

Abstract
Oxidative stress aggravates brain injury following ischemia/reperfusion (I/R). We previously showed that ubiquilin-1 (Ubqln1), a ubiquitin-like protein, improves proteostasis and protects brains against oxidative stress and I/R induced brain injury. Here, we demonstrate that a small molecule compound, L-2-oxothiazolidine-4-carboxylic acid (OTC) that functions as a precursor of cysteine, upregulated Ubqln1 and protected cells against oxygen glucose deprivation-induced cell death in neuronal cultures. Further, the administration of OTC either at 1 hour prior to ischemia or 3 hours after the reperfusion significantly reduced brain infarct injury and improved behavioral outcomes in a stroke model. Administration of OTC also increased glutathione (GSH) level and decreased superoxide production, oxidized protein, and neuroinflammation levels in the penumbral cortex after I/R in the stroke mice. Furthermore, I/R reduced both Ubqln1 and the glutathione S-transferase protein levels, whereas OTC treatment restored both protein levels, which was associated with reduced ubiquitin-conjugated protein level. Interestingly, in the Ubqln1 knockout (KO) mice, OTC treatment showed reduced neuroprotection and increased ubiquitin-conjugated protein level when compared to the similarly treated non-KO mice following I/R, suggesting that OTC-medicated neuroprotection is, at least partially, Ubqln1-dependent. Thus, OTC is a potential therapeutic agent for stroke and possibly for other neurological disorders and its neuroprotection involves enhanced proteostasis.
Keywords: stroke, oxidative stress, proteostasis, cysteine precursor, ubiquilin-1
Introduction
Stroke is a major cause of cerebrovascular disease worldwide, and is associated with a high morbidity and mortality among patients [1]. Ischemic stroke is caused by cerebral thrombosis or embolism in a blood vessel [2], resulting in neuronal cells death and behavioral impairments. To date, tissue-type plasminogen activator (tPA) that breaks down the blood clots and thereby restores the blood flow is still the only Food and Drug Administration-approved treatment for the disorder. However, the utilization of tPA in ischemic stroke has narrow therapeutic time window [3–5]. Once stroke occurs, there is no other effective therapy available to attenuate brain tissue damage. As oxidative stress is a key mediator of cerebral ischemia/reperfusion (I/R)-caused injury, anti-oxidant therapy has long been considered as a means to reduce ischemic stroke-caused brain injury [6]. Despite numerous strategies and attempts to reduce oxidative stress, the clinical outcomes have been failed to translate those successes from bench to bedside. Thus, there is an urgent need to identify new therapeutic alternatives.
Glutathione (GSH), also known as the thiol tripeptide γ-L-glutamyl-L-cysteinyl-glycine, is the most important antioxidant synthesized in cells. It serves as a major mechanism for intracellular antioxidant defense against oxidative damage and maintaining redox homeostasis [7]. When GSH detoxifies free reactive oxygen species (ROS), GSH itself is converted to glutathione-disulfide (GSSG) (oxidized GSH) [8]. The depletion of GSH impairs mitochondrial ATP production and induces cell death signaling pathways [9]. Many studies have shown that glutathione antioxidant system plays an important role in cerebral ischemia [10]. GSH levels are decreased in diseases with oxidative stress, including stroke [11] and a low level of GSH in the brain can increase the risk for stroke [12, 13]. Conversely, restoring GSH levels is associated with reduced neuronal cell death and enhanced animal functional recovery following stroke [14]. Therefore, stabilizing intracellular GSH level is important for neuronal survival and functional recovery following ischemic stroke. However, GSH does not pass the brain-blood barrier easily [15]. To boost GSH level for neuroprotection, an alternative strategy should be employed.
Ubiquilin-1 (Ubqln1) is a ubiquitin-like protein that functions as a ubiquitin receptor that interacts with polyubiquitinated (polyUb) proteins and delivers them to the proteasome for degradation [16], thereby facilitating misfolded protein degradation [17]. In addition to its involvement in the proteasome degradation pathway, Ubqln1 is also reported to function as a molecular chaperone for specific proteins [18, 19] and involved in the autophagy-mediated protein degradation pathway [20, 21]. Our data have suggested that overexpression (OE) of Ubqln1 protects mouse brains against oxidative stress and ischemia/reperfusion (I/R) caused injury, whereas knockout of Ubqln1 exacerbates the injury [22], indicating that Ubqln1 is a therapeutic target. Accordingly, induction of Ubqln1 OE by small molecule compounds may be beneficial to the brains following I/R. To date, however, GM1 ganglioside that is neuroprotective in acute ischemic stroke [23] remains to be the only compound identified to upregulate Ubqln1 [24].
Here, we report identification of L-2-oxothiazolidine-4-carboxylic acid (OTC), a synthetic cysteine precursor, as both GSH booster and Ubqln1 inducer. We determined whether OTC protects neuroprotection in both in vitro and in vivo models of ischemic stroke. We also investigated whether OTC upregulates Ubqln1 and antioxidant enzymes glutathione S-transferase (GST) [25] in a mouse stroke model and whether OTC-mediated neuroprotection is through upregulation of Ubqln1 and improved proteostasis. Our data strongly support that OTC is an appealing therapeutic agent in ischemic stroke or possibly other neurological disorders.
Materials and Methods
Cell cultures, OTC and oxygen-glucose deprivation (OGD) treatments
Wild-type mouse striatal neural cell line (Coriell Institute for Medical Research, Camden, NJ) that was previously utilized in our experiments [26] were cultured in 12-well plates in the complete medium containing the Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (Life Technologies). After 24 h, the cells were treated with different concentrations (0, 5, 10, 20, 50, or 100 μM) of OTC to examine the effect of OTC on Ubqln1 expression. Alternatively, the cells were used for assessing the effect of OTC on OGD-caused neuronal death by replacing the complete medium with a glucose-free Hank’s Balance Salt Solution (HBSS) containing either 0, 5, 10, 20, 50, or 100 μM OTC. The cells were incubated in an oxygen-free chamber filled with 95% N2 and 5% CO2 at 37°C for 14 h prior to ATP assay.
Primary cortical neurons were prepared from postnatal day 0 of wild-type C57BL/6J mice and the procedures were approved by the Institutional Animal Care and Use Committee of the University of South Dakota. Briefly, cerebral cortex was collected and digested with 0.05% trypsin/EDTA at 37°C for 15 min. After centrifugation at 1000 × g for 3 min at room temperature, the supernatant was discarded. The tissue pellets were pipetted up and down several times in the primary neuronal culture medium (Neurobasal medium supplemented with 2% B27 supplement, 2 mM L-glutamine and penicillin/streptomycin) and filtered through a 70 μm nylon cell strainer. The cells were cultured in poly-ornithine-coated 12-well plates in the primary neuronal culture medium at 37oC in a 5% CO2 incubator for 7days before being used to determine the effect of OTC on Ubqlnl expression or on OGD-caused neuronal death, which was similar to the experiments with striatal neural cells described above.
RT-PCR
Following 20 μM OTC or vehicle treatment of striatal cells for 16 h, mRNA isolation was performed by using the TRI Reagent (Molecular Research Center, Cincinnati, OH, USA). Briefly, 1 ml TRI Reagent was added into 10 cm2 of culture dish. The lysate was incubated with 0.2 ml chloroform for 15 min and then centrifuged at 12,000 × g for 15 min at 4oC. The aqueous was collected and isopropanol was used to precipitate RNA. The RNA was washed with 75% ethanol before RNA concentration was determined. Reverse transcription to cDNA was performed with the RevertAid first strand cDNA synthesis kit (Thermo Fisher Scientific). All PCR reactions were conducted with a 2720 Thermo Cycle (Applied Biosystems, ThermoFisher Scientific). The primer set used to assess Ubqln1 expression was as follows. Ubqln1 forward primer: 5’ CGTCCTTAGTGAGCAGTT 3’; Ubqln1 reverse primer: 5’ CTGCATTTGTTGGAGGAAAG 3’. RT-PCR of 18S ribosome (18S-RIB) RNA was used as a loading control using the following primers. 18S-RIB forward primer: 5’-CTCAACACGGGAAACCTCAC-3’; 18S-RIB reverse primer: 5’-TGCCAGAGTCTCGTTCGTTAT-3’. The band intensities of the RT-PCR products were analyzed by using Image J software (NIH).
ATP measurement
ATP measurement was performed to evaluate cell viability using an ATP Bioluminescence assay kit CLS II (Sigma) as previously described [22].
Animals
Adult C57BL/6J male mice (8–12 weeks of age; mean body weight, 25 g) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Ubqlnl knockout (KO) mice and floxed mice (f/f) were generated in our lab and have been previously described [22]. Mice were housed in a 12-hour (h) light/dark cycle with access to food and water ad libitum throughout the experimental period. Sample size calculations and power analysis were performed using the statistical software Stata (StataCorp LP, College Station, TX, USA). Animals were randomly separated into the vehicle group, OTC treatment group, or sham group using an online tool (http://www.graphpad.com/quickcalcs/). All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of South Dakota and were in accordance with the National Institute of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Transient middle cerebral artery occlusion (MCAO)
The mice (2–3 months of age; mean body weight, about 25 g) were anesthetized with isoflurane anesthesia machine (E-Z Anesthesia) and subjected either to MCAO or sham operation. Transient focal cerebral ischemia was induced by transiently occluding the MCA using a modified intraluminal filament technique as previously reported [27, 28]. In brief, male mice were anesthetized with 2% isoflurane and temperature was maintained at 37.0 ± 0.3°C. After 1 h of MCAO, the occluding filament was withdrawn to allow blood reperfusion. Mice neurological score was evaluated using a 5-point scale as previously described [27]. Only the animals with scores between 2 and 3 were included in the experiments.
Administration of OTC
Drug treatment experiments were carried out in a blinded manner such that the experimenters did not know the identity of each agent. Different dosages of OTC (50, 100 or 150 mg/kg) were administered 1 h prior to or after MCAO via intravenous (i.v.) tail vein injection. Alternatively, OTC (100 mg/kg) was also administered at 3 h or 6 h after MCAO, in order to gain insight into the appropriate therapeutic window. Saline injections served as control.
2,3,5-Triphenyltetrazolium chloride (TTC) staining
At 48 h following I/R, mice brains were rapidly removed and stored at −20°C for 15 min to slightly harden the tissue. The brains were sliced into 2- mm thick coronal sections and then incubated with 2% 2,3,5- triphenyltetrazolium chloride (TTC, Sigma) to evaluate the infarct volume, as described previously [22]. The infarct volume was manually analyzed using the Image J software (National Institute of Health).
Neurobehavior assessment
Behavioral tests were performed to assess whether OTC promotes functional recovery after stroke. Mice were examined at 1, 3, 5 and 7 days following MCAO and received daily OTC injection (100 mg/kg). The modified neurological severity scores (mNSS) was graded on a scale of 0 to 18 (0, normal neurological behavior score; 18, maximal neurological deficit; 13–18, severe injury; 7–12, moderate injury; 1–6 mild injury) [29].
Measurement of reduced glutathione (GSH) and oxidized glutathione (GSSG)
Determination of GSH and GSSG levels were performed by a described method [30] with modification. Briefly, after 24 h following MCAO, the animals were sacrificed and brain tissues from the penumbral cortex were harvested. The tissues were homogenized in 1 mL buffer containing 0.1 M sodium phosphate, 5 mM EDTA (pH 8.0) and 1% HPO3. The homogenates were centrifuged at 10,000 × g at 4°C for 30 min to obtain supernatant. For the GSH assay, a total of 20 μg protein for each sample was incubated in a reaction buffer containing 0.1 M sodium phosphate buffer (pH 8.0), 5 mM EDTA and 1 mg/ml O-phthaldehyde for 15 min at room temperature, before fluorescence intensity was measured (excitation/emission, 350 nm/420 nm). For the GSSG assay, samples were incubated first with 0.04 M N-ethyleimide (NEM) for 30 min to interact with GSH present in samples. Then, a total 20 μg of the protein sample was incubated with 0.1 N NaOH, 5 mM EDTA, and 1 mg/ml O-phthaldehyde for 15 min at room temperature. Fluorescence was measured using the same conditions as described above.
Superoxide detection
After 24 h following MCAO, the animals were anesthetized and brains were quickly removed after perfusion with saline and fixed with 4% paraformaldehyde followed by dehydration with 10%, 20%, and 30% gradient sucrose solution. Coronal sections at the caudate level were cut into 10 μm thickness on a cryostat (Leica) at −20°C. Tissue slides were incubated with 5 μM dihydroethidium (DHE) for 30 min at 37 °C, and then washed with PBS. DHE was imaged via a fluorescence microscopy (Carl Zeiss). The intensity of DHE fluorescence in brain tissue was analyzed by Image-Pro Plus 6.0 (Media Cybernetics, Inc. Rockville, MD).
Western Blot Analysis
The brain samples (penumbral cortex) were collected at 24 h post-stroke and were used for Western blot analysis according to previously described method [22]. Primary antibodies used in the studies include anti-Ubqln1 (mouse) antibody (1:1000, Abcam), anti-Ubqln1 (human) antibody (1:1000, Life Technology), anti-GST antibody (1:1000, Cell Signaling), anti-polyUb antibody (1:1000, Cell Signaling), anti-K48-linked polyUb antibody (1:1000, Cell Signaling), and anti-actin (1:1000, Santa Cruz Biotechnology). Detection was performed using appropriate secondary antibodies conjugated with the infrared dyes (1:5000, LI-COR Inc., Lincoln, NE, USA). Protein band intensities were measured using an Odyssey scanner (LI-COR) and quantified using UN-SCAN-IT gel 6.1 software (Silk Scientific Inc., Orem, Utah, USA).
Detection of oxidative protein damage
To detect changes in oxidative modification of proteins, the penumbral cortex was homogenized in ice-cold extraction buffer containing protease and phosphatase inhibitors. Protein carbonyl groups were measured using the oxidized protein Western blot detection kit according to the manufacturer’s instructions (Abcam) [31].
Experimental design and statistical analysis
Number of animals and cell cultures for each experiment is specified in the figure legends and was justified using power analysis. Only male mice were used for Figures 3, 4, 5 and 6 and equal proportions of male and female pups were used for the experiments included in Figures 1 and 2. GraphPad Prism Statistical Software version 7.0 (La Jolla, CA, USA) was used for statistical analysis and graphical display. All numerical data were presented as mean ± SD or SEM. Significant differences between two groups were analyzed using Student’s t-test and those more than two groups were analyzed using one-way or two-way ANOVA followed by Tukey’s post hoc or Sidak multiple comparison analysis. p < 0.05 was considered statistically significant.
Figure 3.
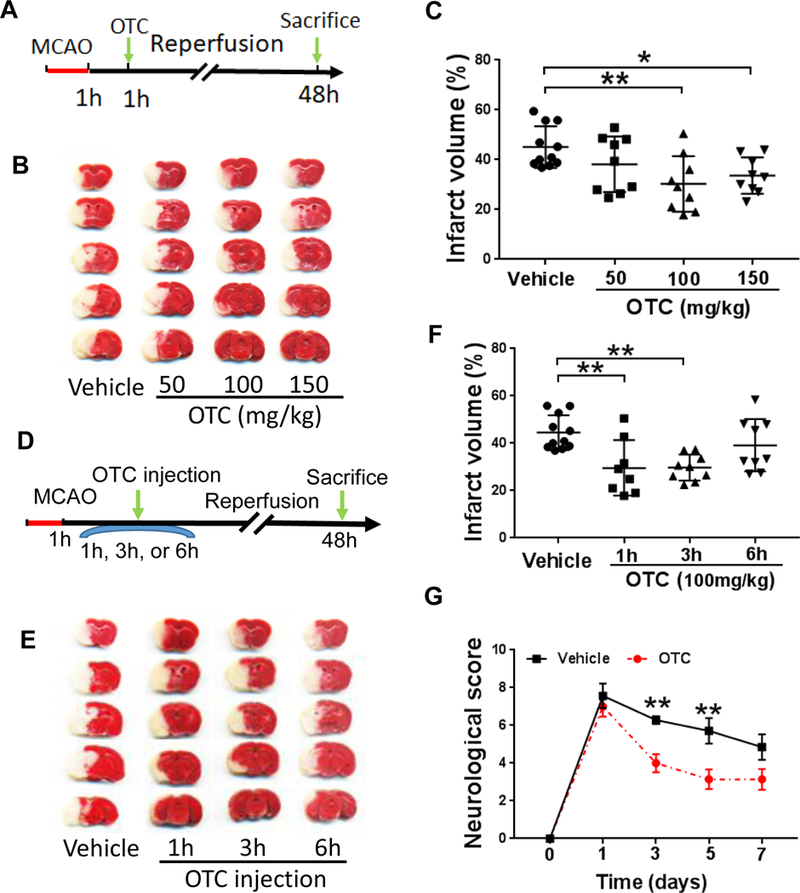
OTC reduces infarct volume and promotes functional recovery after MCAO
A. Diagram of the experimental design. OTC (50,100 or 150 mg/kg) was administered via the tail vein injection at 1 hour after reperfusion. Animals were sacrificed at 48 hours after ischemia and the brains were then isolated for TTC staining.
B. TTC staining of mouse brains after MCAO.
C. Quantitative analysis of TTC staining of mouse brains. Data are shown as mean ± SD. N = 9 mice for each OTC treated group; n = 11 for the vehicle treated group. p = 0.0090, F(3, 34) = 4.517, one-way ANOVA with Tukey’s multiple-comparison test: p Vehicle vs otc (100mg/kg) = 0.0043, p Vehicle vs OTC (150mg/kg) = 0.0304.
D. Diagram of the experimental design. OTC (100 mg/kg) was administered via the tail vein injection at 1 hour, 3 hours or 6 hours after ischemia. Animals were sacrificed at 48 hours and brains were then isolated for TTC staining.
E. Representative images of TTC-stained mouse brains post-treated with 1GG mg/kg OTC or vehicle at different time points respectively.
F. Quantitative analysis of infarct volume of the mice brain. (n = 9 mice per condition, except for mice treated with vehicle for which n = 11 and those treated with OTC n = B). p = 0.0017, F(3, 33) = 6.314, one-way ANOVA with Tukey’s multiple-comparison test: p Vehicle vs 1 hour injection = 0.0065, p Vehicle vs 3 hours injection) = 0.0052.
G. Behavior test of mice after MCAO. OTC-treated mice show better neurobehavioral performance than the vehicle-treated mice. Data are shown as mean ± SEM, n = 7 for each group; p = G.G536, F(4, 60) = 2.477, two-way ANOVA with Sidak’s multiple-comparison test. For day 3, p Vehicle vs OTC = 0.0094, day 5, p Vehicle vs OTC = 0.0027. **p < 0.01.
Figure 4.
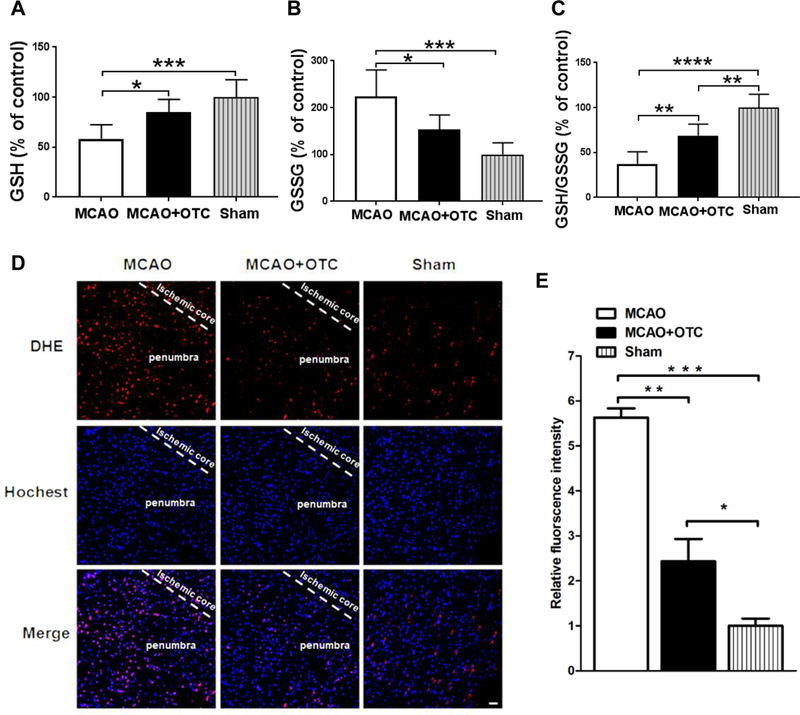
OTC treatment decreased the production of superoxide fluorescence and enhanced GSH antioxidant system in the penumbral cortex after I/R
A. GSH levels were significantly decreased in the penumbral cortex 24 h after ischemic stroke. OTC treatment restored depleted GSH levels. Data are expressed as percentage of control (sham); n = 6 per group. p = 0.0008, F(2, 15) = 11.83, one-way ANOVA with Tukey’s multiple-comparison test: p MCAO vs MCAO + OTC = 0.0208, p MCAO vs sham = 0.0006. Bar heights represent mean. Error bars indicate SD.
B. Elevated GSSG levels were antagonized by OTC treatment. Data are expressed as percentage of control (sham). N = 6, p = 0.0003, F(2, 15) = 14.47, one-way ANOVA with Tukey’s multiple-comparison test: p MCAO vs MCAO + OTC = 0.0215, p MACO vs sham = 0.0002. Bar heights represent mean. Error bars indicate SD.
C. The GSH/GSSG ratio was significantly reduced following MCAO, whereas OTC administration ameliorated the reduction. Data are expressed as percentage of control (sham). n = 6, p <0.0001, F(2, 15) = 31.62, one-way ANOVA with Tukey’s multiple-comparison test: p MCAO vs MCAO + OTC = 0.0031, p MCAO vs sham < 0.0001, p MCAO + OTC vs Sham = G.GG35. Bar heights represent mean. Error bars indicate SD.
D. Fluorescence micrographs of the brain slice in sham, vehicle and OTC groups. Scale bar, 20 μm.
E. Semi-quantitation of DHE fluorescence intensity. Data are expressed as relative fluorescence intensity to sham mice. n = 3, p = 0.0001, F(2, 6) = 53.96, one-way ANOVA with Tukey’s multiple-comparison test: p MCAO vs MCAO + OTC = 0.0010, p MCAO vs sham = 0.0001, p MCAO + OTC vs Sham = 0.0455. Bar heights represent mean. Error bars indicate SD.
Figure 5.
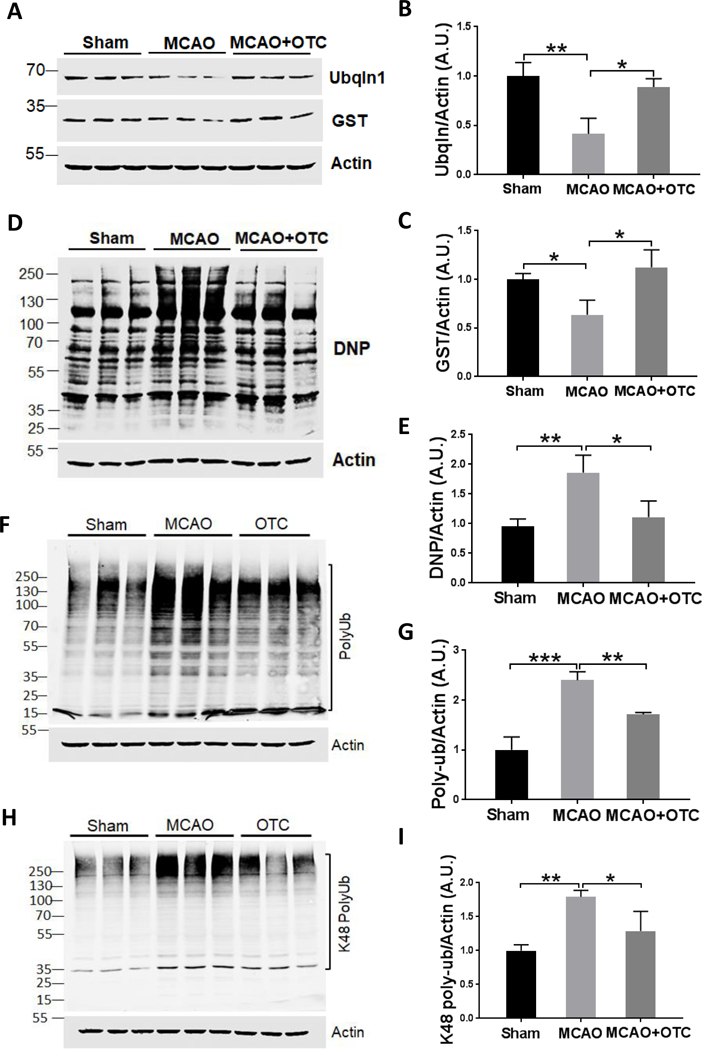
OTC treatment increases the expression of Ubqlnl and decreases the oxidized and polyUb protein level in the brain after MCAO
A. Western blot analysis showed that OTC increased the expression levels of Ubqlnl and GST in the brain of mice after MCAO.
B & C. Quantitative analysis of Ubqlnl (B) and GST (C) level shown in (A). Data are expressed as fold change relative to sham mice after normalization to β-actin.B. Ubqln/actin: p = 0.00134, F(2, 6) = 17.01, one-way ANOVA with Tukey’s multiple-comparison test: p Sham vs MCAO = 0.0037, p MCAO vs MCAO + OTC = 0.0102.C. GST/actin: p = 0.0127, F(2,6) = 9.85, one-way ANOVA with Tukey’s multiple-comparison test: p Sham vs MCAO = 0.012, p MCAO vs MCAO + OTC) = 0.0125. Bar heights represent mean. Error bars indicate SD.
D. OTC decreased the oxidized protein generation in the brain of mice. Protein carbonyl contents, as reflected by the DNP (Dinitrophenyl) level, are shown by western blotting.
E. Quantification of carbonyl contents. Data are expressed as fold change relative to sham mice after normalization to actin. p = 0.0081, F(2, 6) = 11.91, one-way ANOVA with Tukey’s multiple-comparison test: p sham vs MCAO = 0.0092, p MCAO vs MCAO+OTC = 0.0213. Bar heights represent mean. Error bars indicate SD.
F. Western blot results showed that OTC reduces the polyUb protein accumulation in the brains of mice.
G. Quantification of polyUb protein levels. Data are expressed as fold change relative to sham mice after normalization to actin. p = 0.0002, F(2, 6) = 44.69, one-way ANOVA with Tukey’s multiple-comparison test. p sham vs MCAO = 0.0002, p MCAO vs MCAO + OTC = 0.0085. Bar heights represent mean. Error bars indicate SD.
H. Western blot results showing that OTC reduces the K48-linked polyUb protein accumulation in the brains of mice.
I. Quantitative analysis of K48-linked polyUb protein levels. Data are expressed as fold change relative to sham mice after normalization to actin. p =0.0049, F(2, 6) = 14.64, one-way ANOVA with Tukey’s multiple-comparison test. p sham vs MCAO = 0.0042, p MCAO vs MCAO + OTC = 0.0338. Bar heights represent mean. Error bars indicate SD.
Figure 6.
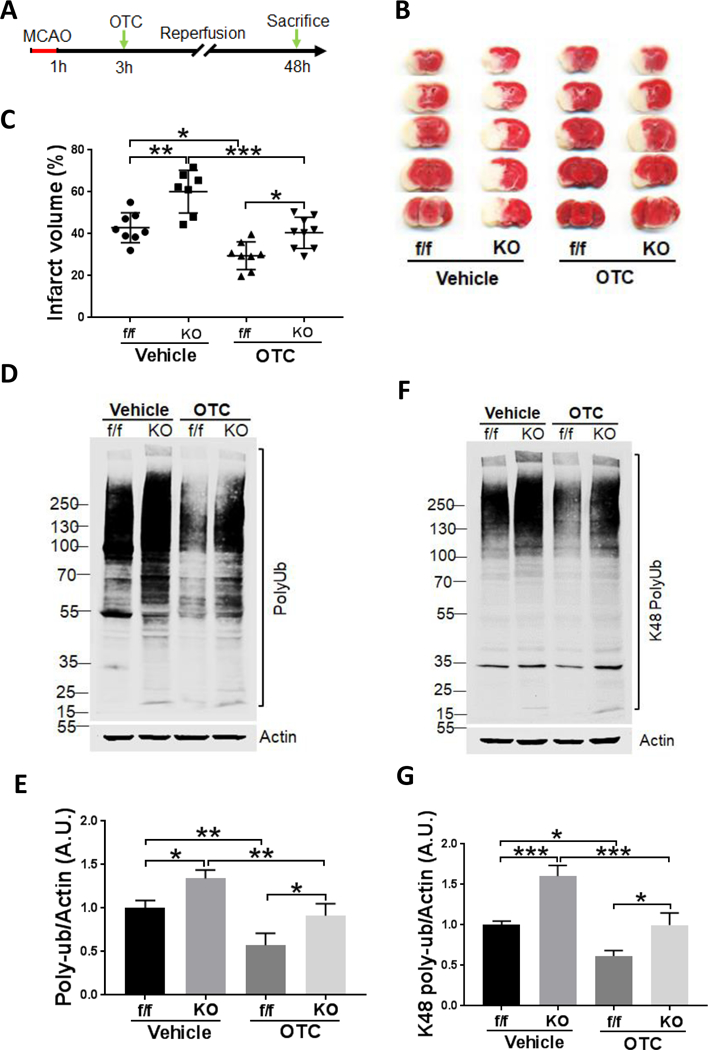
OTC-mediated neuroprotection in mouse brain at least partially relies on Ubqln1 expression
A. Schematic procedure for MCAO and administration of OTC in the Ubqln1 KO and floxed control (f/f) mice.
B. TTC staining of mice brain.
C. Quantitative analysis of brain infarct area. N = 8 mice per condition (except for KO mice treated with vehicle, n = 7, and the KO mice treated with OTC, n = 9). p < 0.0001, F(3, 28) = 19.17, one-way ANOVA with Tukey’s multiple-comparison test: p f/f (Vehicle) vs KO (Vehicle) = 0.0012, p f/f (Vehicle) vs f/f (OTC) = 0.0105, p KO (Vehicle) vs KO (OTC) = 0.0002, p f/f (OTC) vs KO (OTC) = 0.0374. Data are shown as mean ± SD.
D. Western blot analysis of polyUb protein levels in KO and non-KO (floxed, f/f) mice following I/R and OTC treatments.
E. Quantitation of polyUb protein levels shown in (D). Data are expressed as fold change relative to vehicle-treated f/f mice after normalization to actin. p = 0.0003, F(3, 8) = 21.48, one-way ANOVA with Tukey’s multiple-comparison test: p f/f (Vehicle) vs KO (Vehicle) = 0.0310, p f/f (Vehicle) vs f/f (OTC) = 0.0095, p KO (Vehicle) vs KO (OTC) = 0.0090, p f/f (OTC) vs KO (OTC) = 0.0325. Data are shown as mean ± SD.
F. Western blot analysis of K48-linked polyUb protein levels in KO and non-KO (floxed, f/f) mice following I/R and OTC treatments.
G. Quantitation of K48-linked polyUb protein levels shown in (F). Data are expressed as fold change relative to vehicle-treated f/f mice after normalization to actin. p < 0.0001, F(3, 8) = 43.18, one-way ANOVA with Tukey’s multiple-comparison test: p f/f (Vehicle) vs KO (Vehicle) = 0.0006, p f/f (Vehicle) vs f/f (OTC) = 0.0102, p KO (Vehicle) vs KO (OTC) = 0.0006, p f/f (OTC) vs KO (OTC) = 0.0102. Data are shown as mean ± SD.
Figure 1.
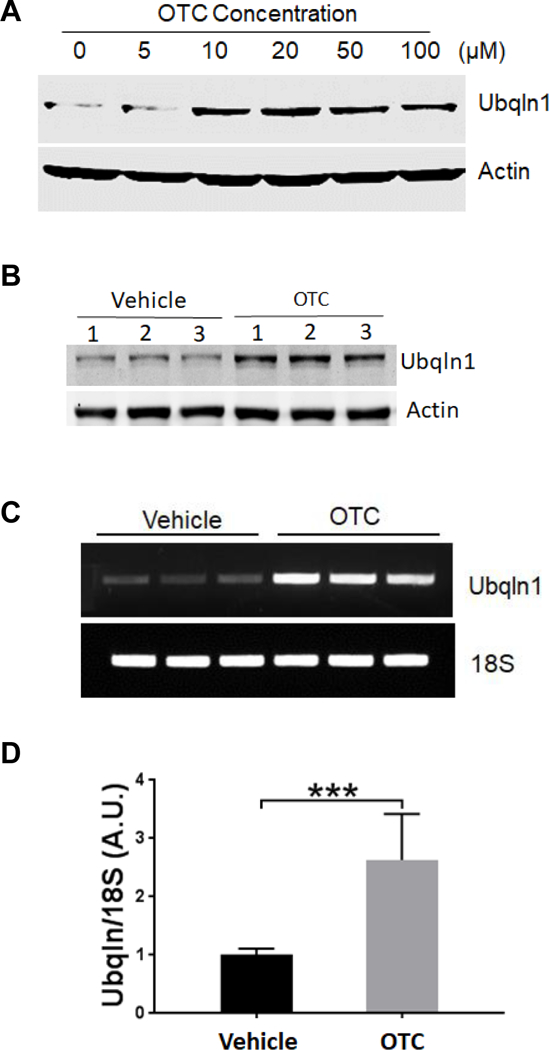
OTC upregulates Ubqlnl expression in neuronal cultures
A. Dose-dependent effect of OTC on Ubqlnl expression in striatal neural cell culture. The cell was treated with the indicated concentrations of OTC for 16 h before collected for Western blot analysis of Ubqlnl protein levels. Actin was used as a loading control.
B. OTC upregulates Ubqlnl expression in primary mouse cortical neuronal cultures. The neurons were treated with 25 μM OTC for 16 h before collected for Western blot analysis of Ubqlnl protein levels.
C. OTC upregulates Ubqlnl mRNA. A semi-quantitation of RT-PCR was utilized to measure Ubqlnl mRNA level from striatal neural cells treated with vehicle or 20 μM of OTC for 16 h.
D. Quantitation of (C). Data are shown as mean ± SD; N = 3. ***p < 0.00l (two-tailed paired Student’s t-test).
Figure 2.
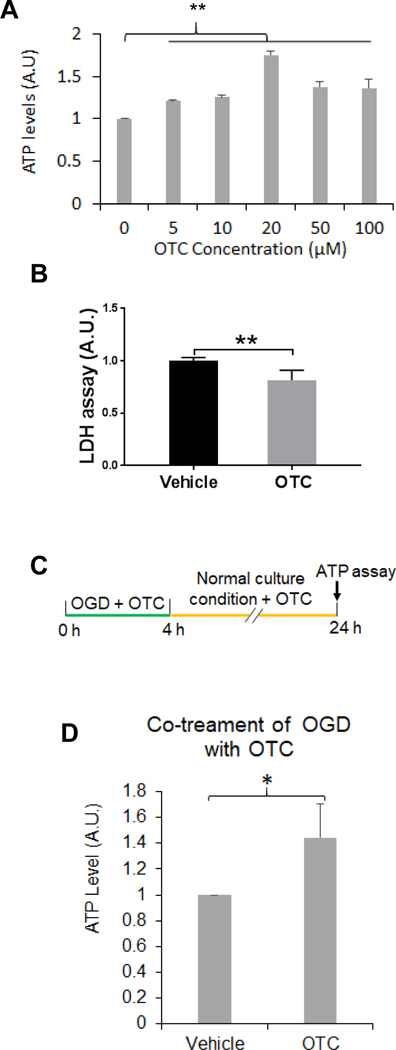
OTC protects neuronal cultures against OGD-caused cell injury
A. OTC increases striatal cell viability following OGD treatment. The striatal cells were incubated with a buffer free of glucose containing the indicated concentrations of OTC in a chamber free of oxygen. After l4 h, the cells were collected for measuring ATP levels. Data are shown as mean ± SD. N = 3; ** p < 0.0l, F(5, 12) = 66.67, one-way ANOVA with Tukey’s multiple-comparison test: p 0 μM vs 5 μM = 0.003, p 0 μM vs l0 μM = 0.0007, p 0 μM vs 20 μM < 0.000l.
B. OTC decreases OGD-induced LDH release. Striatal cells were treated with OGD in presence of 20 μM of OTC or vehicle and after 14 h, LDH release assay was performed. Data are shown as mean ± SD, n = 3; ** p < 0.01 (two-tailed paired Student’s t-test).
C. Diagram of the experimental design for co-treatment of OGD with OTC as shown in (D). Primary mouse cortical neurons were treated with OGD along with 20 μM OTC and after 4 h, the cells were cultured in the normal condition and continued to receive 20 μM OTC treatment until 24 h before ATP was measured.
D. Co-treatment of OGD with OTC increases neuronal viability in primary mouse cortical neuronal culture. Data are shown as mean ± SD, n = 3; p = 0.0413, t(4) = 2.966, two-tailed unpaired Student’s t-test.
Results
OTC upregulates Ubqln1
Our previous data have indicated that OE of Ubqln1 protects mouse brains against oxidative stress and ischemia/reperfusion (I/R) caused injury, whereas knockout of Ubqln1 exacerbates the injury [22]. Therefore, Ubqln1 is a therapeutic target and induction of Ubqln1 OE by small molecule compounds may be beneficial to the brains following I/R. To test this possibility, we performed a cell-based luciferase reporter assay to screen small molecule drug library of the NIH Clinical Collection (http://www.nihclinicalcollection.com) Set 1 containing a total of 446 compounds in order to upregulate Ubqln1 (data not shown). Among several final hits, one was a cysteine precursor, the OTC. To verify this, we treated the striatal cell culture with an increasing concentration of OTC and after 16 h, Ubqln1 protein levels were assessed by Western blot analysis. As shown in Fig. 1A, OTC exhibited a dose-dependent effect on inducing Ubqln1 expression. This result was further confirmed in the primary mouse cortical neuronal culture (Fig. 1B), which was approximately composed of 68% neurons and 32% non-neurons (*p < 0.05, Student’s t-test, Supplemental Figs. S-1A & S-1B). The upregulation of Ubqln1 was likely due to increased transcription of Ubqln1 gene, since our semi-quantitative RT-PCR results showed a remarked elevation of Ubqln1 mRNA level (Figs. 1C & 1D). Further, we performed an affinity purification assay by covalently conjugating OTC to the agarose beads. After incubation of immobilized OTC with mouse brain tissue lysates, the eluated proteins were subjected to mass spectrometry analysis. As shown in Supplemental Table-1, we identified numerous transcriptional factors. These data indicate that OTC upregulates Ubqln1 possibly through enhancing Ubqln1 transcription.
OTC increases cell viability following OGD treatment in cell cultures
Since our previous results have suggested that upregulation of Ubqln1 mediates neuroprotection, we therefore determined whether OTC treatment influences OGD-induced cell viability by culturing the striatal cells with different concentrations of OTC under an OGD condition. After 14 h following OGD, OTC enhanced cell viability (Fig. 2A) and reduced OGD-caused neuronal injury, as reflected by decreased LDH release (Fig. 2B). This was also confirmed in the primary mouse cortical neuronal culture (Figs. 2C & 2D), in which mouse cortical neurons were co-treated with both OGD and OTC (Fig. 1C). Furthermore, co-treatment of the neurons with both OGD and OTC also increased Ubqln1 level compared to the control treatment (Supplemental Fig. S-2). Conversely, primary cortical neuronal cultures derived from the Ubqln1 KO mice showed decreased viability following OGD, as reflected by a reduced ATP level compared to the wild-type control neurons (*p < 0.01, Student’s t-test, Supplementary Fig. S-3). Thus, our in vitro experimental results suggest that OTC increases cell viability following OGD treatment in neuronal cultures.
Pre- or post-MCAO treatment of OTC reduces neuronal death in stroke mice
We then examined whether pre-stroke treatment of OTC in a stroke mouse model alleviates brain injury. Accordingly, mice were intravenously (i.v.) injected with 50 mg/kg OTC 1 h prior to the MCAO procedure, and 24 h following I/R the mice were euthanized to assess brain infarct volume. The reason to sacrifice mice at 24 h is due to that infarct area reaches the peak at this time point, which remains stable until day 7 following the ischemia procedure [32]. In accordance with the in vitro results, pre-treatment of OTC significantly attenuated I/R-caused brain injury as shown by reduced infarct volume compared to the vehicle treatment (data not shown). To further verify whether post-ischemic treatment of OTC is also neuroprotective following MCAO, animals were treated (i.v.) with different doses of OTC at 1 h after ischemia (Fig. 3A). When the mice were treated with OTC at a dose of 100 mg/kg or 150 mg/kg, there was a significant reduction of infarct volume when compared to the vehicle-treated mice (*p < 0.05, Figs. 3B & 3C, one-way ANOVA with Tukey’s multiple-comparison test). Furthermore, mice were also treated with OTC (100 mg/kg) at 1, 3 or 6 h after ischemia to determine an optimal time window (Fig. 3D). Administration of OTC (100 mg/kg) at 3 h following ischemia is still sufficient to suppress I/R-induced brain injury (*p < 0.05, Figs. 3E & 3F, one-way ANOVA with Tukey’s multiple-comparison test), but this effect was lost at 6 h after ischemia (p > 0.05, Figs. 3E & 3F, one-way ANOVA with Tukey’s multiple-comparison test). Therefore, treatment of stroke mice with OTC (100 mg/kg) i.v. at 3 h after ischemia was utilized in all of the following in vivo experiments.
To determine whether OTC treatment improves animal functional recovery, mice were assessed by a modified neurological score system following the MCAO procedure [29, 33]. As shown in Fig. 3G, mice treated with OTC showed more rapidly functional recovery than those treated with vehicle: OTC significantly enhanced functional recovery after 3 days, which persisted until day 7, suggesting that post-treatment of OTC improves animal functional recovery following ischemic stroke. These results indicate that OTC ameliorates I/R-induced neuronal injury and promotes functional recovery in mice when administered within an appropriate window of time.
OTC increases the reduced GSH level and decreases the production of superoxide and GSSG levels in the mouse brain after MCAO
As OTC is a cysteine precursor and cysteine is essential for the biosynthesis of GSH, the most important antioxidant in the cells [34, 35], we next measured GSH and GSSG (oxidized GSH) levels from the penumbral cortex at 24 h after ischemia. We found that following ischemia, GSH significantly decreased in vehicle-treated mice, whereas OTC treatment rescued the reduced GSH (*p < 0.05, ***p < 0.001, Fig. 4A, one-way ANOVA with Tukey’s multiple-comparison test). Conversely, GSSG levels increased in the MCAO group, which was reversed by administration of OTC (*p < 0.05, ***p < 0.01, Fig. 4B, one-way ANOVA with Tukey’s multiple-comparison test). Consistently, the GSH/GSSG ratio, an indicator of cellular health [36], was decreased by ischemic stroke, but OTC treatment significantly restored that decreased GSH/GSSG ratio (**p < 0.01, ***p < 0.001, Fig. 4C, one-way ANOVA with Tukey’s multiple-comparison test). The increased level of GSH in the OTC-treated mouse brains after stroke was also reflected by decreased superoxide level. In the sham group of mice, the level of superoxide measured by the dihydroethidium (DHE) staining was low and few DHE positive cells were observed (Figs. 4D & 4E). However, a significant increase of red fluorescence intensity was detected in the peri-infarct region in both the OTC and vehicle-treated groups when compared with the sham group. OTC treatment markedly reduced the superoxide production when compared with vehicle treatment (*p < 0.05, **p < 0.01, *** p < 0.001, Figs. 4D & 4E, one-way ANOVA with Tukey’s multiple-comparison test). Moreover, OTC treatment also reduced the numbers of both GFAP- and Iba1-positive cells (*p < 0.05, student’s t-test, Supplementary Figs. S-4A, S-4B & S-4C), indicating that OTC might attenuate ischemic stroke-induced neuroinflammation. Thus, administration of OTC increases GSH level and decreases the free radical level and neuroinflammation in the brain following I/R. OTC treatment increases Ubqln1 level and improves proteostasis in mouse brain following I/R As OTC upregulates Ubqln1 expression in neuronal cell cultures, we next determined whether this also occurs in mouse brains. As shown in Figs. 5A & 5B, Ubqln1 was decreased in vehicle-treated mouse brains (MCAO group) following I/R (**p < 0.01, one-way ANOVA with Tukey’s multiple-comparison test), while OTC upregulated the level of Ubqln1 when compared with the sham group (*p < 0.05, one-way ANOVA with Tukey’s multiple-comparison test). Similar to Ubqln1, the anti-oxidant enzyme GST was also significantly decreased in MCAO group animals as compared with the sham group animals (*p < 0.05, Figs. 5A & 5C, one-way ANOVA with Tukey’s multiple-comparison test). However, OTC treatment completely restored GST level (*p < 0.05, Figs. 5A & 5C, one-way ANOVA with Tukey’s multiple-comparison test). Interestingly, Ubqln1 was found to co-immunoprecipitate with GST (Supplementary Fig. S-5A). OE of Ubqln1 appeared to stabilize GST level in cell culture (Supplementary Figs. S-5B, S-5C & S-5D). Additionally, we also examined the level of protein carbonyl content, a marker of protein oxidation [37]. Compared with sham group, the level of protein carbonyl content was significantly increased in the vehicle group after 24 h following I/R. In contrast, the level of protein carbonyl content in OTC treated mice was almost same to the sham group (**p < 0.01, Figs. 5D & 5E, one-way ANOVA with Tukey’s multiple-comparison test). In accordance with these results, OTC also decreased polyUb protein level (**p < 0.01, Figs. 5F & 5G; one-way ANOVA with Tukey’s multiple-comparison test). The polyUb proteins containing Ub-lysine (K)-48-linkage, which functions as the canonical signal targeting proteins for proteasomal degradation pathway [38], were also reduced (Figs. 5H & 5I). These data indicate that OTC upregulates Ubqln1 and GST in mouse brains and improves proteostasis following I/R.
OTC-mediated neuroprotection following MCAO is, at least partially, dependent on Ubqln1
Since OTC promotes GSH biosynthesis and upregulates Ubqln1 level, we finally determined to what extent OTC-conferred neuroprotection is due to upregulation of Ubqln1. We therefore investigated whether KO of Ubqln1 in mice alters OTC-induced neuroprotection following MCAO. We used both Ubqn1 KO and the Ubqln1-floxed (f/f) mice [22] and treated them with OTC (100 mg/kg) at 3 h after MCAO (Fig. 6A). The KO mouse brains showed similar anatomical structures compared to the WT mouse brains (Supplementary Fig. S-6). Following MCAO, Ubqln1 KO mouse brains showed an equal blood flow reduction as the floxed mouse brains, whereas during reperfusion, blood flow in both types of mice returned to a similar value (Supplementary Fig. S-7). Among the vehicle treated mice, TTC staining showed that the infarct volume of KO mice was significantly increased when compared with that of the f/f mice (**p < 0.01; Figs. 6B and 6C, one-way ANOVA with Tukey’s multiple-comparison test). OTC treatment further decreased I/R-caused infarct volume in both in f/f and KO mice when compared with the vehicle treated f/f and KO mice, respectively (*p < 0.05, ***p < 0.001; Figs. 6B and 6C, one-way ANOVA with Tukey’s multiple-comparison test). However, the infarct volume of the KO mice treated with OTC showed a significant increase when compared with that of f/f mice receiving the same treatment (*p < 0.05; Figs. 6B and 6C, one-way ANOVA with Tukey’s multiple-comparison test), indicative of requiring Ubqln1 expression. Interestingly, the polyUb protein level in mice was positively associated with neuronal injury. KO mouse brains showed a significantly increased-level of polyUb protein when compared to the f/f mouse upon either vehicle or OTC treatment (Figs. 6D & 6E). K48-linked polyUb protein levels in different groups of mice also showed the similar pattern as the polyUb (Figs. 6F & 6G). These results indicated that OTC-mediated neuroprotection in I/R is through both Ubqln1-dependent and Ubqln1-independent mechanisms that involve proteostasis.
Discussion
In this study, we demonstrated that OTC, a cysteine precursor, upregulates Ubqln1 and protects OGD- and I/R-induced neuronal death in vitro and in vivo, respectively. The most important results observed here are that the i.v. administration of OTC, even at 3 h after MCAO, is still neuroprotective against I/R-induced neuronal injury, suggesting that this neuroprotective effect is in part dependent on Ubqln1 mediated proteostasis. Moreover, we also observed that OTC increases GSH, Ubqln1 and GST levels. Thus, OTC may be a potential therapeutic agent for stroke and possibly for other neurological disorders through upregulating the antioxidants, GSH, GST and Ubqln1, and therefore improving proteostasis.
The neuroprotective effects of OTC on ischemic stroke mice have dual implications in human health. On one hand, the discovery that pre-ischemic treatment of OTC in mice reduces I/R-caused neuronal injury provides a scientific basis for OTC to be used to prevent stroke-caused health problem. For instance, it may be necessary to further study whether diets supplemented with OTC prevent or alleviate ischemic stroke-caused health problems in humans. On the other hand, as our data shown that post-ischemic treatment of OTC, even at 3 h after MCAO, not only reduces neuronal injury but also promotes brain functional recovery, this provides strong evidence for OTC to be used in treating stroke patients. However, ischemic stroke is highly complicated and heterogeneous [39], but we here only utilized young male mice in our experiments. It remains unknown whether OTC confers a similar neuroprotective effect on aged ischemic brains or on the permanent occlusion condition, as a large number of human ischemic stroke patients are in these conditions. Thus, it is necessary to perform clinical studies to determine the therapeutic efficacy of OTC in different ischemic stroke conditions. Given the nature of OTC being a cysteine precursor, it should be safe to conduct the study in the patients.
After I/R, the blood-brain barrier is disrupted [40] and thereby, OTC, a small molecule compound, should be able to readily penetrate the barrier and infiltrate into the brain, where it is converted into cysteine, an essential substrate for the biosynthesis of GSH, an antioxidant [41]. Cysteine is a sulfur-containing amino acid that contributes to the sulfhydryl group in the GSH molecule, the most important small molecular antioxidant in the cells [7]. This makes cysteine the limiting factor for GSH biosynthesis. However, GSH level decreases in the brain following ischemia [42], which is closely related to increased ROS, peroxynitrite, lipid peroxides and reactive hydroxyl radicals [43] and leads to neuronal cell death [44, 45]. In accordance with the previous observation of reduced GSH level in the brain after I/R, our results showed that OTC treatment restored GSH levels and maintained a proper GSH/GSSG ratio in the penumbral cortex of MCAO mice. These results were further supported by reduced superoxide and oxidized protein levels in the OTC-treated mouse brain following I/R, indicating that OTC reduces oxidative stress-caused protein damages.
Our data also support that OTC also upregulates other two antioxidant proteins, Ubqln1 [22] and GST [46], while both protein levels were decreased in the penumbral cortex following I/R in the vehicle-treated mice. The increased GST caused by OTC is likely through Ubqln1, as our in vitro studies showed that Ubqln1 appears interacting with GST and OE of Ubqln1 increased GST level. Moreover, our results from the Ubqln1 KO mouse studies also proved that Ubqln1, at least partially, mediates the neuroprotective effect exerted by OTC in the brain of mice after MCAO.
Accordingly, our data support that OTC plays dual roles following I/R. On one hand, it increases the level of the small molecular antioxidant, GSH. On the other hand, it upregulates Ubqln1 that in turn increases the level of the antioxidant enzyme, GST. Additionally, Ubqln1 itself enhances the degradation of misfolded proteins [17] and protects neurons against oxidative stress- and I/R-caused injury via enhancing the proteostasis in the brain [22, 26]. Taken together, these different effects would result in improved proteostasis and neuroprotection (Fig. 7). Given the fact that ischemic stroke-induced protein damage and aggregation is associated with neuronal injury [47, 48], maintaining proteostasis may be extremely important for neuronal survival and recovery following I/R. Our data strongly support this possibility, as deletion of Ubqln1 gene, which involves the ubiquitin-proteasome pathway of protein degradation, causes increased neuronal injury and accumulation of damaged proteins following I/R.
Figure 7.
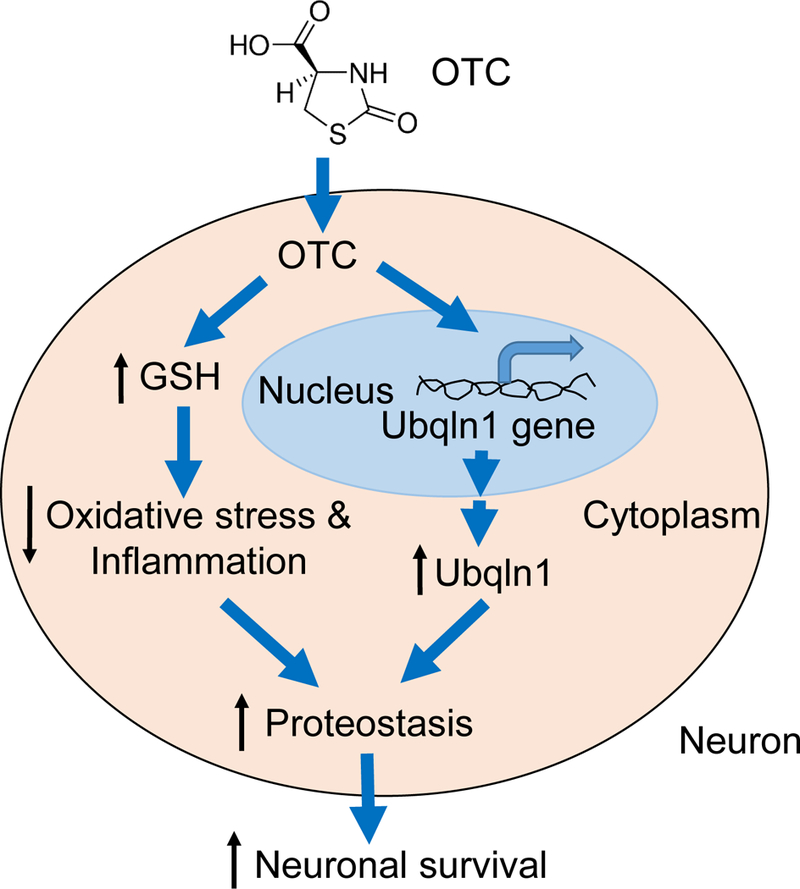
Hypothetical model for the effects of OTC-induced neuroprotection in mouse brains after MCAO. OTC plays dual roles after I/R, by increase of GSH level and by upregulation of Ubqlnl protein level. This leads to alleviated oxidative stress and improved proteostasis, resulting in neuroprotection.
In summary, we demonstrate that OTC upregulates Ubqln1 and protects neurons from OGD- and I/R-caused cell death in vitro and in vivo, respectively. Post-ischemic administration of OTC in a stroke mouse model even at 3 h following the I/R still decreases brain damage and improves functional recovery, suggesting that OTC may be used together with the tPA in the stroke patients. Moreover, we provide evidence showing that the mechanism of this protective effect is through an increase of GSH and upregulation of Ubqln1, resulting in reduced levels of oxidative stress and oxidized proteins. Our study suggests that OTC is potentially a therapeutic agent for the prevention and treatment of cerebral ischemic damage and possible other neurological disorders.
Supplementary Material
Acknowledgments
We would like to thank the Physiology Core Facility at the University of South Dakota (USD) Division of Basic Biomedical Sciences for access to equipment and assistance with data analysis, Mr. Doug Jennewein from the USD-IT Research Computing for help in the database installation and servers operation, and Mrs. Daniela Paez from the USD proteomics Core Facility for assistance in processing and data organization.
Funding: This study was funded by the National Institute of Neurological Disorders and Stroke under research grant NS088084 and by the National Institute of General Medical Sciences under the grant P20GM103443.
Footnotes
Compliance with Ethical Standards
Ethical approval: All applicable international, national, and/or institutional guidelines for the care and use of animals were followed in the study. This article does not contain any studies with human participants performed by any of the authors.
Conflict of interest: The authors declare that no conflict of interest exists.
References
- 1.Donnan GA, Fisher M, Macleod M, Davis SM: Stroke. Lancet 2008, 371(9624):1612–1623. [DOI] [PubMed] [Google Scholar]
- 2.Kasner SE, Grotta JC: Ischemic stroke. Neurol Clin 1998, 16(2):355. [DOI] [PubMed] [Google Scholar]
- 3.Barber PA, Zhang J, Demchuk AM, Hill MD, Buchan AM: Why are stroke patients excluded from TPA therapy? An analysis of patient eligibility. Neurology 2001, 56(8):1015–1020. [DOI] [PubMed] [Google Scholar]
- 4.Cronin CA: Intravenous Tissue Plasminogen Activator for Stroke: A Review of the Ecass Iii Results in Relation to Prior Clinical Trials. J Emerg Med 2010, 38(1):99–105. [DOI] [PubMed] [Google Scholar]
- 5.Armstead WM, Ganguly K, Kiessling JW, Riley J, Chen XH, Smith DH, Stein SC, Higazi AAR, Cines DB, Bdeir K et al. : Signaling, delivery and age as emerging issues in the benefit/risk ratio outcome of tPA For treatment of CNS ischemic disorders. JNeurochem 2010, 113(2):303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi HL, Liu KJ: Cerebral tissue oxygenation and oxidative brain injury during ischemia and reperfusion. Front Biosci-Landmrk 2007, 12:1318–1328. [DOI] [PubMed] [Google Scholar]
- 7.Forman HJ, Zhang H, Rinna A: Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 2009, 30(1–2):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC: Mitochondrial Glutathione, a Key Survival Antioxidant. AntioxidRedox Sign 2009, 11(11):2685–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Redza-Dutordoir M, Averill-Bates DA: Activation of apoptosis signalling pathways by reactive oxygen species. Biochim BiophysActa 2016, 1863(12):2977–2992. [DOI] [PubMed] [Google Scholar]
- 10.Schulz JB, Lindenau J, Seyfried J, Dichgans J: Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 2000, 267(16):4904–4911. [DOI] [PubMed] [Google Scholar]
- 11.Choi J, Liu RM, Kundu RK, Sangiorgi F, Wu W, Maxson R, Forman HJ: Molecular mechanism of decreased glutathione content in human immunodeficiency virus type 1 Tat-transgenic mice. J Biol Chem 2000, 275(5):3693–3698. [DOI] [PubMed] [Google Scholar]
- 12.Namba K, Takeda Y, Sunami K, Hirakawa M: Temporal profiles of the levels of endogenous antioxidants after four-vessel occlusion in rats. J Neurosurg Anesth 2001, 13(2):131–137. [DOI] [PubMed] [Google Scholar]
- 13.Park EM, Choi JH, Park JS, Han MY, Park YM: Measurement of glutathione oxidation and 8-hydroxy-2 ‘-deoxyguanosine accumulation in the gerbil hippocampus following global ischemia. Brain Res Protoc 2000, 6(1–2):25–32. [DOI] [PubMed] [Google Scholar]
- 14.Yabuki Y, Fukunaga K: Oral Administration of Glutathione Improves Memory Deficits Following Transient Brain Ischemia by Reducing Brain Oxidative Stress. Neuroscience 2013, 250:394–407. [DOI] [PubMed] [Google Scholar]
- 15.Smeyne M, Smeyne RJ: Glutathione metabolism and Parkinson’s disease. Free Radic Biol Med 2013, 62:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko HS, Uehara T, Tsuruma K, Nomura Y: Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett 2004, 566(1–3): 110–114. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Monteiro MJ: Ubiquilin interacts and enhances the degradation of expanded-polyglutamine proteins. Biochem Biophys Res Commun 2007, 360(2):423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stieren ES, El Ayadi A, Xiao Y, Siller E, Landsverk ML, Oberhauser AF, Barral JM, Boehning D: Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J Biol Chem 2011, 286(41):35689–35698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itakura E, Zavodszky E, Shao S, Wohlever ML, Keenan RJ, Hegde RS: Ubiquilins Chaperone and Triage Mitochondrial Membrane Proteins for Degradation. Mol Cell 2016, 63(1):21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J, Fang S, Cuervo AM, Nixon RA, Monteiro MJ: Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet 2010, 19(16):3219–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee DY, Arnott D, Brown EJ: Ubiquilin4 is an adaptor protein that recruits Ubiquilin1 to the autophagy machinery. EMBO Rep 2013, 14(4):373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Lu L, Hettinger CL, Dong G, Zhang D, Rezvani K, Wang X, Wang H: Ubiquilin-1 protects cells from oxidative stress and ischemic stroke caused tissue injury in mice. J Neurosci 2014, 34(8):2813–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oppenheimer S: GM1 ganglioside therapy in acute ischemic stroke. Stroke 1990, 21(5):825. [DOI] [PubMed] [Google Scholar]
- 24.Liu Z, Ruan Y, Yue W, Zhu Z, Hartmann T, Beyreuther K, Zhang D: GM1 up-regulates Ubiquilin 1 expression in human neuroblastoma cells and rat cortical neurons. Neurosci Lett 2006, 407(1):59–63. [DOI] [PubMed] [Google Scholar]
- 25.Ahmad A, Khan MM, Javed H, Raza SS, Ishrat T, Khan MB, Safhi MM, Islam F: Edaravone ameliorates oxidative stress associated cholinergic dysfunction and limits apoptotic response following focal cerebral ischemia in rat. Mol Cell Biochem 2012, 367(1–2):215–225. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Qiao F, Wang H: Enhanced Proteostasis in Post-ischemic Stroke Mouse Brains by Ubiquilin-1 Promotes Functional Recovery. Cell Mol Neurobiol 2016, 37(7):1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu L, Wang H: Transient focal cerebral ischemia upregulates immunoproteasomal subunits. Cell Mol Neurobiol 2012, 32(6):965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Min JW, Lu L, Freeling JL, Martin DS, Wang H: USP14 inhibitor attenuates cerebral ischemia/reperfusion-induced neuronal injury in mice. J Neurochem 2017, 2017,140(5) 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen JL, Zhang CL, Jiang H, Li Y, Zhang LJ, Robin A, Katakowski M, Lu M, Chopp M: Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cerebr BloodFMet 2005, 25(2):281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hissin PJ, Hilf R: A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem 1976, 74(1):214–226. [DOI] [PubMed] [Google Scholar]
- 31.Gilliam LAA, Lark DS, Reese LR, Torres MJ, Ryan TE, Lin CT, Cathey BL, Neufer PD: Targeted overexpression of mitochondrial catalase protects against cancer chemotherapy-induced skeletal muscle dysfunction. Am J Physiol-Endoc M 2016, 311(2):E293–E301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu F, Schafer DP, McCullough LD: TTC, fluoro-Jade B and NeuN staining confirm evolving phases of infarction induced by middle cerebral artery occlusion. J Neurosci Methods 2009, 179(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen JL, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M: Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke 2001, 32(11):2682–2688. [DOI] [PubMed] [Google Scholar]
- 34.Terrill JR, Boyatzis A, Grounds MD, Arthur PG: Treatment with the cysteine precursor l-2-oxothiazolidine-4-carboxylate (OTC) implicates taurine deficiency in severity of dystropathology in mdx mice. Int JBiochem Cell Biol 2013, 45(9):2097–2108. [DOI] [PubMed] [Google Scholar]
- 35.Bolling AK, Solhaug A, Morisbak E, Holme JA, Samuelsen JT: The dental monomer hydroxyethyl methacrylate (HEMA) counteracts lipopolysaccharide-induced IL-1beta release-Possible role of glutathione. Toxicol Lett 2017, 270:25–33. [DOI] [PubMed] [Google Scholar]
- 36.Owen JB, Butterfield DA: Measurement of oxidized/reduced glutathione ratio. Methods Mol Biol 2010, 648:269–277. [DOI] [PubMed] [Google Scholar]
- 37.Chevion M, Berenshtein E, Stadtman ER: Human studies related to protein oxidation: protein carbonyl content as a marker of damage. Free Radic Res 2000, 33 Suppl:S99–108. [PubMed] [Google Scholar]
- 38.Grice GL, Nathan JA: The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci 2016, 73(18):3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sommer CJ: Ischemic stroke: experimental models and reality. Acta Neuropathol 2017, 133(2):245–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang GY, Betz AL: Reperfusion-induced injury to the blood-brain barrier after middle cerebral artery occlusion in rats. Stroke 1994, 25(8):1658–1664; discussion 1664–1655. [DOI] [PubMed] [Google Scholar]
- 41.Yu X, Long YC: Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci Rep 2016, 6:30033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh S, Das N, Mandal AK, Dungdung SR, Sarkar S: Mannosylated Liposomal Cytidine 5 ‘ Diphosphocholine Prevent Age Related Global Moderate Cerebral Ischemia Reperfusion Induced Mitochondrial Cytochrome C Release in Aged Rat Brain. Neuroscience 2010, 171(4):1287–1299. [DOI] [PubMed] [Google Scholar]
- 43.Ansari MA, Ahmad AS, Ahmad M, Salim S, Youscuf S, Ishrat T, Islam F: Selenium protects cerebral ischemia in rat brain mitochondria. Biol Trace Elem Res 2004, 101(1):73–86. [DOI] [PubMed] [Google Scholar]
- 44.Ansari MA, Joshi G, Huang QZ, Opii WO, Abdul HM, Sultana R, Butterfield DA: In vivo administration of D609 leads to protection of subsequently isolated gerbil brain mitochondria subjected to in vitro oxidative stress induced by amyloid beta-peptide and other oxidative stressors: Relevance to Alzheimer’s disease and other oxidative stress-related neurodegenerative disorders. Free Radical Bio Med 2006, 41(11):1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie CS, Lovell MA, Markesbery WR: Glutathione transferase protects neuronal cultures against four hydroxynonenal toxicity. Free Radical Bio Med 1998, 25(8):979–988. [DOI] [PubMed] [Google Scholar]
- 46.Yang Y, Wang JY, Li Y, Fan CX, Jiang S, Zhao L, Di SY, Xin ZL, Wang BD, Wu GL et al. : HO-1 Signaling Activation by Pterostilbene Treatment Attenuates Mitochondrial Oxidative Damage Induced by Cerebral Ischemia Reperfusion Injury. Mol Neurobiol 2016, 53(4):2339–2353. [DOI] [PubMed] [Google Scholar]
- 47.Ge P, Luo Y, Liu CL, Hu B: Protein aggregation and proteasome dysfunction after brain ischemia. Stroke 2007, 38(12):3230–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo T, Park Y, Sun X, Liu C, Hu B: Protein misfolding, aggregation, and autophagy after brain ischemia. Transl Stroke Res 2013, 4(6):581–588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.