

Abstract
We developed a strategy to transform olefins into homoallylic nitriles by a mechanism that combines copper catalysis with alkyl nitrile radicals. The radicals are easily generated from alkyl nitriles in the presence of a mild oxidant, di-terf-butyl peroxide. This cross-dehydrogenative coupling between simple olefins and alkylnitriles bears advantages to the conventional use of halides and toxic cyanide reagents. With this method, we showcase the facile synthesis of a flavoring agent, natural product, and polymer precursor from simple olefins.
Keywords: copper, alkenes, alkylnitriles, cross-dehydrogenative coupling, radicals
Graphical Abstract
Functionalizing alkenes for pennies: A copper catalyzed cross-dehydrogenative coupling of unactivated olefins with alkylnitriles was developed by dual sp3 C–H bond cleavage.
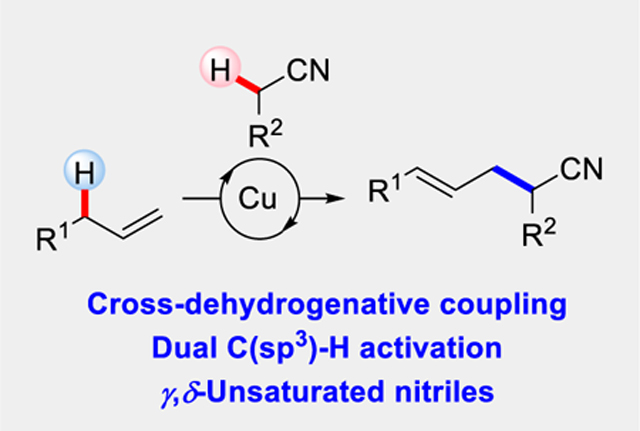
While radicals play a key role in biochemistry,[1] their potential for use in organic synthesis remains vast, with new concepts emerging,[2] including applications in cross-coupling.[3] By combining Cu-catalysis with radicals, Heck-type transformations have been achieved, including allylic trifluoromethylation,[4] arylation,[5] and alkylation.[6] These radical transformations enable bond constructions previously impossible and provide an attractive approach for olefin synthesis (Figure 1). Inspired by versatility of nitriles,[7] we designed a strategy for transforming simple olefins into γ,δ-unsaturated nitriles by taming the reactivity of a cyanoalkyl radical. Rather than requiring functionalized halides and toxic cyanide reagents, this transformation enables olefin feedstocks to be coupled with alkyl nitriles to generate homoallylic nitriles in a single-step, using an earth-abundant metal-catalyst (Figure 1).[8]
Figure 1.

Allylic cyanoalkylation using alkylnitrile.
The nitrile functional group is common in both materials[9] and medicines,[10] while being a useful handle for elaboration.[7] As shown in Figure 1, we proposed a cross-dehydrogenative coupling (CDC)[11] between an olefin and acetonitrile.[12] Initial oxidation of an alkylnitrile forms the corresponding cyanoalkyl radical, which can add to an olefin to give alkyl radical A.[13–16] Radicals such as A have been implicated in olefin hydrocyanoalkylations[13] and bifunctionalizations, such as oxycyanoalkylation[14] and cyanoalkylation/arylation.[15,16] In the presence of a copper(II) catalyst, Koichi first showed that radicals can be trapped to generate an alkylcopper(III) intermediate B.[17] Koichi’s kinetic studies suggest that alkyl radicals can be trapped by copper with rate constants in excess of 106 M−1s−1.[17] Theoretical studies on the CF3 allylic functionalization invoke a triflate counterion-assisted elimination.[4b] On the basis of these studies, we reasoned that the appropriate counterion would be critical for controlling regio-and stereochemistry in the final elimination.
With this mechanistic hypothesis in mind, we focused on the copper-catalyzed allylic cyanoalkylation of 1-dodecene in acetonitrile, using di-terf-butyl peroxide (DTBP) as the oxidant.
DTBP is a convenient and inexpensive radical initiator in synthetic and polymer chemistry, commonly used for generating radicals from acetonitrile.[13–16] [It might make more sense to move Zhu’s work here because you are saying that he also used copper and peroxides. Zhu demonstrated that Cu/peroxide can generate cyanoalkyl radicals from alkylnitriles, which can then add to alkenes through an intermolecular process.[14a,b,d,e,16] In Zhu’s work, the generated alkyl radical is typically trapped to afford hydrocyanoalkylations and bifunctionalizations, such as xxxx.. But we imagine diverting intermediate A to achieve dehydrogenative olefin-functionalization.
In the absence of copper, treatment of 1-dodecene with DTBP afforded the known hydrocyanoalkylation product 4a in 25% yield, with no desired cyanoalkene 3a. Copper(I) and copper(II) complexes bearing weak counterions provided 4a as the major product (28–70% yields) (Table 1), in accordance with reported studies on hydrocyanoalkylation.[13] Catalysts used by Zhu were not effective in our proposed allylic cyanoalkylation.[14a,b,d,e,16] In contrast, the (thiophene-2-carbonyloxy)copper(I) (CuTc) (previously used as catalyst in allylic trifluoromethylation[4b]) provided the cyanoalkene 3a as the major product in 30% yield. In comparison to copper(I) acetate, we found that copper(II) acetate showed higher efficiency and chemoselectivity by providing 3a in 47% yield, >20:1 regioselectivity. By replacing acetate with more basic pivalate, the desired alkene was obtained in 65% yield, >20:1 regioselectivity. Other oxidants such as terf-butyl hydroperoxide (TBHP) and dicumyl peroxide (DCP) were ineffective. Using an electron-rich benzonitrile derivative as an additive further improved efficiency, presumably by improving catalyst solubility. In the presence of one equivalent of veratronitrile, 3a was obtained in 90% yield, >20:1 rr and 4:1 E/Z. Only trace amount of 4a was observed (<5% yield). These results support the notion that a carboxylate counterion facilitates the elimination and enables >20:1 regioselectivity to provide the γ,δ-unsaturated nitrile. A syn-elimination affords the E-isomer as the major product.[18]
Table 1:
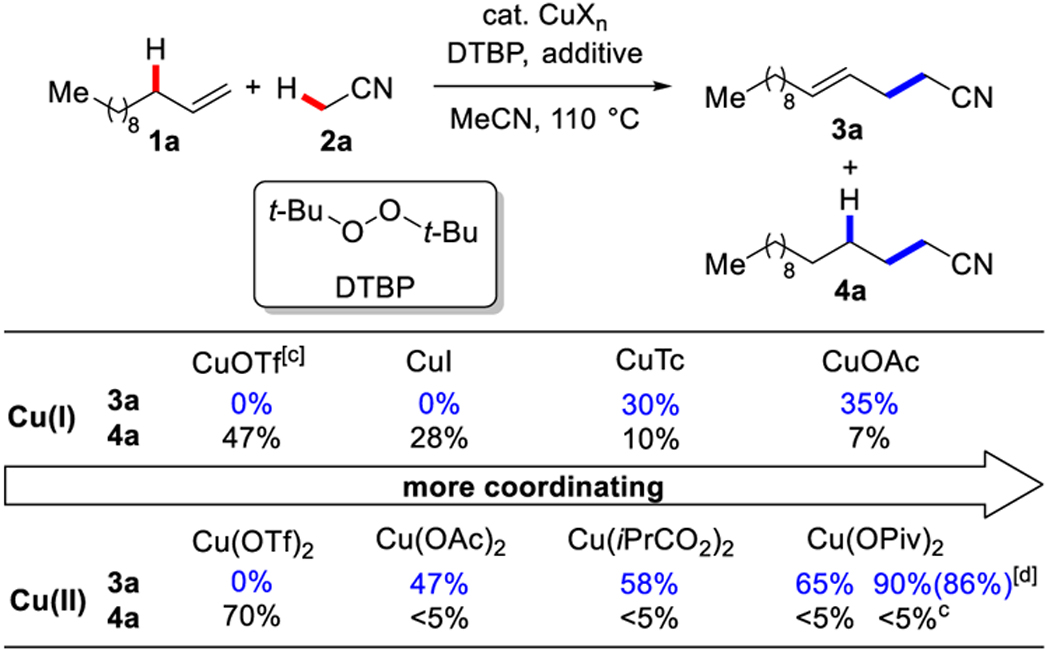 |
Reaction conditions: 1a (0.20 mmol), CuXn (20 mol%) and DTBP (0.80 mmol) in acetonitrile 2a (1.5 mL), 110 °C, 6 h.
Yields were determined by NMR analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. Isolated yield is shown in parentheses.
(CuOTf)2 PhMe (10 mol%).
With veratronitrile (0.20 mmol). A 4:1 E/Z ratio was determined by NMR analysis of the unpurified reaction mixture.
With this protocol, we elaborated a wide-range of terminal olefins (Scheme 1). Unactivated linear terminal olefins gave the corresponding γ,δ-unsaturated nitriles (3a-c) in 80–86% yields with >20:1 rr and 4:1 E/Z ratio. For the substrates bearing ester (3d, 3e), amide (3f), cyano (3g) and ether (3h) groups, regioselective CDC reactions with acetonitrile provided the corresponding products in 75–82% yields. Increasing the steric hindrance at the 4-position of the olefins slightly decreased the yields but increased the E/Z ratios of the products (3i 7:1 E/Z; 3j 11:1 E/Z; 3k >20:1 E/Z). With a tert-butyl group at the 3-position, we observed >20:1 regioselectivity and >20:1 E/Z selectivity (3k). The regioselectivity is unaffected by the increased steric hindrance at the 4-position of the olefins. 3-Aryl substituted substrates gave the corresponding nitriles (3l-n) in 40–46% yields with >20:1 E/Z selectivity. A substrate with an electron-withdrawing group on the phenyl ring (3n) showed slightly higher reactivity than the one with an electron-donating group (3m). Trisubstituted-alkenyl nitriles were synthesized in 50–77% yields from 3,3- or 1,1-disubstituted olefins (3o-r and 3t). A series of nitriles were also tested as coupling partners and solvent. Propionitrile and butyronitrile show decreased reactivity compared to acetonitrile, most likely due to steric effects and a lower solubility of the copper catalyst in these nitriles (3v, 3w). Only trace amount of hydrocyanoalkylation product 4 were observed with the olefins shown in Scheme 1. With facile access to various nitriles, we next focused on applying them as building blocks.
Scheme 1.

Allylic cyanoalkylation of terminal olefins. Reaction conditions: 1 (0.20 mmol), Cu(OPiv)2 (20 mol%), DTBP (0.80 mmol) and veratronitrile (0.20 mmol) in alkylnitrile 2 (1.5 mL), 110 °C, 6 h. E/Z ratios determined by NMR analysis of the unpurified reaction mixture are shown in parentheses. [a] 3t1 and 3t2 were isolated as a mixture. [b] 24 h.
Due to the versatility of the nitrile group, we can now use simple olefins to access a range of motifs, including an industrial flavor agent, a natural product, and a polymer precursor (Scheme 2). For example, treatment of 3b with TMSCl in ethanol provided the pear-flavored agent, ethyl 4-decenoate 7, in 85% yield.[19] The 4-alkyl γ-lactones are members of a large family of natural flavors, widely used in food industry.[20] From the same compound 3b, γ-decalactone 8 was obtained in 73% yield by a one-pot, hydrolysis and intramolecular hydroacyloxylation. Our strategy provides an efficient route to fatty acids. For example, lyngbic acid, isolated from the marine cyanophyte Lyngbya majuscule,[21] exhibits antimicrobial activity.[22] By hydrolysis of the cyano group in compound 3h, lyngbic acid 9 can be obtained in 87% yield. Ru-catalyzed hydrogenation of 3d provided the nylon 9 precursor 10 in 75% yield.[23]
Scheme 2.

Applications of the γ,δ-unsaturated nitriles.
Next, we examined internal olefins (Scheme 3). With (E)-5-decene, the reaction gave cyanoalkene 3x in 62% yield with with >20:1 rr and 12:1 E/Z ratio after 24 hours (Scheme 3a). With (Z)-5-decene, (E)-isomer 3x was obtained as the major product in a similar yield and E/Z selectivity as the (E)-olefin substrate (60% yield, 11:1 E/Z) (Scheme 3b). The C–C bonds were formed at the 5-position of the substrates. No 3-propylnon-4-enenitrile 5 was observed from potential allylic radical F or π-allylcopper intermediate G by allylic C–H bond activation (Scheme 3c). We saw no carbocation rearrangement type products 6, which would arise from carbocation intermediate H (Scheme 3d).[24] Nor were these 1,2-hydride shift products detected in experiments yielding compounds 3o-q shown in Scheme 1. These observations suggest that an allylic radical or carbocation are most likely not key intermediates in our cross-coupling.
Scheme 3.

Allylic cyanoalkylation of internal olefins.
To gain more insight into the mechanism, we performed a radical trapping and radical clock experiments (Scheme 4). The allylic cyanoalkylation reactions were suppressed in the presence of radical inhibitors TEMPO or BHT (Scheme 4a). In addition, compound 12 was obtained in 60% yield from (1-cyclopropylvinyl)benzene 11 via sequential ring-opening of cyclopropylmethyl radical intermediate and cyclization (Scheme 4b).[13b] These observations suggest that a radical pathway is involved in this cross-coupling (Scheme 5).
Scheme 4.

Intermediate trapping and radical clock experiments.
Scheme 5.

Proposed rationale for regioselectivity.
Addition of the cyanoalkyl radical to the olefin generates radical intermediate A. To explain the regioselectivity, we propose that π-bonding of cyano group to copper(III)[25] shields the H at the β-position to direct the pivalate to abstract the H at the δ-position.
In summary, we have developed a copper catalyzed cross-dehydrogenative coupling of unactivated olefins with alkylnitriles by dual sp3 C–H bond cleavage. High chemo- and regioselectivity for an E2-type elimination was achieved by (1) the pivalate counterion and (2) the directing effect of cyano group. By using a catalyst derived from earth abundant salts, we can access 4-alkenylnitriles from simple olefins. Both terminal and internal olefins can be transformed into γ,δ-unsaturated nitrile, versatile synthetic building blocks. These studies contribute to the emerging use of radicals for catalytic cross-coupling.
Supplementary Material
Acknowledgements
Funding was provided by the National Science Foundation (CHE-1465263) and UC Irvine. We are grateful to Eli Lilly for a Grantee Award.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) Halliwell B, Free Radical in Biochemistry and Medicine, in Molecular Biology and Biotechnology, (Eds.: Meyers RA), VCH, New York, 1995, 327; [Google Scholar]; b) Hermes-Lima M, Oxygen in Biology and Biochemistry: Role of Free Radicals, in Functional Metabolism: Regulation and Adaptation, (Eds.:Storey KB), Wiley, Hoboken, 2004, 319. [Google Scholar]
- [2].a) For a recent review, see Yan M, Lo JC, Edwards JT, Baran PS, J. Am. Chem. Soc 2016, 138, 12692; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) For recent examples, see: Ghosh I, Ghosh T, Bardagi JI, Konig B, Science 2014, 346, 725; [DOI] [PubMed] [Google Scholar]; c) Lo JC, Gui J, Yabe Y, Pan C-M, Baran PS, Nature 2014, 516, 343; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jeffrey JL, Terrett JA, MacMillan DWC, Science 2015, 349, 1532; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gui J, Pan C-M, Jin Y, Qin T, Lo JC, Lee BJ, Spergel SH, Mertzman MF, Pitts WJ, La Cruz TE, Schmidt MA, Darvatkar N, Natarajan SR, Baran PS, Science 2015, 348, 886; [DOI] [PubMed] [Google Scholar]; f) Müller DS, Untiedt NL, Dieskau AP, Lackner GL, Overman LE, J. Am. Chem. Soc 2015, 137, 660; [DOI] [PubMed] [Google Scholar]; g) Nawrat CC, Jamison CR, Slutskyy Y, MacMillan DWC, Overman LE, J. Am. Chem. Soc 2015, 137 11270; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR, Nature 2016, 539, 268; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Murphy JJ, Bastida D, Paria S, Fagnoni M, Melchiorre P, Nature 2016, 532, 218; [DOI] [PubMed] [Google Scholar]; j) Brill ZG, Grover HK, Maimone TJ, Science 2016, 352, 1078; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Musacchio AJ, Lainhart BC, Zhang X, Naguib SG, Sherwood TC, Knowles RR, Science 201, 355, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) For recent examples, see: Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS, J. Am. Chem. Soc 2011, 133, 18566; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Biswas S, Weix DJ, J. Am. Chem. Soc 2013, 135, 16192; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tellis JC, Primer DN, Molander GA, Science 2014, 345, 433; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC, Science 2014, 345, 437; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Johnston CP, Smith RT, Allmendinger S, MacMillan DWC, Nature 2016, 536, 322; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang W. Zhang, McCann SD, Wang D, Chen P, Stahl SS, Liu G, Science 2016, 353, 1014; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC, Science 2016, 352, 1304; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Shields BJ, Doyle AG, J. Am. Chem. Soc 2016, 138, 12719; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Green SA, Matos JLM, Yagi A, Shenvi RA, J. Am. Chem. Soc 2016, 138, 12779. [DOI] [PubMed] [Google Scholar]
- [4].a) Parsons AT, Buchwald SL, Angew. Chem. Int. Ed 2011, 50, 9120; Angew. Chem. 2011, 123, 9286; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu J, Fu X, Luo D-F, Jiang Y-Y, Xiao B, Liu Z-J, Gong T-J, Liu L, J. Am. Chem. Soc 2011, 133, 15300; [DOI] [PubMed] [Google Scholar]; c) Wang X, Ye Y, Zhang S, Feng J, Xu Y, Zhang Y, Wang J, J. Am. Chem. Soc 2011, 133, 16410; [DOI] [PubMed] [Google Scholar]; d) Chu L, Qing F-L, Org. Lett 2012, 14, 2106; [DOI] [PubMed] [Google Scholar]; e) Beniazza R, Molton F, Duboc C, Tron A, McClenaghan ND, Lastécouères D, Vincent J-M, Chem. Commun 2015, 51, 9571. [DOI] [PubMed] [Google Scholar]
- [5].Phipps RJ, McMurray L, Ritter S, Duong HA, Gaunt MJ, J. Am. Chem. Soc 2012, 134, 10773. [DOI] [PubMed] [Google Scholar]
- [6].a) Zhu Y, Wei Y, Chem. Sci 2014, 5, 2379; [Google Scholar]; b) Liu D, Liu C, Li H, Lei A, Chem. Commun 2014, 50, 3623; For other Cu-catalyzed allylic alkylation, see: [DOI] [PubMed] [Google Scholar]; c) Bao H, Bayeh L, Tambar UK, Angew. Chem. Int. Ed 2014, 53, 1664; Angew. Chem. 2014, 126, 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Rappoport Z, The Chemistry of the Cyano Group, Wiley, London, 1970; [Google Scholar]; b) Fatiadi AJ, Supplement C: The Chemistry of Triple-Bonded Functional Groups, Part 2, in The Chemistry of Functional Groups, (Eds.: Pappoport Z, Patai S), Wiley, London, 1983, 1057; [Google Scholar]; c) Fleming FF, Wang Q, Chem. Rev 2003, 103, 2035; [DOI] [PubMed] [Google Scholar]; d) López R, Palomo C, Angew. Chem. Int. Ed 2015, 54, 13170. [DOI] [PubMed] [Google Scholar]
- [8].a) For recent examples, see: Guo X-X, Gu D-W, Wu Z, Zhang W, Chem. Rev 2015, 155, 1622; [DOI] [PubMed] [Google Scholar]; b) Miao J, Ge H, Eur. J. Org. Chem 2015. 7859. [Google Scholar]
- [9].Hu P, Chai J, Duan Y, Liu Z, Cui G, Chen L, J. Mater. Chem. A, 2016, 4, 10070. [Google Scholar]
- [10].a) Fleming F, Nat. Prod. Rep 1999, 16, 597; [Google Scholar]; b) Fleming FF, Yao L, Ravikumar PC, Funk L, Shook BC, J. Med. Chem 2010, 53, 7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) For selected reviews, see: Li C-J, Acc. Chem. Res 2009, 42, 335; [DOI] [PubMed] [Google Scholar]; b) Scheuermann CJ, Chem. Asian J 2010, 5, 436; [DOI] [PubMed] [Google Scholar]; c) Liu C, Zhang H, Shi W, Lei A, Chem. Rev 2011, 111, 1780; [DOI] [PubMed] [Google Scholar]; d) Yeung CS, Dong VM, Chem. Rev 2011, 111, 1215; [DOI] [PubMed] [Google Scholar]; e) Li B-J, Shi Z-J, Chem. Soc. Rev 2012, 41, 5588; [DOI] [PubMed] [Google Scholar]; f) Girard SA, Knauber T, Li C-J, Angew. Chem. Int. Ed 2014, 53, 74. [DOI] [PubMed] [Google Scholar]
- [12].a) For cross-dehydrogenative coupling of acetonitriles by dual sp3 C–H bond cleavage, see: Liu Y, Yang K, Ge H, Chem. Sci 2016, 7, 2804;28660057 [Google Scholar]; b) Zhang W, Yang S, Shen Z, Adv. Synth. Catal 2016, 358, 2392. [Google Scholar]
- [13].a) Bruno JW, Marks TJ, Lewis FD, J. Am. Chem. Soc 1981, 103, 3608; [Google Scholar]; b) Bruno JW, Marks TJ, Lewis FD, J. Am. Chem. Soc 1982, 104, 5580; [Google Scholar]; c) Sonawane HR, Bellur NS, Shah VG, J. Chem. Soc., Chem. Commun 1990, 1603; [Google Scholar]; d) Li Z, Xiao Y, Liu Z-Q, Chem. Commun 2015, 51, 9969. [DOI] [PubMed] [Google Scholar]
- [14].a) Bunescu A, Wang Q, Zhu J, Org. Lett 2015, 17, 1890; [DOI] [PubMed] [Google Scholar]; b) Chatalova-Sazepin C, Wang Q, Sammis GM, Zhu J, Angew. Chem. Int. Ed 2015, 54, 5443; Angew. Chem. 2015, 127, 5533; [DOI] [PubMed] [Google Scholar]; c) Chu X-Q, Xu X-P, Meng H, Ji S-J, RSC Adv 2015, 5, 67829; [Google Scholar]; d) Ha TM, Chatalova-Sazepin C, Wang Q, Zhu J, Angew. Chem. Int. Ed 2016, 55, 9249; Angew. Chem. 2016, 128, 9395; [DOI] [PubMed] [Google Scholar]; e) Ha TM, Wang Q, Zhu J, Chem. Commun 2016, 52, 11100. [DOI] [PubMed] [Google Scholar]
- [15].a) Li J, Wang Z, Wu N, Gao G, You J, Chem. Commun 2014, 50, 15049; [DOI] [PubMed] [Google Scholar]; b) Tang S, Zhou D, Li Z-H, Fu M-J, Li J, Sheng R-L, Li S-H, Synthesis 2015, 47, 1567. [Google Scholar]
- [16].Bunescu A, Wang Q, Zhu J, Angew. Chem. Int. Ed 2015, 54, 3132; Angew. Chem. 2015, 127, 3175. [DOI] [PubMed] [Google Scholar]
- [17].a) Kochi JK, J. Am. Chem. Soc 1962, 84, 3271; [Google Scholar]; b) Kochi JK, Bemis A, Jenkins CL, J. Am. Chem. Soc 1968, 90, 4616; [Google Scholar]; c) Jenkins CL, Kochi JK, J. Am. Chem. Soc 1972, 94, 843; [Google Scholar]; d) Jenkins CL, Kochi JK, J. Am. Chem. Soc 1972, 94, 856; [Google Scholar]; e) Kochi JK, Acc. Chem. Res 1974, 7, 351. [Google Scholar]
- [18].Carey FA, Sunburg RJ, Advanced Organic Chemistry: Reaction and Synthesis, 5th Ed, Part B, Springer, New York, 2010. [Google Scholar]
- [19].McDonald S, Schulze M, Peltz M, Bolliet D, Burroughs L, Perfumer & Flavorist 2011, 36, 24. [Google Scholar]
- [20].a) Schreier P, Drawert F, Kerényi Z, Junker AZ, Lebensm. Unters. Forsch 1976, 161, 249; [DOI] [PubMed] [Google Scholar]; b) Lübke M, Guichard E, Tromelin A, Quere JLL, J. Agric. Food. Chem 2002, 50, 7094. [DOI] [PubMed] [Google Scholar]
- [21].Cardellina JJ, Dalietos D, Marner F, Mynderse JS, Moore RE, Phytochemistry 1978, 17, 2091. [Google Scholar]
- [22].Gerwick WH, Reyes S, Alvarado B, Phytochemistry 1987. 26, 1701. [Google Scholar]
- [23].Abel GA, Nguyen KO, Viamajala S, Varanasi S, Yamamoto K, RSC Adv 2014, 4, 55622. [Google Scholar]
- [24].Whitmore FC, J. Am. Chem. Soc 1932, 54, 3274. [Google Scholar]
- [25].a) Bullock RM, Headford CEL, Kegley SE, Norton JR, J. Am. Chem. Soc 1985, 107, 727; [Google Scholar]; b) Michelin RA, Mozzon M, Bertani R, Coord. Chem. Rev 1996, 147, 299. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.