

Abstract
Pathogenic variants in the TRAPPC6B gene were recently found to be associated in three consanguineous families, with microcephaly, epilepsy, and brain malformations. Here, we report on a 3.5-year-old boy, born to consanguineous Lebanese parents, who presented with developmental delay, lactic acidosis, postnatal microcephaly, and abnormal brain magnetic resonance imaging. By whole exome sequencing, a novel homozygous likely pathogenic variant in exon 1 of the TRAPPC6B gene (c.23T > A; [p.Leu8*]) was identified. A review of the clinical description and literature is discussed, pointing out the phenotypic heterogeneity associated with mutations in this gene.
Keywords: Lebanon, whole exome sequencing, TRAPPC6B
Introduction
Intellectual disability (ID) and global developmental delay are genetically heterogeneous, and in many cases, it is difficult to arrive at a diagnosis in affected children. A specific molecular diagnosis can aid in deciding potential treatment options, has prognostic implications, and helps families in planning future pregnancies through prenatal and preimplantation genetic diagnosis. 1 However, with more than 800 separate genes involved in specific genetic types of monogenic ID, 2 such a diagnosis is extremely difficult using conventional means. In recent years, chromosomal microarrays (CMA) and more recently, whole exome sequencing (WES) techniques have been used successfully in the molecular diagnosis of ID and global development delay disorders. 3
Here, we report a Lebanese patient with global developmental delay and intellectual disability who is homozygous for a novel nonsense mutation of the TRAPPC6B gene. This is the third report so far of a variant in this gene causing nonsyndromic ID.
Case Report
The patient, a boy, was born at term after a C-section to healthy consanguineous Lebanese parents ( Fig. 1A ) after an uncomplicated pregnancy. At birth, his weight was 3,500 g, his length 51 cm, and his head circumference 35 cm. At the age of 3 months, the parents noticed that he was unable to make eye contact, unable to focus on objects or follow movements, and was not grabbing things with his hands.
Fig. 1.
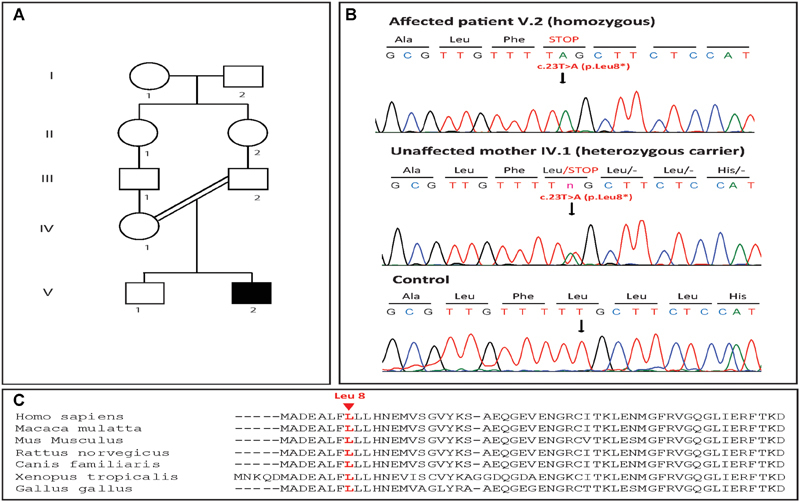
( A ) Pedigree of the studied family. Blackened symbols indicate affected individuals. ( B ) Chromatograms from the patient (V.2), one parent (IV.1) and a control showing the homozygous mutation detected in the family; the c.23T > A (p.Leu8*) nonsense mutation in the TRAPPC6B gene (NM_001079537). ( C ) Alignment of TRAPPC6B amino acid sequences of the protein region comprising the mutation under investigation between different species using CLUSTAL 2.1. Leucine 8 of TRAPPC6B (NP_001073005), highlighted in red, is highly conserved across different species.
At first examination at the age of 15 months, he was hypotonic, could sit when helped, and was saying two monosyllabic words, but interacted well. His head circumference was 43.3 cm (<3rd centile); length 74 cm (15th centile); and weight 10.2 Kg (25th centile). He showed axial hypotonia, microcephaly, and a beaked nose. Brain MRI showed a mild periventricular delay in myelination, while brain spectroscopy showed a small lipid peak.
Paraclinical work-up showed lactic acidosis (39.1 mg/dL), normal chromatography of amino acids in blood and of organic acids in urine, and normal thyroid stimulating hormone (TSH), coenzyme Q10 and total and free carnitine blood levels. The lactate level was repeated at 1 year 8 months and was at 29.1 mg/dL. The patient was started on L-carnitine and coenzyme Q10 and Vitamin B2.
By the age of 20 months, he started to stand-up correctly and to walk assisted and started to show better verbal interaction. At 2 years of age, he started walking alone and showed normal muscle tone. At this point in time, he was able to say five words. Six months later, he started climbing stairs and opening doors easily, and was able to say up to 10 clear words. At 3 and a half years, his occipito-frontal circumference (OFC) was 46.5 cm (<3rd centile), his height 96.5 cm (25th centile), and his weight 15 kg (25th centile). He could say more than 20 words and was able to express himself correctly with gestures.
Brain MRI showed mild enlargement of the body of both lateral ventricles, loss of periventricular white matter with high signal intensity on T2-weighted fluid-attenuated inversion recovery (T2/FLAIR), and thinning of the corpus callosum.
Molecular Analysis
Informed consent for genetic analysis was obtained from the family in compliance with national ethics regulation. Genomic DNA was isolated from the patient's blood sample using standard techniques.
Approximately 37 Mb (214,405 exons) of the consensus coding sequences (CCS) were enriched from fragmented genomic DNA by more than 340,000 probes designed against the human genome (Nextera Rapid Capture Exome; Illumina, CA, United States). The generated library was sequenced on an Illumina NextSeq platform (Illumina) to an average coverage depth of 70 to 100×. An end to end in-house bioinformatics pipeline, including base calling, primary filtering of low quality reads and probable artifacts, and annotation of variants was applied. All disease-causing variants reported in HGMD, ClinVar or CentoMD (class 1) as well as all variants with minor allele frequency (MAF) of less than 1% in ExAc database were considered. Evaluation was focused on exons and intron boundaries ± 20.
Whole exome sequencing revealed the presence of a homozygous variant in the patient in the TRAPPC6B gene. This NM_001079537.1:c.23T > A substitution was predicted to result in an early truncation of the protein (p.Leu8*). Sanger sequencing confirmed the presence of this variant in homozygous state in the patient and heterozygous state in the mother ( Fig. 1B ).
Discussion
Members of the TRAPP (transport protein particle) family of protein form multisubunit complexes that have been shown to function principally in membrane trafficking, autophagy, and mitosis. 4 5 The 12 human TRAPP proteins associate in two separate complexes, TRAPP II and TRAPP III, with a common core of proteins in each complex. Mutations in individual TRAPP genes have been shown to be associated with a diverse range of disorders, ranging from skeletal dysplasia ( TRAPPC2 ), and muscular dystrophy ( TRAPPC11 ) to neurodevelopmental delay and brain abnormalities. 6 The latter phenotype has been reported in patients with mutations in TRAPPC9 , 7 8 TRAPPC6A , 9 TRAPPC6B , 10 and TRAPPC2L 11 genes. The TRAPPC6B protein demonstrates an α/β-plait topology, capable of forming both homo and heterodimers. 12 In fact, a TRAPPC6-BET3 heterodimer could very likely function as the starting point of the TRAPP complex assembly at the Golgi membrane. 12 13
Mutations in the TRAPPC6B gene have so far been reported in only a handful of families worldwide. The first of these was a substitution mutation involving the splice site (c.150–2A > G), which was reported in three unrelated Egyptian consanguineous families affected by neurodevelopmental disorder with microcephaly, epilepsy, and brain atrophy (NEDMEBA; OMIM: 617862). 10 This is an extremely rare autosomal recessive disorder characterized by global developmental delay, severe intellectual disability and speech delay, microcephaly, stereotypic autistic traits, and early-onset generalized seizures. In addition, another report identified a separate homozygous nonsense mutation (c.124C > T) in this gene in a patient with intellectual disability in the absence of any obvious dysmorphic features or comorbidities. 14 Similar to the Egyptian patients, the patient reported in our study also shows a phenotype of microcephaly from birth, global developmental delay, intellectual disability, severe speech delay, global hypotonia, and thin corpus callosum on MRI. Interestingly, our patient did not show any autistic features or seizures ( Table 1 ).
Table 1. Clinical comparison between all patients reported to date with TRAPPC6B mutations .
| Patient | Age at diagnosis (y) | Microcephaly | ID | Autistic features | Hypotonia | Ataxia | Seizures | Lactic acidosis | MRI findings | TRAPPC6B mutation | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 8 | + | + | + | + | + | + | NA | NA | p.E28VfsTer11 | 10 |
| 2 | 3 | + | + | + | + | + | + | NA | NA | p.E28VfsTer11 | 10 |
| 3 | 12 | + | + | + | + | ++ | + | NA | Cortical atrophy, thin corpus callosum, cerebellar atrophy; all progressive | p.E28VfsTer11 | 10 |
| 4 | 10 | + | + | + | + | ++ | + | NA | Cortical atrophy, thin corpus callosum, cerebellar atrophy | p.E28VfsTer11 | 10 |
| 5 | 10 | + | + | + | + | + | + | NA | Thin corpus callosum | p.E28VfsTer11 | 10 |
| 6 | 2 | + | + | + | + | + | + | NA | Cortical atrophy, thin corpus callosum | p.E28VfsTer11 | 10 |
| 7 | NA | – | + | – | – | – | – | – | NA | p.Arg42Ter | 14 |
| 8 | 3.5 | + | + | − | + | − | – | + | Thinning of corpus callosum, periventricular leucomalacia | p.Leu8Ter | Present study |
Abbreviation: ID, intellectual disability.
Note: + indicates presence of indicated sign, ++ indicates presence of sign with increased severity, − indicates absence of indicated sign, and NA indicates nonavailability of information.
The c.23T > A substitution identified by WES in our study creates a premature stop codon resulting in a severely truncated protein. Mutation Taster classifies it as disease causing with a prediction probability of 1, indicating the highest security of prediction. combined annotation dependent depletion (CADD) predicted it to be deleterious with a phred-like scaled C-score of 37. The variant was not available on dbSNP, GnomAD, or ClinVar. In addition, the leucine residue affected here is fairly conserved across species ( Fig. 1C ), all of which point toward a likely pathogenic status for the variant. Both NEDMEBA mutations reported previously in this gene result in truncated proteins (p. Glu28Valfs*11 and p. Arg42*), albeit longer than the 8-residue peptide our variant generates. Functional studies on one of these variants using a zebrafish model showed a reduced head size, as well as neuronal hyperexcitability. 10 Considering that the variant we report here results in a severely truncated polypeptide, the lack of generalized seizures in our patient is noteworthy. However, all previously reported NEDMEBA patients reported with multifocal seizures have been older children, with the youngest being 8 years of age. At the time of reporting, our patient was 3.5 years of age, and he will be monitored in follow-up visits for generalized and focal seizures.
With the progressive reduction in the cost of high throughput molecular diagnostic techniques, their use in the clinic is increasingly becoming more and more common. This is especially true in the diagnosis of rare, monogenic neurological disorders. 15 16 Clear molecular diagnoses of these rare disorders will not only assist in understanding the pathogenesis of these disorders, but also help the families in planning for both managing the condition and future pregnancies.
Conflict of Interest None declared.
Ethical Approval
Informed and written consent was provided by the parents. The study protocol was approved by the institutional ethics committee and was in agreement with the guidelines of the Declaration of Helsinki.
References
- 1.Vasudevan P, Suri M. A clinical approach to developmental delay and intellectual disability. Clin Med (Lond) 2017;17(06):558–561. doi: 10.7861/clinmedicine.17-6-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiurazzi P, Pirozzi F. Advances in understanding - genetic basis of intellectual disability. F1000 Res. 2016;5:5. doi: 10.12688/f1000research.7134.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vickers R R, Gibson J S. A review of the genomic analysis of children presenting with developmental delay/intellectual disability and associated dysmorphic features. Cureus. 2019;11(01):e3873. doi: 10.7759/cureus.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sacher M, Jiang Y, Barrowman J et al. TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. EMBO J. 1998;17(09):2494–2503. doi: 10.1093/emboj/17.9.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J J, Lipatova Z, Segev N. TRAPP complexes in secretion and autophagy. Front Cell Dev Biol. 2016;4:20. doi: 10.3389/fcell.2016.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sacher M, Shahrzad N, Kamel H, Milev M P. TRAPPopathies: An emerging set of disorders linked to variations in the genes encoding transport protein particle (TRAPP)-associated proteins. Traffic. 2019;20(01):5–26. doi: 10.1111/tra.12615. [DOI] [PubMed] [Google Scholar]
- 7.Mochida G H, Mahajnah M, Hill A D et al. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am J Hum Genet. 2009;85(06):897–902. doi: 10.1016/j.ajhg.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hnoonual A, Graidist P, Kritsaneepaiboon S, Limprasert P. Novel compound heterozygous mutations in the TRAPPC9 gene in two siblings with autism and intellectual disability . Front Genet. 2019;10:61. doi: 10.3389/fgene.2019.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohamoud H S, Ahmed S, Jelani M et al. A missense mutation in TRAPPC6A leads to build-up of the protein, in patients with a neurodevelopmental syndrome and dysmorphic features. Sci Rep. 2018;8(01):2053. doi: 10.1038/s41598-018-20658-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marin-Valencia I, Novarino G, Johansen A et al. A homozygous founder mutation in TRAPPC6B associates with a neurodevelopmental disorder characterised by microcephaly, epilepsy and autistic features . J Med Genet. 2018;55(01):48–54. doi: 10.1136/jmedgenet-2017-104627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milev M P, Graziano C, Karall D et al. Bi-allelic mutations in TRAPPC2L result in a neurodevelopmental disorder and have an impact on RAB11 in fibroblasts . J Med Genet. 2018;55(11):753–764. doi: 10.1136/jmedgenet-2018-105441. [DOI] [PubMed] [Google Scholar]
- 12.Kümmel D, Müller J J, Roske Y, Misselwitz R, Büssow K, Heinemann U. The structure of the TRAPP subunit TPC6 suggests a model for a TRAPP subcomplex. EMBO Rep. 2005;6(08):787–793. doi: 10.1038/sj.embor.7400463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kümmel D, Müller J J, Roske Y, Henke N, Heinemann U. Structure of the Bet3-Tpc6B core of TRAPP: two Tpc6 paralogs form trimeric complexes with Bet3 and Mum2. J Mol Biol. 2006;361(01):22–32. doi: 10.1016/j.jmb.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 14.Harripaul R, Vasli N, Mikhailov A et al. Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry. 2018;23(04):973–984. doi: 10.1038/mp.2017.60. [DOI] [PubMed] [Google Scholar]
- 15.Klein C J, Foroud T M. Neurology individualized medicine: when to use next-generation sequencing panels. Mayo Clin Proc. 2017;92(02):292–305. doi: 10.1016/j.mayocp.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Foo J N, Liu J J, Tan E K. Whole-genome and whole-exome sequencing in neurological diseases. Nat Rev Neurol. 2012;8(09):508–517. doi: 10.1038/nrneurol.2012.148. [DOI] [PubMed] [Google Scholar]