

Abstract
Protein phosphatase 2A (PP2A) is a heterotrimeric protein serine/threonine phosphatase that regulates a diverse range of cellular activities. The PPP2R1A gene on chromosome 19 (19q13.41) encodes the α isoform of the scaffolding subunit of the PP2A holoenzyme, which functions to link the catalytic subunit to the regulatory subunit. Here we present a case of a newborn boy with a novel PPP2R1A gene mutation (c.548G>A; p.Arg183Gln) with severe lateral and third ventriculomegaly, hypoplastic corpus callosum, and pontocerebellar hypoplasia. To our knowledge, this is the sixth case reported in the literature, thus expanding the phenotype of this rare genetic condition.
Keywords: protein phosphatase 2A, PPP2R1A, ventriculomegaly
Introduction
Protein phosphatase 2A (PP2A) is a heterotrimeric protein serine/threonine phosphatase that regulates a diverse range of cellular activities. It is made up of a catalytic subunit (C), a scaffolding subunit (A), and variable regulatory subunits (B). 1 PP2A has been associated with several cellular processes ubiquitously, including DNA replication, cell proliferation, tumor suppression, and Wnt signaling. 2 The PPP2R1A gene on chromosome 19 (19q13.41) encodes the α isoform of the scaffolding subunit (Aα), which functions to link the catalytic subunit to the regulatory subunit (B56δ is one of several). Mutations in PPP2R1a have the potential to disrupt B56δ-dependent dephosphorylation activity. 3 Aα is expressed in many different tissue types, but B56δ is highly expressed in the brain, 4 5 which becomes important when considering phenotypic manifestations of altered protein function.
The United Kingdom Deciphering Developmental Disorders study 6 identified three de novo mutations in the PPP2R1A gene (among 1,133 parent-child trios sequenced), and Houge et al described an additional two cases. Here, we review prior cases and report a sixth case, thus expanding the phenotype of this genetic mutation.
Case Report
A baby boy was born through a scheduled cesarean section at 38 weeks to a 34-year-old mother due to multiple known congenital anomalies. Pregnancy was uncomplicated until 18 weeks of gestation, when a routine fetal survey demonstrated bilateral ventriculomegaly and hypoplastic aortic arch. Fetal magnetic resonance imaging (MRI) at 19 weeks and 5 days confirmed bilateral ventriculomegaly and an enlarged third ventricle. Amniocentesis revealed an unremarkable chromosomal microarray and karyotype, as well as negative L1CAM (L1 cell adhesion molecule) sequencing (for X-linked hydrocephalus). Routine prenatal testing was unremarkable, including negative cytomegalovirus and Toxoplasma polymerase chain reaction studies. The infant initially required respiratory support with continuous positive airway pressure but was stabilized on room air by 7 minutes of life. Birth weight was 2.8 kg (11.83th percentile) and head circumference was 36.8 cm (96.4th percentile). The infant was admitted to the neonatal intensive care unit for further evaluation and monitoring.
There were no prenatal exposures to medications or substances (aside from prenatal vitamins), and there was no significant travel history. Family history was significant for maternal miscarriage at 8 weeks and stillborn birth in the maternal grandmother, details of which are unknown. There was no family history of developmental delay or neurologic disorders.
The infant's examination was notable for the following dysmorphisms: down-slanting palpebral fissures, low-set ears, anteverted nares, high asymmetric palate, mild retrognathia, pectus excavatum, bilateral fifth finger clinodactyly, increased gap between the third and fourth toes, hyperextensible fingers, right-sided inguinal hernia, and low truncal tone.
An MRI of the brain was performed on the first day of life, which was significant for severe ventriculomegaly involving the lateral and third ventricles, as well as pontocerebellar hypoplasia. A repeat study was performed on the third day of life to evaluate the patency of the cerebral duct for surgical planning. This redemonstrated the prior findings and revealed cerebrospinal fluid signal through the duct, though with some suggestion of stenosis. Bilateral choroid plexus cysts were present and both dangling in the left lateral ventricle. No intact corpus callosum was noted midline, and commiserated fibers could be confirmed only anteriorly ( Fig. 1 ). A right occipital ventriculoperitoneal shunt was placed on the fifth day of life. Ophthalmic examination revealed optic nerve hypoplasia. The infant was treated with Keppra for seizure activity. Given prenatal findings of aortic arch abnormalities, serial echocardiograms were performed through closure of the ductus arteriosus and demonstrated a hypoplastic transverse aortic arch with no arch obstruction.
Fig. 1.
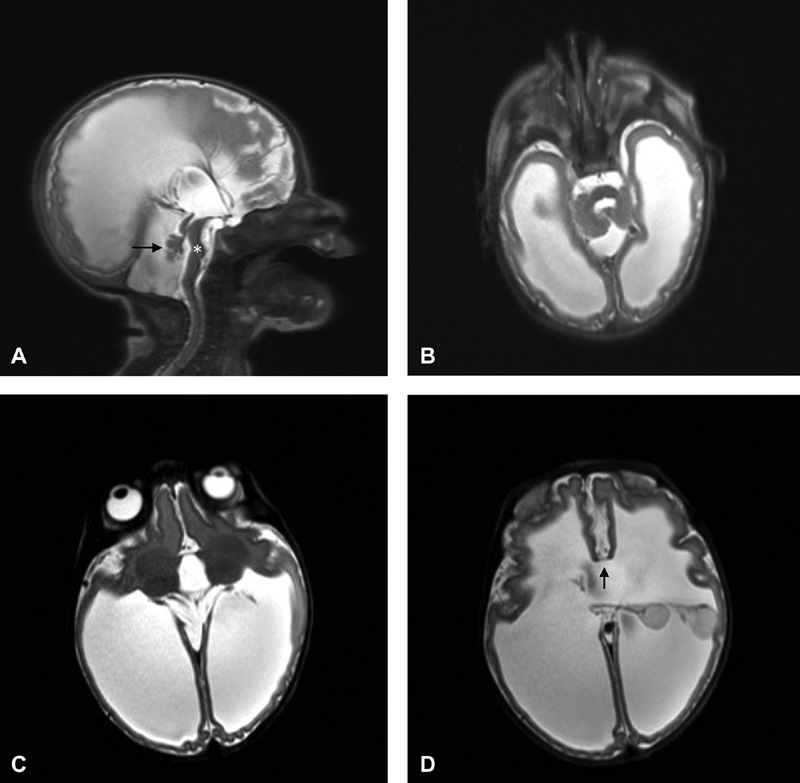
( A ) Sagittal HASTE image shows severe ventriculomegaly, pontine hypoplasia ( asterisk ), and dysplastic vermis (arrow). ( B ) Axial HASTE (HAlf fourier Single-shot Turbo spin Echo) image demonstrates severe ventriculomegaly with very thin, abnormally smooth, and packed cortex suspicious for polymicrogyria. ( C ) Axial HASTE image redemonstrating severe ventriculomegaly, as well as abnormal rotation of the thalami and poor definition of the posterior limb of the internal capsules that may represent incomplete separation of the thalami from the putamina. ( D ) Axial HASTE image is notable for floating choroid plexuses “on stalks” both flipped into the left lateral ventricle, only scant commissurated callosal fibers noted anteriorly (arrow), and no callosal fibers posteriorly.
To assist in diagnosis and prognostication, a karyotype and microarray were sent and were normal (46, XY). Trio whole exome sequencing was performed and revealed a de novo missense mutation in the PPP2R1A gene (c.548G>A; p.Arg183Gln).
The patient was discharged home from the hospital at 47 days of life with feeds provided by the family through a nasogastric tube (swallow study demonstrated significant aspiration risk). He was cared for at home by his family and was followed closely by the palliative care team. His course was complicated by recurrent aspiration pneumonias, and the patient died of sepsis and respiratory failure at 114 days of life.
Discussion
We describe a newborn boy with a novel PPP2R1A gene mutation and clinical features including several facial dysmorphic features, severe lateral and third ventriculomegaly, hypoplastic corpus callosum, pontocerebellar hypoplasia, optic nerve hypoplasia, and a hypoplastic transverse aortic arch. To our knowledge, this is the sixth case reported in the literature expanding the phenotype of this rare genetic condition.
Of the five cases described in earlier studies 3 (1–11 years of age), all cases were a result of a normal pregnancy. Hypotonia, developmental delay, and severe intellectual disability were present in all cases after birth. Epilepsy was present in four of five cases. MRI findings were significant for hypoplasia or agenesis of the corpus callosum in all cases. Ventriculomegaly was present in two of five cases. Common dysmorphic features included an elongated face with tented upper lip, mild hypertelorism, and down-slanting palpebral fissures. Additional clinical features include scoliosis (two of five cases) and cortical visual impairment (two of five cases). All pathogenic variants reported to date are missense variants, suggesting a dominant negative mechanism of disease. 3
In comparison to the previously described cases, our case overlaps in many ways but perhaps exhibits a more severe manifestation of the phenotype ( Table 1 ). Interestingly, the amino acid affected in our case (183) is one following in sequence to the amino acid reported in three of the cases (182). PP2A is composed of 15 repeat HEAT motifs, which allow the scaffolding subunit to link to the regulatory and catalytic subunits. 7 Our mutation, along with four of five others reported, is present in HEAT domain 5, which links to B regulatory subunits. 8 All prior mutations reported have been shown to affect PP2A holoenzyme formation, surprisingly from either hindered C subunit interaction or defective catalytic activity with or without maintained B56δ interaction. 3 By replacing arginine (basic side chain) with glutamine (polar, uncharged side chain) as in our case, the protein formation may be more disrupted and less able to maintain its scaffolding effect as compared with amino acid changes that lead to no side change difference. For example, in the only patient in whom epilepsy was not present (c.536C>T;Pro179Leu), proline (aliphatic side chain) was replaced by leucine (aliphatic side chains). This may explain why severity of ventriculomegaly in our case is striking, as well as its association with pontocerebellar hypoplasia. Phenotypically, our patient had abnormalities of his finger and toes, as well as pectus excavatum. Aortic arch abnormalities are yet to be reported. Further analysis of additional patients is required to make further conclusions.
Table 1. Description of reported cases.
| Case | Age, sex at evaluation | DNA change | Protein change | CNS findings | Non-CNS findings | Source |
|---|---|---|---|---|---|---|
| 1 | 3.5 y, F | c.536C>T | p.Pro179Leu | Agenesis of the corpus callosum, Epilepsy not present | Cortical visual impairment | Houge et al |
| 2 | 4 y, F | c.544C>T | p.Arg182Trp | Hypoplasia of the corpus callosum and enlarged ventricles, epilepsy present | Scoliosis | Houge et al |
| 3 | 11 y, M | c.544C>T | p.Arg182Trp | Hypoplasia of the corpus callosum, epilepsy present | Scoliosis, hip dysplasia, need for gastrostomy | Houge et al |
| 4 | 1 y, F | c.544C>T | p.Arg182Trp | Agenesis of the corpus callosum, enlarged ventricles, delayed myelination, epilepsy present | Cortical visual impairment, absent uterus/vagina, unilateral kidney agenesis, unilateral postaxial polydactyly, Heterozygote for a TMEM67 splice mutation | Houge et al |
| 5 | 5 y, M | c.773G>A | p.Arg258His | Hypoplasia of the corpus callosum and delayed myelination, epilepsy present | Entropion of eyelids, obstipation, hyperactivity | Houge et al |
| 6 | Newborn, M, deceased at 114 d | c.548G>A | p.Arg183Gln | Hypoplasia of the corpus callosum, pontocerebellar hypoplasia, and severe ventriculomegaly, epilepsy present | Fifth finger clinodactyly, pectus excavatum, inguinal hernia, hypoplastic aortic arch, optic nerve hypoplasia | Present case |
Abbreviations: CNS, central nervous system; F, female; M, male.
Of note, our patient was also heterozygous for a genetic variant in the SEMA3E gene, which was inherited from the proband's mother who is also heterozygous. This gene encodes semaphorin 3E, which is involved in embryonic development. Semaphorin activity in the neural crest has been linked specifically to development of the human heart. 9 Given the mother's normal phenotype, this variant was felt to be not pathogenic in this case. However, without further analysis, we cannot exclude the possibility that this variation in combination with the PPP2R1A mutation may contribute to the more severe phenotype observed in our patient when compared with the previously reported cases.
It should also be noted that mutations in PPP2R5D , which encodes the regulatory B56δ subunit of PP2A, present in a clinically similar manner. There are 11 cases reported in the literature, all of which are phenotypically brain restricted. 3 Houge et al notes that based on the 1,133 cases of moderate-to-severe intellectual disability studied through the United Kingdom Deciphering Developmental Disorders study, 5 the prevalence of PP2A subunit mutations may be as high as 0.6% (when copy number variants were not present).
Conclusion
In conclusion, we report the clinical features of a newborn infant with a de novo missense mutation in the PPP2R1A gene. While there are only 6 reported cases in the literature, the findings of the United Kingdom Deciphering Developmental Disorders study 6 may suggest that this gene should be considered in children with hypotonia, severe intellectual disability, and MRI findings of ventriculomegaly, immature development of the corpus callosum, and pontine hypoplasia.
Footnotes
Conflict of Interest None declared.
References
- 1.Seshacharyulu P, Pandey P, Datta K, Batra S K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013;335(01):9–18. doi: 10.1016/j.canlet.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virshup D M. Protein phosphatase 2A: a panoply of enzymes. Curr Opin Cell Biol. 2000;12(02):180–185. doi: 10.1016/s0955-0674(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 3.Houge G, Haesen D, Vissers L E et al. B56δ-related protein phosphatase 2A dysfunction identified in patients with intellectual disability. J Clin Invest. 2015;125(08):3051–3062. doi: 10.1172/JCI79860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCright B, Rivers A M, Audlin S, Virshup D M. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem. 1996;271(36):22081–22089. doi: 10.1074/jbc.271.36.22081. [DOI] [PubMed] [Google Scholar]
- 5.Louis J V, Martens E, Borghgraef P et al. Mice lacking phosphatase PP2A subunit PR61/B'delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3beta. Proc Natl Acad Sci U S A. 2011;108(17):6957–6962. doi: 10.1073/pnas.1018777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fitzgerald T W, Gerety S S, Jones W Det al. Large-scale discovery of novel genetic causes of developmental disorders Nature 2015519(7542):223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groves M R, Hanlon N, Turowski P, Hemmings B A, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell. 1999;96(01):99–110. doi: 10.1016/s0092-8674(00)80963-0. [DOI] [PubMed] [Google Scholar]
- 8.Xu Y, Chen Y, Zhang P, Jeffrey P D, Shi Y. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol Cell. 2008;31(06):873–885. doi: 10.1016/j.molcel.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lalani S R, Safiullah A M, Molinari L M, Fernbach S D, Martin D M, Belmont J W. SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet. 2004;41(07):e94. doi: 10.1136/jmg.2003.017640. [DOI] [PMC free article] [PubMed] [Google Scholar]