

Abstract
Autosomal recessive variants in the adenosine deaminase, tRNA specific 3 ( ADAT3 ) gene cause a syndromic form of intellectual disability due to a loss of ADAT3 function. This disorder is characterized by developmental delay, intellectual disability, speech delay, abnormal brain structure, strabismus, microcephaly, and failure to thrive. A small subset of individuals with ADAT3 deficiency have other structural birth defects including atrial septal defect, patent ductus arteriosus, hypospadias, cryptorchidism, and micropenis. Here, we report a sibling pair with novel compound heterozygous missense variants that affect a conserved amino acid in the deaminase domain of ADAT3. These siblings have many of the features characteristic of this syndrome, including, intellectual disability, hypotonia, esotropia, failure to thrive, and microcephaly. Both had gastroesophageal reflux disease (GERD), feeding problems, and aspiration requiring thickening of feeds. Although they have no words, their communication abilities progressed rapidly when they began to use augmentative and alternative communication (AAC) devices. One of these siblings was born with an anterior congenital diaphragmatic hernia, which has not been reported previously in association with ADAT3 deficiency. We conclude that individuals with ADAT3 deficiency should be monitored for GERD, feeding problems, and aspiration in infancy. They may also benefit from the use of AAC devices and individualized educational programs that take into account their capacity for nonverbal language development. Additional studies in humans or animal models will be needed to determine if ADAT3 deficiency predisposes to the development of structural birth defects.
Keywords: ADAT3, mental retardation autosomal recessive 36, syndromic intellectual disability, congenital diaphragmatic hernia, augmentative and alternative communication devices
Introduction
The adenosine deaminase, tRNA specific 3 (ADAT3) gene encodes a member of a family of adenosine deaminases that convert adenosine to inosine in the anticodon region of tRNAs. 1 ADAT3 functions in a heterodimer with ADAT2 to generate inosine in the first nucleotide of the anticodon at position 34, which is also referred to as the “wobble position.” 2 The location of inosine at this position in the tRNA allows pairing with either adenine, cytosine, or uracil in the original codon. 3
In 2013, Alazami et al reported eight consanguineous Arab families with an autosomal recessive form of intellectual disability and strabismus (OMIM 615286). 4 Additional features seen in affected individuals included growth failure, microcephaly, tone abnormalities, epilepsy, and central nervous system (CNS) abnormalities. All affected members of these families were homozygous for a c.382G>A, p.Val128Met ADAT3 variant (NM_138422.1), which leads to a single amino acid change affecting a hook that protrudes from the surface of the protein. Haplotype analysis revealed evidence of a founder effect within this population. With reference to the most current version of this transcript (NM_138422.4), which encodes the longer isoform of ADAT3, this variant is described as c.430G>A, p.Val144Met. For consistency, all variants will be described based on transcript NM_138422.4 throughout the remainder of this manuscript.
Subsequently, El-Hattab et al described affected individuals from consanguineous Saudi and Yemeni families all of whom were homozygous for the same p.Val144Met ADAT3 variant. 5 They expanded the previously reported phenotype by noting dysmorphic facial features, behavioral problems, recurrent otitis media, and growth hormone deficiency. Sharkia et al described a consanguineous sibling pair from an Arab community in Israel who were also homozygous for the p.Val144Met ADAT3 variant. 6 They noted features consistent with those described by El-Hattab et al along with gait difficulties, balance problems, teeth abnormalities, neuropathy, and contractures of the hand, wrist, and fingers.
Salehi Chaleshtori et al described an Iranian girl with who was homozygous for a c.99_106dupGAGCCCGG, Glu36Glyfs*44 ADAT3 variant. 7 Although her phenotype overlapped those of individuals described with the p.Val128Met ADAT3 variant, her intellectual disability was classified as mild rather than moderate to severe as described in previous reports, and her language development was delayed but not severely affected.
Here, we report a sibling pair with compound heterozygous missense changes affecting a conserved amino acid in the deaminase domain of ADAT3.
Materials and Methods
Editorial Policies and Ethical Considerations
Parents provided informed consent, and Subjects 1 and 2 were enrolled in a research study approved by the institutional review board of Baylor College of Medicine (protocol H-22769). This study was conducted in accordance with the ethical standards of the institutional committee on human research and were in keeping with international standards.
Exome Sequencing
Exome sequencing was performed on a clinical basis at GeneDx ( https://www.genedx.com/ ) on DNA extracted from Subject 2's whole blood. The mean depth of coverage was 177X with 98.7% of the exome being covered at 10× or greater. For the ADAT3 gene, 100% of the coding region was covered at 10X or higher. Subject 2's ADAT3 variants were confirmed, and their inheritance pattern was determined, by Sanger sequence of whole blood DNA from Subject 2 and his parents. Subsequently, Sanger sequencing performed on a clinical basis at Baylor Genetics ( https://www.baylorgenetics.com/ ) confirmed that Subject 1 had inherited the same ADAT3 variants as Subject 2. These analyses were performed on DNA extracted from Subject 1's whole blood.
Results
Clinical Descriptions
Subject 1
Subject 1 is a 12-year, 1-month-old female of European decent. She was born at 34 6/7 weeks gestation and was delivered vaginally after an induction for preeclampsia and intrauterine growth restriction (IUGR). Her birth weight was 1.575 kg (3rd centile) and her length was 38.1 cm (<1st centile, z-score −2.67). She was initially apneic but responded well to less than thirty seconds of positive pressure, bag mask ventilation. Her Apgar scores were 2 and 8 at 1 and 5 minutes respectively. She did not require intubation or supplemental oxygen. The placenta was noted to be much smaller than expected for gestational age and birth weight consistent with preterm preeclampsia. A head ultrasound performed at two days of age was normal. She was discharged to home from the neonatal intensive care unit on day of life 29.
At 5 months of age, she was evaluated for poor feeding and choking episodes occasionally accompanied by cyanosis. An upper gastrointestinal and small bowel series revealed intestinal malrotation and she was scheduled for a laparoscopic Ladd's procedure. During this procedure, she was found to have a moderately sized anterior diaphragmatic hernia that was covered by a membranous sac. The hernia sac was resected followed by primary closure of the diaphragmatic defect.
Despite this surgery, she continued to have choking spells. She was diagnosed with gastroesophageal reflux disease (GERD), and a swallow study revealed discoordination of sucking and swallowing with aspiration of thin liquids but not of thick liquids or purees. As a result, parents thickened liquids until she was three years of age. She was also noted to have developmental delay, hypotonia, failure to thrive, microcephaly, torticollis, and position-related plagiocephaly, which was treated with a helmet between 8 and 9 months of age. She underwent a tonsillectomy at 3 years and 11 months of age for obstructive apnea, and pressure equalization tubes were placed at 4 years of age. She has strabismus which was treated with patching, and myopia for which she currently wears glasses.
Due to developmental delays and hypotonia, she was enrolled in a variety of therapies starting at 6 months of age. Her motor milestones included reaching for objects at 4 to 5 months, holding her head up at 5 to 6 months, rolling over from her back to her stomach at 9 to 10 months, transferring objects from one hand to the other at 10 months, sitting unsupported at 12 months, crawling at 2 ½ years, and pulling up to stand and walking at 3 years of age.
Her language development was more severely affected. She cooed between 3 and 4 months of age but did not progress in her verbal language skills. With the help of therapy, she developed and used approximately 10 signs. However, her poor fine motor skills made it difficult for her to master more advanced signs. Beginning at 5 years of age, her parents began using laminated pictures as a means of communication. By 7 years of age, her vocabulary had expanded to a point to which the use of laminated pictures became impractical and she began using an electronic augmentative and alternative communication (AAC) device. Currently, she is able to form complete sentences using this device and routinely scrolls though five levels of categorical-based folders to select specific words she wishes to use.
Although she does not have any spoken words, she reads on a developmental reading assessment (DRA) level 14, which corresponds to a second grade level. She knows approximately 85 sight words and is learning to type on a keyboard. She is currently in the fourth grade with half of her time being spent in mainstream classes and half in special education classes.
At her most recent physical examination, performed when she was 11 years and 7 months of age, her height was 119.1 cm (< 1st centile, z-score of −2.74), her weight was 25.2 kg (<1st centile, z-score of −3.90), and her head circumference was 49.5 cm (<2nd centile, z-score of −2.05). She had a myopathic face, a high forehead, a bulbous nasal tip, a right-sided epicanthal fold, hypertelorism, a long, smooth philtrum, mild 5th finger clinodactyly and relatively flat feet ( Figs. 1 and 2 ). She was able to move about the room without difficulty but walked with her knees bent due to bilateral contractures. She was generally hypotonic. She showed no fear of strangers and was socially engaging. She used her electronic AAC device to explain her desires and demonstrated an understanding of verbal responses and commands.
Fig. 1.

Facial features and pedigree of Subjects 1 and 2. ( A , B ) Subject 1 when she was 12 years, 1 month old. She had a myopathic face, high forehead, bulbous nasal tip, right-sided epicanthal fold, hypertelorism, long, smooth philtrum, and a hemangioma on her left cheek. ( C , D ) Subject 2 when he was 7 years, 10 months old. He had a myopathic face, high forehead, bulbous nasal tip, hypertelorism, down-slanting palpebral fissures, and a long smooth philtrum. ( E ) Pedigree showing subjects 1 and 2 along with the ADAT3 variants they inherited from their unrelated mother and father. ADAT3, adenosine deaminase, tRNA specific 3 gene.
Fig. 2.

Photographs of Subject 1 (S1) over time. Photos taken at 6 months (m), 1, 2, 3, 4, 5, 7, and 8 years (y) of age.
Subject 2
Subject 2 is the 7-year, 10-month-old full brother of Subject 1. He was born at 35 weeks gestation and was delivered vaginally after induction for maternal preeclampsia and IUGR. His birth weight was 1680 g (3rd centile), his length was 42 cm (5th centile), and his head circumference was 31 cm (27th centile). Apgar scores were 6 at 1 minute, 9 at 5 minutes, and 9 at 10 minutes. He had transient asymptomatic hypoglycemia and was transferred to the neonatal intensive care unit where he remained hemodynamically stable and on room air until he was discharged at 21 days of age.
He was diagnosed with GERD at 3 months of age. A swallow study obtained at 8 months of age revealed aspiration with thin liquids but no evidence of aspiration with thickened liquids or purees. He displayed reduced posterior oral containment and a delayed swallow, which contributed to the aspiration events. A reevaluation at 1 year and 7 months of age, found that he had oral phase dysphagia with reduced chewing pattern of solid consistency. He was treated with feeding therapy and thickening of liquids until 3 years, 4 months of age.
He held his head up at 6 months of age, rolled over at 1 year of age, sat unsupported at 15 months of age, did not crawl, cruised at 24 months of age, and walked independently at 3 years of age. Although he cooed and babbled, his speech did not progress, and he currently has no spoken words. At approximately 4 years of age, he started learning to indicate his desires using laminated pictures, and at 6 years of age he transitioned to an electronic AAC device. He continues to use this device and routinely scrolls through two levels of categorical-based folders to select words he wishes to use. He can identify letters and knows approximately 15 to 20 sight words. He is currently in the first grade and spends half of his time in mainstream classes and half of his time in special education classes. He continues to receive occupational and speech therapy. He was also diagnosed with esotropia that was treated with patching. He currently wears glasses for myopia with astigmatism.
At his most recent physical examination, performed when he was 7 years, 4 months of age, his height was 111.5 cm (1st centile), his weight was 19.3 kg (5th centile), and his head circumference was 51.5 cm (39th centile). He had a myopathic face, a high forehead, a bulbous nasal tip, hypertelorism, down-slanting palpebral fissures, and a long smooth philtrum ( Figs. 1 and 3 ). His clinical cardiac examination was normal. He was generally hypotonic, but he moved about the room without difficulty. He used his electronic (AAC) device to communicate and responded appropriately to verbal commands.
Fig. 3.
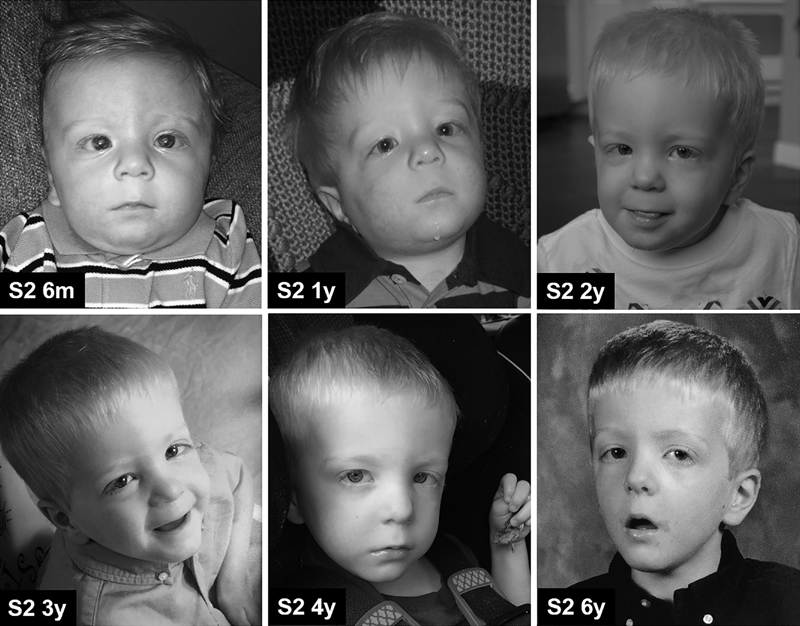
Photographs of Subject 2 (S2) over time. Photos taken at 6 months (m), 1, 2, 3, 4, and 6 years (y) of age.
Diagnostic and Molecular Studies
Evaluations performed for Subject 1 included standard newborn screening tests, a hearing evaluation, a chromosome analysis on blood and fibroblasts, an array-based copy number variant analysis (CMA V8,0; Baylor Genetics), a cytomegalovirus urine screen, 7-dehydrocholesterol level, a creatine kinase level, an acylcarnitine analysis, a renal ultrasound, a brain magnetic resonance imaging (MRI) performed at 10 months of age, an echocardiogram, cerebrospinal fluid (CSF) neurotransmitter studies, a skeletal survey, urinary glycosaminoglycan analyses, thyroid hormone (free T4) and thyroid stimulating hormone (TSH) levels, and carbohydrate deficient transferrin studies all of which were normal.
Similarly, a high-resolution chromosome analysis performed on cultured lymphocytes obtained from Subject 2 revealed a normal 46,XY chromosomal compliment and an array-based copy number variant analysis (CMA Oligo V8.1.1, Baylor Genetics) was normal. Standard newborn screening tests and routine hearing screens were also normal.
Clinical exome sequencing was performed on Subject 2 and revealed compound heterozygous variants in ADAT3; a maternally inherited c.587C>T, p.Ala196Val variant and a paternally inherited c.586_587delinsTT, p.Ala196Leu variant [NM_138422.4] ( Fig. 4 ). Both of these variants are considered damaging by Sorting Intolerant from Tolerant (SIFT) ( http://sift.jcvi.org/ ), probably damaging by PolyPhen-2 ( http://genetics.bwh.harvard.edu/pph2/ ) and disease causing by MutationTaster ( http://www.mutationtaster.org/ ). These results were confirmed by Sanger sequencing, and subsequently the same ADAT3 variants were identified in Subject 1 by Sanger sequencing on a clinical basis.
Fig. 4.
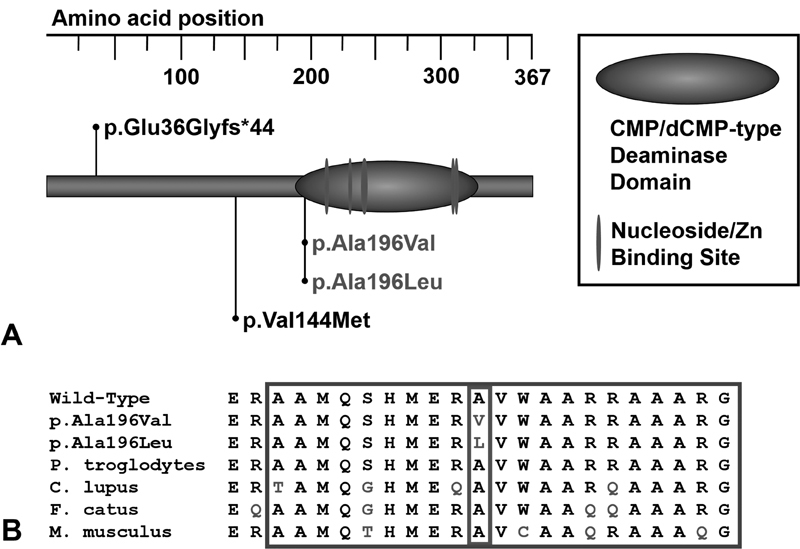
ADAT3 variants associated with ADAT3 deficiency. ( A ) The locations of the ADAT3 variants reported in individuals with ADAT3 deficiency are shown in relation to the protein domains of ADAT3. Variants found in Subjects 1 and 2 are shown in gray. Previously described pathogenic variants are shown in black. ( B ) The variants seen in Subjects 1 and 2 affect a conserved residue located in the CMP/dCMP-type deaminase domain of ADAT3 (small outline). The large outline indicates residues around these changes that are part of the ADAT3's CMP/dCMP-type deaminase domain. ADAT3, adenosine deaminase, tRNA specific 3; CMP/dCMP, cytidine monophosphate/deoxycytidine monophosphate.
In the gnomAD database ( https://gnomad.broadinstitute.org/ ), the c.587C>T, p.Ala196Val variant is seen seven times in the heterozygous state with allele frequencies of 2/7820 Latino, 4/34678 European (non-Finnish), 1/9246 African, and 7/77984 total. No individuals were homozygous for this variant in the gnomAD database. The c.586_587delinsTT, p.Ala196Leu variant is not seen in the gnomAD database.
Discussion
Here we describe two siblings who carry putatively deleterious, compound heterozygous missense variants in ADAT3; a maternally inherited c.587C>T, p.Ala196Val variant and a paternally inherited c.586_587delinsTT, p.Ala196Leu variant [NM_138422.4]. These changes affect the same conserved amino acid in the CMP/dCMP-type deaminase domain of ADAT3 ( Fig. 4 ) and both lead to the substitution of the alanine at that position to a hydrophobic amino acid (valine or leucine). Both variants are considered pathogenic based on our in silico analyses and the phenotypic overlap seen between Subjects 1 and 2 and other individuals with ADAT3 deficiency ( Table 1 ). Taken together, this suggests that this area of the CMP/dCMP-type deaminase domain, and more specifically the alanine at position 196, plays a critical role in the function of ADAT3.
Table 1. Phenotypes documented in individuals with ADAT3 deficiency.
| Clinical features | Alazami et al 4 | El-Hattab et al 5 | Sharkia et al 6 | Salehi Chalestori et al 7 | Subject 1 | Subject 2 | Number (%) |
|---|---|---|---|---|---|---|---|
| Brain function/structure | |||||||
| Intellectual disability | 24/24 | 15/15 | 2/2 | + | + | + | 44/44 (100%) |
| Spasticity | 7/24 | 6/15 | − | n/d | + | − | 14/43 (33%) |
| Epilepsy | 3/24 | 3/15 | − | n/d | − | − | 6/43 (14%) |
| Brain anomalies | 9/15 | 9/13 | 2/2 | n/d | − | n/d | 20/31 (65%) |
| ADHD/hyperactivity | 1/24 | 2/15 | n/d a | + | − | − | 4/42 (10%) |
| Aggressive | n/d | 4/15 | n/d a | n/d | − | − | 4/17 (23%) |
| Hypotonia | 10/24 | 6/15 | 2/2 | n/d | + | + | 20/43 (47%) |
| Eyes/vision | |||||||
| Esotropia/strabismus | 22/24 | 10/15 | 2/2 | − | + | + | 36/44 (82%) |
| Myopia | n/d | 1/15 | n/d | n/d | + | + | 3/17 (18%) |
| Hearing loss | n/d | 1/15 | n/d | n/d | − | − | 1/17 (6%) |
| Growth/GI | |||||||
| IUGR | n/d | 1/15 | − | − | + b | + b | 3/19 (16%) |
| Failure to thrive/short stature | 22/24 | 11/15 | 2/2 | n/d | + | + | 37/43 (86%) |
| Microcephaly | 11/24 | 11/15 | 2/2 | + | + | − | 26/44 (59%) |
| GERD | n/d | 2/15 | n/d | n/d | + | + | 4/17 (24%) |
| Skeletal/joints | |||||||
| Joint contractures | n/d | 1/15 | 2/2 | n/d | + | − | 4/19 (21%) |
| Talipes/vertical talus | 2/24 | 2/15 | − | n/d | − | − | 4/43 (9%) |
| Physical features | |||||||
| Prominent forehead | n/d | 5/15 | 2/2 | n/d | − | − | 7/19 (37%) |
| High forehead | n/d | 4/15 | 1/2 | n/d | + | + | 7/19 (37%) |
| Slanted palpebral fissures | n/d | 4/15 | 2/2 | n/d | − | + | 7/19 (37%) |
| Epicanthus | n/d | 4/15 | 2/2 | n/d | + | − | 7/19 (37%) |
| Hypertelorism/telecanthus | n/d | 4/15 | 2/2 | n/d | + | + | 8/19 (42%) |
Abbreviations: −, phenotype not reported; +, phenotype reported; ADHD, attention deficit hyperactivity disorder; GI, gastrointestinal; GERD, gastroesophageal reflux disease; IUGR, intrauterine growth restriction; n/d, evaluation not performed or not reported.
Sharkia et al indicated that their patients had behavioral problems similar to those previously documented by El-Hattab et al, but did not delineate them further.
In the setting of preeclampsia.
Note: Abnormalities documented only in a single patient: impulsivity, autism, hearing loss, excessive dental caries, atrial septal defect, patent ductus arteriosus, congenital diaphragmatic hernia, recurrent gastroenteritis, chronic constipation, medullary nephrocalcinosis, polyhydramnios, hypospadias, cryptorchidism, micropenis, rocker bottom feet, and decreased sweating.
Hypotonia is a common feature described in individuals with ADAT3 deficiency and can predispose infants to GERD, feeding difficulties, and aspiration. 8 9 Subjects 1 and 2 have generalized hypotonia, were diagnosed with GERD, had feeding difficulties, and were treated with thickening of liquids due to aspiration of thin liquids identified by swallow studies. 10 Surprisingly, GERD and feeding difficulties have only been documented in a small subset of individuals with ADAT3 deficiency ( Table 1 ). It is possible that these phenotypes may be underdiagnosed or underreported in this disorder.
Speech delay is also a common feature identified in individuals with ADAT3 deficiency. Subjects 1 and 2 did not progress beyond cooing and babbling and have no spoken words. However, they communicate effectively with the use of electronic AAC devices. Subject 1 is reading at a second grade level and Subject 2 can identify letters and knows 15 to 20 sight words. This suggest that individuals with ADAT3 deficiency may have greater expressive language and communication abilities than would be suggested by their verbal language development. Hence, they may benefit from the use of AAC devices and individualized educational programs that take into account their capacity for nonverbal language development. 11 12
Subject 1 had a moderately sized anterior diaphragmatic hernia that was covered by a membranous sac. 13 Diaphragmatic hernias have not been documented in other individuals with ADAT3 deficiency. Other structural birth defects that have been described in a small subset of individuals with ADAT3 deficiency include atrial septal defect, patent ductus arteriosus, hypospadias, cryptorchidism, and micropenis ( Table 1 ). Additional studies, in humans and/or animal models, will be needed to determine if ADAT3 deficiency predisposes to the development of these and other structural birth defects.
Acknowledgments
The authors thank family members for participating in this research.
Funding Statement
Funding D.A.S. reports grants from National Institutes of Health, during the conduct of the study. This work was funded in part by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD; R01 HD093660).
Footnotes
Conflict of Interest None declared.
References
- 1.Torres A G, Piñeyro D, Filonava L, Stracker T H, Batlle E, Ribas de Pouplana L. A-to-I editing on tRNAs: biochemical, biological and evolutionary implications. FEBS Lett. 2014;588(23):4279–4286. doi: 10.1016/j.febslet.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 2.Gerber A P, Keller W.An adenosine deaminase that generates inosine at the wobble position of tRNAs Science 1999286(5442):1146–1149. [DOI] [PubMed] [Google Scholar]
- 3.Crick F H. Codon–anticodon pairing: the wobble hypothesis. J Mol Biol. 1966;19(02):548–555. doi: 10.1016/s0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- 4.Alazami A M, Hijazi H, Al-Dosari M S et al. Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J Med Genet. 2013;50(07):425–430. doi: 10.1136/jmedgenet-2012-101378. [DOI] [PubMed] [Google Scholar]
- 5.El-Hattab A W, Saleh M A, Hashem A et al. ADAT3-related intellectual disability: further delineation of the phenotype. Am J Med Genet A. 2016;170A(05):1142–1147. doi: 10.1002/ajmg.a.37578. [DOI] [PubMed] [Google Scholar]
- 6.Sharkia R, Zalan A, Jabareen-Masri A, Zahalka H, Mahajnah M. A new case confirming and expanding the phenotype spectrum of ADAT3-related intellectual disability syndrome. Eur J Med Genet. 2018:S1769-7212(18)30574-3. doi: 10.1016/j.ejmg.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Salehi Chaleshtori A R, Miyake N, Ahmadvand M, Bashti O, Matsumoto N, Noruzinia M. A novel 8-bp duplication in ADAT3 causes mild intellectual disability. Hum Genome Var. 2018;5:7. doi: 10.1038/s41439-018-0007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Engel-Hoek L, de Groot I JM, de Swart B JM, Erasmus C E. Feeding and swallowing disorders in pediatrci neuromuscular diseases: an overview. J Neuromuscul Dis. 2015;2(04):357–369. doi: 10.3233/JND-150122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seddon P C, Khan Y. Respiratory problems in children with neurological impairment. Arch Dis Child. 2003;88(01):75–78. doi: 10.1136/adc.88.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan D R, Larson K, Davidson K, May K, Rahbar R, Rosen R L. Feeding interventions are associated with improved outcomes in children with laryngeal penetration. J Pediatr Gastroenterol Nutr. 2019;68(02):218–224. doi: 10.1097/MPG.0000000000002167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson K M, Hennig S. The state of research and practice in augmentative and alternative communication for children with developmental/intellectual disabilities. Ment Retard Dev Disabil Res Rev. 2007;13(01):58–69. doi: 10.1002/mrdd.20133. [DOI] [PubMed] [Google Scholar]
- 12.Light J, McNaughton D, Caron J. New and emerging AAC technology supports for children with complex communication needs and their communication partners: state of the science and future research directions. Augment Altern Commun. 2019;35(01):26–41. doi: 10.1080/07434618.2018.1557251. [DOI] [PubMed] [Google Scholar]
- 13.Kardon G, Ackerman K G, McCulley D J et al. Congenital diaphragmatic hernias: from genes to mechanisms to therapies. Dis Model Mech. 2017;10(08):955–970. doi: 10.1242/dmm.028365. [DOI] [PMC free article] [PubMed] [Google Scholar]