

Abstract
Interactions between cell surface glycans and glycan binding proteins (GBPs) have a central role in immune response, pathogen-host recognition, cell-cell communication, and myriad other biological processes. Because of the weak association between GBPs and glycans in solution, multivalent and cooperative interactions in the dense glycocalyx have an outsized role in directing binding affinity and selectivity. However, a major challenge in glycobiology is that few experimental approaches exist for examining and understanding quantitatively how glycan density affects avidity with GBPs, and there is a need for new tools that can fabricate glycan arrays with the ability to vary their density controllably and systematically in each feature. Here we use thiol-ene reactions to fabricate glycan arrays using a recently developed photochemical printer that leverages a digital micromirror device and microfluidics to create multiplexed patterns of immobilized mannosides, where the density of mannosides at each feature was varied by dilution with the inert spacer allyl alcohol. Association between these immobilized glycans and FITC-labelled concanavalin A (ConA) – a tetrameric GBP that binds to mannosides multivalently – was measured by fluorescence microscopy. We observed that fluorescence decreased nonlinearly with increasing spacer concentration in the features, and we present a model that relates average mannoside-mannoside spacing to the abrupt drop-off in ConA binding. Applying these recent advances in microscale photolithography to the challenge of mimicking the architecture of the glycocalyx could lead to a rapid understanding of how information is trafficked on the cell surface.
Keywords: multivalency, glycan microarrays, microfluidics, thiol-ene, photochemistry, surface science
Graphical Abstract
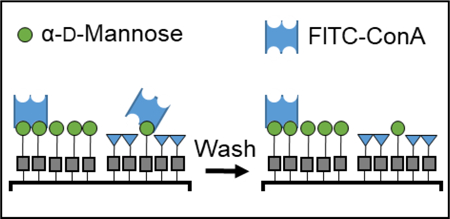
Variable density glycan microarrays were used to study the multivalent binding of lectins.
Introduction
Many biological processes, from immune response to host–pathogen interactions, are mediated by recognition between glycan binding proteins (GBPs) and glycans within the glycocalyx – the dense layer of glycolipids, glycoproteins, and glycopolymers on the surface of every eukaryotic cell.1-4 In this environment, multivalency, whereby contacts form between a GBP and multiple glycans simultaneously, plays an outsized role in determining substrate specificity and binding avidity.5-9 As a result of multivalency, avidity increases up to 106 M−1 are commonly observed between GBPs and substrates that present multiple glycans in close enough proximity to allow for multipoint binding, a phenomenon termed the “cluster glycoside effect”.10–13 Despite its central role in glycobiology, quantitative measures of multivalency, particularly in substrates that are designed to mimic the presentation of glycans on the cell surface, remains a significant and unresolved experimental challenge because of difficulties associated with surface immobilization chemistry, the paucity of lithographic methods that can print glycans with control over density, and unsatisfactory models for anticipating the impact of multivalent interactions on the surface.
Creating substrates that can systematically vary glycan presentation requires control over surface immobilization chemistry, while employing printing technologies that are compatible with delicate organic materials, like glycans. As such, strategies for controlling glycan surface presentation must consider both the printing method and the immobilization chemistry. The surface chemistry used to prepare the Consortium for Functional Glycomics glycan array, for example, involves pin-printing amino-functionalized glycans onto NHS-activated surfaces.14, 15 Other common immobilization chemistries involve functionalizing a substrate with a monolayer of N-hydroxysuccinimidyl ester to covalently immobilize 3’-amine oligonucleotides conjugated with glycans,16 epoxy–activated substrates that react with amino-functionalized glycans,17, 18 thiol-ene photochemical click reactions,19–23 patterning fluorous derivatized glycans onto commercially available Teflon/epoxy coated microscope slides,24 and immobilizing lipid-linked oligosaccharides onto nitrocellulose substrates.25, 26 Alternatively, the Gildersleeve Group conjugates glycans to bovine serum albumin (BSA), creating “neoglycoproteins” that are then themselves immobilized to epoxide coated glass substrates, where the glycan density can be manipulated by either mixing with non-conjugated BSA or varying the density of glycans within each neoglycoprotein.27–29 The patterning of glycans into arrays is typically accomplished by creating spots using an inkjet or pin-printer. 14 With these technologies, solutions of the appropriately functionalized glycans are deposited onto substrates functionalized with the complementary reactivity, resulting in features of covalently immobilized glycans with typical diameters of ~500 μm. These printing technologies are popular because they are non-destructive towards delicate glycans and compatible with the common immobilization reactions. However, other techniques could substantially reduce feature diameter, and thereby increase the number of spots per array and limit the amount of expensive glycan or GBP needed for microarray analysis. Some of these next-generation printing techniques that have been explored recently in the context of glycan microarrays include scanning-probe lithography,22, 30 microcontact printing,21, 31, 32 and photochemical patterning enabled by a digital micromirror device (DMD).20 Ideally, a glycan arraying platform – which constitutes immobilization chemistry and the lithography method for patterning – should be compatible with immobilization chemistry, create features with <100 μm diameters, and possess the ability to control glycan density within each feature.
Varying the density of glycans is particularly important for glycan microarrays because doing so is necessary for investigating how multipoint binding between GBP and a glycan-coated surface affects avidity. To this end, diverse approaches have been adopted to vary glycan density within the features of a microarray. Liang and coworkers, for example, studied the binding of Concanavalin A (ConA) to mannosides and oligomannosides that were immobilized onto a NHS-activated glass substrate that was prepared via robotic pin-printing.33 In this study, they varied the concentration of mannose in the printing solutions from 0.6 to 100 μM, and studied binding to the fluorophore-labelled lectin. They measured increased ConA avidity to the substrate at spots printed with higher concentration mannose solutions. The authors concluded that when the printing solutions drop to 1 μM and 0.6 μM, the mannosides were too widely spaced for ConA to achieve multivalent binding, which was necessary to obtain avidity sufficiently high for the GBP to remain on the surface. Alternatively, Oleyaran and coworkers fabricated variable glycan-density arrays with BSA-neoglycoproteins to investigate how multivalency affects GBP avidity.28 The binding of fluorescently labelled ConA to neoglycoproteins with systematically varying glycan (monomannose and oligomannose) concentration was assessed. The results of this experiment showed that as the ratio between mannose-conjugated neoglycoprotein and unmodified BSA changes from 1:0 to 1:7 in the printing solution, the spacing between glycans increased. Increased spacing between glycans resulted in a decrease of overall multivalent opportunities on the surface, thus decreasing the overall fluorescence of the feature, which they concluded was a consequence of the inability of ConA to bind multivalently as the monomannose density was reduced. However, at low concentrations of the oligomannose, ConA was still able to bind the features since multivalent opportunities persist. Concerned that the glycan monolayers that are typically used in glycan arrays bind GBPs weakly as a result of surface roughness, Godula and coworkers prepared glycopolymer microarrays. Attempting to mimic the natural presentation of mucins, which are highly glycosylated cell-surface proteins, they created heavily glycosylated synthetic brush polymers using reversible addition-fragmentation chain-transfer (RAFT) polymerization and printed them into microarrays.34, 35 Aminooxy labelled glycans were grafted at different densities onto polymer chains of different lengths to examine how density affected avidity. The affinities of four lectins (Soybean agglutinin (SBA), Wisteria floribunda lectin (WFL), Vicia villosa-B-4 agglutinin (VVA), and Helix pomatia agglutin (HPA)) to the glycosylated brush polymers were assessed. SBA, WFL, and VVA all displayed multivalent binding, as dissociation constants (Kd) decreased with increasing glycan grafting density. HPA, however, did not show any change in Kd despite the changes in glycan density, thus indicating that it did not associate to the brush polymer with avidity. Thus, glycan arrays are powerful platforms to assess the impact of multivalency on GBP binding, but despite these important contributions, there is still a need for techniques that can reduce the printing areas and systematically vary surface concentration.
Another challenge that arises in understanding these data is scaling changes in avidity to a molecular level understanding of multivalency, and, to this end, several models have been proposed. In a review on multivalency, Mammen et al. have suggested36 that the enthalpy of the system is dominated by strain on the multivalent ligands, while entropic changes are dependent upon perturbations to lectin conformation. Brewer and coworkers used isothermal titration calorimetry to investigate the thermodynamics of the binding between ConA and multivalent mannosides in solution.37 The results showed that as the number of mannosides on a ligand increases, the avidity to ConA also increases, and that the enthalpy of the system increased linearly with the number of mannosides. Brewer, also emphasized the importance of the entropy of the system by showing how the conformation of the lectin, SBA in this case, affects multivalency on a mucin. Because of the structure of SBA and the conformation when it binds to mucin galactose, SBA can “bind and jump” from one residue to the next.38 Houseman and coworkers argue that it is particularly important to study multivalent interactions on surfaces because of increased control over glycan spacing in monolayers, and immobilized ligands are in an environment that reduces nonspecific binding.39 Here we build upon these investigations to study ConA-mannoside multivalency by employing a new method for printing glycan microarrays, 20 which allows for the density of glycans within an array to be varied systematically. This printer combines microfluidics, a digital micromirror device, and a reactive surface to control the ratio of glycan and spacer within a feature. We use this tool to print features of variable-density mannosides onto a surface and study their binding to solutions with different concentrations of FITC-labelled ConA. Reduced feature dimensions provides a route to take multiple readings at each binding condition to obtain statistically robust data. Finally, we relate changes in avidity to changes in mannoside spacing and suggest that ditopic binding is necessary for the protein to achieve sufficient avidity to remain surface bound. This work presents a versatile new way of studying quantitatively the relationship between surface density and avidity in situations where multivalency is central.
Materials and Methods
Synthesis.
The mannoside, pent-4-enyl α-D-mannopyranoside (α-Man), was prepared as previously reported and characterized by 1H NMR and mass spectrometry, and all spectroscopic data were consistent with the literature data.20
Surface preparation.
The thiol-terminated Si/SiOx slides were prepared following previously reported literature procedures.20, 40 Si/SiO2 wafers were purchased from Nova Electronic Materials (USA), (3-mercaptopropyl)trimethoxysilane was purchased from Gelest (USA), and all other chemicals were purchased from VWR and used as received. The wafers were cleaned by submerging them in piranha solution (3:1 H2SO4:H2O2) for 15 min and then taken out and rinsed with MilliQ H2O. The substrates were dried under a stream of air. Once dried, the slides were placed in a 120 mL PhMe solution containing 4.5 mL of (3-mercaptopropyl)trimethoxysilane, which was heated to 37 °C in a H2O bath for 4 h. The substrates were then rinsed with PhMe, a mixture of PhMe and EtOH, and then EtOH. Finally, the glass slides were cured in an oven at 105 °C for 18 h and stored in MeOH at 4 °C until used.
Printer design and microarray fabrication.
A TERA Fab E-Series printer was modified to print glycan microarrays. The system is equipped with an LED (405 nm) light source that reflects off a DMD that passes through an objective to focus the pattern onto the substrate, which is mounted on a piezoelectric stage. Patterns were designed on Adobe Illustrator and converted to BMP files for the DMD software. DMF solutions of the mannoside and allyl alcohol were made at a concentration of 200 mM and 100 mM of the photoinitiator diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO). The solution was charged in a syringe and placed in a syringe pumps (New Era Pump Systems Inc., USA) with PEEK tubing to flow the solution through the fluid cell. Eleven different 0.5 mL solutions were prepared for printing, where the concentration of alkene (glycan + spacer) remained at 200 mM, but the mole fraction, χ, of glycan varied from 1 to 0.01. Syringe pump flow rates were set to 5 μL/min during exposure time. A syringe with DMF was loaded into the syringe pump and set with a flow rate of 100 μL/min for 5 min in between each exposure time to wash away the precious glycan solution in the fluid cell. After immobilization was completed for all prints, the substrate was rinsed in 200 proof EtOH and then sonicated in DMF for 10 min. The surface of the array was then passivated by immersing it in a 1% w/v BSA solution in PBS (10 mM PBS, pH= 7.4) for 30 min.
Lectin binding and analysis.
A previously described microfluidic incubation chip 20 was used to introduce the lectin solutions to the glycan microarrays. FITC-labeled ConA was purchased from Vector Laboratories Inc. and was used as received. DMF solutions were prepared (PBS buffer, 10 mM, MgCl2 0.9 mM, CaCl2 0.5 mM, pH= 7.4, 0.01% Tween20) with lectin concentrations of 0.5 – 0.001 mg/mL. The ConA solutions were injected into the microarray using a custom built microfluidic chip with 250 μm-wide channels that can introduce 11 different lectin solutions to the microarray simultaneously. The surface was incubated for 16 hr at room temperature with 8 different ConA solutions (0.5 – 0.001 mg/mL, 4.8 μM – 9.6 nM) and 3 channels containing only PBS buffer as controls. Following the incubation, the array was washed by immersing the chip into a buffer solution (PBS buffer, 10 mM, MgCl2 0.9 mM, CaCl2 0.5 mM, pH= 7.4, 0.01% Tween20) for 10 min, and this washing was repeated with fresh solution 3 times. Binding was analyzed with an Olympus BX60 fluorescence microscope (540–585 nm long pass filter). Images were analyzed with ImageJ 41 to measure the fluorescence of each feature, with fluorescence reported as normalized fluorescence (NF = feature fluorescence/background fluorescence) from the average measurement of 3 different features.
Results and Discussion
Here we investigate the multivalent binding of ConA with mannoside-patterned glycan microarrays (Figure 1). ConA is a plant lectin that has two states: as a homotetramer that exists at pH > 7, as a homodimer when the pH < 6, and both states exists in equilibria when the pH is 6‒7. The monomeric subunit of ConA consists of 237 amino acids with a mass of 25.5 kDa and has three binding sites. Of the three sites, two are for metal ions, typically Ca2+ and Mn2+, and the third for the glycan. ConA first binds to the metal ions, which opens up the binding site for the glycan. ConA preferentially binds mannosides, and also binds glucosides, but with a lower affinity.42 To study the effect of multivalency on ConA-mannoside avidity, a glycan microarray was prepared where the glycan density was varied systematically in each feature. This array was prepared with a new photochemical printer described recently by our group.20 Briefly, the printer integrates a TERA-Fab E Series printer, which consists of a DMD (1024 × 768 mirrors) and an LED (405 nm, 32 mW/cm2), that are coupled with a piezoelectric stage that supports the substrate (Figure 1A). A CPU coordinates the spatiotemporal delivery of light onto the substrate and the movement of the stage. Onto the TERA-Fab E Series stage, we have mounted a fluid cell40 where photochemical reactions occur on the functionalized substrate and in solution, and microfluidics deliver and remove reagents from the fluid cell (Figure 1B‒C). We have demonstrated the capabilities of this photochemical printer by preparing multiplexed grafted-from brush polymer arrays43 and multiplexed glycan microarrays20.
Figure 1.
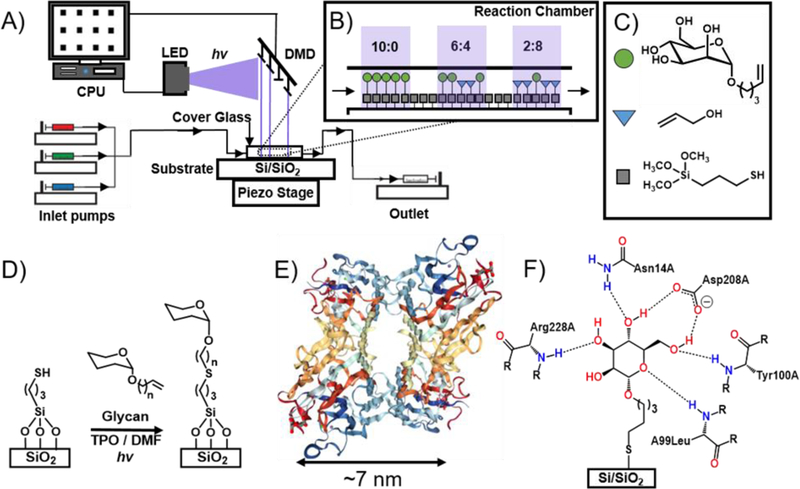
A) The printing platform combines a digital micromirror device, piezoelectric stage, and microfluidics to perform photochemical surface reactions. B) The surface density is manipulated by varying mole fraction, χ, of α-Man (green circles) to allyl alcohol spacer (blue triangles). C) The three molecules involved in the surface functionalization. D) Thiol-ene photochemical surface chemistry is used for immobilization of α-Man and allyl alcohol. E) Image from the RCSB PDB (rcsb.org) of PDB ID 3QLQ42 shows the ribbon structure of ConA in its tetrameric form; each monomer binds two metal ions and one glycan (mannoside). Spacing between each glycan binding site is approximately 7 nm. F) The bonding interactions presumed to occur between an immobilized mannoside and ConA.
Here, the thiol-ene photochemical click reaction was used to immobilize different ratios of the alkene-labelled mannoside, pent-4-enyl-α-D-mannopyranoside (α-Man), and spacer, allyl alcohol, in different ratios to form features on the substrate (Figure 1D). The thiol-ene reaction is a popular reaction for surface immobilization of biological probes because it is biorthogonal, proceeds rapidly and in high yield, and with few byproducts.44–46 Previously, ourselves and others have shown it can be used to immobilize dyes40, glycans20 and glycopolymers,22 confirming its compatibility for preparing microarrays, and we have studied the reaction kinetics to determine the reaction time required to proceed to completion. Here we perform the thiol-ene reaction in the printer described above to create glycan arrays by mounting a Si/SiO2 wafer that is functionalized with 3-(mercaptopropyl) trimethoxysilane into the fluid cell. Microfluidics introduced solutions containing different ratios of the alkene-functionalized α-Man and the spacer allyl alcohol (total concentration α-Man + allyl alcohol = 200 mM) in DMF with the photosensitizer diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO, 100 mM). The ratio of α-Man : allyl alcohol was varied from 1 to 0.01. Features of 45 × 45 μm were prepared by irradiating the surface in the presence of the printing solution for 20 min at 8 mW/cm2 intensity, and 32 feature of each α-Man : allyl alcohol ratio were printed. Between solutions, the substrate was washed with DMF for 5 min at a flow rate of 100 μL/min to minimize contamination. Once the printing was completed, the substrate was washed with EtOH, sonicated in DMF for 10 min, then unreacted areas of the surface were passivated by immersing it into a 1% (w/v) bovine serum albumin (BSA) solution. BSA was selected as a passivating agent because it has been used by us20, 22, 30 and others16, 47 to prevent non-specific adsorption of GBPs to surfaces.
The binding of ConA to the variable density glycan microarray was measured using fluorescence microscopy. These experiments provided fluorescence values for 8 different ConA concentrations to glycans patterned at 11 different α-Man : allyl alcohol ratios. The lectin concentrations range from 4.8 μM to 9.6 nM. The fluorescence data for the binding of a 240 nM solution of FITC-ConA onto the different features is presented in Figure 2A. The binding data were assembled into a heat map to represent changes in fluorescence with changes in both glycan density and lectin concentration (Figure 2B), which illustrates that fluorescence intensity decreases with decreasing ConA concentration and with mannoside χ in the printed feature. Finally, plotting of these fluorescence data show (Figure 2C) a non-linear decrease with decreasing α-Man mole fraction (χ = [glycan] / ([glycan] + [spacer])), with a drop-off to background at χ = 0.2. This nonlinear decrease in binding, with decreasing χ, is consistent with other studes of the multivalent binding of ConA in microarrays.27, 33 No fluorescence above the background was observed in the control channels that contained only buffer solution.
Figure 2.
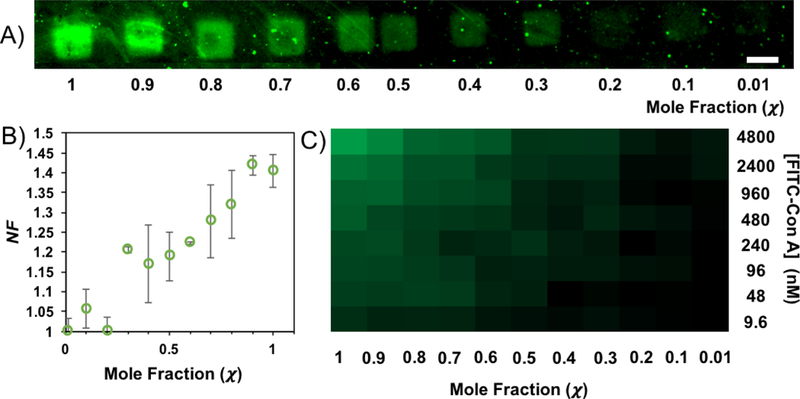
A) Fluorescence image of the printed features of α-Man following exposure to ConA concentration at 240 nM shows a decrease in NF as mole fraction, χ, decreases. The scale bar is 45 μm. B) Data showing the decrease of NF as the χ of glycan decreases in each feature. The error bars are one standard deviation from the mean. C) A heat map representing binding at all glycan χ and ConA concentrations.
The α-Man-ConA fluorescence data was used to determine the avidity (Kd) at each different ConA concentration and each χ by applying a Langmuir isotherm model (Eq. 1), which is commonly used to quantify binding in microarrays.27, 33, 48
| Eq. 1 |
In this model, [L] is the concentration of the lectin, F is the observed fluorescence counts, and Fmax is the maximum fluorescence observed when ConA binds to α-Man (Fmax was observed at [ConA] = 4.8 μM and χ = 1). The binding data for all 88 different conditions (8 ConA concentrations x 11 χ) are presented in Table 1, and Kd values range from 2700 to 43 nM. The following trends are seen when calculating the Kd for each binding event. The observed Kd is dependent upon both χ and ConA concentration. For all ConA concentrations, fluorescence measurements for χ ≤ 0.2 were identical to background, and no features were observed, indicating that no specific binding was occurring or that fluorescence signal was too low to measure by our analytical methods. At the lowest ConA concentration (9.6 nM), the difference between χ = 1 to χ = 0.01 in Kd is only 5 nM, which became statistically similar to the background, although patterns were still observable at χ=0.3, and so we conclude that binding cannot be measured accurately with our fluorescence method at this concentration. We observed that as glycan concentration decreased, Kd increased. For example, at 4.8 μM ConA (highest [ConA]), Kd increases 10-fold from 270 nM (χ = 1) to 2200 nM (χ = 0.3). Similarly, in the study done by Wong et al.,33 as the printing concentrations of monomannose decreases, the Kd increases, concluding that the spacing is too large for ConA to bind multivalently, which is in good agreement with our results. Alternatively, if χ is held constant, Kd increases with increasing [ConA]. For example, at χ = 0.8 the Kd range from 710 nM ([ConA] = 4.8 μM) to 28 nM ([ConA] = 48 nM). At high [ConA], the Kd values are higher than previously reported, however, as the concentration of [ConA] decreases the Kd values begin to resemble what is reported by Gildersleeve27 (69 nM) and Wong33 (80 nM). There is an aberrant data point when [ConA] is 2400 nM at χ = 0.9 that does not seem to follow the binding trends. We believe that the unexpectedly low fluorescence readout in this area was the result of improper immobilization of α-Man during the printing of the features, which could have been caused by poor thiol monolayer formation or contamination in that area during printing.
Table 1.
Dissociation constants, Kd, determined by fluorescence data and Eq. 1 for each binding event between ConA and immobilized mannose at different mole fractions, χ. Errors are reported as one standard deviation from the mean taking from three different features in the array.
| [ConA] nM |
||||||||
|---|---|---|---|---|---|---|---|---|
| 4800 | 2400 | 960 | 480 | 240 | 96 | 48 | 9.6 | |
| χ | Kd (nM) | |||||||
| 1 | 270 ± 50 | 180 ± 42 | 240 ± 54 | 18 ± 2 | 110 ± 78 | 52 ± 5 | 43 ± 15 | NA |
| 0.9 | 270 ± 37 | 4 ± 1 | 350 ± 66 | 41 ± 15 | 100 ± 49 | 49 ± 11 | 28 ± 5 | NA |
| 0.8 | 710 ± 150 | 490 ± 110 | 400 ± 77 | 88 ± 26 | 130 ± 6 | 56 ± 14 | 29 ± 6 | NA |
| 0.7 | 910 ± 256 | 650 ± 190 | 420 ± 91 | 91 ± 39 | 140 ± 34 | 71 ± 8 | 37 ± 3 | NA |
| 0.6 | 380 ± 68 | 550 ± 200 | 460 ± 170 | 140 ± 57 | 160 ± 44 | 74 ± 31 | 39 ± 8 | NA |
| 0.5 | 2100 ± 426 | 1100 ± 290 | 570 ± 120 | 170 ± 100 | 170 ± 33 | 64 ± 14 | 51 ± 11 | NA |
| 0.4 | 1800 ±370 | 1100 ± 100 | 660 ± 140 | 190 ± 61 | 180 ± 7 | 74 ± 6 | 66 ± 3 | NA |
| 0.3 | 2200 ± 490 | 1100 ± 340 | 550 ± 78 | 190 ± 85 | 200 ± 37 | 76 ± 10 | 70 ± 5 | NA |
| 0.2 | NB | NB | NB | NB | NB | NB | NB | NB |
| 0.1 | NB | NB | NB | NB | NB | NB | NB | NB |
| 0.01 | NB | NB | NB | NB | NB | NB | NB | NB |
NB = No binding NA = Features undetectable
The changes in avidity can be explained by considering the differing ability of ConA to participate in multivalent binding as glycan density is modulated. Here we modify a simple model for surface density developed by Oyelaran that is based on average glycan spacing to explain the observed avidity trends. In this model, neoglycoprotein-coated surfaces are assumed to be two-dimensional arrays of ligands, and that these ligands are evenly spaced across the surface.28 To estimate average spacing, we must first determine the density of molecules on the surface, which here we assume to be ~1013 molecules·cm–2 with a range of 0.1 – 0.6 molecules·nm–2, which is based on previous work from our group on immobilizing electroactive probes onto functionalized substrates.30 At this grafting density, there are from 2 × 108 – 1 × 109 bound molecules per 45 × 45 μm feature. The estimated average distance is found by dividing the feature area (2.025 × 109 nm2) by the number of molecules in that feature, and taking the square root to find the distance. Using the lowest density estimate, we find that at χ = 1, the estimated average distance between mannosides is 1 nm. As χ decreases, the spacing between mannosides increases, such that at χ = 0.2, the calculated average distance between mannosides is 7.1 nm, which is slightly larger than the distance between binding sites in ConA (7 nm), rendering multivalent binding nearly impossible, which is in good agreement with our data. Although this model is only a rough approximation, it does illuminate why binding decreases so dramatically at this critical threshold of χ = 0.2. Assuming that at least two ConA-α-Man contacts are required for ConA to remain bound to the surface, at these low values of χ, this is not possible, and, as such, the protein is easily washed off the surface (Figure 3A). Calculations were also repeated assuming that there are 0.6 molecules·nm–2. Both calculations were plotted (Figure 3B) to show the range of potential spacing between immobilized glycans at different mole fractions and how they related to changes in Kd.
Figure 3.
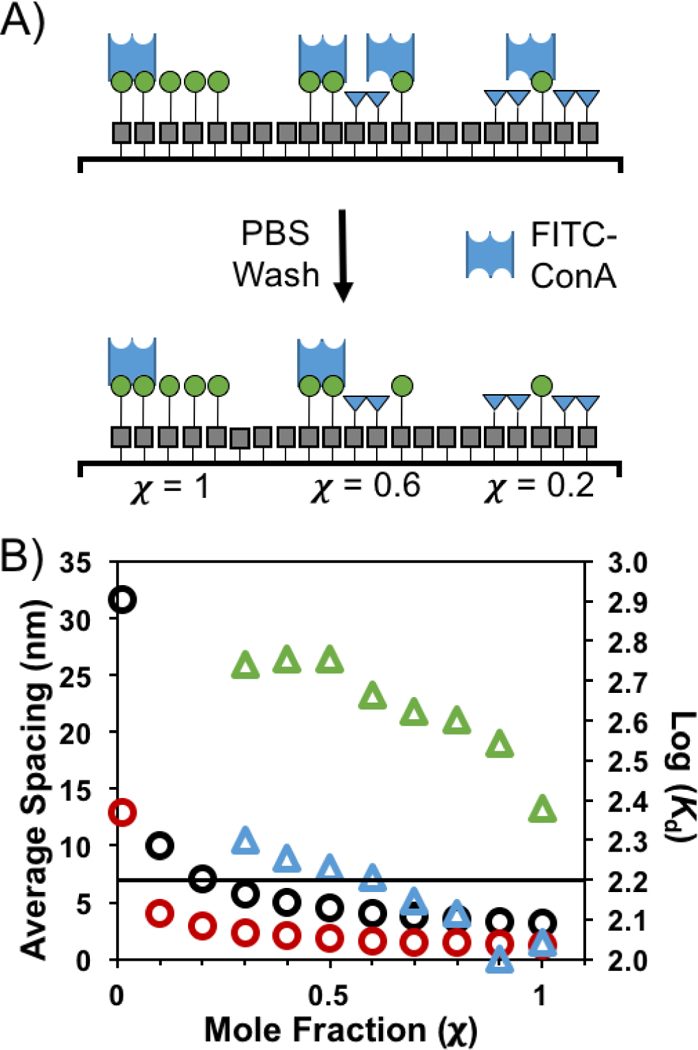
A) Model demonstrating how reducing χ decreases ConA recognition. As the number of glycans on the surface decreases, the average spacing between glycans becomes larger than the distance between ConA binding sites. This resulting monovalent binding is not sufficiently strong to survive washing. B) The estimated average spacing between mannosides as a function of χ are represented by the circles. The black circles assume a molecular packing of 0.1 molecules·nm–2, and the red circles represent changes in spacing assuming a molecular packing of 0.6 molecules·nm–2. The triangles are the log(Kd) values from Table 1 for [ConA] of 960 nM (green) and 240 nM (blue). The black line corresponds to an average spacing of 7 nm, which is the distance between glycan-binding pockets in tetrameric ConA.
Conclusions
Here we used a new photochemical printer combined with microfluidics to fabricate glycan microarrays with systematically varying glycan density. By varying the ratio of α-Man : allyl alcohol in the printing solution used during the photochemical immobilization, the average spacing between glycans was controlled. The association between the glycans in this microarray and FITC-ConA was studied in a microfluidic chip, where solutions of varying [ConA] were exposed to the features printed at varying χ. Fluorescence microscopy analysis of the resulting array provided Kds for 8 different ConA concentrations and 11 different χ. In addition to increasing Kd with decreasing χ, we observed an abrupt decrease in fluorescence at χ = 0.2. We explain this phenomenon using a model that considers the average spacing between glycans, and conclude that at χ = 0.2, the glycan spacing is too great for the ConA to bind the surface multivalently. We believe that this versatile new printing and analysis strategy could help glycobiologists investigate quantitatively how multivalency affects association, and thereby understand how infomation is trafficked on the cell surface and other biointerfaces.
ACKNOWLEDGMENTS
This work was supported by funding from the Air Force Office of Scientific Research (FA9550–17-1–0356), the Department of Defense (MURI 15RT0675), a SEED grant from the City University of New York Advanced Science Research Center, and a SEED grant from the Professional Staff Congress of the City University of New York. A Research Centers in Minority Institutions Program grant from the National Institute of Health Disparities (MD007599) of the National Institutes of Health (NIH), which supports the infrastructure at Hunter College is also acknowledged.
Footnotes
The authors declare no competing financial interests.
REFERENCES
- 1.van Kooyk Y and Rabinovich GA, Nat. Immunol, 2008, 9, 593. [DOI] [PubMed] [Google Scholar]
- 2.Van Breedam W, Pöhlmann S, Favoreel HW, de Groot RJ and Nauwynck HJ, FEMS Microbiol. Rev, 2014, 38, 598–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith AE and Helenius A, Science, 2004, 304, 237–242. [DOI] [PubMed] [Google Scholar]
- 4.Wang PG and Bertozzi CR, Glycochemistry: principles, synthesis, and applications, Marcel Dekker, Inc., New York, 2001. [Google Scholar]
- 5.Müller C, Despras G and Lindhorst TK, Chem. Soc. Rev, 2016, 45, 3275–3302. [DOI] [PubMed] [Google Scholar]
- 6.Jiménez Blanco JL, Ortiz Mellet C and García Fernández JM, Chem. Soc. Rev, 2013, 42, 4518–4531. [DOI] [PubMed] [Google Scholar]
- 7.Fasting C, Schalley CA, Weber M, Seitz O, Hecht S, Koksch B, Dernedde J, Graf C, Knapp E-W and Haag R, Angew. Chem. Int. Ed, 2012, 51, 10472–10498. [DOI] [PubMed] [Google Scholar]
- 8.Jayaraman N, Chem. Soc. Rev, 2009, 38, 3463–3483. [DOI] [PubMed] [Google Scholar]
- 9.Renaudet O and Roy R, Chem. Soc. Rev, 2013, 42, 4515–4517. [DOI] [PubMed] [Google Scholar]
- 10.Lundquist JJ and Toone EJ, Chem. Rev, 2002, 102, 555–578. [DOI] [PubMed] [Google Scholar]
- 11.Dimick SM, Powell SC, McMahon SA, Moothoo DN, Naismith JH and Toone EJ, J. Am. Chem. Soc, 1999, 121, 10286–10296. [Google Scholar]
- 12.Cecioni S, Imberty A and Vidal S, Chem. Rev, 2015, 115, 525–561. [DOI] [PubMed] [Google Scholar]
- 13.Bernardi A, Jiménez-Barbero J, Casnati A, De Castro C, Darbre T, Fieschi F, Finne J, Funken H, Jaeger K-E, Lahmann M, Lindhorst TK, Marradi M, Messner P, Molinaro A, Murphy PV, Nativi C, Oscarson S, Penadés S, Peri F, Pieters RJ, Renaudet O, Reymond J-L, Richichi B, Rojo J, Sansone F, Schäffer C, Turnbull WB, Velasco-Torrijos T, Vidal S, Vincent S, Wennekes T, Zuilhof H and Imberty A, Chem. Soc. Rev, 2013, 42, 4709–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heimburg-Molinaro J, Song X, Smith DF and Cummings RD, Curr. Protoc. Protein. Sci, 2011, 64, 12.10.11–12.10.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song X, Heimburg-Molinaro J, Cummings RD and Smith DF, Curr. Opin. Chem. Biol, 2014, 18, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chevolot Y, Bouillon C, Vidal S, Morvan F, Meyer A, Cloarec J-P, Jochum A, Praly J-P, Vasseur J-J and Souteyrand E, Angew. Chem. Int. Ed, 2007, 46, 2398–2402. [DOI] [PubMed] [Google Scholar]
- 17.de Boer AR, Hokke CH, Deelder AM and Wuhrer M, Anal. Chem, 2007, 79, 8107–8113. [DOI] [PubMed] [Google Scholar]
- 18.Song X, Xia B, Lasanajak Y, Smith DF and Cummings RD, Glycoconj. J, 2008, 25, 15–25. [DOI] [PubMed] [Google Scholar]
- 19.Wojcik F, Lel S, O’Brien AG, Seeberger PH and Hartmann L, Beilstein J. Org. Chem, 2013, 9, 2395–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valles DJ, Naeem Y, Carbonell C, Wong AM, Mootoo DR and Braunschweig AB, ACS Biomater. Sci. Eng, 2019, Revisions [DOI] [PubMed] [Google Scholar]
- 21.Wendeln C, Heile A, Arlinghaus HF and Ravoo BJ, Langmuir, 2010, 26, 4933–4940. [DOI] [PubMed] [Google Scholar]
- 22.Bian S, Zieba SB, Morris W, Han X, Richter DC, Brown KA, Mirkin CA and Braunschweig AB, Chem. Sci, 2014, 5, 2023–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neumann K, Conde-González A, Owens M, Venturato A, Zhang Y, Geng J and Bradley M, Macromolecules, 2017, 50, 6026–6031. [Google Scholar]
- 24.Ko K-S, Jaipuri FA and Pohl NL, J. Am. Chem. Soc, 2005, 127, 13162–13163. [DOI] [PubMed] [Google Scholar]
- 25.Fukui S, Feizi T, Galustian C, Lawson AM and Chai W, Nat. Biotechnol, 2002, 20, 1011. [DOI] [PubMed] [Google Scholar]
- 26.Feizi T and Chai W, Nat. Rev. Mol. Cell Biol, 2004, 5, 582–588. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Li Q, Rodriguez LG and Gildersleeve JC, J. Am. Chem. Soc, 2010, 132, 9653–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oyelaran O, Li Q, Farnsworth D and Gildersleeve JC, J. Proteome Res, 2009, 8, 3529–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Campbell C, Li Q and Gildersleeve JC, Mol. Biosyst, 2010, 6, 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bian S, He J, Schesing KB and Braunschweig AB, Small, 2012, 8, 2000–2005. [DOI] [PubMed] [Google Scholar]
- 31.Wendeln C, Rinnen S, Schulz C, Arlinghaus HF and Ravoo BJ, Langmuir, 2010, 26, 15966–15971. [DOI] [PubMed] [Google Scholar]
- 32.Godula K, Rabuka D, Nam KT and Bertozzi CR, Angew. Chem. Int. Ed, 2009, 48, 4973–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang P-H, Wang S-K and Wong C-H, J. Am. Chem. Soc, 2007, 129, 11177–11184. [DOI] [PubMed] [Google Scholar]
- 34.Godula K, Rabuka D, Nam KT and Bertozzi CR, Angew. Chem. Int. Ed, 2009, 48, 4973–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Godula K and Bertozzi CR, J. Am. Chem. Soc, 2012, 134, 15732–15742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mammen M, Choi S-K and Whitesides GM, Angew. Chem. Int. Ed, 1998, 37, 2754–2794. [DOI] [PubMed] [Google Scholar]
- 37.Dam TK, Roy R, Das SK, Oscarson S and Brewer CF, J. Biol. Chem, 2000, 275, 14223–14230. [DOI] [PubMed] [Google Scholar]
- 38.Dam TK, Gerken TA and Brewer CF, Biochemistry, 2009, 48, 3822–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houseman BT and Mrksich M, Chem. Biol, 2002, 9, 443–454. [DOI] [PubMed] [Google Scholar]
- 40.Carbonell C, Valles DJ, Wong AM, Tsui MW, Niang M and Braunschweig AB, Chem, 2018, 4, 857–867. [Google Scholar]
- 41.Schneider CA, Rasband WS and Eliceiri KW, Nat. Methods, 2012, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trastoy B, Bonsor DA, Pérez-Ojeda ME, Jimeno ML, Méndez-Ardoy A, García Fernández JM, Sundberg EJ and Chiara JL, Adv. Funct. Mater, 2012, 22, 3191–3201. [Google Scholar]
- 43.Carbonell C, Valles DJ, Wong AM, Touve M, Gianneschi NC and Braunschweig AB, Nat. Comm. 2019, Revisions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoyle CE and Bowman CN, Angew. Chem. Int. Ed, 2010, 49, 1540–1573. [DOI] [PubMed] [Google Scholar]
- 45.Jonkheijm P, Weinrich D, Köhn M, Engelkamp H, Christianen PCM, Kuhlmann J, Maan JC, Nüsse D, Schroeder H, Wacker R, Breinbauer R, Niemeyer CM and Waldmann H, Angew. Chem, 2008, 120, 4493–4496. [DOI] [PubMed] [Google Scholar]
- 46.Lowe AB, Polym. Chem, 2014, 5, 4820–4870. [Google Scholar]
- 47.Smith EA, Thomas WD, Kiessling LL and Corn RM, J. Am. Chem. Soc, 2003, 125, 6140–6148. [DOI] [PubMed] [Google Scholar]
- 48.Kuno A, Uchiyama N, Koseki-Kuno S, Ebe Y, Takashima S, Yamada M and Hirabayashi J, Nat. Methods, 2005, 2, 851. [DOI] [PubMed] [Google Scholar]