

Abstract
Background:
To evaluate the methylation levels of human telomerase reverse transcriptase (hTERT) promoter three CpG island (CGIs) regions and its prognostic impact in Chinese patients with acral and mucosal melanoma.
Methods:
Bioinformatics software was used to analyze hTERT gene promoter. Fresh frozen tissues were taken from 14 patients with melanoma (6 acral melanoma and 8 mucosal melanoma) and 14 pigmented nevus as control subjects (14 acral pigmented nevus). Bisulfite sequencing PCR (BSP) combined TA clone sequencing was used to assess the methylation levels of hTERT promoter CGIs regions. The relative expression level of hTERT mRNA was measured by quantitative real-time polymerase chain reaction (qRT-PCR).
Results:
CGIs-1 (-1392–-1098 bp), CGIs-2 (-945–-669 bp), and CGIs-3 (-445–-48 bp) were selected for our study. Our results indicated that the methylation levels of hTERT promotor CGIs regions in melanoma were greater than pigmented nevus (CGIs-1: 69.3 ± 18.7% vs 46.8 ± 20.4%, t = 3.048 P = .005; CGIs-2: 73.8 ± 14.7% vs 55.6 ± 16.0%, t = 3.120 P = .004; CGIs-3: 5.8 ± 2.2% vs 2.2 ± 1.3%, t = 5.164 P < .001). The relative expression level of hTERT in melanoma was greater than in pigmented nevus (50.39 ± 9.16 vs 26.10 ± 7.25, t = 7.778, P < .001). Linear regression analysis showed that the methylation level of CGIs-2 in melanoma was positively correlated with the relative expression level of hTERT mRNA (R2 = .490, F = 13.478, P = .003). Combined with the analysis of clinicopathological features, the methylation level of CGIs-2 in melanoma with lymph node metastasis was greater than in melanoma without lymph node metastasis, and the methylation level of CGIs-2 increased with TNM staging.
Conclusion:
CGIs-2 methylation level was associated with the relative expression level of hTERT mRNA, lymph node metastasis and TNM staging, suggesting that CGIs-2 hypermethylation might be used to evaluate the prognosis in Chinese patients with acral and mucosal melanoma.
Keywords: BSP, hTERT, melanoma, methylation, mRNA, promoter
1. Introduction
Melanoma is a malignant tumor derived from melanocytes. It is mostly found in the skin and mucosa, and it is the most deadly malignant tumor of the skin. The mechanism by which it occurs and develops is still unclear. In recent years, the incidence of melanoma has increased annually. The 5-year survival rate for melanoma diagnosed early is 90% after surgery,[1] but some melanomas are not diagnosed until after metastasis has occurred. Although many advances have been made in the treatment of metastatic melanoma, the prognosis of metastatic melanoma is still poor.[1] For this reason, early diagnosis and prognosis assessment have important effects on melanoma. Clinical and radiographic of melanoma has no obvious specificity. The gold standard for diagnosis and judgment malignant degree of melanoma is histopathological examination. However, due to the lack of clear molecular diagnostic factors, the accuracy of histopathological examination largely depends on the skill level and experience of pathologists, which makes it easy to misdiagnose or overdiagnosis clinically.[1–2] For this reason, it is necessary to find new biomarkers for diagnosis and prognosis evaluation.
The occurrence and development of melanoma are the result of the joint action of multiple genes and multiple steps. Currently, various studies have shown that the most important factor is the activation of relevant oncogenes and the decrease or disappearance of the expression of tumor suppressor genes, which fosters proliferation and the invasion ability of cancer cells. The telomerase catalytic subunit encoded by the human telomerase reverse transcriptase (hTERT) gene is the core enzyme that determines telomerase activity. It also plays a role in cell immortalization, tumorigenesis, and development. Early studies have shown that hTERT promoter region mutations is similar to BRAF V600E and NRAS mutations, which can lead to the activation of proto-oncogenes and play an important role in the malignant transformation of melanocytes. They are associated with the prognosis of melanoma.[3–4] Although the study reports that approximately 50% of melanomas have mutations in the hTERT gene promoter region, recent studies have shown that the mutations rate in hTERT gene promoter region in acral and mucosal melanoma were 9.3% and 7.5%, respectively.[5–6] In this way, hTERT gene promoter region mutations may not be associated with the prognosis of acral and mucosal melanoma. In addition to mutations, one recent study found that the methylation level of hTERT promoter region is a potential diagnostic marker to distinguish benign melanocytic neoplasms from melanoma. It is related to the prognosis of melanoma,[7] but no study have been conducted on the methylation levels of different CpG island (CGIs) regions in the hTERT promoter of melanoma. However, some researchers believe that the hTERT promoter region is partially hypermethylated, with a hypermethylated region around -600 bp upstream of the transcription start site, and the core promoter region around the transcription start site is a hypomethylated region, the hypomethylation of the core promoter region is critical for hTERT mRNA expression level.[8] The role of methylation of the hTERT promoter region in tumors in hTERT mRNA expression level is controversial. It is therefore necessary to study the relationship between the methylation levels of different CGIs regions in the hTERT promoter of melanoma and the mRNA relative expression level of hTERT and analyze the potential value of the methylation levels of CGIs in the hTERT promoter region as a biomarker in the prognosis evaluation of melanoma.
2. Materials and methods
2.1. hTERT promoter region CGIs selection
The original sequence of the hTERT promoter region (accession AF 098956) was found in GenBank and entered into Methyl Primer Express v1.0 software. Parameters were set according to the definition of CpG island, namely, CG content >50%, observed value/predicted value (CpG observed/expected) >60%, and design primer sequences.
2.2. Tissue sample
From 2014 to 2016, fresh frozen tissue samples were collected from 14 patients with melanoma (6 acral melanoma and 8 mucosal melanoma, 7 men and 7 women, with an average age of 61.7 ± 12.5 years) and 14 patients with pigmented nevus (14 acral pigmented nevus, 7 men and 7 women, with an mean age of 54.1 ± 15.3 years) at the Department of Dermatology, People's Hospital of Xinjiang Uygur Autonomous Region. All of the patients were diagnosed by two pathologists on the basis of HE staining and IHC. None of the patients underwent any radiotherapy, chemotherapy, or other treatment prior to surgery, and samples were stored at −80°C. The study was approved by the Ethics Committee of the People's Hospital of Xinjiang Uygur Autonomous Region and written consent was obtained from all patients before the operation.
2.3. Genomic DNA extraction and bisulfite sequencing PCR (BSP)
The extraction of DNA was carried out using a DNA extraction kit (TianGen, Beijing, China) in accordance with the manufacturer's instructions. Then, bisulfite conversion of DNA was manipulated with a DNA EpiTect Bisulfite Kit (Qiagen, 59104, Germany) according to the manufacturer's instructions and purified reaction products (The reaction system consisted of 20 μL sample DNA, 85 μL Bisulfite Mix, 15 μL DNA Protect Buffer, 20 μL ddH2O, total volume 140 μL; thermal cycle conditions: denaturation 95°C for 5 minutes, incubation at 60°Cfor 25 minutes, denaturation at 95°C for 5 minutes, incubation at 60°C for 85 minutes, denaturation at 95°C for 5 minutes, incubation at 60°C for 175 minutes, followed by holding at 20°C). The unmethylated cytosine (C) in the genome was converted into uracil (U), and the methylated cytosine remained unchanged. Detection of methylated DNA after purification, the reaction system consisted of 2 μL sample DNA, 5 μL of 10 x Ex HiFi Buffer, 5 μL of GC Enhancer, 5 μL of each primer, 4 μL of dNTP, 1 μL of HiFi-ase, 23 μL of ddH2O, and a total volume of 50 μL. PCR reaction conditions: 95°C for 5 minutes, 95°C for 35 seconds, 45°C for 35 seconds, 72°C for 45 seconds, 40 cycles; 72°C for 10 minutes, and 4°C for 5 minutes. PCR products were detected using 1% agarose gel electrophoresis.
2.4. TA cloning
Purification of PCR products, the reaction system consisted of 1 μL sample cDNA, 2.5 μL of 10 x Ex HiFi Buffer, 2.5 μL of GC Enhancer, 2.5 μL of each primer, 2 μL of dNTP, 0.5 μL of HiFi-ase, and 11.5 μL of ddH2O, to a total volume of 25 μL. PCR reaction conditions were as follows 40 cycles of 95°C for 5 minutes, 95°C for 35 seconds, 45°C for 35 seconds, 72°C for 45 seconds, 40 cycles; 72°C for 10 minutes, and 4°C for 5 minutes. PCR products were separated and detected by 1% agarose gel electrophoresis. Then, the PCR products were recovered by thin agarose gel DNA recovery kit (GENERAY Biotechnology, gk2043–50, China). Purified PCR then was linked into PMD 19-T vectors (TaKaRa, Dalian, China) to transform DH5a receptor cells, and 10 positive monoclonal flora were selected for sequencing (Wuhan Barfield Biotechnology Service Co., Ltd.). Sequencing results were analyzed using a quantification tool for methylation analysis (QUAM, quantification tool for methylation analysis).
2.5. Quantitative real-time PCR
Total RNA was extracted using a Qiagen RNA extraction kit (Qiagen, Germany), according to the manufacturer's instructions, and cDNA was reverse transcribed using total RNA as a template. PCR was carried out using cDNA as template. The reaction system consisted of 2 μL cDNA, 2.5 μL of 10× Buffer, 2 μL of dNTP, 1.5 μL of 25 mmol/L MgCl2, 0.2 μL of Taq enzyme, 15 μL of ddH2O, 1 μL of each primer, and a total volume of 25 μL. PCR reaction conditions: 95°C for 10 minutes, 95°C for 15 seconds, 62°C for 20 seconds, 40 cycles. Fluorescence signals were collected during each cycle to produce amplification curves and melting curves. The cycle threshold (Ct) of each sample was recorded. The expression level of hTERT mRNA relative internal reference was expressed as 2−ΔCt, where ΔCt = Ct (hTERT)–Ct (β-actin).
2.6. Statistical analysis
Statistical calculations were performed using SPSS 25.0 software (IBM, NY, USA), and all of the data were expressed as mean ± standard deviation. The differences between groups were analyzed by two independent samples t tests and one-way ANOVA, and the correlation analysis was performed using linear regression analysis. P < .05 was considered to be statistically significant.
3. Results
3.1. Prediction and screening of CGIs in hTERT promoter region
According to the software predictions, 3 CGIs regions located in the transcriptional regulatory region were selected: CGIs-1 (-1392–-1098 bp, the methylation in this region has rarely been reported), CGIs-2 (-945–-699 bp, located around 600 bp upstream of the transcription site) and CGIs-3 (-445–-48 bp, including the core promoter region), and CG site location were identified (Fig. 1). The primer sequences of BSP and qRT-PCR were shown in Table 1.
Figure 1.
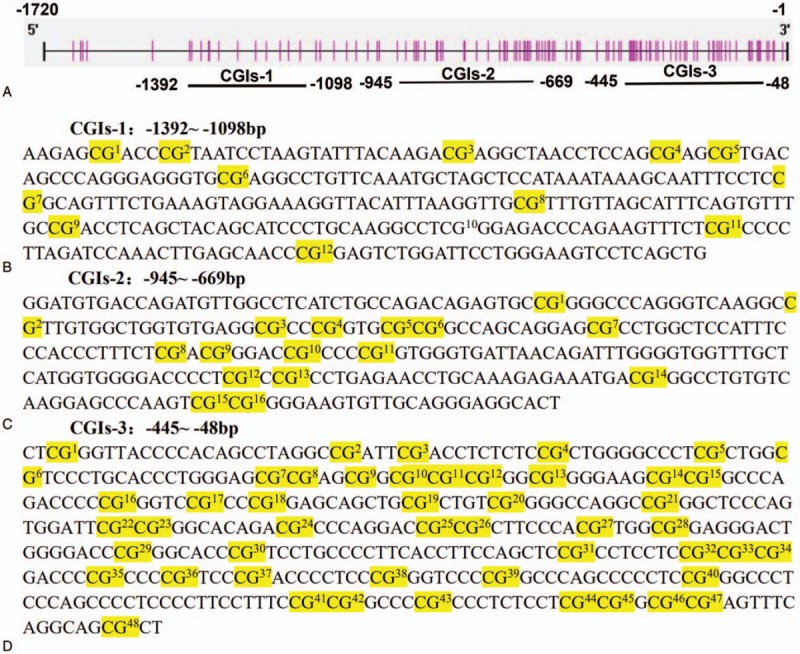
(A) The location of the hTERT promoter region and 3 CGIs regions; (B), (C), and (D) are the 3 CGIs regions and CG sites.
Table 1.
Primer sequences of 3 CGIs regions of BSP and qRT-PCR.

3.2. Analysis of BSP and TA clone sequencing results
The results of agarose gel electrophoresis of partial BSP products were shown in Fig. 2. The results of partial TA clone sequencing were shown in Fig. 3. Black circles represented methylated CG sites and white circles represented unmethylated CG sites. Changes in methylation were detected in 38 CG sites, including all of the CG sites of CGIs-2. Only the second CG site in CGIs-1 showed no change in methylation in all samples. Only 11 CG sites out of the CGIs-3 showed changes in methylation. Statistical analysis showed that the methylation levels of the 3 CGIs regions in the melanoma group were greater than those in the pigmented nevus group (CGIs-1: 69.3 ± 18.7% vs 46.8 ± 20.4%, t = 3.048 P = .005; CGIs-2: 73.8 ± 14.7% vs 55.6 ± 16.0%, t = 3.120 P = .004; CGIs-3: 5.8 ± 2.2% vs 2.2 ± 1.3%, t = 5.164 P < .001) (two independent samples t tests) (Fig. 4A).
Figure 2.
Results of 1% agarose gel electrophoresis of BSP products of melanoma, (A) CGIs-1, (B) CGIs-2, (C) CGIs-3.
Figure 3.
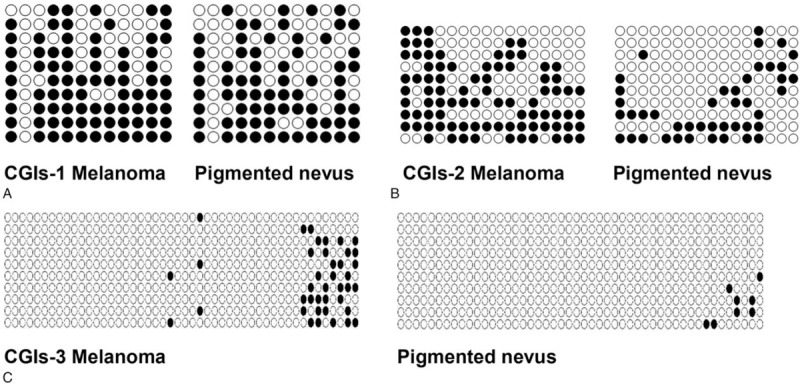
TA cloning results for melanoma sample one and pigmented nevus sample one: each row represents a bacterial clone, each circle represents a CG site, white represents unmethylated CG sites, and black represents methylated CG Sites, each column representing 10 bacterial clones of one CG site; methylation level = methylated CG site / (methylated CG site + unmethylated CG site).
Figure 4.
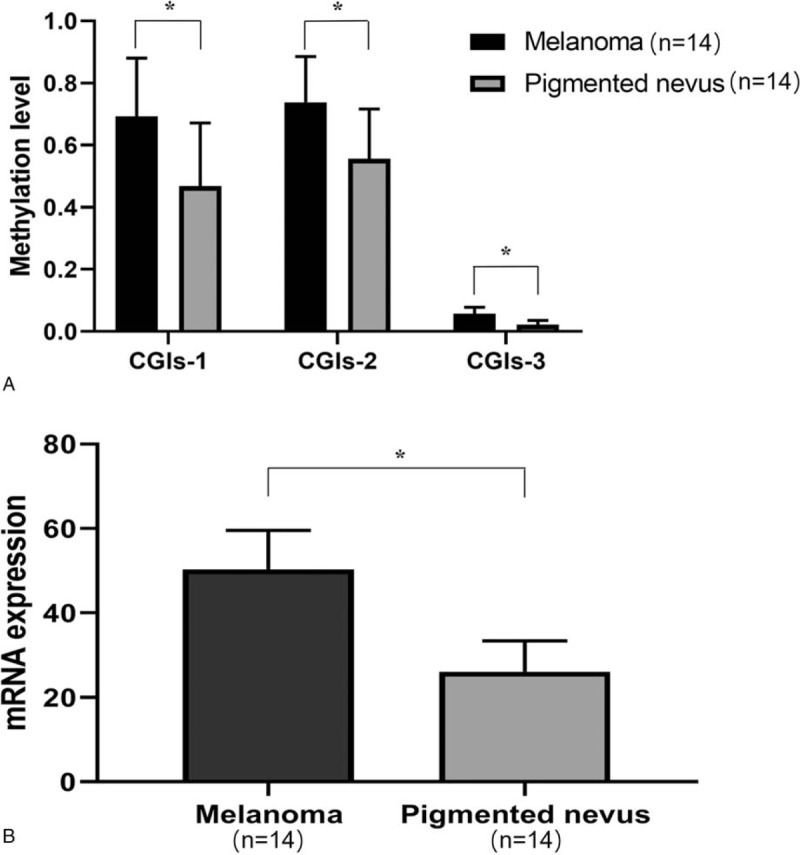
(A) Methylation level analysis of melanoma and pigmented nevus, (B) Relative expression level of hTERT mRNA of melanoma and pigmented nevus, ∗P < .05.
3.3. The relative expression level of hTERT mRNA and its correlation with CGIs methylation level
The relative expression of hTERT mRNA in melanoma (50.39 ± 9.16) was higher than that in the pigmented nevus group (26.10 ± 7.25), and the difference was statistically significant (t = 7.778, P < .001) (2 independent samples t test) (Fig. 4B). Linear regression analysis showed that hTERT mRNA relative expression level was positively correlated with the CGIs-2 methylation level (R2 = .490, F = 13.478, P = .003) in the melanoma group and not correlated with CGIs-1 or CGIs-3 methylation levels.
3.4. Correlation analysis of CGIs methylation level and clinicopathological features in melanoma
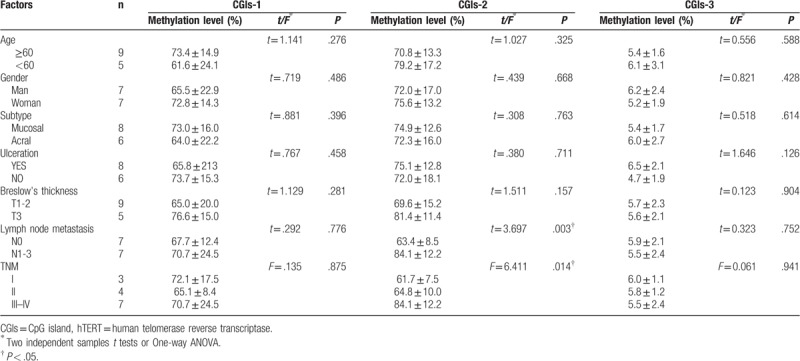
Clinicopathological features included age, gender, subtype, ulceration, Breslow thickness, lymph node metastasis, and TNM staging. In our study, the methylation levels of the 3 CGIs regions were not correlated with age, gender, subtype, ulceration and Breslow's thickness (P > .05), and the CGIs-2 methylation level was positively correlated with lymph node metastasis and TNM staging (P < .05) (Two independent samples t tests or One-way ANOVA) (Table 2).
Table 2.
Correlation analysis of the methylation levels of three CGIs regions in hTERT promoter and clinicopathological features in melanoma.
4. Discussion
Among Chinese, the most common type of melanoma is acral melanoma, followed by mucosal melanoma, both of which are non-UV-mediated melanoma.[9] Acral melanoma exhibits unique clinical features, mainly affecting the palms of the hands and the soles of the feet. Mucosal melanoma mainly affecting the mucosa of the head, neck, anus, rectum, and vulva vaginal. DNA methylation plays an important role in the formation and development of tumor by regulating cell cycle, apoptosis, invasion, immune recognition, and DNA repair. Unlike genetic damage, methylation is a reversible process that may be corrected with epigenetic drugs.[10] In recent years, more and more studies have suggested that DNA methylation can be used as a biomarker for tumor diagnosis, treatment, risk stratification, and evaluation of prognosis .[11–18] In this study, the methylation level of the hTERT promoter region in melanoma tissues was detected by BSP combined with the TA clonal sequencing method, and the methylation status of each CpG site in the target fragment was detected with high reliability and accuracy.[19]
The binding of transcription factors to transcription factor binding sites in gene promoter regions is the key to the regulation of gene expression.[8] Binding of transcriptional activators (c-Myc) and repressors (WT1 and CTCF) to the hTERT promoter may be controlled by DNA methylation, in which methylated CGIs prevent their binding to the target sites, leading to hTERT activation.[20] Our study found that the group of melanoma in the three CGIs regions of hTERT promoter had a methylation level that was higher than pigmented nevus, and the methylation level of CGIs-3 was significantly lower than CGIs-1 and CGIs-2 (Tab. 2). Moreover, the relative expression level of hTERT mRNA in the melanoma group was higher than in the pigmented nevus group (50.39 ± 9.16 vs 26.10 ± 7.25, t = 7.778, P < .001). Linear regression analysis showed the relative expression level of hTERT mRNA in the melanoma group was positively correlated with the methylation level of CGIs-2 (R2 = .49 F = 13.478, P = .003), indicating that the methylation of the hTERT promoter region may be one of the pathways of hTERT activation.
Early studies on the methylation level of the hTERT promoter region in breast cancer, believed that hypermethylation of the promoter region at the upstream of hTERT transcription starting point −600 bp might be related to the high expression of hTERT mRNA. Fan et al. [7] found that the hTERT promoter region at −667 to −482 bp upstream of the transcriptional start was hypermethylation of melanoma, and positively correlated with the hTERT mRNA expression level. In this study, CGIs-2 was located at −945 to −669 bp upstream of the site of the start of transcription. The methylation level of melanoma group was significantly higher than pigmented nevus group and positively correlated with hTERT mRNA relative expression level (R2 = .490 F = 13.478, P = .003). Combined with clinicopathological features, the methylation level of CGIs-2 was found to be positively correlated with lymph node metastasis and TNM staging (P < .05). This indicates that CGIs-2 hypermethylation may be indicative of late tumor TNM staging and poor prognosis in melanoma. Therefore, CGIs-2 is expected to be a suitable biomarker for the prognosis of melanoma.
Zinn et al[21] by studying different tumor cell lines (such as colon cancer, lung cancer, breast cancer) and the levels of methylation of different segments of the hTERT promoter region, found that hTERT core promoter region (transcription start −150 to +150 bp) hypomethylation is the key to its expression. However, Masood et al[22] performed a methylation analysis and found that the hTERT core promoter region in breast cancer and normal tissues both have hypomethylation status. They concluded that hypomethylation of the breast cancer hTERT core promoter region does not control the mechanism underlying its mRNA expression. In this study, the CGIs-3 contained hTERT in a core promoter region. Unlike the CGIs-1 and CGIs-2, most of the CGIs-3 CG sites in melanoma and pigment nevus organization did not show changes in methylation, only 11 CG loci showed detectable changes in methylation. The non-methylated status of CGIs-3 in pigmented nevus does not support the suggestion that demethylation is required for hTERT gene expression. In addition, the methylation level of CGIs-3 in the melanoma was not correlated with the relative expression of hTERT mRNA, clinicopathologic factors. In this way, whether CGIs-3 methylation can be used to distinguish melanoma from nevus requires further study. The role of CGIs-1 methylation level in tumorigenesis and development has only rarely been reported, possibly due to its lack of transcription factor binding sites. The level of CGIs-1 methylation was not found to be correlated with hTERT mRNA relative expression level or clinicopathologic factors. Therefore, whether CGIs-1 methylation can be used as a biomarker for the prognosis evaluation of melanoma needs further study.
In conclusion, CGIs methylation of the hTERT promoter region may be a suitable biomarker of the relative expression level of hTERT mRNA, which is of great significance for evaluation of prognosis in the Chinese patients with acral and mucosal melanoma. However, due to the small number of tissue samples collected, it is not clear whether methylation level is an independent prognostic factor of melanoma. For this reason, the role of the hTERT promoter CGIs methylation level and the relative expression level of hTERT mRNA in the prognosis evaluation of the Chinese patients with acral and mucosal melanoma must be verified in further studies with large sample groups.
Author contributions
Conceptualization: Haixia Xu, Weijia Wang, Tingting Li, Xiaojing Kang.
Data curation: Haixia Xu, Weijia Wang, Tingting Li.
Formal analysis: Haixia Xu.
Funding acquisition: Xiaojing Kang.
Investigation: Weijia Wang, Xiaojing Kang.
Methodology: Weijia Wang, Xiaojing Kang.
Project administration: Juan Zhao, Xiaojing Kang.
Resources: Juan Zhao, Xiaojing Kang.
Software: Haixia Xu, Tingting Li.
Supervision: Xiaojing Kang.
Writing – original draft: Haixia Xu, Juan Zhao, Xiaojing Kang.
Writing – review & editing: Haixia Xu, Juan Zhao, Xiaojing Kang.
Footnotes
Abbreviations: BSP = bisulfite sequencing PCR, CGIs = CpG island, hTERT = human telomerase reverse transcriptase.
How to cite this article: Xu H, Wang W, Zhao J, Li T, Kang X, College XC, University AM, Urumqi, Dermatology Do, Region PH. Aberrant hTERT promoter methylation predicts prognosis in chinese patients with acral and mucosal melanoma. Medicine. 2019;98:43(e17578).
This work was funded by the Natural Science Foundation of Xinjiang Uygur Autonomous Region (grant no. 2016D01C101) and the International Science and Technology Cooperation Project of Xinjiang Uyghur Autonomous Region (grant no. 20146022).
The authors declare that they have no conflicts of interest.
References
- [1].Schadendorf D, Fisher DE, Garbe C, et al. Melanoma. Nat Rev Dis Primers 2015;1:15003. [DOI] [PubMed] [Google Scholar]
- [2].Han L, Yuan Y, Zheng S, et al. The pan-cancer analysis of pseudogene expression reveals biologically and clinically relevant tumour subtypes. Nat Commun 2014;5:3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kang X, Zeng Y, Liang J, et al. Aberrations and clinical significance of BRAF in malignant melanoma: a series of 60 cases in Chinese Uyghur. Medicine 2018;97:e9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shain AH, Bastian BC. From melanocytes to melanomas. Nat Rev Cancer 2016;16:345–58.. [DOI] [PubMed] [Google Scholar]
- [5].Vazquez Vde L, Vicente AL, Carloni A, et al. Molecular profiling, including TERT promoter mutations, of acral lentiginous melanomas. Melanoma Res 2015;26:93–9.. [DOI] [PubMed] [Google Scholar]
- [6].Öztürk Sari Ş, Yilmaz İ, Taşkin OÇ, et al. BRAF, NRAS, KIT, TERT, GNAQ/GNA11 mutation profile analysis of head and neck mucosal melanomas: a study of 42 cases. Pathology 2017;49:55–61.. [DOI] [PubMed] [Google Scholar]
- [7].Fan Y, Lee S, Gang W, et al. Telomerase expression by aberrant methylation of the TERT promoter in melanoma arising in giant congenital nevi. J Invest Dermatol 2016;136:339–42.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kyo S, Takakura M, Fujiwara T, et al. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci 2008;99:1528–38.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Merkel EA, Gerami P. Malignant melanoma of sun-protected sites: a review of clinical, histological, and molecular features. Lab Invest 2017;97:630–5.. [DOI] [PubMed] [Google Scholar]
- [10].Fratta E, Sigalotti L, Covre A, et al. Epigenetics of melanoma: implications for immune-based therapies. Immunotherapy 2013;5:1103–16.. [DOI] [PubMed] [Google Scholar]
- [11].Yu W, Qin X, Jin Y, et al. Tianshengyuan-1 (TSY-1) regulates cellular Telomerase activity by methylation of TERT promoter. Oncotarget 2016;8:7977–88.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu Y, Li G, Dong H, et al. Telomerase reverse transcriptase methylation predicts lymph node metastasis and prognosis in patients with gastric cancer. Onco Targets Ther 2016;11:279–86.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kit OI, Vodolazhskiy DI, Kolesnikov EN, et al. Epigenetic markers of esophageal cancer: DNA methylation. Biomed Khim 2016;62:520–6.. [DOI] [PubMed] [Google Scholar]
- [14].Castelobranco P, Choufani S, Mack S, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol 2013;14:534–42.. [DOI] [PubMed] [Google Scholar]
- [15].Li JJ, Zheng JR, Wang YZ. The correlations between DNA methylation and polymorphisms in the promoter region of the human telomerase reverse transcriptase (hTERT) gene with postoperative recurrence in patients with thyroid carcinoma (TC). World J Surg Oncol 2017;6:15:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kumar A, Nilednu P, Kumar A, et al. Epigenetic perturbation driving asleep telomerase reverse transcriptase: possible therapeutic avenues in carcinoma. Tumour Biol 2017;39:1010428317695951. [DOI] [PubMed] [Google Scholar]
- [17].Sood S, Patel FD, Ghosh S, et al. Epigenetic alteration by DNA Methylation of ESR1, MYOD1 and hTERT gene promoters is useful for prediction of response in patients of locally advanced invasive cervical carcinoma treated by chemoradiation. Clin Oncol 2015;27:720–7.. [DOI] [PubMed] [Google Scholar]
- [18].Can AS, Bilal U, Vinay T, et al. Reactivation of telomerase in cancer. Cell Mol Life Sci 2016;73:1659–70.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jiang LL, Xie JK, Cui JQ, et al. Promoter methylation of yes-associated protein (YAP1) gene in polycystic ovary syndrome. Medicine 2017;96:e5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Leão R, Apolónio JD, Lee D, et al. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: clinical impacts in cancer. J Biomed Sci 2018;25:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zinn RL, Pruitt K, Eguchi S, et al. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res 2007;67:194–201.. [DOI] [PubMed] [Google Scholar]
- [22].Masood S, El-Gabry E, Zhang C, et al. DNA Methylation of the hTERT gene in breast cancer revisited: diagnostic and clinical implications. Lab Med 2016;47:293–9.. [DOI] [PubMed] [Google Scholar]