through its discard activity, Prp43p functions to repress cryptic 3′ splice sites and thereby promote fidelity (Mayas et al. 2010). Because Prp43p functions to discard substrates at multiple stages of splicing, in addition to releasing the excised intron at the end of a splicing cycle (Arenas and Abelson 1997; Martin et al. 2002), Prp43p functions as a general terminator of splicing. Interestingly, the roles of Prp22p and Prp43p in rejecting and discarding suboptimal substrates have been repurposed in a range of ascomycetes fungi for the biogenesis of telomerase RNA, which corresponds to a released 5′ exon intermediate in these species (Kannan et al. 2013, 2015; Qi et al. 2015).
Prp43p requires a cofactor of the G-patch protein family (Aravind and Koonin 1999; Robert-Paganin et al. 2015) for efficient ATPase and RNA unwinding activity. Further, distinct G-patch proteins serve to activate Prp43p in different processes, such as splicing and ribosome biogenesis (Lebaron et al. 2005, 2009; Tsai et al. 2005; Boon et al. 2006; Combs et al. 2006; Leeds et al. 2006; Pandit et al. 2006; Tanaka et al. 2007; Chen et al. 2014b; Heininger et al. 2016). In splicing, the conserved G-patch protein Ntr1p/Spp382p activates Prp43p and forms the NTR (NTC-Related) complex with Prp43p, Cwc23p, and Ntr2p, a factor that is found primarily in fungi and plants. Ntr1p, Cwc23p, and Ntr2p also help to recruit Prp43p to the spliceosome (Tsai et al. 2005). The regulation of recruitment and activation is important, given that Prp43p acts as a general terminator of splicing (Arenas and Abelson 1997; Martin et al. 2002; Pandit et al. 2006; Koodathingal et al. 2010; Mayas et al. 2010; Chen et al. 2013). Ntr2p and/or a portion of Ntr1p appear to enforce this regulation, because the G-patch domain of Ntr1p alone in conjunction with Prp43p enables disassembly of spliceosomal complexes otherwise refractory to termination (Fourmann et al. 2016, 2017).
As for many members of the SF2 family of ATPases, a long-term goal has been to determine the physiological RNA target of Prp43p. Cross-linking in vivo has revealed that Prp43p interacts directly with rRNA and snoRNA as well as U6 snRNA, implicating these RNAs as targets in ribosome biogenesis and splicing, respectively (Bohnsack et al. 2009). However, a recent in vitro study has implicated the U2-branch site duplex as the target for Prp43p in splicing (Fourmann et al. 2016). A structure of the budding yeast spliceosome poised for disassembly is consistent with Prp43p targeting either U6 or the branch site duplex (Wan et al. 2017), whereas a structure of the analogous human spliceosomal intermediate is more consistent with U6 as the target (Zhang et al. 2019). However, neither structure reveals a direct interaction between Prp43p and a specific RNA.
In this study, we have gained insight into the mechanism of Prp43p action through an investigation of the function of the 3′ end of U6. We have found that the 3′ end of U6 is required for spliceosome disassembly and intron release as well as for the discard of a rejected, suboptimal substrate. These functions parallel the functions of Prp43p (Arenas and Abelson 1997; Martin et al. 2002; Mayas et al. 2010; Chen et al. 2013). Further, the requirements for the 3′ end of U6 in intron turnover parallel the requirements for Prp43p—both require 2′ hydroxyls and neither requires a specific sequence (Martin et al. 2002; Tanaka and Schwer 2006). Indeed, Prp43p cross-links in vitro to the 3′ end of U6. Our observations imply a mechanism for spliceosome disassembly and intron release in which Prp43p acts as a winch that pulls on the 3′ end of U6 to disrupt interactions between the catalytic U6 snRNA and the spliceosome.
Results
The 3′ end of U6 is required for excised intron release and spliceosome disassembly
In vivo, the 3′ domain of U6 is essential for growth in yeast (Bordonné and Guthrie 1992) and plays a critical role in stabilizing U6 snRNA through the recruitment of the La homolog protein Lhp1p and subsequently the Lsm2-8 complex. Unlike Lhp1p, the Lsm complex tolerates the terminal, cyclic 2′, 3′ phosphate of U6 in metazoans and the terminal 3′ monophosphate in yeast, both of which result from 3′ end maturation during U6 biogenesis, whereas Lhp1p binds the UUUOH terminus of all RNA polymerase III transcripts (Wolin and Cedervall 2002) and the Lsm complex binds primarily to the GUUUU terminal sequence of U6 (Bordonné and Guthrie 1992; Wolff and Bindereif 1995; Achsel et al. 1999; Pannone et al. 2001; Licht et al. 2008; Zhou et al. 2014; Didychuk et al. 2017; Montemayor et al. 2018). The 3′ end of U6 has additionally been shown in vitro to play an early role in splicing by promoting the annealing of U6 to U4, again through recruitment of the Lsm complex and also through the subsequent recruitment of Prp24p (Vidal et al. 1999; Rader and Guthrie 2002; Ryan et al. 2002; Licht et al. 2008). Surprisingly, the 3′ end of U6 has also been implicated in vitro in playing a late role in splicing—in the turnover of the excised lariat intron long after Prp24p has dissociated from U6 during U4/U6 annealing and long after the Lsm2-8 complex has dissociated during spliceosome activation. Specifically, in whole-cell extract from budding yeast, either reconstituting extract with 3′ end-truncated U6 or targeting the 3′ end of U6 for RNase H-mediated cleavage with a DNA oligonucleotide complementary to the last 31 nt of U6 stabilized the excised, lariat intron product of splicing, preventing its turnover in such reactions (Fabrizio et al. 1989; Ryan et al. 2002). Truncation of the 3′ end of U6 by as few as 23 nt (nt 90–112) was sufficient to stabilize the excised intron (Ryan et al. 2002). This region of U6 participates in two structures—U2/U6 helix II (nt 92–102) and a component of the intramolecular telestem (nt 92–95)—and also binds to the Lsm2-8 complex (nt 108–112).
To distinguish more finely the region of U6 essential for intron turnover, we targeted RNase H cleavage even closer to the 3′ end of U6 in budding yeast extract, using an oligonucleotide complementary to the last 18 nt of U6 (AS 95–112) or a chimeric DNA/2′-O-methyl oligonucleotide (rAS 103–106) that directed RNase H to cleave 7–10 nt from the 3′ end (Fig. 1A). Both oligonucleotides resulted in a substantial accumulation of excised, lariat intron, as reflected by an increase in the molar ratio of excised intron to mRNA from 0.32 ± 0.05 to 0.91 ± 0.06 or to 0.88 ± 0.10, respectively (Fig. 1B). In both cases, the native U6 was truncated predominantly to nucleotide 103 or 104 at the 3′ end of U2/U6 helix II, presumably through the action of a 3′ exonuclease (cf. Ryan et al. 2002). Thus, we conclude that the last ∼10 nt of U6 are essential for turnover of the excised, lariat intron, a region outside of U2/U6 helix II and the U6 telestem but including the binding site for the Lsm2-8 complex.
To determine whether the 3′ end of U6 is also required for turnover of the excised, lariat intron in vivo, we tested a series of deletions in this region for growth phenotypes and stabilization of the excised intron. Because the Lsm2-8 binding site is essential in vivo, we investigated deletions just upstream of the binding site. Specifically, we deleted 2, 3, and all 5 nt in the region of U6 between U2/U6 helix II and the Lsm2-8 binding site; further, given that helix II is not required for growth or excised intron turnover (Field and Friesen 1996; see below), we also deleted 6, 8, 10, and 12 nt extending into this region (Fig. 1C). At 30°C, the 2-, 3-, 5- and 6-nt deletions grew as wild type, the 8-nt deletion grew less well, and the 10- and 12-nt deletions were lethal; intriguingly, at 16°C the 6-nt deletion showed a mild cold-sensitive phenotype, suggesting a defect in a structural rearrangement (Supplemental Fig. S1D–F. Northern analysis confirmed that the U6 deletions were shorter than full-length U6 (Fig. 1D; Supplemental Fig. S1H). None of the viable deletions altered the levels of unspliced or spliced U3A snoRNA, indicating no significant defect in splicing this model substrate; in contrast, the 5-, 6-, and 8-nt deletions increased the levels of excised, lariat U3A intron by three to sevenfold (Fig. 1D; Supplemental Fig. S1G), indicating a role for the 3′ end of U6 in excised intron turnover in vivo, as in vitro.
Turnover of the excised lariat intron by exonucleases requires debranching, which requires release from the spliceosome (Martin et al. 2002). The spliceosome releases the excised, lariat intron in two ATP-dependent steps. First, the DEAH ATPase Prp22p releases the mRNA (Company et al. 1991; Schwer and Gross 1998; Wagner et al. 1998), which then allows the DEAH ATPase Prp43p to subsequently disassemble the spliceosome and release the excised lariat intron (Arenas and Abelson 1997; Martin et al. 2002). To determine the stage at which the 3′ end of U6 is required for excised intron turnover, we analyzed in vitro splicing reactions by glycerol gradients, which separate released splicing products from spliceosome-bound products. In splicing reactions in which the 3′ end of U6 was cleaved with an antisense oligo, the stabilized, excised lariat intron did not migrate at the top of the gradient, demonstrating that the 3′ end of U6 does not somehow impact debranching directly, after release of the intron from the spliceosome. Instead, the stabilized, excised lariat intron comigrated with spliceosomes deep in the gradient, implicating a direct or upstream role for the 3′ end of U6 in release of the intron from the spliceosome (Supplemental Fig. S1A,B).
To account fully for the excised, lariat intron released from the spliceosome, we precluded turnover of these lariats by assembling splicing reactions in dbr1Δ extract lacking the debranchase Dbr1p. In these extracts, cleavage of the 3′ end of U6 shifted the migration of the excised lariat intron from shallow fractions of the gradient to deep, spliceosomal fractions, establishing that the 3′ end of U6 is required for release of the excised lariat intron from the spliceosome (Fig. 2A, cf. first and second panels; Fig. 2B, right panel); northern blotting showed that truncated U6 also comigrates with the spliceosome deep in the gradient, confirming that U6 lacking ∼10 nt at the 3′ end incorporates into spliceosomes (Supplemental Fig. S1C). This requirement of the 3′ end of U6 in excised intron release parallels the requirement for Prp22p and Prp43p in intron release; specifically, intron release was reduced by adding to splicing reactions recombinant Prp22p having a dominant-negative K512A mutation and was impeded by adding recombinant Prp43p having a dominant-negative Q423E mutation, as expected (Fig. 2A, cf. the first panel with the third and fourth panels; Fig. 2B, right panel; Schwer and Gross 1998; Leeds et al. 2006). Cleavage of the 3′ end of U6 did not shift the migration of mRNA, in contrast to the excised intron, from shallow fractions to spliceosome-containing fractions, indicating that the 3′ end of U6 is not required for mRNA release (Fig. 2A,B). This lack of a requirement for the 3′ end of U6 in mRNA release contrasts with a requirement for Prp22p in mRNA release and parallels the lack of a requirement for Prp43p; indeed, whereas the Prp22p mutation retained mRNA in spliceosome-containing fractions, the Prp43p mutation did not, as expected (Fig. 2A,B). These data therefore demonstrate that the 3′ end of U6 is required after mRNA release, specifically at the stage of intron release and spliceosome disassembly; these data thus implicate components of the NTR complex, including Prp43p, as factors that mediate the requirement for the 3′ end of U6 in spliceosome disassembly.
Figure 2.
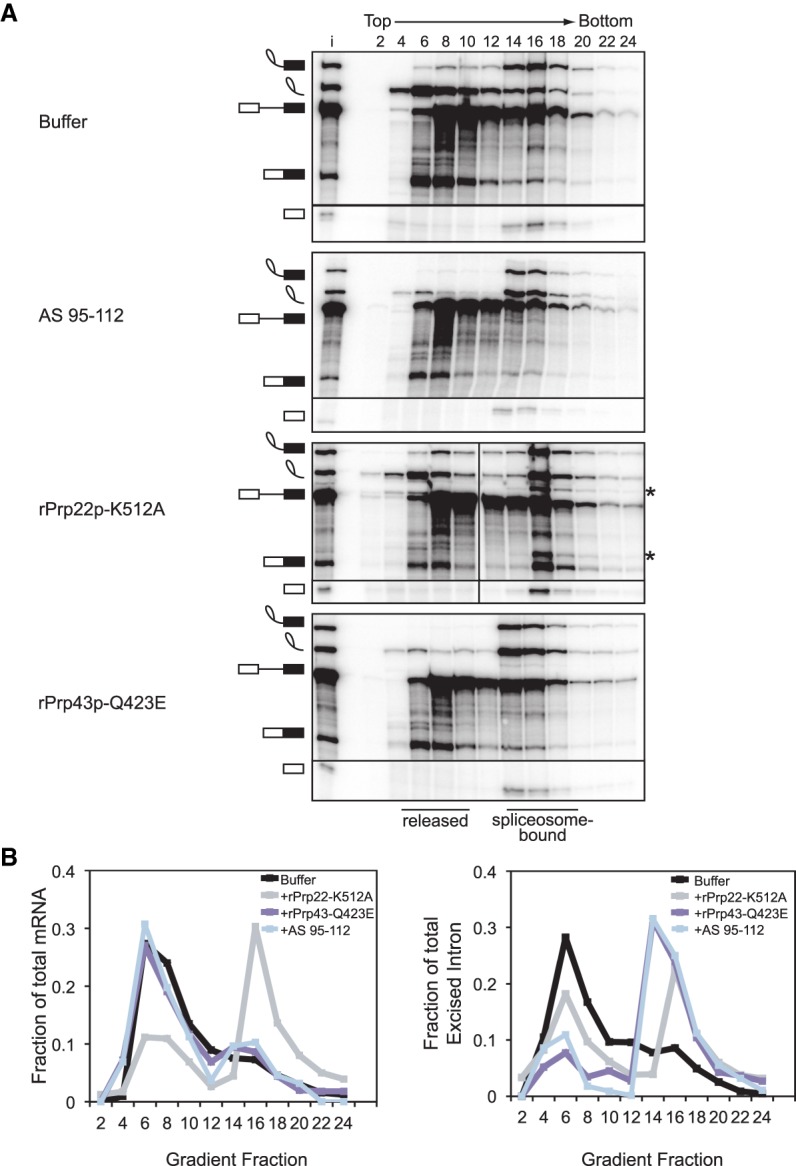
The 3′ end of U6 is required for release of the excised intron. (A) Truncation of the 3′ end of U6 by ∼10 nt impedes release of the excised intron from the spliceosome. Radiolabeled ACT1 pre-mRNA was spliced in mutant dbr1Δ (yJPS799) extracts subjected to RNase H cleavage directed by DNA oligo AS 95–112 (see Fig. 1A) or supplemented with buffer, mutated rPrp22p-K512A, or mutated rPrp43p-Q423E. Splicing reactions were fractionated on a glycerol gradient; input (i) and fraction numbers are indicated above the top panel. Fractions containing released or spliceosome-bound splicing species are highlighted below the bottom panel. The dividing line in the rPrp22p-K512A panel indicates an empty lane that was removed for consistency between panels. The asterisks identify excised intron and mRNA resulting from splicing at an upstream, suboptimal 3′ splice site due to a loss of fidelity resulting from the Prp22p-K512A mutant (Mayas et al. 2006). See also Supplemental Figure S1. Note that the modest nature of the shift for the excised intron in the rPrp22p-K512A gradient was unexpected, based on the literature and our own experiments (e.g., Supplemental Fig. 1), and may reflect the mutant dbr1Δ extracts used. (B) Quantitation of mRNA (left) or excised intron (right) from gradients in A. Data are normalized as a fraction of total mRNA or total excised intron within each gradient.
Note that these and subsequent experiments indicate that truncation of the 3′ end of U6 can also lead to a mild decrease in the efficiency of 5′ splice site cleavage (e.g., Fig. 2A; Supplemental Fig. S1A) (for a discussion of possible causes, see legend to Supplemental Fig. S1A).
The 3′ end of U6 is required for fidelity and discard of a suboptimal lariat intermediate
In addition to its role during canonical disassembly following exon ligation, Prp43p—likely in the context of the NTR complex (Chen et al. 2013; Su et al. 2018)—also contributes to splicing fidelity by promoting, through spliceosome disassembly, discard of suboptimal substrates such as pre-mRNA rejected by Prp16p and intermediates rejected by Prp22p (Koodathingal et al. 2010; Mayas et al. 2010). Thus, if the requirement for the 3′ end of U6 in spliceosome disassembly and release of a canonical intron reflects a requirement for Prp43p, then the 3′ end of U6 should also be required for Prp43p-dependent discard of suboptimal substrates and splicing fidelity. To test these predictions, we first assayed for the requirement of the 3′ end of U6 in the discard of splicing intermediates after Prp22p-mediated rejection of a suboptimal 3′ splice site, again by glycerol gradient analysis of in vitro splicing reactions in dbr1Δ extract. Specifically, we utilized a UBC4 pre-mRNA containing a suboptimal, UgG 3′ splice site, which is rejected by Prp22p before the stage of exon ligation and then discarded as 5′ exon and lariat intermediate by Prp43p (Mayas et al. 2006, 2010). As expected, with full-length U6 a substantial fraction of the rejected and discarded UgG lariat intermediate migrated in fractions near the top of the gradient (Fig. 3A, top panel; Fig. 3B). In contrast, with truncated U6, the UgG lariat intermediate migrated primarily in deeper, spliceosome-containing fractions, indicating a defect in discard of the rejected intermediate (Fig. 3A, bottom panel; Fig. 3B). This defect parallels an equivalent defect conferred by the Q423E mutation in Prp43p (Fig. 3A, middle panel; Fig. 3B; Mayas et al. 2010). Thus, like Prp43p, the 3′ end of U6 is required for discard of a suboptimal intermediate.
Figure 3.
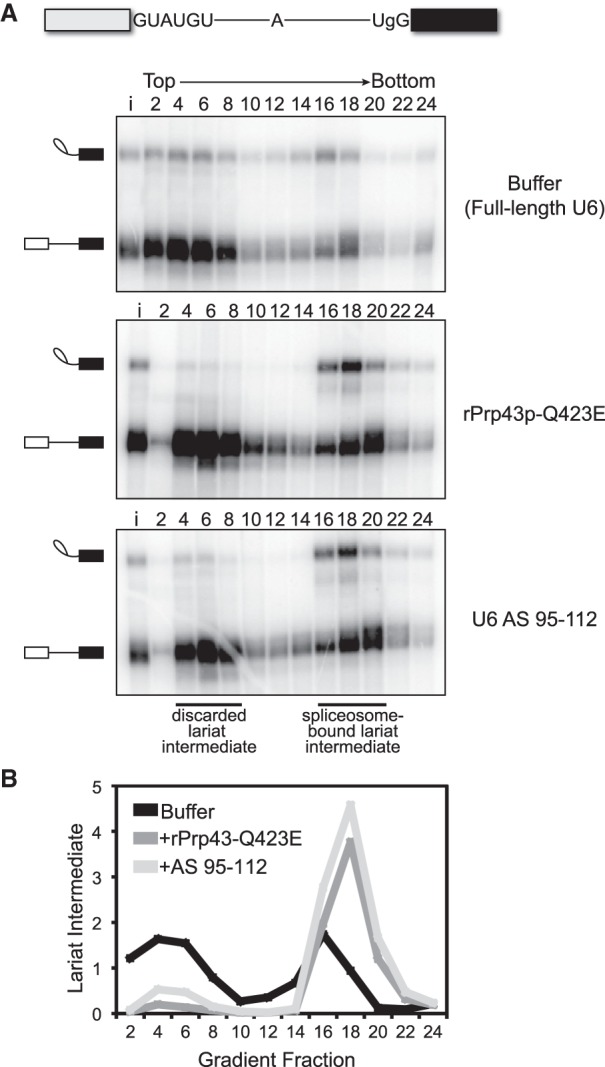
The 3′ end of U6 is required in vitro for Prp43p-dependent discard of a suboptimal substrate. (A) As with the rPrp43p-Q423E mutant (Mayas et al. 2010), 3′-truncated U6 impedes discard of a suboptimal lariat intermediate. Radiolabeled UBC4 pre-mRNA having a suboptimal UgG 3′ splice site was spliced in mutant dbr1Δ (yJPS799) extracts with buffer, with rPrp43p-Q423E, or after RNase H cleavage of U6 in extract with DNA oligo AS 95–112 (see Fig. 1A). Splicing reactions were fractionated on a glycerol gradient; input (i) and fraction numbers are indicated above the top panel. The migration of spliceosome-bound and discarded lariat intermediate is indicated. See also Supplemental Figure S2. (B) Quantitation of lariat intermediate levels from glycerol gradients in A. Data are normalized to input levels of lariat intermediate.
Through its role in discard, Prp43p promotes the fidelity of splicing by minimizing formation of cryptic mRNAs at the stage of exon ligation (Mayas et al. 2010). For example, with a UBC4 pre-mRNA having a UgG 3′ splice site mutation, Prp43p not only discards the suboptimal lariat intermediate but also minimizes splicing at a cryptic 3′ splice site, 6 nt upstream of the suboptimal, UgG 3′ splice site (Supplemental Fig. S2). We found that in DBR1 wild-type extract, this proofreading function of Prp43p was also dependent on the 3′ end of U6. Specifically, truncation of U6 with oligonucleotide AS 95–112 led to stabilization of the suboptimal lariat intermediate and increased production of cryptic mRNA from the UgG substrate, comparable to the increase conferred by recombinant mutant Prp43p-Q423E (Supplemental Fig. S2). Thus, a requirement for the 3′ end of U6 again parallels a requirement for Prp43p. In sum, these data illustrate a striking correlation between the requirement for Prp43p and the 3′ end of U6 in disassembly, discard, and fidelity.
The 3′ end of U6 is required after binding of the NTR complex
Given the strong correlation between the requirements for Prp43p and the 3′ end of U6, we investigated the mechanism by which the 3′ end of U6 functions in spliceosome disassembly and intron release by first determining whether nucleotides at the 3′ end of U6 are required before or after recruitment of the NTR complex to the spliceosome, following release of mRNA by Prp22p. Specifically, we stalled splicing of ACT1 pre-mRNA at the intron release stage by cleavage of U6 with oligonucleotide AS 95–112 or with dominant-negative rPrp43p-Q423E as a positive control for NTR complex association (Small et al. 2006) or with dominant-negative rPrp22p-K512A as a negative control for NTR complex association (Chen et al. 2013). We then tested for binding of the NTR complex to spliceosomes by assaying for coimmunoprecipitation of excised intron with the NTR complex, using antibodies to Ntr1p, Ntr2p, or Prp43p (James et al. 2002; Tanaka et al. 2007). When U6 was truncated, immunoprecipitation of Ntr1p, Ntr2p, or Prp43p enriched for excised intron relative to other splicing species, especially pre-mRNA and mRNA (Fig. 4, top panel, cf. lane 3 with lanes 6,9,11), as observed for the immunoprecipitation of Ntr1p and Ntr2p in reactions stalled by rPrp43p-Q423E (Fig. 4, cf. lanes 2,5,8); in contrast, such enrichment was not observed in reactions stalled by rPrp22p-K512A, despite the accumulation of comparable levels of excised intron (Fig. 4, cf. lanes 4,7,10,12). For splicing reactions stalled by the truncation of U6, we confirmed by northern that immunoprecipitation of the NTR complex not only coimmunoprecipitated excised intron but also truncated U6 (Fig. 4, bottom panel, lanes 6,9,11). These data indicate that the 3′ end of U6 is not required for the recruitment of the NTR complex, indicating that this region of U6 plays a role downstream from NTR binding to promote intron release and spliceosome disassembly; note that whereas Cwc23p has also been shown to complex with Ntr1p and Ntr2p, Cwc23p is not required for binding of Ntr1p and Ntr2p to intron-lariat spliceosomes or for release of excised intron (Pandit et al. 2006; Su et al. 2018), so the 3′ end of U6 cannot play a role in recruiting Cwc23p to promote spliceosome disassembly.
Figure 4.

The 3′ end of U6 is required after the binding of Prp43p, Ntr1p, and Ntr2p to the spliceosome. Denaturing PAGE analysis of in vitro splicing reactions after immunoprecipitation with anti-Ntr1p, -Ntr2p, or -Prp43p antibodies (top, middle panels). Radiolabeled ACT1 pre-mRNA was spliced in yeast extracts (yJPS1448) that were subjected to RNase H cleavage with DNA oligo AS 95–112 or supplemented with buffer, mutated rPrp22p-K512A, or mutated rPrp43p-Q423E. Immunoprecipitation was not performed with anti-Prp43p antibodies on reactions supplemented with rPrp43p-Q423E because of complicating effects of excess recombinant protein. Twenty percent of each reaction was analyzed as input. Cleavage and coimmunoprecipitation of U6 was monitored by northern blot with a radioactive probe directed to nucleotides 28–54 of U6 (bottom panel); “M” indicates a marker lane. The vertical dividing line in all three panels indicates a control lane that was omitted for clarity; the additional vertical dividing line in the northern blot indicates an extra marker lane that was omitted for clarity. See also Supplemental Figure S3 for tests for association of other factors, Lsm3p and Prp24p, with the spliceosome during disassembly.
Although the requirement for the 3′ end of U6 after NTR binding suggests that the 3′ end may impact the function of Prp43p directly, at other stages of splicing the 3′ end interacts with the Lsm2-8 complex and Prp24p, so these factors could, in principle, play roles in spliceosome disassembly and intron release. Although Prp24p dissociates after U4/U6 annealing and the Lsm proteins dissociate after spliceosome activation (Jandrositz and Guthrie 1995; Chan et al. 2003), it is not entirely clear when these factors reassociate with U6 in the splicing cycle, and a recent crystal structure of a U6 snRNP complex suggested the possibility that Prp24p and by implication the Lsm2-8 complex may promote displacement of U2 from U6 during spliceosome disassembly (Montemayor et al. 2014). Thus, we tested for association of these factors with spliceosomes at the stage of disassembly and intron release by stalling spliceosomes with the rPrp43p-Q423E mutant and assaying for association of the Lsm complex or Prp24p by immunoprecipitation directly from splicing reactions (Supplemental Fig. S3). As expected, Lsm3p coimmunoprecipitated U4, U5, and U6, reflecting association with the tri-snRNP, under all conditions tested, and Lsm3p coimmunoprecipitated pre-mRNA when stalled by low ATP at the stage of U4 release; in contrast, immunoprecipitation of Lsm3p did not enrich for excised intron relative to pre-mRNA or mRNA, as compared with a control without antibody, whereas immunoprecipitation of Ntr2p did (Supplemental Fig. S3A). Similarly, Prp24p coimmunoprecipitated U6 snRNA, as expected, but did not coimmunoprecipitate accumulated excised intron (Supplemental Fig. S3B). In contrast, Ntr1p of the NTR complex did coimmunoprecipitate accumulated excised intron (Supplemental Fig. S3B). Although we cannot rule out occlusion of epitopes, these data provide no evidence that either the Lsm2-8 complex or Prp24p associates with spliceosomes poised for disassembly; supporting these findings, purified spliceosomes poised for disassembly have not revealed the presence of Prp24p or Lsm proteins by mass spectrometry or the requirement of trans-acting factors for disassembly after NTR association (Small et al. 2006; Tsai et al. 2007; Fourmann et al. 2013; Chen et al. 2014a; Wan et al. 2017; Zhang et al. 2019). Together, these data provide no evidence for a role of Prp24p or the Lsm2-8 complex in spliceosome disassembly.
The conserved sequence of the 3′ end of U6 is not required for disassembly
Given the parallels between the requirements of Prp43p and the 3′ end of U6 in spliceosome disassembly and given the association of the NTR complex with spliceosomes stalled by truncated U6, we proceeded to test a model in which the 3′ end of U6 serves as a substrate of Prp43p. Because SF1 and SF2 ATPases bind fundamentally to the backbone of nucleic acid and generally without sequence specificity (Fairman-Williams et al. 2010), with some exceptions (e.g., Prabu et al. 2015), this model predicts that the conserved sequence at the 3′ end of U6, required for Lsm2-8 complex binding (Achsel et al. 1999; Vidal et al. 1999; Zhou et al. 2014) is not required for spliceosome disassembly. To test this prediction, we mutated the U-rich sequence at the 3′ end of U6 to AG- or AC-rich sequences, calculated by mFold (Zuker 2003) to form minimal to no secondary structure (100_AG and 100_AC; Fig. 5A). Because the three uracils at nucleotides 100–102 form base pairs with U2 as part of U2/U6 helix II, we also included a variant that did not alter these nucleotides (103_AG) (Fig. 5A). Significantly, none of these sequence variants compromised intron turnover (Fig. 5B), establishing that spliceosome disassembly does not require a specific sequence at the 3′ end of U6, thus establishing functional evidence that intron turnover does not require the sequence-dependent Lsm2-8 complex (Achsel et al. 1999; Zhou et al. 2014). Moreover, these data are consistent with a role for Prp43p in acting on the 3′ end of U6.
Figure 5.

Spliceosome disassembly does not require the sequence of the 3′ end of U6 but does require the 2′ hydroxyls of the 3′ end. (A) U6 3′ end sequences are depicted showing mutations used in B. “WT” corresponds to full-length, unmodified, transcribed U6 snRNA. U6 Δ97–112 and 97–112D were constructed by transcribing nucleotides 1–96 of U6 and then either using as is (Δ97–112) or first ligating to a DNA oligo (97–112D). The remaining variants were constructed by ligating segments of synthetic RNA. (B) Disassembly does not require specific sequence of the 3′ end of U6. Denaturing PAGE analysis of radiolabeled in vitro splicing reactions using ACT1 pre-mRNA in extracts (ySCC1) reconstituted (Recon.) with the indicated U6 variants. U6 Δ97–112 and 97–112D correspond to positive controls that block disassembly and stabilize excised intron, as shown in D. Quantitation of intron turnover is shown below the gel, wherein the ratio of excised intron to mRNA was calculated and normalized relative to the wild-type U6 control in lane 3; values with a substantial increase of twofold or greater over wild type are indicated in bold, underlined. (C) U6 3′ end sequences are depicted showing deoxy substitutions used in D. Underlined bases are DNA. “WT” corresponds to full-length, unmodified, transcribed U6 snRNA; for the remaining U6 variants, the first 96 nt were transcribed and then used as is (Δ97–112) or first ligated to synthetic oligonucleotides corresponding to unmodified RNA (97–112R), DNA (97–112D), or RNA–DNA chimeras (97–104D, 105–112D). (D) Disassembly requires the 2′ hydroxyls of the 3′ end of U6. Denaturing PAGE analysis of splicing reactions using ACT1 pre-mRNA in ySCC1 extracts reconstituted with the indicated U6 variants. See also Supplemental Figures S4, S5. (E) U6 3′ end sequences are depicted showing deoxy substitutions used in F. U6 variants are shown and were constructed as in C. (F) Disassembly only requires 2′ hydroxyls in the last ∼16 nt of the 3′ end of U6. Denaturing PAGE analysis of splicing reactions using ACT1 pre-mRNA in extracts (yJPS866) reconstituted with the indicated U6 variants. (G) Prp43p unwinds a duplex with a short RNA, but not DNA, single-stranded overhang in vitro. Unwinding of a 24-bp RNA/DNA duplex with a 10-nt single-stranded RNA overhang (red) or an 8-nt DNA substitution (blue) was assayed in the presence of rPrp43p, its activator rNtr1p (1–120), and ATP (T) or ADP (D), as a negative control. (H) Prp43p-mediated unwinding is impeded by DNA at the proximal end and middle of a duplex but not at the distal end. Unwinding of a 24-bp RNA/DNA duplex with or without an 8-nt DNA substitution and with a 16-nt single-stranded RNA overhang was assayed in the presence of rPrp43p, its activator rNtr1p (1–120), and ATP (T) or ADP (D), as a negative control. (Red) RNA, (blue) DNA. Quantification is shown for the average of two replicate experiments; error bars indicate the range of values; unwinding was calculated as the fraction of released oligo relative to the total oligo—single-stranded and duplexed. Color coding in the left panel serves as a key for the right panel. (mod.) Modification.
The 2′ hydroxyls of the 3′ end of U6 are required for spliceosome disassembly
Whereas Prp43p does not depend on a specific sequence for binding to nucleic acid, it does depend on the 2′ hydroxyls of RNA both for binding to model substrates and for nucleic acid–dependent ATPase activity (Martin et al. 2002; Tanaka and Schwer 2006). We therefore tested whether spliceosome disassembly similarly depends on the 2′ hydroxyls of RNA. Relative to U6 snRNA controls, U6 substituted in the last 16 nt with DNA (97–112D) accumulated excised intron relative to mRNA, similar to 3′ truncated U6 (Δ97–112) or the rPrp43p-Q423E mutant (Fig. 5C,D, cf. lanes 3–7). This requirement for RNA at the 3′ end of U6 is also consistent with a role for Prp43p action at the 3′ end of U6.
To define precisely the region of U6 that must be RNA to promote intron turnover, we varied the length and position of DNA substitutions and assayed for intron turnover (Fig. 5C–F). Consistent with our finding that truncation of as few as 9 or 10 nt from the 3′ end results in a defect in intron turnover (Fig. 1B), as few as 8 nt of DNA at the 3′ end blocked intron turnover as robustly as rPrp43p-Q423E (Fig. 5C,D, cf. lanes 4 and 8); 6 or 4 nt of DNA at the 3′ end of U6 were insufficient to similarly compromise intron turnover (Supplemental Fig. S4). Interestingly, placing the 8-nt DNA block 8 nt upstream of the 3′ end and overlapping with U2/U6 helix II (Fig. 5C,D, lane 9) still strongly inhibited intron turnover, indicating that 8 nt of RNA at the 3′ end of U6 are not sufficient for spliceosome disassembly. In contrast, 8-nt DNA blocks 16, 20, or 34-nt upstream of the 3′ end of U6 permitted intron turnover, indicating that RNA is not required in the loop region of the U6 ISL or in the bulge between the ISL and U2/U6 helix II (Supplemental Fig. S5A; see below). To refine the 5′ boundary of where RNA is required in the 3′ end of U6 for intron turnover, we tiled 8-nt blocks of DNA in 1-nt steps 9, 10, and 11 nt from the 3′ end. These substitutions conferred diminishing consequences for intron turnover, with the impact of DNA substitutions on intron turnover dropping off markedly between nucleotides 96 and 97 (Fig. 5E,F, cf. 96–103D vs. 97–104D, lanes 9 and 10). Thus, the dependence of intron turnover and spliceosome disassembly on RNA at the 3′ end of U6 is confined to a short stretch of 16 nt. Given evidence that small, 8-nt DNA blocks can inhibit DEAH box ATPases (Semlow et al. 2016), these data are consistent with a model in which an RNA-dependent helicase, such as Prp43p, binds to single-stranded RNA at the 3′ end of U6 and translocates upstream in a 2′-hydroxyl-dependent fashion to disrupt interactions, such as U2/U6 helix II.
This model is consistent with our analysis of Prp43p/Ntr1p activity on idealized unwinding substrates. Specifically, we found that a 10-nt ssRNA overhang, analogous to the ssRNA at the 3′ end of U6, was sufficient to support unwinding of a 24-bp duplex and that an 8-nt ssDNA substitution at the 3′ end of the overhang impeded unwinding and did so by impeding binding of Prp43p to the substrate (Fig. 5G; Supplemental Fig. S5B), consistent with an interpretation in which the 105–112D substitution impedes recruitment of Prp43p to U6. Additionally, we found that an 8-nt DNA substitution at the proximal end or in the middle of a duplex permitted binding of Prp43p/Ntr1p to the substrate but reduced unwinding more than fivefold (Fig. 5H; Supplemental Fig. S5B), consistent with an interpretation in which the 97–104D substitution specifically impedes unwinding of U2/U6 helix II by Prp43p.
We also found that a DNA substitution at the distal end of the duplex did not impede unwinding (Fig. 5H, lanes 16–19), paralleling the failure of DNA substitutions upstream of 97–104D but within U2/U6 helix II to impair spliceosome disassembly. These results suggest that these noninhibitory DNA blocks either (1) impede translocation by Prp43p along a region that is not required for unwinding (because unwinding has already occurred) or (2) allow translocation of Prp43p, because unwinding has already occurred, and DNA only inhibits translocation during unwinding or while performing work beyond translocation. Thus, we cannot rule out at this time a model in which Prp43p translocates along U6 beyond U2/U6 helix II.
The 3′ end of U6 interacts directly with the DEAH box ATPase Prp43p
To test explicitly whether the 3′ end of U6 may serve as a substrate for Prp43p, we assayed for a direct interaction between U6 and Prp43p by site-specific cross-linking. Although a recent in vitro study implicated Prp43p as interacting with pre-mRNA but not snRNAs (Fourmann et al. 2016), a previous study reported interactions between Prp43p and U6 using CRAC (UV cross-linking and analysis of cDNA) in vivo (Bohnsack et al. 2009); nevertheless, these U6 sites, captured at steady state, did not reveal an interaction at the 3′ end of U6 and instead provided evidence for interactions centered just downstream from the 5′ stem loop and on the 3′ side of the ISL, which is flanked by regions that spliceosome disassembly did not require to be RNA (Supplemental Fig. S5A). Thus, we performed site-specific UV cross-linking with U6 containing a 32P label upstream of three consecutive, photoactivatable 4-thio-U (s4U) residues incorporated into the 3′ end of U6 (Fig. 6A). We depleted U6 from Prp19p-HA tagged yeast extract, reconstituted with s4U-substituted U6 (*s4U_104–106 U6), assembled splicing reactions with a Cy5-labeled, ACT1 pre-mRNA substrate (Supplemental Fig. S6B) and then, after the reactions were irradiated with UV light, enriched for spliceosome-bound U6 by immunoprecipitation with anti-HA antibodies.
Figure 6.

The 3′ end of U6 snRNA interacts directly with Prp43p at the disassembly stage of splicing. (A) The 3′ end sequence of *s4U(104–106) U6, used for cross-linking, is shown with the location of 4-thio-U modifications (s4U) and the single 32P label (*p). (B,C) U6 cross-links to Prp43p in a splicing-dependent manner. Splicing of Cy5-ACT1 pre-mRNA was performed under the indicated conditions in extracts (ySCC1) reconstituted with *s4U(104–106) U6 and supplemented where indicated with either rPrp43p-Q423E, GST-tagged rPrp43p-Q423E, or rPrp16p-K379A; splicing reactions were monitored by PAGE (Supplemental Fig. S6B); control reactions were performed in the absence of extract as indicated. After cross-linking, spliceosomes were immunoprecipitated (IP) under native conditions by HA-tagged Prp19p, and then cross-links between U6 and spliceosomal proteins were analyzed by SDS-PAGE; control reactions were analyzed directly. Migration of protein size markers are indicated to the left, and the positions of U6 cross-linked to rPrp43p-Q423E or rPrp43p-Q423E-GST in the absence of extract are indicated to the right. An asterisk indicates migration of rPrp43p-Q423E or rPrp43p-Q423E-GST cross-linked to U6 in the absence of extract. See also Supplemental Figures S6, S7. (D) The 3′ end sequence of *s4U(104–106) U6, used for cross-linking, is shown with the location of 4-thio-U modifications (s4U), the single 32P label (*p), and DNA substitutions (underlined), where “R” indicates the U6 construct with RNA only at the 3′ end, and “D” indicates the U6 construct with the indicated DNA substitutions at the 3′ end. See also Supplemental Figure S7. (E) U6 cross-links to Prp43p in an RNA-dependent manner. Splicing, cross-linking, and immunoprecipitation were executed and analyzed as in B and C except that the U6 variants shown in D were utilized. The vertical lines in panels C and E represent lanes that were omitted for clarity.
In the absence of added rPrp43p-Q423E, U6 forms a number of prominent cross-links (Fig. 6B, lane 1). In the presence of rPrp43p-Q423E, U6 formed an additional, prominent cross-link at ∼125 kDa, which corresponds to the molecular weight of Prp43p plus U6 snRNA (87.5 kDa + 37 kDa; Fig. 6B, cf. lane 1 with lanes 4 and 5). Robust formation of this ∼125-kDa species depended on both ACT1 pre-mRNA and ATP (Fig. 6B, cf. lanes 2 and 3 with 4 and 5), as well as UV and s4U (Fig. 6C; Supplemental Fig. S7). In the presence of rPrp43p-Q423E tagged with GST, the ∼125-kDa cross-link disappeared and a new cross-link appeared at ∼150 kDa (Fig. 6B, lane 6), roughly equivalent to the increase in size expected for a GST tag (26 kDa). Supporting that these species reflect cross-linking of U6 to Prp43p, the mobilities of the ∼125- and ∼150-kDa cross-links correlated to the mobilities of rPrp43p-Q423E and rPrp43p-Q423E-GST, respectively, cross-linked to U6 in the absence of yeast extract (Fig. 6B, cf. lanes 4 and 5 with lane 7, and lane 6 with lane 8).
Having identified U6 cross-links in spliceosomes stalled prior to intron release, we next tested whether U6 cross-linked to Prp43p explicitly and, if so, whether such cross-linking occurred specifically at the 3′ end of U6. From the same cross-linking reactions described above, we pulled down His6-tagged Prp43p using Ni-NTA beads under denaturing conditions and then cleaved the 3′ end away from the remainder of U6 using RNase H targeted with an oligo complementary to nucleotides 48–93 (Supplemental Fig. S6C,D). In the presence of rPrp43p-Q423E, the 3′ fragment of U6 was cross-linked in a species that migrated at ∼100 kDa, which corresponds roughly to the molecular weight of the fragment plus Prp43p (∼6.3 kDa + 87.5 kDa); this cross-link comigrates with a cross-link between the 3′ fragment of U6 and rPrp43p-Q423E formed in the absence of extract (Supplemental Fig. S6C). As with the ∼125-kDa species in the native spliceosome immunoprecipitates, substantial formation of this ∼100-kDa cross-link depended on both ACT1 transcript and ATP, and the cross-link shifted to a higher molecular weight of ∼125 kDa when extracts were instead supplemented with GST-tagged rPrp43p-Q423E (Supplemental Fig. S6C). These cross-linking data confirm that U6 interacts directly with Prp43p and establish that U6 does so at its 3′ end.
To confirm that the interaction between U6 and Prp43p occurs at a late stage of splicing, we tested for dependence of cross-linking on earlier steps in splicing. When we allowed spliceosome assembly but impeded spliceosome activation by using a low concentration of ATP (0.1 µM) (Tarn et al. 1993), U6 no longer cross-linked efficiently to rPrp43p-Q423E, as compared with reactions with high concentrations of ATP (Fig. 6C, cf. lanes 3 and 4). When we permitted spliceosome activation but stalled splicing after the first chemical step of splicing with an ATPase-deficient Prp16p mutant (K379A) (Schneider et al. 2002), U6 again failed to cross-link efficiently to rPrp43p-Q423E (Fig. 6C, cf. lanes 3 and 5; Supplemental Fig. S7, cf. lanes 5 and 6). These data confirm that U6 interacts with Prp43p at a late stage in the splicing cycle.
Given that intron turnover requires that the 3′ end of U6 be RNA and not DNA (Fig. 5), to test whether Prp43p interacts with the 3′ end of U6 in a functional manner, we tested whether this interaction required RNA. Specifically, we assayed for cross-linking between Prp43p and U6 snRNA in which the last 16 nt (97–112) were replaced with DNA, with the exception of the three s4U-substituted nucleotides at 104–106 (Fig. 6D). This DNA substitution compromised cross-linking of Prp43p-Q423E to U6, indicating that the interaction of Prp43p with the 3′ end of U6 requires RNA (Fig. 6E, cf. lanes 3 and 4).
Because the region of U6 that must be RNA to promote intron turnover overlaps with U2/U6 helix II, we considered whether the sole function of the 3′ end of U6 at this stage is to enable Prp43p-mediated unwinding of U2/U6 helix II, which is present in the intron lariat spliceosome (ILS) (Wan et al. 2017; Zhang et al. 2019). If so, we would predict that disruption of U2/U6 helix II would bypass the requirement for Prp43p. We thus tested whether mutation of U6 nucleotides 93 to 102 to poly(C) (Fig. 7A) would suppress the intron turnover defect of rPrp43p-Q423E. This U6 variant did not suppress the intron turnover defect conferred by rPrp43p-Q423E (Fig. 7B), indicating that disruption of helix II is not sufficient for intron release and spliceosome disassembly. Together, our data implicate a model in which Prp43p disrupts interactions with U6 more broadly by pulling on the 3′ end of U6 to build up tension and thereby to disrupt interactions by acting at a distance (Fig. 7C; see Discussion).
Figure 7.

A model for Prp43p function on the 3′ end of U6 involving disruption of structures beyond U2/U6 helix II. (A) U2/U6 helix II is depicted in the context of wild-type U6 or U6 with mutations that would disrupt base-pairing to U2. (B) Disruption of U2/U6 helix II does not bypass the requirement for Prp43p. In vitro splicing reactions were executed using radiolabeled ACT1 pre-mRNA in extracts (yJPS866) reconstituted with wild-type or helix II mutant U6 (illustrated in A) and with or without rPrp43p-Q423E, and then analyzed by denaturing PAGE. Quantitation of intron turnover is shown below the gel, wherein the ratio of excised intron to mRNA was calculated and normalized relative to the wild-type U6 control in lane 1; values with a substantial increase of twofold or greater over wild type are indicated in bold, underlined. (C) A mechanistic model for Prp43p function in splicing termination. See text for details.
Discussion
Whereas identifying the functions of RNA-dependent SF2 ATPases in splicing has been relatively straightforward (Cordin and Beggs 2013), determining their targets and precise mechanisms has proven more challenging. Although we know the DEAH box ATPase Prp43p plays a critical role in terminating splicing, both to discard rejected suboptimal substrates and to release an excised optimal intron (Arenas and Abelson 1997; Martin et al. 2002; Pandit et al. 2006; Tsai et al. 2007; Koodathingal et al. 2010; Mayas et al. 2010), the mechanism of Prp43p action has been unclear (Fourmann et al. 2016; Wan et al. 2017; Zhang et al. 2019). We have discovered that the requirement for Prp43p in terminating splicing parallels a requirement for the 3′ end of U6 in the discard of a rejected lariat intermediate and in the release of an excised intron (Figs. 1–3). The 3′ end of U6 is not required for the binding of the Prp43p-bound NTR complex to the spliceosome but rather a downstream step (Fig. 4). Consistent with a direct role for Prp43p at the 3′ end of U6 in splicing termination, Prp43p activity and splicing termination show the same nucleic acid preferences (Fig. 5). Indeed, Prp43p cross-links to the 3′ end of U6 at a late stage of splicing in a manner that requires RNA nucleotides at the 3′ end (Fig. 6). Our data suggest a model in which Prp43p translocates along U6, leading to spliceosome disassembly through a pulling mechanism, as recently defined for two other DEAH box ATPases in splicing, Prp16p and Prp22p (Semlow et al. 2016).
The first suggestion of a direct spliceosomal substrate for Prp43p derived from an in vivo cross-linking study, which revealed cross-linking of Prp43p to U6 snRNA (Bohnsack et al. 2009) in addition to rRNAs and snoRNAs, consistent with the additional role for Prp43p in ribosome biogenesis (Combs et al. 2006; Leeds et al. 2006; Lebaron et al. 2009). Subsequently, in vitro biochemical experiments suggested that Prp43p targets U2 snRNP for displacement from pre-mRNA and does so by interacting with pre-mRNA (Fourmann et al. 2016). Although a recent cryo-EM structure of the yeast spliceosome at the disassembly stage did indicate proximity of Prp43p to the pre-mRNA intron, the structure revealed the closest proximity is to the 3′ domain of U6, although direct interactions were not observed (Wan et al. 2017). A more recent cryo-EM structure of the human spliceosome also indicated proximity of Prp43p to the 3′ domain of U6, although again no direct interactions were observed and Prp43p was no closer than 40Å from U6 (Zhang et al. 2019). Our data provide direct and functional evidence that Prp43p targets the 3′ end of U6 at the disassembly stage.
Evidence for two alternative models for Prp43p raises the question as to whether Prp43p functions in multiple capacities, as demonstrated for the RNA helicase DbpA, for example (Karginov and Uhlenbeck 2004). On the one hand, the model that Prp43p displaces U2 snRNP from pre-mRNA is founded on compelling data (Fourmann et al. 2016). First, the data supporting the model derive from experiments that leveraged a variant of Prp43p that is sufficient to disassemble the ILS in the absence of Ntr1p and Ntr2p, a variant that is constitutively activated by fusion of Prp43p to the G-patch domain of Ntr1p (Prp43p_Ntr1GP). Second, the model derives from direct and specific cross-linking from Prp43p_Ntr1GP to both pre-mRNA and to U2 snRNP proteins. Indeed, hPrp43p has been implicated as a component of the U2 snRNP—both in splicing (Will et al. 2002) and U7 biogenesis (Friend et al. 2007). Third, for all three spliceosomal intermediates tested, Prp43p_Ntr1GP-mediated disassembly consistently released the U2 snRNP. On the other hand, the model and supporting data are difficult to reconcile with several observations in the literature. First, the exclusive cross-linking of Prp43p_Ntr1GP to pre-mRNA does not align with the specific cross-linking of Prp43p to U6 in vivo (Bohnsack et al. 2009). Second, the data lead to a model in which Prp43p translocates 5′ to 3′ along the pre-mRNA to displace U2, but the DExH families of RNA-dependent ATPases translocate in the 3′ to 5′ direction (Gilman et al. 2017), and although earlier work provided some evidence for 5′ to 3′ translocation by Prp43p (Tanaka and Schwer 2006; Tanaka et al. 2007; Lebaron et al. 2009), more recent data using unwinding substrates with unstructured, single-stranded tails has established a strong preference for Prp43p in translocating in the 3′ to 5′ direction (He et al. 2017). Additionally, the structure of the human spliceosome at the disassembly stage revealed proteins bound to the intron upstream of the U2-branchsite interaction, proteins that may preclude unwinding of this interaction by Prp43p (Zhang et al. 2019). Third, the cross-linking and disassembly data do not reflect the interaction and activity of Prp43p on spliceosomes poised for disassembly but rather the interaction and activity of Prp43p_Ntr1GP on spliceosomal intermediates that have not been identified as physiological targets for Prp43p either in vivo or in splicing extracts; for example, whereas the Bact complex is targeted by the Prp43p_Ntr1GP fusion for disassembly (Fourmann et al. 2016), this complex is not targeted by the NTR complex (Chen et al. 2013), presumably because Hsh155p sterically blocks Ntr2p binding (Rauhut et al. 2016; Yan et al. 2016). Further, whereas the B complex is targeted by the Prp43p_Ntr1GP fusion for disassembly (Fourmann et al. 2016), and Prp43p and Ntr1p have been suggested to discard at this stage of spliceosome activation in vivo (Pandit et al. 2006), even before dissociation of the Lsm2-8 complex from the 3′ end of U6 (see below), the NTR complex is not sufficient to disassemble the B complex in vitro (Fourmann et al. 2016). Additional work will be required to resolve these discrepancies and to determine whether Prp43p functions through one mode or multiple modes at the disassembly stage or through distinct modes at different stages of splicing.
Our data define a model in which Prp43p releases a substrate by disassembling the spliceosome through translocation along U6, starting at the 3′ end (Fig. 7C). In particular, our cross-linking, binding, and DNA substitution data (Figs. 5, 6; Supplemental Figs. S5–S7) indicate that Prp43p first binds the single-stranded tail of U6 and then translocates into U2/U6 helix II, disrupting this interaction. Unwinding of this helix likely contributes to spliceosome disassembly not just by initiating release of U2 from U6 but also by favoring formation of the 5′ stem loop of U2, which is mutually exclusive with U2/U6 helix I, a structure that includes the catalytic triad and juxtaposes the 5′ splice site and branch site sequences (Madhani and Guthrie 1992). Unwinding of U2/U6 helix II is not sufficient, however (Fig. 7A,B). In one model, limited translocation along the 3′ end of U6 would not only displace U2 but also build up tension that would destabilize upstream interactions, such as the catalytic U6 ISL and interactions between the ISL and Prp8p, a central splicing cofactor uniquely conserved from spliceosomes to self-splicing group II introns (Piccirilli and Staley 2016); curiously, a U6 double mutant that stabilizes U2/U6 helix II and the U6 ISL accumulates a novel U2/U6 complex, which may reflect impedance of Prp43p function (Burke et al. 2012). Destabilization of interactions at the heart of the spliceosome may then trigger disassembly of the remainder of the spliceosome, although both in vivo and sensitive in vitro assays have provided evidence for a role for the DExH box ATPase Brr2p as well (Small et al. 2006; cf. Fourmann et al. 2013). A role for Prp43p in disrupting interactions by pulling on an RNA to build up tension and thereby disrupting interactions at a distance parallels recent evidence that the related DEAH box ATPases Prp16p and Prp22p pull on the substrate to dislodge regions of the substrate from the catalytic core of the spliceosome (Semlow et al. 2016). Consistent with this limited-pulling model, we have not yet identified regions of U6 upstream of U2/U6 helix II, in which DNA substitutions inhibit Prp43p function (Fig. 5; Supplemental Fig. S5). However, these DNA substitutions fall in primarily ssRNA regions. In an alternative model, Prp43p translocates more extensively along U6, performing disruptive work at regions yet to be defined. Consistent with this model, in vivo cross-linking identified interactions between Prp43p and upstream regions of U6 (Bohnsack et al. 2009). Defining the complete path of translocation and regions of disruptive work will require further studies.
Our functional data together with recent cryoEM structural data (Wan et al. 2017; Zhang et al. 2019) support a model by which Prp43p is targeted to its substrate and activated for spliceosome disassembly. In this model, Prp43p is localized in proximity to the 3′ end of U6 by binding to the NTC component Syf1p, and Prp43p is activated by binding of the conserved factor Ntr1p as well as Ntr2p in yeast to the spliceosome (Tsai et al. 2005; Boon et al. 2006; Tanaka et al. 2007), because binding appears to juxtapose the activating G-patch domain of Ntr1p with Prp43p; binding of Ntr1p and Ntr2p to the spliceosome may also derepress the NTR complex by reconfiguring interactions within the NTR complex (Fourmann et al. 2016, 2017). This model also rationalizes the action of Prp43p and the NTR complex at other stages of splicing, because Syf1p remains in proximity to the 3′ end of U6 in all spliceosomal complexes observed by cryoEM to-date (Bact, B*, C, C*, P, and ILS) (Wan et al. 2019; Yan et al. 2019). Although the NTR complex functions as a general splicing terminator (Pandit et al. 2006; Koodathingal et al. 2010; Mayas et al. 2010; Chen et al. 2013), the NTR complex is restricted from acting at several stages of splicing. As noted, the binding and activation model rationalizes the inactivity of the NTR complex against the Bact complex and the C* and P complex, because Ntr2p binds to a pocket in Prp8 that is mutually exclusive with binding of Hsh155p, in the Bact complex, and of Prp22p, in the C* and P complexes (Wan et al. 2017; Zhang et al. 2019). Additionally, the structure of the C complex rationalizes the inactivity of the NTR complex against this complex; Prp16p binds to the spliceosome in this complex in a manner that is mutually exclusive with the binding of Ntr1p and Ntr2p. The binding and/or positioning of Hsh155p, Prp16p, and Prp22p likely signals productively engaged spliceosomes, whereas the prolonged absence of these factors likely signals unproductive stalled spliceosomes, rendering them subject to NTR-mediated discard.
Interestingly, at the stage that Syf1p becomes stably bound to the spliceosome, as a component of the NTC complex, the Lsm2-8 complex dissociates from the 3′ end of U6 (Chan et al. 2003). Given the requirements for the 3′ end of U6 in spliceosome disassembly and the interaction of Prp43p with the 3′ end of U6, it is unlikely that Prp43p is active for disassembly of the spliceosome by translocation along U6 while the Lsm2-8 complex remains bound to U6. Thus, coincident with spliceosome activation in the transition from the B complex to Bact complex, both the docking site for Prp43p (Syf1p) and a target for Prp43p (3′ end of U6) become, in principle, available. In this manner, as the spliceosome becomes competent for catalysis, it also becomes primed for disassembly and termination, likely to ensure fidelity and recycling.
Materials and methods
Strains
In vitro splicing was performed in whole-cell extracts of the strains listed in Table 1.
Table 1.
Strains used in this study

To make strains yJPS866 and yJPS867, strains GLS618 and GLS616 (Rader and Guthrie 2002) were transformed with either [pSE362-PRP24] or [pSE362-PRP24-3HA], respectively, and the PRP24-URA3 plasmid was shuffled out on solid 5-fluoroorotic acid (5-FOA) medium (0.67% yeast nitrogen base, 0.2% drop-out mix uracil, 2% glucose, 50 µg/mL uracil, 0.1% 5-FOA, and 2% agar).
To make strain yJPS1448, strain BY4741 (Open Biosystems) was transformed with the plasmid (PRP19-URA3), and then PRP19 was replaced with LEU2, yielding the intermediate strain yJPS1276. Next, a V5 tag was integrated just downstream from LSM3 using KANMX6 as a marker, yielding the intermediate strain yJPS1431. Finally, the plasmid [PRP19-URA3] was replaced with the plasmid [PRP19-HIS] (bJPS2442), yielding yJPS1448.
To generate the yeast strain harboring HA-tagged Prp8p used in Supplemental Figure S1, we transformed yJPS1481 with plasmid pJU204 (pSE362-PRP8-3HA; a gift from C. Guthrie) and shuffled out PRP8-URA3 on 5-FOA and then streaked for single colonies on rich media.
The yeast strains harboring U6 deletion mutations were derived from the parental Saccharomyces cerevisiae strain yJPS1589, which is derived from YHM118 (Madhani and Guthrie 1994b); deleted for multiple genes including SNR6; and maintained with a URA3-marked SNR6 plasmid; yJPS1589 will be described in detail elsewhere. The plasmid pJPS12 that expresses wild-type SNR6 from pSE358 (TRP1, CEN), described in Madhani and Guthrie (1992), was used as the parental plasmid for all site directed mutagenesis, by QuikChange (Agilent), to generate the described U6 deletions, which were confirmed by Sanger sequencing of SNR6, yielding the following plasmids.
pJPS2951: U6Δ2 has UU at position 105–106 deleted and was generated using 5′-CCGTTTTACAAAGAGATTTATCGTTTTTTTTTTATCCTCGAG-3′ and 5′-CTCGAGGATAAAAAAAAAACGATAAATCTCTTTGTAAAACGG-3′
pJPS2952: U6Δ3 has UUC at position 105–107 deleted and was generated using 5′-CCGTTTTACAAAGAGATTTATGTTTTTTTTTTATCCTCGAGTG-3′ and 5′-CACTCGAGGATAAAAAAAAAACATAAATCTCTTTGTAAAACGG-3′
pJPS2953: U6Δ5 has AUUUC at position 103–107 deleted and was generated using 5′-CCGTTTTACAAAGAGATTTGTTTTTTTTTTATCCTCGAG-3′ and 5′-CTCGAGGATAAAAAAAAAACAAATCTCTTTGTAAAACGG-3′
pJPS2954: U6Δ6 has UAUUUC at position 102–107 deleted and was generated using 5′-CCGTTTTACAAAGAGATTGTTTTTTTTTTATCCTCG-3′ and 5′-CGAGGATAAAAAAAAAACAATCTCTTTGTAAAACGG-3′
pJPS2955: U6Δ8 has UUUAUUUC at position 100–107 deleted and was generated using 5′-GGATGAACCGTTTTACAAAGAGAGTTTTTTTTTTATCCTCGAGTG-3′ and 5′-CACTCGAGGATAAAAAAAAAACTCTCTTTGTAAAACGGTTCATCC-3′
pJPS2956: U6Δ10 has GAUUUAUUUC at position 98–107 deleted and was generated using 5′-GAACCGTTTTACAAAGAGTTTTTTTTTTATCCTCG-3′ and 5′-CGAGGATAAAAAAAAAACTCTTTGTAAAACGGTTC-3′
pJPS2957: U6Δ12 has GAGAUUUAUUUC at position 96–107 deleted and was generated using 5′-GAACCGTTTTACAAAGTTTTTTTTTTATCCTCG-3′ and 5′-CGAGGATAAAAAAAAAACTTTGTAAAACGGTTC-3′.
Plasmids encoding the U6 deletion mutations were transformed into yJPS1589. Assessment of the viability of resulting strains was performed after streaking transformants on solid 5-FOA medium to counter-select against the URA3 plasmid expressing wild-type SNR6. Yeast growth assays of viable deletions were performed by spotting yeast from overnight cultures, grown in YPDA and serially diluted in 10-fold increments, with a frogger on solid rich medium (YPDA; 1% yeast extract, 2% peptone, 2% glucose, 0.004% adenine sulfate and 2% agar) at the indicated temperatures for the indicated days. The prp43-Q423N mutant (yJPS575), which accumulates excised lariat intron, has been described in Leeds et al. (2006).
Yeast transformations were performed using the LiOAc method.
Preparation of recombinant proteins
Recombinant, dominant-negative His6-tagged rPrp43p-Q423E was overexpressed from pJPS1384 (Mayas et al. 2010) in BL21 CodonPlus (DE3) RIL (Agilent) cells as described elsewhere (Semlow et al. 2016); the induced cells were lysed using a French press; recombinant protein was enriched via a Ni+2-nitroloacetic acid column (Ni-NTA; QIAGEN), as described previously (Edwalds-Gilbert et al. 2000); and the eluted protein was fractionated on a 15%–30% glycerol gradient, from which peak fractions were isolated, essentially as described elsewhere (Martin et al. 2002; Schneider et al. 2002; Semlow et al. 2016). Recombinant, dominant-negative rPrp22p-S635A, rPrp22p-K512A, and rPrp16p-K379A were expressed as described previously (Semlow et al. 2016).
To add an amino-terminal GST tag to Prp43p-Q423E, the GST sequence from the pGEX-6P plasmid (GE Healthcare) was amplified using primers 5′-GGCAGCAGCCATCATCATCATCATC
ACAGCAGCGGCCTGATGTCCCCTATACTAGGTTATTGG-3′ and 5′-TCTTCTTTTGGAACCCA
TCATATGGCTGCCGCGCGGCACGGGCCCCTGGAACAGA-3′, which contain sequence that overlaps with the region between the His6-tag and amino terminus of Prp43p (underlined) from pJPS1384, with the remaining sequence overlapping with pGEX-6P. The product strands of this PCR amplification were subsequently used as megaprimers to perform overlap extension PCR cloning into pJPS1384 using Phusion DNA polymerase (Bryksin and Matsumura 2010). The insert was verified via Sanger sequencing, and the GST-tagged rPrp43p-Q423E protein was expressed and purified as described above.
Recombinant Prp43p and Ntr1p (1–120) proteins used for in vitro unwinding and EMSA experiments were expressed and purified as described previously (He et al. 2017).
In vitro splicing, RNase H digestion, and U6 depletion/reconstitution
Wild-type ACT1 pre-mRNA was transcribed from a HindIII-linearized pBST7ACTΔ6 plasmid (Mayas et al. 2006) using T7 RNA polymerase (Stevens and Abelson 2002) in the presence of 32P-UTP or, in the case of cross-linking experiments, Cy5-UTP. UBC4 pre-mRNA having either a wild-type UAG or mutated UgG 3′ splice site were transcribed from PCR-generated DNA templates prepared by two rounds of PCR amplification. In the first round, pJPS2125 was amplified using primer 5′-GAACTAAGTGATCTAGAAAGG-3′ paired with either 5′-AACATGAAGTAGGTGGATCTCTAGTTCAATAGCAT-3′ for wild-type UAG or 5′-AACATGAAGTAGGTGGATCTCCAGTTCAATAGCAT-3′ for mutated UgG (underlined). The first-round PCR products were then used as templates for the second round of amplification in which primer 5′-TAATACGACTCACTATAGGGAGAACTAAGTGATCT-3′ was paired with either the UAG or UgG primers listed above to introduce a T7 promoter for transcription (Mayas et al. 2010).
Yeast whole-cell extracts were prepared using the liquid nitrogen method as described elsewhere (Umen and Guthrie 1995), with modifications (Mayas et al. 2006). In vitro splicing reactions were performed under standard splicing conditions (Mayas et al. 2006) using 2 mM ATP (except where indicated) and 0.4–4 nM 32P-body-labeled ACT1 or UBC4 (having either a wild-type UAG or mutated UgG 3′ splice site) pre-mRNA substrate for 15–30 min at 20°C. Where noted, splicing reactions were supplemented with recombinant, dominant-negative rPrp43p-Q423E, rPrp43p-Q423E-GST, rPrp22p-K512A, rPrp22p-S635A, or rPrp16p-K379A in buffer D (20 mM HEPES [pH 7.9], 0.2 mM EDTA, 50 mM KCl, 20% v/v glycerol, 0.5 mM DTT), at a final concentration of ∼20–100 ng/µL (0.2–1 µM). Following in vitro splicing, RNA products were extracted using phenol/chloroform and separated on 6% (for ACT1 substrate) or 15% (for UBC4 substrates) denaturing polyacryamide gels.
For experiments testing the consequences of 3′ end cleavage of endogenous U6, a DNA oligo (or DNA-2′-O-methyl chimeric oligo) complementary to the targeted region was incubated at a concentration of ∼10–20 µM with splicing reactions lacking ATP and substrate at 30°C for 30 min (Fabrizio et al. 1989; Lapham et al. 1997). Following cleavage of U6 nucleotides via endogenous RNase H activity in the extract, an aliquot was removed to evaluate cleavage via northern blotting using a radiolabeled d1 oligo probe complementary to U6 nucleotides 28–54 (Small et al. 2006). Reactions were then supplemented with the appropriate amounts of ATP and splicing substrate and splicing was carried out as described above.
Depletion of endogenous U6 and reconstitution with T7-transcribed or synthetic wild-type U6, chimeric RNA–DNA U6 or s4U-substituted U6 was performed essentially as described (Fica et al. 2013, 2014). All observations are validated by redundant experiments and/or experiments that were reproduced at least twice.
Northern blotting
Roughly 50 mL of yeast cultures were grown in YPDA to an OD600 of ∼ 0.7, and then total RNA was extracted using hot acidic phenol. A total of 20 µg RNA was separated on a denaturing 8% polyacrylamide gel in 1xTBE and transferred semidry to an Amersham Hybond-N membrane (GE Healthcare). To visualize pre-U3A and excised U3A intron, mature U3A, or U6, membranes were probed with 32P-labeled oligos 5′-AGCTGCTGCAATGGTTG-3′, 5′-TTCCTATAGAAATGATCCTATGAAG-3′, or 5′-ATCTCTGTATTGTTTCAAATTGACCAA-3′, respectively, in Amersham Rapid-Hyb buffer (GE Healthcare) and washed twice with 5X SSC, diluted from 20X SSC (3 M NaCl, 0.3 M sodium citrate), and 0.1% SDS when probing for pre-U3A and excised U3A intron and washed once more with 1X SSC and 0.1% SDS when probing for mature U3A or U6. Membranes were exposed to a phosphor screen and signal was detected using a Storm PhosphorImager (Molecular Dynamics).
Debranching of excised lariat intron
A total of 20 µg of RNA was incubated with 2 µM recombinant yDbr1p (expressed and purified as described in Khalid et al. 2005) in 50 mM Tris-HCl pH 7, 25 mM NaCl, 0.01% Triton X-100, 0.1 mM EDTA, 2.5 mM DTT, 0.15% glycerol, and 2 mM MnCl2 in a final volume of 10 µL for 30 min at 30°C.
Glycerol gradients
Splicing reactions in a volume of 100 µL were assembled (following RNase H-directed cleavage of the 3′ end of endogenous U6, where indicated; see method above) in the presence of 2 nM radiolabeled ACT1 or UBC4 pre-mRNA and incubated at 20°C for 30 min. Following incubation, 5 µL was removed and quenched as an input control, and an additional 5 µL was removed for evaluating cleavage of U6 via northern blot. The remaining reaction volume was then loaded onto hand-poured, 11-mL 15%–40% glycerol gradients (20 mM HEPES [pH 7.9], 100 mM KCl, 2 mM MgCl2) and centrifuged at 37,000 rpm for 12–14 h (Mayas et al. 2010). Aliquots of 440 µL were withdrawn from the top of the gradients, and RNA was extracted and fractionated on 6% (ACT1 pre-mRNA) or 15% (UBC4 pre-mRNA) denaturing polyacrylamide gels. For 15% gels, the gels were removed from plates and exposed wet to PhosphorImager screens overnight in the cold room. See Data analysis below for description of gel imaging and analysis.
Immunoprecipitations and affinity pull-downs
For immunoprecipitation of Prp43p-, Ntr1p-, Ntr2p-, and Prp24p-associated spliceosomes or snRNPs under native conditions (James et al. 2002), 25-µL splicing reactions were assembled (with or without RNase-H directed cleavage of U6; see method above) and incubated at 20°C for 30 min. After removing a 5-µL aliquot as an input control, the remaining reaction volume was diluted 1:4 with IPP150 (10 mM Tris-HCl, pH 8, 150 mM NaCl, and 0.1% NP-40 substitute (Fluka)) and added to 5–7 µL of protein A sepharose beads (Sigma) in a 50% slurry from the vendor that had been washed in IPP150 and conjugated to ∼5–10 µg of affinity-purified or protein A-purified anti-Prp43p, anti-Ntr1p, or anti-Ntr2p antibodies (gifts from B. Schwer) and incubated with nutation for 1 h at 4°C. After incubation, beads were washed twice with 200 µL IPP150 and then twice with 500 µL of IPP150 for Prp43p, Ntr1p, and Ntr2p immunoprecipitation (IP) or just twice with 500 µL of IPP150 for all other IPs. RNA was extracted from the beads using phenol/chloroform.
For immunoprecipitation of spliceosomes or snRNPs from extract of the yeast strains PRP19-HA, LSM3-V5, or PRP24-3HA, 6–12 µg anti-HA antibodies (i.e., 12CA5, University of Chicago Monoclonal Antibody Facility) or 6 µg anti-V5 antibodies (R960-25, Invitrogen) per reaction were first conjugated to protein A sepharose beads in a 50% slurry that was first washed in IPP150. Immunoprecipitation was carried out as described above.
For affinity pull-downs of His6-tagged rPrp43p-Q423E or His6-GST-tagged rPrp43p-Q423E from UV-cross-linked splicing reactions under denaturing conditions, 5 µL of a 50% slurry of Ni-NTA beads were first washed in 20 volumes of wash buffer I (6 M guanidine•HCl, 50 mM [Tris pH 7.8], 300 mM NaCl, 0.1% NP-40 substitute, 10 mM imidazole, 5 mM betamercaptoethanol [BME]) (Granneman et al. 2009). To the washed beads suspended in 50 µL of wash buffer I, 20 µL of UV-cross-linked splicing reaction was added and incubated for 3 h with nutation at 4°C. Following incubation, beads were washed twice with 250 µL of wash buffer I and then washed twice with 250 µL of wash buffer II (50 mM Tris [pH 7.8], 50 mM NaCl, 0.1% NP-40 substitute, 10 mM imidazole, and 5 mM BME) and treated with RNase H and analyzed by SDS-PAGE as described below. RNA was extracted from the beads using phenol/chloroform.
Synthesis of U6 for in vitro reconstitution and cross-linking
The 5′ piece of chimeric U6 snRNAs was constructed either by transcribing the first 96 nt of U6 with T7 polymerase (Stevens and Abelson 2002) or by ligating the first 91 nt of U6 in two sequential splint-mediated ligation steps from three pieces of synthetic RNA (Fica et al. 2014) or synthetic RNA substituted with blocks of DNA (Dharmacon). Synthetic oligonucleotides were first deprotected and gel purified prior to ligation. U6 oligonucleotides corresponding to U6 residues 97–112 or residues 92–112, made from 5′ phosphorylated synthetic RNA or synthetic RNA with various blocks of nucleotides substituted with DNA (Dharmacon or Integrated DNA Technologies) were then joined to U6 1–96 or U6 1–91, respectively, via a final splinted ligation. All ligation steps were performed with T4 DNA ligase (New England Biolabs) and DNA splints (Integrated DNA Technologies) with 20 nt complementary to each side of the ligation junction in a 1:1:1 ratio of 5′ RNA:3′ RNA:splint. RNA pieces and splint were first annealed in the presence of 1X TEN buffer (10 mM Tris-HCl, pH 7.5; 1 mM EDTA; 66 mM NaCl) by heating to 90°C in a PCR block, and then slowing cooling to 18°C at a rate of 1°C per minute. T4 DNA ligase buffer, RNase inhibitor at 5% of the reaction volume (RNasin, Promega), and T4 DNA ligase (4000 units per 5-µL reaction) were then added and incubated at 37°C for 4–6 h. Reactions were separated on a denaturing polyacrylamide gel (7M urea), and ligation products were identified by UV-shadowing briefly at 254 nm, excised, eluted overnight at 4°C in 1X TE (10 mM Tris-HCl, pH 7.5; 1 mM EDTA), and ethanol precipitated.
Note that for designing U6 snRNA with mutations in the region of U2/U6 helix II, we avoided poly(A) and poly(G) because of their tendency to form secondary interactions (i.e., base-stacking and quartets, respectively). For one DEAH-box helicase, MLE, a preference for poly(U) as well as poly(CU) has been observed (Prabu et al. 2015), so we chose not to use poly(C) as a comparison.
For cross-linking substrates, a synthetic oligonucleotide corresponding to U6 97–112, with 4-thio-U (s4U) substitutions at positions 104 to 106 (Dharmacon), was first deprotected and phenol-chloroform-extracted and then 5′ end radiolabeled with [γ-32P]ATP (PerkinElmer, 6000 Ci/mmol) using T4 PNK (New England Biolab or Thermo Fisher) according to the manufacturer's protocol. Following PNK treatment, the oligo was purified using a G-25 spin column, phenol-chloroform-extracted, ethanol-precipitated, and resuspended in 1X TE. The radiolabeled oligo was then ligated as described above to the previously assembled U6 1–96 in a ratio of 1.4:1:1.25 (U6 1–96:U6 97–112:splint), with the exception that ligation products were identified from a PhosphorImager scan of the wet gel instead of UV-shadowing.
In vitro unwinding and binding experiments
Substrates for unwinding and EMSA experiments were made by annealing RNA oligos (purchased from Sigma-Aldrich) to a 24mer DNA oligo containing a Quasar 670 (Cy5) fluorophore at the 3′ end (Biosearch Technologies) that was purified as described previously (He et al. 2017).
RNA oligos:
RNA-1: 5′GGCACCAACACAAAACACAUCUACACUCAACAAU
RNA-2: 5′GGCACCAACACAAAACACAUCUACAC[dT][dC][dA][dA][dC][dA][dA][dT]
RNA-3: 5′GGCACCAACACAAAACACAUCUACACUCAACAAUCACUUU
RNA-4: 5′ GGCACCAACACAAAAC[dA][dC][dA][dT][dC][dT][dA][dC]ACUCAACAAUCACUUU
RNA-5: 5′ GGCACCAA[dC][dA][dC][dA][dA][dA][dA][dC]ACAUCUACACUCAACAAUCACUUU
RNA-6: 5′ [dG][dG][dC][dA][dC][dC][dA][dA]CACAAAACACAUCUACACUCAACAAUCACUUU
The sequence of the RNA oligos were designed as previously described (He et al. 2017) to avoid secondary structures.
DNA oligos:
Cy5 labeled oligo: 5′GTAGATGTGTTTTGTGTTGGTGC C-Cy5
Unlabeled oligo: 5′GTAGATGTGTTTTGTGTTGGTGCC
Capture oligo: 5′ GGCACCAACACAAAACACATCTAC
Unwinding reactions contained 4 nM substrate, 100 nM rPrp43p and 200 nM of its cofactor rNtr1p (1–120) in 40 mM Tris-HCl (pH 7.0), 0.5 mM MgCl2, and 2 mM DTT, and in addition 80 nM unlabeled oligo to prevent reannealing of the Cy5 labeled oligo to the RNA oligo after unwinding. Unwinding was initiated by the addition of 1 mM ATP/MgCl2, and reactions were incubated on ice; alternatively, 1 mM ADP/MgCl2 was added as a control. Samples having a volume of 10 µL were removed at various time points and quenched with 5 µL of 3× loading buffer (5% Ficoll-type 400, 25% glycerol, 1.5X TBE, 20 mM EDTA, 0.5% SDS, 0.1% NP40 [Sigma-Aldrich 74385] and 160× excess of capture oligo) and analyzed on a native 10% polyacrylamide gel in 0.5X TBE (44.5 mM Tris-borate, 1 mM EDTA) containing 0.1% SDS. Binding reactions (10 µL) for elecromobility shift assay (EMSA) reactions were assembled identically to unwinding reactions except that the 1 mM ATP/MgCl2 was omitted. Reactions were incubated on ice for 10 min, after which 5 µL of loading buffer (50% glycerol, 0.2% Triton X-100, 20 mM EDTA) was added, and reactions were analyzed on an 8% polyacrylamide gel in 0.25X TBE.
In Figure 5G, RNA-1 and RNA-2 were annealed with the Cy5-labeled oligo to generate substrate-1 and substrate-2, respectively. In Figure 5H, RNA-3 to RNA-6 were annealed with the Cy5-labeled oligo to generate substrate-3 to substrate-6, respectively.
UV cross-linking
Splicing reactions were first reconstituted with s4U-substituted, 32P-radiolabeled U6 snRNA (see above) at 15 fmol/µL reaction. Reactions were assembled with 2 nM ACT1 pre-mRNA body-labeled with Cy5. Aliquots of each reaction were removed and analyzed on a splicing gel. The remainder of the reaction (∼30 µL) was then pipetted onto a parafilm-covered aluminum block on ice and exposed to UV light (365 nm) at a distance of ∼3 cm for 20 min in a darkened room under an aluminum foil tent at 4°C (Sontheimer 1994). Reactions were then subjected to immunoprecipitation under native conditions using anti-HA antibodies or to affinity pull-down under denaturing conditions using Ni-NTA beads. In the case of the denaturing pull-down, after the final wash step (see method above) samples were digested with RNase H (Thermo Scientific) targeting U6 nucleotides 28–54 and 48–93 after incubation with 2.5 mM EDTA at 65°C for 5 min to inactivate residual DNase I from the U6 depletion. All reactions were quenched with 6X SDS loading buffer and fractionated on an 8% tris-glycine SDS–polyacrylamide gel along with a protein ladder (BioRad, Precision Plus Dual Color Standard). A photograph of the dried protein gel was overlaid with the scanned phosphorimage of the gel to estimate the molecular weights of bands on the phosphorimage.
Primer extension analysis of UBC4 splicing reactions
Following slow-annealing to ∼500 fmol of 32P-radiolabeled primer oJPS239, RNA from splicing reactions (∼20 fmol pre-mRNA in 10.5 µL splicing reaction) was analyzed by primer extension (15 µL reaction volume total) using AMV reverse transcriptase (Promega) as previously described (Mayas et al. 2010). Products were analyzed on an 8% denaturing polyacrylamide sequencing gel.
Data analysis
Gels were dried, except where indicated, and exposed to storage PhosphorImager screens (Amersham Biosciences) for 24–48 h and scanned on a Typhoon PhosphorImager (Amersham Biosciences). For splicing gels of cross-linking reactions or gels of in vitro unwinding reactions and EMSA experiments, which used either Cy5-lableled splicing substrate or Cy5-lableled unwinding substrates, respectively, the gel was scanned wet within the two gel plates using the red laser on a Typhoon PhosphorImager. Bands were quantified using TotalLab Quant software (version 12.2, TotalLab) using an automated rolling ball background subtraction algorithm.
Supplementary Material
Acknowledgments
We thank B. Schwer for antibodies, C. Guthrie for strains, and D. Semlow for expressing and purifying recombinant Prp22p and Prp16p proteins. We also thank Yi Zeng for help with data analysis and members of the Staley lab for comments on the manuscript. This work was supported by a National Institutes of Health (NIH) National Research Service Award (NRSA) Individual Postdoctoral Fellowship (F32GM10192) awarded to R.T. and an NIH Research Project Grant (R01GM062264) awarded to J.P.S.
Author contributions: R.T. and J.P.S. designed the overall study; R.T. performed all experiments except for the in vivo experiments and in vitro unwinding and binding experiments, which were performed by K.H.N.; data analysis and interpretation was performed by R.T. and J.P.S., except for data analysis for in vivo and unwinding experiments, which was performed by K.H.N. and J.P.S. The manuscript was written, reviewed, and edited by R.T. and J.P.S., with contributions by K.H.N.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.328294.119.
References
- Achsel T, Brahms H, Kastner B, Bachi A, Wilm M, Lührmann R. 1999. A doughnut-shaped heteromer of human Sm-like proteins binds to the 3′-end of U6 snRNA, thereby facilitating U4/U6 duplex formation in vitro. EMBO J 18: 5789–5802. 10.1093/emboj/18.20.5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. 1999. G-patch: a new conserved domain in eukaryotic RNA-processing proteins and type D retroviral polyproteins. Trends Biochem Sci 24: 342–344. 10.1016/S0968-0004(99)01437-1 [DOI] [PubMed] [Google Scholar]
- Arenas JE, Abelson JN. 1997. Prp43: an RNA helicase-like factor involved in spliceosome disassembly. Proc Natl Acad Sci 94: 11798–11802. 10.1073/pnas.94.22.11798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai R, Yan C, Wan R, Lei J, Shi Y. 2017. Structure of the post-catalytic spliceosome from Saccharomyces cerevisiae. Cell 171: 1589–1598.E8. 10.1016/j.cell.2017.10.038 [DOI] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Liu W-T, Dybkov O, Will CL, Hartmuth K, Urlaub H, Kastner B, Stark H, Lührmann R. 2017. Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 542: 318–323. 10.1038/nature21079 [DOI] [PubMed] [Google Scholar]
- Bohnsack MT, Martin R, Granneman S, Ruprecht M, Schleiff E, Tollervey D. 2009. Prp43 bound at different sites on the pre-rRNA performs distinct functions in ribosome Synthesis. Mol Cell 36: 583–592. 10.1016/j.molcel.2009.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon KL, Auchynnikava T, Edwalds-Gilbert G, Barrass JD, Droop AP, Dez C, Beggs JD. 2006. Yeast ntr1/spp382 mediates prp43 function in postspliceosomes. Mol Cell Biol 26: 6016–6023. 10.1128/MCB.02347-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordonné R, Guthrie C. 1992. Human and human—yeast chimeric U6 snRNA genes identify structural elements required for expression in yeast. Nucleic Acids Res 20: 479–485. 10.1093/nar/20.3.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryksin AV, Matsumura I. 2010. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. BioTechniques 48: 463–465. 10.2144/000113418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SM, Guthrie C. 1993. A mechanism to enhance mRNA splicing fidelity: the RNA-dependent ATPase Prp16 governs usage of a discard pathway for aberrant lariat intermediates. Cell 73: 1377–1391. 10.1016/0092-8674(93)90363-U [DOI] [PubMed] [Google Scholar]
- Burke JE, Sashital DG, Zuo X, Wang YX, Butcher SE. 2012. Structure of the yeast U2/U6 snRNA complex. RNA 18: 673–683. 10.1261/rna.031138.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SP, Kao DI, Tsai WY, Cheng SC. 2003. The Prp19p-associated complex in spliceosome activation. Science 302: 279–282. 10.1126/science.1086602 [DOI] [PubMed] [Google Scholar]
- Chen H-C, Tseng C-K, Tsai R-T, Chung C-S, Cheng S-C. 2013. Link of NTR-mediated spliceosome disassembly with DEAH-box ATPases Prp2, Prp16, and Prp22. Mol Cell Biol 33: 514–525. 10.1128/MCB.01093-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Shulha HP, Ashar-Patel A, Yan J, Green KM, Query CC, Rhind N, Weng Z, Moore MJ. 2014a. Endogenous U2·U5·U6 snRNA complexes in S. pombe are intron lariat spliceosomes. RNA 20: 308–320. 10.1261/rna.040980.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-L, Capeyrou R, Humbert O, Mouffok S, Kadri YA, Lebaron S, Henras AK, Henry Y. 2014b. The telomerase inhibitor Gno1p/PINX1 activates the helicase Prp43p during ribosome biogenesis. Nucleic Acids Res 42: 7330–7345. 10.1093/nar/gku357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs DJ, Nagel RJ, Ares M, Stevens SW. 2006. Prp43p is a DEAH-box spliceosome disassembly factor essential for ribosome biogenesis. Mol Cell Biol 26: 523–534. 10.1128/MCB.26.2.523-534.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Company M, Arenas J, Abelson J. 1991. Requirement of the RNA helicase-like protein PRP22 for release of messenger RNA from spliceosomes. Nature 349: 487–493. 10.1038/349487a0 [DOI] [PubMed] [Google Scholar]
- Cordin O, Beggs JD. 2013. RNA helicases in splicing. RNA Biol 10: 83–95. 10.4161/rna.22547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didychuk AL, Montemayor EJ, Carrocci TJ, DeLaitsch AT, Lucarelli SE, Westler WM, Brow DA, Hoskins AA, Butcher SE. 2017. Usb1 controls U6 snRNP assembly through evolutionarily divergent cyclic phosphodiesterase activities. Nat Commun 8: 497 10.1038/s41467-017-00484-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwalds-Gilbert G, Kim DH, Kim SH, Tseng YH, Yu Y, Lin RJ. 2000. Dominant negative mutants of the yeast splicing factor Prp2 map to a putative cleft region in the helicase domain of DExD/H-box proteins. RNA 6: 1106–1119. 10.1017/S1355838200992483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, McPheeters DS, Abelson J. 1989. In vitro assembly of yeast U6 snRNP: a functional assay. Genes Dev 3: 2137–2150. 10.1101/gad.3.12b.2137 [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Urlaub H, Lührmann R. 2009. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell 36: 593–608. 10.1016/j.molcel.2009.09.040 [DOI] [PubMed] [Google Scholar]
- Fairman-Williams ME, Guenther U-P, Jankowsky E. 2010. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol 20: 313–324. 10.1016/j.sbi.2010.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica SM, Nagai K. 2017. Cryo-electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat Struct Mol Biol 24: 791–799. 10.1038/nsmb.3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica SM, Tuttle N, Novak T, Li N-S, Lu J, Koodathingal P, Dai Q, Staley JP, Piccirilli JA. 2013. RNA catalyses nuclear pre-mRNA splicing. Nature 503: 229–234. 10.1038/nature12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica SM, Mefford MA, Piccirilli JA, Staley JP. 2014. Evidence for a group II intron-like catalytic triplex in the spliceosome. Nat Struct Mol Biol 21: 464–471. 10.1038/nsmb.2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica SM, Oubridge C, Galej WP, Wilkinson ME, Bai X-C, Newman AJ, Nagai K. 2017. Structure of a spliceosome remodelled for exon ligation. Nature 542: 377–380. 10.1038/nature21078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field DJ, Friesen JD. 1996. Functionally redundant interactions between U2 and U6 spliceosomal snRNAs. Genes Dev 10: 489–501. 10.1101/gad.10.4.489 [DOI] [PubMed] [Google Scholar]
- Fourmann JB, Schmitzova J, Christian H, Urlaub H, Ficner R, Boon KL, Fabrizio P, Lührmann R. 2013. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev 27: 413–428. 10.1101/gad.207779.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourmann J-B, Dybkov O, Agafonov DE, Tauchert MJ, Urlaub H, Ficner R, Fabrizio P, Lührmann R. 2016. The target of the DEAH-box NTP triphosphatase Prp43 in Saccharomyces cerevisiae spliceosomes is the U2 snRNP-intron interaction. Elife 5: e15564 10.7554/eLife.15564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourmann J-B, Tauchert MJ, Ficner R, Fabrizio P, Lührmann R. 2017. Regulation of Prp43-mediated disassembly of spliceosomes by its cofactors Ntr1 and Ntr2. Nucleic Acids Res 45: 4068–4080. 10.1093/nar/gkw1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend K, Lovejoy AF, Steitz JA. 2007. U2 snRNP binds intronless histone pre-mRNAs to facilitate U7-snRNP-dependent 3′ end formation. Mol Cell 28: 240–252. 10.1016/j.molcel.2007.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X-D, Ares M. 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15: 689–701. 10.1038/nrg3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galej WP, Wilkinson ME, Fica SM, Oubridge C, Newman AJ, Nagai K. 2016. Cryo-EM structure of the spliceosome immediately after branching. Nature 537: 197–201. 10.1038/nature19316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman B, Tijerina P, Russell R. 2017. Distinct RNA-unwinding mechanisms of DEAD-box and DEAH-box RNA helicase proteins in remodeling structured RNAs and RNPs. Biochem Soc Trans 45: 1313–1321. 10.1042/BST20170095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman S, Kudla G, Petfalski E, Tollervey D. 2009. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc Natl Acad Sci 106: 9613–9618. 10.1073/pnas.0901997106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang J, Wan R, Yan C, Shi Y. 2015. Structural basis of pre-mRNA splicing. Science 349: 1191–1198. 10.1126/science.aac8159 [DOI] [PubMed] [Google Scholar]
- He Y, Staley JP, Andersen GR, Nielsen KH. 2017. Structure of the DEAH/RHA ATPase Prp43p bound to RNA implicates a pair of hairpins and motif Va in translocation along RNA. RNA 23: 1110–1124. 10.1261/rna.060954.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heininger AU, Hackert P, Andreou AZ, Boon K-L, Memet I, Prior M, Clancy A, Schmidt B, Urlaub H, Schleiff E, et al. 2016. Protein cofactor competition regulates the action of a multifunctional RNA helicase in different pathways. RNA Biol 13: 320–330. 10.1080/15476286.2016.1142038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliker AK, Staley JP. 2004. Multiple functions for the invariant AGC triad of U6 snRNA. RNA 10: 921–928. 10.1261/rna.7310704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James S-A, Turner W, Schwer B. 2002. How Slu7 and Prp18 cooperate in the second step of yeast pre-mRNA splicing. RNA 8: 1068–1077. 10.1017/S1355838202022033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandrositz A, Guthrie C. 1995. Evidence for a Prp24 binding site in U6 snRNA and in a putative intermediate in the annealing of U6 and U4 snRNAs. EMBO J 14: 820–832. 10.1002/j.1460-2075.1995.tb07060.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R, Hartnett S, Voelker RB, Berglund JA, Staley JP, Baumann P. 2013. Intronic sequence elements impede exon ligation and trigger a discard pathway that yields functional telomerase RNA in fission yeast. Genes Dev 27: 627–638. 10.1101/gad.212738.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R, Helston RM, Dannebaum RO, Baumann P. 2015. Diverse mechanisms for spliceosome-mediated 3′ end processing of telomerase RNA. Nat Commun 6: 6104 10.1038/ncomms7104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov FV, Uhlenbeck OC. 2004. Interaction of Escherichia coli DbpA with 23S rRNA in different functional states of the enzyme. Nucleic Acids Res 32: 3028–3032. 10.1093/nar/gkh640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid MF, Damha MJ, Shuman S, Schwer B. 2005. Structure-function analysis of yeast RNA debranching enzyme (Dbr1), a manganese-dependent phosphodiesterase. Nucleic Acids Res 33: 6349–6360. 10.1093/nar/gki934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koodathingal P, Novak T, Piccirilli JA, Staley JP. 2010. The DEAH box ATPases Prp16 and Prp43 cooperate to proofread 5′ splice site cleavage during pre-mRNA splicing. Mol Cell 39: 385–395. 10.1016/j.molcel.2010.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapham J, Yu YT, Shu MD, Steitz JA, Crothers DM. 1997. The position of site-directed cleavage of RNA using RNase H and 2′-O-methyl oligonucleotides is dependent on the enzyme source. RNA 3: 950–951. [PMC free article] [PubMed] [Google Scholar]
- Lebaron S, Froment C, Fromont-Racine M, Rain J-C, Monsarrat B, Caizergues-Ferrer M, Henry Y. 2005. The splicing ATPase prp43p is a component of multiple preribosomal particles. Mol Cell Biol 25: 9269–9282. 10.1128/MCB.25.21.9269-9282.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebaron S, Papin C, Capeyrou R, Chen Y-L, Froment C, Monsarrat B, Caizergues-Ferrer M, Grigoriev M, Henry Y. 2009. The ATPase and helicase activities of Prp43p are stimulated by the G-patch protein Pfa1p during yeast ribosome biogenesis. EMBO J 28: 3808–3819. 10.1038/emboj.2009.335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeds NB, Small EC, Hiley SL, Hughes TR, Staley JP. 2006. The splicing factor Prp43p, a DEAH box ATPase, functions in ribosome biogenesis. Mol Cell Biol 26: 513–522. 10.1128/MCB.26.2.513-522.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht K, Medenbach J, Lührmann R, Kambach C, Bindereif A. 2008. 3′-cyclic phosphorylation of U6 snRNA leads to recruitment of recycling factor p110 through LSm proteins. RNA 14: 1532–1538. 10.1261/rna.1129608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Li X, Zhang L, Jiang J, Hill RC, Cui Y, Hansen KC, Zhou ZH, Zhao R. 2017. Structure of the yeast spliceosomal postcatalytic P complex. Science 358: 1278–1283. 10.1126/science.aar3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C. 1992. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell 71: 803–817. 10.1016/0092-8674(92)90556-R [DOI] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C. 1994a. Dynamic RNA-RNA interactions in the spliceosome. Annu Rev Genet 28: 1–26. 10.1146/annurev.ge.28.120194.000245 [DOI] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C. 1994b. Randomization-selection analysis of snRNAs in vivo: evidence for a tertiary interaction in the spliceosome. Genes Dev 8: 1071–1086. 10.1101/gad.8.9.1071 [DOI] [PubMed] [Google Scholar]
- Martin A, Schneider S, Schwer B. 2002. Prp43 is an essential RNA-dependent ATPase required for release of lariat-intron from the spliceosome. J Biol Chem 277: 17743–17750. 10.1074/jbc.M200762200 [DOI] [PubMed] [Google Scholar]
- Mayas RM, Maita H, Staley JP. 2006. Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat Struct Mol Biol 13: 482–490. 10.1038/nsmb1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayas RM, Maita H, Semlow DR, Staley JP. 2010. Spliceosome discards intermediates via the DEAH box ATPase Prp43p. Proc Natl Acad Sci 107: 10020–10025. 10.1073/pnas.0906022107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montemayor EJ, Curran EC, Liao HH, Andrews KL, Treba CN, Butcher SE, Brow DA. 2014. Core structure of the U6 small nuclear ribonucleoprotein at 1.7-Å resolution. Nat Struct Mol Biol 21: 544–551. 10.1038/nsmb.2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montemayor EJ, Didychuk AL, Yake AD, Sidhu GK, Brow DA, Butcher SE. 2018. Architecture of the U6 snRNP reveals specific recognition of 3′-end processed U6 snRNA. Nat Commun 9: 1–11. 10.1038/s41467-018-04145-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgur S, Buchwald G, Falk S, Chakrabarti S, Prabu JR, Conti E. 2015. The conformational plasticity of eukaryotic RNA-dependent ATPases. FEBS J 282: 850–863. 10.1111/febs.13198 [DOI] [PubMed] [Google Scholar]
- Pandit S, Lynn B, Rymond BC. 2006. Inhibition of a spliceosome turnover pathway suppresses splicing defects. Proc Natl Acad Sci 103: 13700–13705. 10.1073/pnas.0603188103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannone BK, Kim SD, Noe DA, Wolin SL. 2001. Multiple functional interactions between components of the Lsm2-Lsm8 complex, U6 snRNA, and the yeast La protein. Genetics 158: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirilli JA, Staley JP. 2016. Reverse transcriptases lend a hand in splicing catalysis. Nat Struct Mol Biol 23: 507–509. 10.1038/nsmb.3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabu JR, Müller M, Thomae AW, Schüssler S, Bonneau F, Becker PB, Conti E. 2015. Structure of the RNA helicase MLE reveals the molecular mechanisms for uridine specificity and RNA-ATP coupling. Mol Cell 60: 487–499. 10.1016/j.molcel.2015.10.011 [DOI] [PubMed] [Google Scholar]
- Pyle AM. 2008. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys 37: 317–336. 10.1146/annurev.biophys.37.032807.125908 [DOI] [PubMed] [Google Scholar]
- Qi X, Rand DP, Podlevsky JD, Li Y, Mosig A, Stadler PF, Chen JJ-L. 2015. Prevalent and distinct spliceosomal 3′-end processing mechanisms for fungal telomerase RNA. Nat Commun 6: 6105 10.1038/ncomms7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader SD, Guthrie C. 2002. A conserved Lsm-interaction motif in Prp24 required for efficient U4/U6 di-snRNP formation. RNA 8: 1378–1392. 10.1017/S1355838202020010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan PL, Guthrie C. 1998. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr Biol 8: 847–855. 10.1016/S0960-9822(07)00345-4 [DOI] [PubMed] [Google Scholar]
- Rauhut R, Fabrizio P, Dybkov O, Hartmuth K, Pena V, Chari A, Kumar V, Lee C-T, Urlaub H, Kastner B, et al. 2016. Molecular architecture of the Saccharomyces cerevisiae activated spliceosome. Science 353: 1399–1405. 10.1126/science.aag1906 [DOI] [PubMed] [Google Scholar]
- Robert-Paganin J, Réty S, Leulliot N. 2015. Regulation of DEAH/RHA helicases by G-patch proteins. Biomed Res Int 2015: 931857. 10.1155/2015/931857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DE, Stevens SW, Abelson J. 2002. The 5′ and 3′ domains of yeast U6 snRNA: Lsm proteins facilitate binding of Prp24 protein to the U6 telestem region. RNA 8: 1011–1033. 10.1017/S1355838202026092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberg MJ, Wu T, Ritchie DB, Fica S, Staley JP, Atta KA, LaPointe P, MacMillan AM. 2013. A conformational switch in PRP8 mediates metal ion coordination that promotes pre-mRNA exon ligation. Nat Struct Mol Biol 20: 728–734. 10.1038/nsmb.2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Hotz H-R, Schwer B. 2002. Characterization of dominant-negative mutants of the DEAH-box splicing factors Prp22 and Prp16. J Biol Chem 277: 15452–15458. 10.1074/jbc.M112473200 [DOI] [PubMed] [Google Scholar]
- Schwer B, Gross CH. 1998. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J 17: 2086–2094. 10.1093/emboj/17.7.2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti MM, Swanson MS. 2016. RNA mis-splicing in disease. Nat Rev Genet 17: 19–32. 10.1038/nrg.2015.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, Staley JP. 2012. Staying on message: ensuring fidelity in pre-mRNA splicing. Trends Biochem Sci 37: 263–273. 10.1016/j.tibs.2012.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, Blanco MR, Walter NG, Staley JP. 2016. Spliceosomal DEAH-Box ATPases remodel pre-mRNA to activate alternative splice sites. Cell 164: 985–998. 10.1016/j.cell.2016.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small EC, Leggett SR, Winans AA, Staley JP. 2006. The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol Cell 23: 389–399. 10.1016/j.molcel.2006.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer EJ. 1994. Site-specific RNA crosslinking with 4-thiouridine. Mol Biol Rep 20: 35–44. 10.1007/BF00999853 [DOI] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. 1998. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92: 315–326. 10.1016/S0092-8674(00)80925-3 [DOI] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. 1999. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol Cell 3: 55–64. 10.1016/S1097-2765(00)80174-4 [DOI] [PubMed] [Google Scholar]
- Stevens SW, Abelson J. 2002. Yeast pre-mRNA splicing: methods, mechanisms, and machinery. Methods Enzymol 351: 200–220. 10.1016/S0076-6879(02)51849-8 [DOI] [PubMed] [Google Scholar]
- Su Y-L, Chen H-C, Tsai R-T, Lin P-C, Cheng S-C. 2018. Cwc23 is a component of the NTR complex and functions to stabilize Ntr1 and facilitate disassembly of spliceosome intermediates. Nucleic Acids Res 46: 3764–3773. 10.1093/nar/gky052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JS, Manley JL. 1995. A novel U2–U6 snRNA structure is necessary for mammalian mRNA splicing. Genes Dev 9: 843–854. 10.1101/gad.9.7.843 [DOI] [PubMed] [Google Scholar]
- Tanaka N, Schwer B. 2006. Mutations in PRP43 that uncouple RNA-dependent NTPase activity and pre-mRNA splicing function. Biochemistry 45: 6510–6521. 10.1021/bi052656g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Aronova A, Schwer B. 2007. Ntr1 activates the Prp43 helicase to trigger release of lariat-intron from the spliceosome. Genes Dev 21: 2312–2325. 10.1101/gad.1580507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn WY, Lee KR, Cheng SC. 1993. Yeast precursor mRNA processing protein PRP19 associates with the spliceosome concomitant with or just after dissociation of U4 small nuclear RNA. Proc Natl Acad Sci 90: 10821–10825. 10.1073/pnas.90.22.10821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai RT, Fu RH, Yeh FL, Tseng CK, Lin YC, Huang YH, Cheng SC. 2005. Spliceosome disassembly catalyzed by Prp43 and its associated components Ntr1 and Ntr2. Genes Dev 19: 2991–3003. 10.1101/gad.1377405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai RT, Tseng CK, Lee PJ, Chen HC, Fu RH, Chang KJ, Yeh FL, Cheng SC. 2007. Dynamic interactions of Ntr1-Ntr2 with Prp43 and with U5 govern the recruitment of Prp43 to mediate spliceosome disassembly. Mol Cell Biol 27: 8027–8037. 10.1128/MCB.01213-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umen JG, Guthrie C. 1995. A novel role for a U5 snRNP protein in 3′ splice site selection. Genes Dev 9: 855–868. 10.1101/gad.9.7.855 [DOI] [PubMed] [Google Scholar]
- Vidal VP, Verdone L, Mayes AE, Beggs JD. 1999. Characterization of U6 snRNA–protein interactions. RNA 5: 1470–1481. 10.1017/S1355838299991355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JD, Jankowsky E, Company M, Pyle AM, Abelson JN. 1998. The DEAH-box protein PRP22 is an ATPase that mediates ATP-dependent mRNA release from the spliceosome and unwinds RNA duplexes. EMBO J 17: 2926–2937. 10.1093/emboj/17.10.2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan R, Yan C, Bai R, Huang G, Shi Y. 2016. Structure of a yeast catalytic step I spliceosome at 3.4 Å resolution. Science 353: 895–904. 10.1126/science.aag2235 [DOI] [PubMed] [Google Scholar]
- Wan R, Yan C, Bai R, Lei J, Shi Y. 2017. Structure of an intron lariat spliceosome from Saccharomyces cerevisiae. Cell 171: 120–132.E12. 10.1016/j.cell.2017.08.029 [DOI] [PubMed] [Google Scholar]
- Wan R, Bai R, Yan C, Lei J, Shi Y. 2019. Structures of the catalytically activated yeast spliceosome reveal the mechanism of branching. Cell 177: 339–351.E13. 10.1016/j.cell.2019.02.006 [DOI] [PubMed] [Google Scholar]
- Wilkinson ME, Fica SM, Galej WP, Norman CM, Newman AJ, Nagai K. 2017. Postcatalytic spliceosome structure reveals mechanism of 3′–splice site selection. Science 358: 1283–1288. 10.1126/science.aar3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Lührmann R. 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, Lührmann R. 2002. Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J 21: 4978–4988. 10.1093/emboj/cdf480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff T, Bindereif A. 1995. Mutational analysis of human U6 RNA: stabilizing the intramolecular helix blocks the spliceosomal assembly pathway. Biochim Biophys Acta 1263: 39–44. 10.1016/0167-4781(95)00085-U [DOI] [PubMed] [Google Scholar]
- Wolin SL, Cedervall T. 2002. The La protein. Annu Rev Biochem 71: 375–403. 10.1146/annurev.biochem.71.090501.150003 [DOI] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, Shi Y. 2016. Structure of a yeast activated spliceosome at 3.5 Å resolution. Science 353: 904–911. 10.1126/science.aag0291 [DOI] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, Shi Y. 2017. Structure of a yeast step II catalytically activated spliceosome. Science 355: 149–155. 10.1126/science.aak9979 [DOI] [PubMed] [Google Scholar]
- Yan C, Wan R, Shi Y. 2019. Molecular mechanisms of pre-mRNA splicing through structural biology of the spliceosome. Cold Spring Harb Perspect Biol 11: a032409 10.1101/cshperspect.a032409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhan X, Yan C, Zhang W, Liu D, Lei J, Shi Y. 2019. Structures of the human spliceosomes before and after release of the ligated exon. Cell Res 29: 274–285. 10.1038/s41422-019-0143-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Hang J, Zhou Y, Wan R, Lu G, Yin P, Yan C, Shi Y. 2014. Crystal structures of the Lsm complex bound to the 3′ end sequence of U6 small nuclear RNA. Nature 506: 116–120. 10.1038/nature12803 [DOI] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.