

Abstract
For the 2018 YPIC Challenge contestants were invited to try to decipher two unknown English questions encoded by a synthetic protein expressed in Escherichia coli. In addition to deciphering the sentence, contestants were asked to determine the 3D structure and detect any post-translation modifications left by the host organism.
We present our experimental and computational strategy to characterize this sample by identifying the unknown protein sequence and detecting the presence of post-translational modifications. The sample was acquired with dynamic exclusion disabled to increase the signal-to-noise ratio of the measured molecules, after which spectral clustering was used to generate high-quality consensus spectra. De novo spectrum identification was used to determine the synthetic protein sequence, and any post-translational modifications introduced by E. coli on the synthetic protein were analyzed via spectral networking. This workflow resulted in a de novo sequence coverage of 70 %, on par with sequence database searching performance. Additionally, the spectral networking analysis indicated that no systematic modifications were introduced on the synthetic protein by E. coli.
The strategy presented here can be directly used to analyze samples for which no protein sequence information is available or when the identity of the sample is unknown. All software and code to perform the bioinformatics analysis is available as open source, and self-contained Jupyter notebooks are provided to fully recreate the analysis.
Keywords: mass spectrometry, proteomics, spectral clustering, de novo, spectral networking
Graphical Abstract
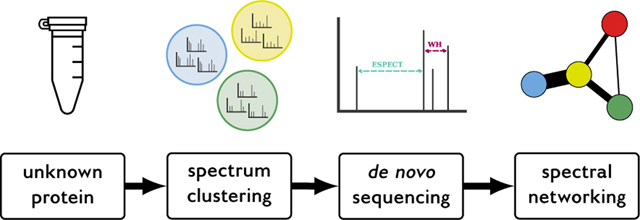
1. Introduction
Mass spectrometry (MS) is a powerful analytical technique to characterize proteins in complex biological samples. The typical strategy to identify unknown tandem mass spectrometry (MS/MS) spectra is via sequence database searching.1 Here, experimental MS/MS spectra are compared to theoretical spectra derived from a protein sequence database for the organism(s) of interest. Alternatively, spectral library searching can be used to identify unknown MS/MS spectra by comparing them against a library of high-quality, previously observed spectra with known peptide sequences2,3 or against simulated spectra generated by recent powerful machine learning techniques that highly accurately predict fragment intensities.4–6
Both of these approaches depend on the availability of a ground truth reference set to which the unknown spectra are compared, either in the form of a sequence database or a spectral library. Alternatively, if such prior information is not available, such as, for example, during antibody sequencing or for non-model organisms whose genome has not been sequenced yet, de novo searching can be used to directly derive peptide sequences from the unknown MS/MS spectra based on the mass differences between pairs of their fragment ion peaks.7
Here, we describe our approach to characterize an unknown protein sample in the context of the 2018 Young Proteomics Investigators Club (YPIC) Challenge. YPIC is an initiative by the European Proteomics Association (EuPA) to connect and support young scientists in proteomics. As part of their activities they have organized scientific challenges in 2017 and in 2018 where participants were invited to analyze mysterious protein samples.8
The 2018 YPIC Challenge consisted of trying to decipher two unknown English questions encoded by a synthetic protein expressed in E. coli. The challenge encouraged participants to fully characterize the protein sample through several subtasks, such as protein sequence identification, detection of post-translational modifications (PTMs), and development of bioinformatic approaches.
Because the sample consisted of an unknown, synthetic, protein and no sequence database was available, we used de novo searching, in combination with spectral clustering, to identify the protein sequence. Additionally, spectral networking was used to discover common mass differences between spectra and detect potential PTMs. Finally, circular dichroism (CD) spectroscopy was used to analyze the protein’s secondary structure.
All bioinformatics software that was used to analyze the data is freely available as open source. Self-contained Jupyter notebooks9 containing all processing steps are available at https://github.com/bittremieux/ypic_challenge_2018, to fully reproduce the bioinformatics analysis.
2. Materials and methods
2.1. 2018 YPIC Challenge description
We received a sample vial containing 12.5 μg of an unknown protein via mail from the organizers of the YPIC Challenge. As per the included product sheet, the synthetic protein was expressed in E. coli by PolyQuant and encoded two concatenated English questions.10 The sentence did not contain the letters ‘B’ and ‘K’, and the letters ‘O’ and ‘U’ were replaced by the letter ‘K’ in the protein. The protein sequence was flanked with ‘MAGR’ in the beginning and ‘LAAALEHHHHHH’ at the end for digestion and purification reasons.
The 2018 YPIC Challenge categories were as follows:
Answer E. coli’s question.
Three-dimensional grammar: Find out how this sentence folds.
Bioinformazing: Develop the coolest bioinformatics approach to decipher the sentence.
Protein punctuation: Look for the biological equivalent of punctuation: PTMs left behind by E. coli.
#Bioreactivity: Can you generate and describe bioreactivity in this Twitter-sized message?
Here we describe our efforts to identify the unknown protein sequence to answer E. coli’s question, and identify any PTMs that are present. An important emphasis is placed on the bioinformatics analysis using freely available software tools, and self-contained Jupyter notebooks9 containing all processing steps are available as open source at https://github.com/bittremieux/ypic_challenge_2018.
2.2. Experimental procedures
2.2.1. Protein sample preparation
The sample was reconstituted with 125 μL 0.1 % formic acid (final concentration 0.1 μg/μL protein). An aliquot (1μg; 10μL) of reconstituted sample was reduced (50mM dithiothreitol), alkylated (150mM iodoacetamide), and digested with Promega trypsin (1 : 50 enzyme—substrate ratio; 0.02 μg trypsin) for 4h at 37 °C with shaking. Digested peptides were concentrated via speed-vac to a final concentration of 0.33fmol/μL.
In addition to the conventional trypsin digest, following a CD spectroscopy solvent swap, the remaining sample was split into three parts and digested with three other proteases: pepsin, chymotrypsin, and Lys-C. The conditions for these reactions follow the trypsin digest conditions above, with the exception of the pepsin digestion which was held at a low pH (pH < 2.0).
2.2.2. LC-MS/MS data acquisition
Peptides were separated with a Waters NanoAcquity UPLC and emitted into a Thermo Q-Exactive HF tandem mass spectrometer. Pulled tip columns were created from 75 μm inner diameter fused silica capillary in-house using a laser pulling device and packed with 2.1μm C18 beads (Dr. Maisch GmbH) to 300 mm. Trap columns were created from 150 μm inner diameter fused silica capillary fritted with Kasil on one end and packed with the same C18 beads to 25 mm. Buffer A was water and 0.1 % formic acid, while buffer B was 98% acetonitrile and 0.1% formic acid. For each injection, 3 μL of each sample was loaded with 5 μL 2% B and eluted using the following program: 0min to 90min 2% to 35% B, 90min to 100 min 35% to 60% B, followed by a 35min washing gradient.
The Thermo Q-Exactive HF was set to positive mode in a top-20 configuration. Precursor scans (300 m/z to 2000 m/z) were collected at 60 000 resolution to hit an automatic gain control (AGC) target of 3 × 106. The maximum inject time was set to 100 ms. Fragment scans were collected at 30 000 resolution to hit an AGC target of 1 × 105 with a maximum inject time of 55 ms. The isolation width was set to 1.6 m/z with a normalized collision energy of 27. Precursors with charge up to +6 that achieved a minimum AGC of 5 × 103 were acquired. Dynamic exclusion was disabled. The digested sample was acquired using this method in technical triplicate.
Intact mass analysis was performed on a 1 μg aliquot of the reconstituted sample (0.1 μg/μL protein in 0.1 % formic acid) by analyzing the reconstituted, reduced, and alkylated (but undigested) sample with the DDA method described above. Intact mass was determined by the MS1 spectrum mass-to-charge and charge values reported in Thermo XCalibur.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium11 via the PRIDE12 partner repository with the dataset identifier PXD014003.
2.2.3. Circular dichroism spectroscopy
Following reconstitution of the protein sample as described above, the original protein sample, minus the 2 μg of protein aliquoted for intact mass and trypsin digestion experiments, was speed vac’d to dryness to change to a CD spectroscopy-compatible buffer. The dried protein sample was reconstituted in 10 mM KPO4 (pH 7.4) to 0.05μg/pL (assuming 12.5 μg original protein per the product sheet and 2 μg used for the initial MS experiments) to meet the CD cuvette minimum volume requirement of 200 μL buffer. Absorbance from 180 nm to 240 nm were acquired on a Jasco J-810 spectropolarimeter.
Analysis of the CD spectra was not necessary, as the sample did not absorb any polarized light and therefore produced no spectra to interpret. Insufficient sample concentration was confirmed by testing absorbance of ultraviolet light at 280 nm and 200 nm. The sample did not display any absorbance of light (polarized or UV).
2.3. Data analysis
Raw files were converted to the MGF format using msconvert (ProteoWizard version 3.0.10141)13 for further processing. During conversion MS/MS spectra were centroided using the vendor algorithm and the precursor m/z and charge was recalculated based on the preceding MS scan.
Next, MS/MS spectra were clustered and consensus spectra were generated using MaRaCluster (version 1.00.1)14 with a similarity p-value threshold of 10−5, precursor mass tolerance 50 ppm, and requiring at least 3 MS/MS spectra per cluster.
After spectral clustering low-quality clusters were removed by only retaining the clusters that represented at least 10 original spectra and whose consensus spectra had precursor charge 2 or 3.
The high-quality consensus spectra were used for de novo spectrum identification and spectral networking. DeNovoGUI (version 1.16.2)15 was used as a unified interface to the Novor (version 1.05.0573),16 DirecTag (version 1.4.66),17 and PepNovo+ (version 3.1)18 de novo search engines. Settings for de novo spectrum identification were precursor mass tolerance 20 ppm; fragment mass tolerance 0.02 Da; and cysteine carbamidomethylation, methionine oxidation, and acetylation of the peptide N-terminus as variable modifications. Peptide-spectrum matches (PSMs) were visualized and manually investigated using DeNovoGUI.
A spectral network was constructed using the high-quality consensus spectra. Prior to matching spectra to each other they were preprocessed using spectrum_utils (version 0.2.1)19 by removing noise peaks with an intensity below 5% of the base peak intensity and at most the 150 most intense peaks were retained. Next, peak intensities were scaled by their square root before being normalized by their norm to have a magnitude of one. The shifted dot product20 was used to match modified spectra to each other with fragment mass tolerance 0.02 Da. Each consensus spectrum formed a node in the spectral network, with an edge between two nodes if the shifted dot product between the two corresponding spectra was greater than or equal to 0.8. Peptide sequences were assigned to nodes in the spectral network if the corresponding consensus spectra could be identified by Novor with a minimum score of 70. Only subgraphs in the spectral network consisting of at least three nodes were considered.
The high-quality consensus spectra produced by spectral clustering were also used for sequence database searching. A fasta database for E. coli was downloaded from UniProt (strain K12; version 2019/06/11), to which the sequence of the synthetic protein was added as an additional entry. The Tide search engine21 (Crux22 version 3.2), was used for spectrum identification. Search settings included cysteine carbamidomethylation as a static modification and methionine oxidation as a variable modification, trypsin cleavage with at most two missed cleavages, precursor mass tolerance 300 Da, and fragment mass tolerance 0.02 Da. Other search settings were kept at their default values. PSMs were split based on whether they corresponded to E. coli proteins or the YPIC protein, and the YPIC PSMs were filtered to a false discovery rate (FDR) threshold of 1 %.23
2.3.1. Code availability
Jupyter notebooks9 containing all processing steps and analyses are available at https://github.com/bittremieux/ypic_challenge_2018. Custom processing was done in Python using open-source Python libraries including NumPy,24 pandas,25 NetworkX,26 Matplotlib,27 Seaborn,28 Pyteomics,29 and spectrum_utils.19 The shifted dot product is implemented as an external C++ module for Python.20
3. Results and discussion
3.1. Confirmation of intact mass
Prior to any peptide analysis, we determined the intact mass of the protein. While the final 2018 YPIC Challenge product sheet notes that the molecular weight of the protein is approximately 16.65 kDa, we received our challenge sample prior to the disclosure of this additional information. An MS1 spectrum of the intact mass confirms that the protein has an approximate mass of 16.4 kDa (figure 1).
Figure 1:

MS1 scan of the intact synthetic protein indicating an approximate intact mass of 16.4 kDa.
3.2. Synthetic protein identification
When analyzing a protein of unknown sequence, one key decision is to determine which digestion enzyme to use. To help inform our decision we simulated the digestion of various corpuses using multiple proteases to determine whether they would generally yield peptides whose lengths are amenable to detection by mass spectrometry (supplementary section 1). This simulation indicated that although tryptic peptides generated from English are typically slightly longer than peptides with a biological origin, they are suitable for MS analysis, leading us to mainly use trypsin for digestion purposes.
Since spectra were collected without dynamic exclusion enabled, molecules that are present in the sample will be selected multiple times for MS/MS measurement while spurious signals will only be measured a limited number of times. A downside of this approach is that the spectral data will contain multiple spectra that are virtually identical to each other as the same peptide is repeatedly measured. To condense the data volume the spectra were clustered with MaRaCluster.14 Spectral clustering groups similar spectra together and creates a single consensus spectrum to represent each spectral cluster, reducing the number of spectra from 110 234 spectra in the original raw files to 380 consensus spectra representing at least ten spectra after spectral clustering (only retaining the spectra with precursor charge 2 or 3).
Next, these consensus spectra were identified. As no sequence database was available for the unknown synthetic protein de novo identification was performed. The Novor,16 DirecTag,17 and PepNovo+18 search engines were used through DeNovoGUI.15 The resulting PSMs were subsequently manually validated, a task that became feasible thanks to the reduction in data volume by the spectral clustering. From the de novo identifications we were able to decode several parts of the unknown synthetic protein:
Start of protein: “Have you ever wondered what the mo[st]” (figures 2a to 2d)
“[…]ns in life ar[e]” (figure 2e)
“[r]espect when it comes to what you” (figures 2f and 2g)
End of protein: “[pro]duce in a cell.” (figure 2h)
Figure 2:
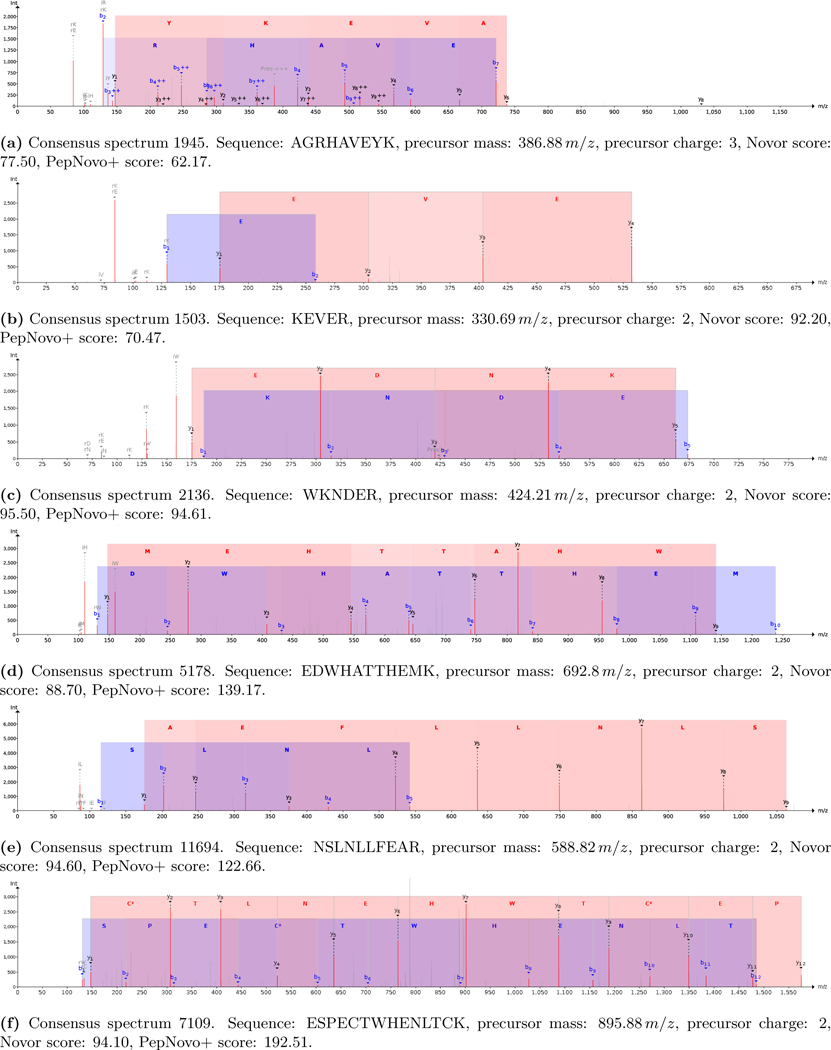
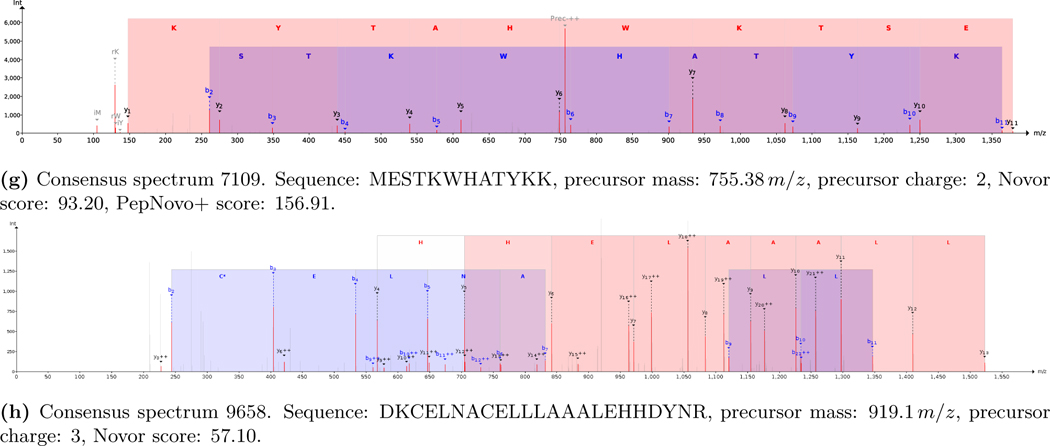
Relevant PSMs decoding the unknown synthetic protein.
Meanwhile, the full synthetic protein sequence, provided by the 2018 YPIC Challenge organizers after the challenge, was: “Have you ever wondered what the most fundamental limitations in life are? Is there a structure to respect when it comes to what you can produce in a cell?”.10 Consequently, the de novo identifications lead to a 70% sequence coverage (98 out of 140 amino acids). This result is in line with sequence coverages that are typically achieved during tryptic analyses of biological samples with a similar complexity. The parts of the protein that remained unidentified are likely caused by specific properties of the corresponding peptides which make them unamenable to identification using mass spectrometry, such as very short peptides after tryptic cleavage or peptides that cannot be properly ionized. We additionally tried to obtain complementary peptides using alternative proteases (pepsin, chymotrypsin, and Lys-C) to increase the sequence coverage. Unfortunately these experiments failed due to the sample loss observed during the preceding CD experiment (section 3.5).
3.3. Spectral networking to detect post-translational modifications
The typical approach to identify potentially modified peptides is by specifying variable modifications during a sequence database search. Similarly, variable modifications can be specified during de novo searching as well. However, de novo searching has to overcome several challenges compared to sequence database searching, including amino acid permutation complexity,7 and the inclusion of variable modifications exacerbates these challenges. Therefore, to maximize the confidence in the obtained de novo identifications only frequent PTMs introduced during sample processing30 were specified to avoid a combinatorial explosion of the search space.
As an alternative strategy to find PTMs we have employed spectral networking.31 A spectral network was constructed by representing each consensus spectrum as a node in a graph and connecting two nodes if their corresponding spectra are highly similar as measured by the shifted dot product20,32 (figure 3). Because the shifted dot product takes mass shifts induced by a modification into account while matching two spectra the spectral network will contain connections between modified peptides and their unmodified counterparts. Subsequently, based on the precursor mass difference between connected spectra in the spectral network and (partial) identifications of the spectra the presence and identity of various modifications, such as PTMs or amino acid substitutions, can be derived (figure 3).
Figure 3:

A spectral network connects (un)modified spectra. The peptide sequence (if known) and the precursor mass and precursor charge are shown for each node in the spectral network. Edges between two nodes are annotated with the corresponding precursor mass difference. The spectral similarity based on the shifted dot product is indicated by the weight of the edge.
The spectral network shows a strong similarity between multiple spectra despite small differences in the identified sequences due to amino acid substitutions (CT ↔ DM, ES ↔ DH). Although the spectrum corresponding to the light red shaded node could not be fully identified through de novo searching, its high similarity to related spectra indicates that it was likely derived from the same peptide. Indeed, a full identification was precluded by the absence of any successfully matched b-ions, while the C-terminal tag “CTWHENLTCK” could still be annotated based on the y-ions.
Connected spectra in the spectral network were manually checked for the presence of PTMs and the most frequently occurring mass differences were referenced to common modifications in Unimod.33 This analysis indicated little to no systematic presence of PTMs. The most frequent mass differences were observed between unidentified spectra of low quality (manual quality assessment), likely derived from small molecular contaminants, and did not correspond to any common modifications. Although a targeted analysis is recommended to conclusively determine the presence or absence of modifications, these results suggest that no PTMs are systematically introduced on the synthetic peptide by E. coli.
3.4. Validation using sequence database searching
We performed a sequence database search to validate the spectrum identifications from the de novo analysis and the spectral networking analysis using the ground truth synthetic protein sequence provided by the YPIC Challenge organizers.
Importantly, while the clustered consensus spectra were searched using a sequence database containing both the synthetic protein and E. coli proteins, FDR filtering was conducted using only the PSMs that matched to the synthetic protein to improve its statistical power.23,34 Out of the 380 consensus spectra 52 spectra were matched to peptides corresponding to the synthetic protein. Interestingly, there were no decoy matches among these 52 PSMs; all decoy matches occurred to low-scoring E. coli PSMs. This strongly indicates that our acquisition strategy to repeatedly sample the same ions, followed by spectral clustering, succeeded in maximally measuring relevant ions and producing high-quality consensus spectra.
Sequence coverage of these 52 PSMs was 65 %, which is slightly below the sequence coverage obtained via de novo searching. This confirms that spectral clustering helped to maximize the signal-to-noise ratio of the consensus spectra, as sequence database searching is typically expected to outperform de novo searching.7 A small caveat in comparing these search results is that the de novo search results were manually validated taking the problem statement into account, i.e. that the sequence should be an English sentence. This expert validation helped to confirm the correct de novo identifications, which is not possible for more general use cases.
Next, we evaluated the spectral networking results compared to the sequence database search results. Because we did not have any prior knowledge about which modifications could be expected to be present in the sample, we performed an open search using a wide precursor mass window to be able to match modified spectra against their unmodified peptide sequences and perform an untargeted PTM analysis. About half of the 52 PSMs have a non-zero mass difference between the spectrum neutral mass and the peptide mass, indicating the presence of modifications (table 1). Most of these mass differences likely correspond to modifications that were introduced during sample handling. Meanwhile, there is little evidence of systematic modifications introduced on the synthetic protein by E. coli, confirming the results obtained via spectral networking.
Table 1:
Mass differences observed during the open search and their likely modifications sourced from Unimod33 (matched to within 20ppm).
| # PSMs | Δm (Da) | Potential modification |
|---|---|---|
| 1 | −18.009 | Pyro-glu from Glu |
| 1 | −17.024 | Loss of ammonia |
| 1 | −9.035 | Arg ⟶ Phe substitution |
| 10 | 0.988 | Deamidation |
| 3 | 3.998 | Trp oxidation to kynurenin |
| 1 | 35.977 | Thr ⟶ His substitution |
| 4 | 127.916 | Unknown modification |
| 1 | 209.022 | Carbamidomethylated DTT modification of Cys |
| 1 | 252.020 | Nitroso Sulfamethoxazole Sulphenamide thiol adduct |
| 2 | 268.048 | Nitroso Sulfamethoxazole semimercaptal thiol adduct |
3.5. Structural analysis using circular dichroism spectroscopy
We attempted CD spectroscopy to estimate the protein’s secondary structure. The CD spectra, however, were inconclusive (data not shown). Based on absorption spectra acquired at the same time as the CD spectra, the concentration of protein in the CD cuvette was negligible. There are several reasons why the CD and absorption spectroscopy experiments might have failed. First, the concentration of protein (0.05 μg/μL) may have been too dilute, considering the range of ideal protein concentration for CD spectroscopy is 0.1 μg/μL to 0.2 μg/μL. Second, the buffer conditions used (10mM KPO4 (pH 7.4)) may not be ideal for the protein’s biochemistry, which would result in poor resolubilization of the protein. Third, the protein may have degraded during −80 °C storage and multiple freeze-thaw cycles during the course of the other experiments. Any one of these reasons may have contributed to the loss of protein observed in this experiment.
4. Conclusion
We have presented our results in identifying an unknown synthetic protein as part of the 2018 YPIC Challenge. Although we did not identify the full synthetic protein, based on a standard trypsin digest we are able to detect spectral evidence covering about two third of the unknown sequence. This is in line with the sequence coverage that is typically obtained during routine tryptic analyses of biological samples with a similar complexity. Although our attempts to use different proteases to increase the sequence coverage failed due to lack of sample material and sample loss that occurred during multiple experiments, we anticipate that this strategy would have generated alternative peptides.35 Additionally, using unconventional digestion strategies such as microwave- assisted digestion to obtain semi-random peptide cleavage,36 might have increased the protein sequence coverage.
Dynamic exclusion is typically enabled in shotgun proteomics to avoid repeatedly sampling the same ion. Instead, we decided not to use dynamic exclusion to maximize the signal-to-noise ratio for the subsequent spectral clustering step. A disadvantage of this strategy, however, is that if a low-abundance peptide co-elutes with a high-abundance peptide the former might not get selected for MS/MS measurement. Considering the long gradient that was used compared to the low sample complexity, enabling dynamic exclusion with a short exclusion time could have been beneficial to measure peptides that are more challenging to ionize. This could potentially have been combined with a narrow isolution window to reduce co-isolation of co-eluting peptides.37
Despite not being able to identify the full protein sequence using de novo searching, we used spectral clustering and spectral networking to investigate the presence of frequent modifications. Based on this analysis we did not see any systematic modifications on the synthetic protein, which was confirmed by an open sequence database search. This corresponds to the lack of notable PTMs in E. coli as well.
Although in this case the sample consisted of a contrived synthetic protein in the context of the 2018 YPIC Challenge, the experimental and computational strategy we have described here can similarly be used to analyze other unknown protein samples that are of more biological interest, such as, for example, antibody sequencing. Notably, our spectral clustering approach can be used to increase the signal-to-noise ratio of spectra prior to de novo identification.38 Additionally, spectral networking is an increasingly popular strategy to analyze small molecules measured by mass spectrometry.39
Finally, we want to conclude by addressing E. coli’s question: “Have you ever wondered what the most fundamental limitations in life are? Is there a structure to respect when it comes to what you can produce in a cell?” Clearly scientific progress continues to push the boundaries of our knowledge on the most fundamental questions in life, including by educational and stimulating challenges such as the 2018 YPIC Challenge tackled here. The unique sample content of this challenge, consisting of a synthetic English sentence expressed as a recombinant protein in E. coli, prompted us to devise a creative analysis strategy. Additionally, it shows that there are few limitations on the information that can be encoded as a protein. We envision that this type of work can boost innovative new applications, such as, for example, using proteins as a data storage medium.40
Supplementary Material
Supplementary section 1: Simulated digestion of English dictionary. Supplementary figure S1: Simulated peptide length.
Acknowledgement
We want to thank YPIC for the organization of their 2018 challenge and for their efforts in unifying the community of young proteomics researchers.
W.B. is a postdoctoral researcher of the Research Foundation – Flanders (FWO). W.B. was supported by a postdoctoral fellowship of the Belgian American Educational Foundation (BAEF). L.K.P. was supported by NIH F31 AG055257.
Footnotes
Supporting information
Supplementary section 1: Simulated digestion of English dictionary. Supplementary figure S1: Simulated peptide length.
References
- (1).Eng JK, Searle BC, Clauser KR, Tabb DL A Face in the Crowd: Recognizing Peptides through Database Search. Molecular & Cellular Proteomics 2011, 10, R111.009522, DOI: 10.1074/mcp.R111.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Griss J. Spectral Library Searching in Proteomics. PROTEOMICS 2016, 16, 729–740, DOI: 10.1002/pmic.201500296. [DOI] [PubMed] [Google Scholar]
- (3).Shao W, Lam H. Tandem Mass Spectral Libraries of Peptides and Their Roles in Proteomics Research. Mass Spectrometry Reviews 2017, 36, 634–648, DOI: 10.1002/mas.21512. [DOI] [PubMed] [Google Scholar]
- (4).Degroeve S, Martens L. MS2PIP: A Tool for MS/MS Peak Intensity Prediction. Bioinformatics 2013, 29, 3199–3203, DOI: 10.1093/bioinformatics/btt544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gessulat S, Schmidt T, Zolg DP, Samaras P,et al. Prosit: Proteome-Wide Prediction of Peptide Tandem Mass Spectra by Deep Learning. Nature Methods 2019, 16, 509–518, DOI: 10.1038/s41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- (6).Tiwary S, Levy R, Gutenbrunner P, Salinas Soto F,et al. High-Quality MS/MS Spectrum Prediction for Data-Dependent and Data-Independent Acquisition Data Analysis. Nature Methods 2019, 16, 519–525, DOI: 10.1038/s41592-019-0427-6. [DOI] [PubMed] [Google Scholar]
- (7).Muth T, Hartkopf F, Vaudel M, Renard BY A Potential Golden Age to Come-Current Tools, Recent Use Cases, and Future Avenues for de Novo Sequencing in Proteomics. PROTEOMICS 2018, 18, 1700150, DOI: 10.1002/pmic.201700150. [DOI] [PubMed] [Google Scholar]
- (8).Dhaenens M. Introducing the YPIC Challenge. EuPA Open Proteomics 2019, DOI: 10.1016/j.euprot.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Thomas K, Benjamin R-K, Fernando P, Brian G,et al. In Positioning and Power in Academic Publishing: Players, Agents and Agendas; IOS Press: 2016, pp 87–90. [Google Scholar]
- (10).Klug H, Sigloch FC English Lessons for E. Coli. EuPA Open Proteomics 2019, DOI: 10.1016/j.euprot.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Deutsch EW, Csordas A, Sun Z, Jarnuczak A,et al. The ProteomeXchange Consortium in 2017: Supporting the Cultural Change in Proteomics Public Data Deposition. Nucleic Acids Research 2017, 45, D1100–D1106, DOI: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M,et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Research 2019, 47, D442–D450, DOI: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chambers MC, Maclean B, Burke R, Amodei D,et al. A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nature Biotechnology 2012, 30, 918–920, DOI: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).The M, Käll L. MaRaCluster: A Fragment Rarity Metric for Clustering Fragment Spectra in Shotgun Proteomics. Journal of Proteome Research 2016, 15, 713–720, DOI: 10.1021/acs.jproteome.5b00749. [DOI] [PubMed] [Google Scholar]
- (15).Muth T, Weilnböck L, Rapp E, Huber CG, et al. DeNovoGUI: An Open Source Graphical User Interface for de Novo Sequencing of Tandem Mass Spectra. Journal of Proteome Research 2014, 13, 1143–1146, DOI: 10.1021/pr4008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ma B. Novor: Real-Time Peptide de Novo Sequencing Software. Journal of The American Society for Mass Spectrometry 2015, 26, 1885–1894, DOI: 10.1007/s13361-015-1204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tabb DL, Ma Z-Q, Martin DB, Ham A-JL, et al. DirecTag: Accurate Sequence Tags from Peptide MS/MS through Statistical Scoring. Journal of Proteome Research 2008, 7, 3838–3846, DOI: 10.1021/pr800154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Frank A, Tanner S, Bafna V, Pevzner P. Peptide Sequence Tags for Fast Database Search in Mass- Spectrometry. Journal of Proteome Research 2005, 4, 1287–1295, DOI: 10.1021/pr050011x. [DOI] [PubMed] [Google Scholar]
- (19).Bittremieux W. Spectrum_utils: A Python Package for Mass Spectrometry Data Processing and Visualization. bioRxiv 2019, DOI: 10.1101/725036. [DOI] [PubMed] [Google Scholar]
- (20).Bittremieux W, Meysman P, Noble WS, Laukens K. Fast Open Modification Spectral Library Searching through Approximate Nearest Neighbor Indexing. Journal of Proteome Research 2018, 17, 3463–3474, DOI: 10.1021/acs.jproteome.8b00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Diament BJ, Noble WS Faster SEQUEST Searching for Peptide Identification from Tandem Mass Spectra. Journal of Proteome Research 2011, 10, 3871–3879, DOI: 10.1021/pr101196n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McIlwain S, Tamura K, Kertesz-Farkas A, Grant CE, et al. Crux: Rapid Open Source Protein Tandem Mass Spectrometry Analysis. Journal of Proteome Research 2014, 13, 4488–4491, DOI: 10.1021/pr500741y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sticker A, Martens L, Clement L. Mass Spectrometrists Should Search for All Peptides, but Assess Only the Ones They Care About. Nature Methods 2017, 14, 643–644, DOI: 10.1038/nmeth.4338. [DOI] [PubMed] [Google Scholar]
- (24).Van der Walt S, Colbert SC, Varoquaux G. The NumPy Array: A Structure for Efficient Numerical Computation. Computing in Science & Engineering 2011, 13, 22–30, DOI: 10.1109/MCSE.2011.37. [DOI] [Google Scholar]
- (25).McKinney W. In Proceedings of the 9th Python in Science Conference, ed. by van der Walt S, Millman J,Austin, Texas, USA, 2010, pp 51–56. [Google Scholar]
- (26).Hagberg AA, Schult DA, Swart PJ In Proceedings of the 7th Python in Science Conference - SciPy ‘08, ed. by Varoquaux G, Vaught T, Millman J,Pasadena, CA USA, 2008, pp 11–15. [Google Scholar]
- (27).Hunter JD Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering 2007, 9, 90–95, DOI: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- (28).Waskom M, Botvinnik O, O’Kane D, Hobson P,et al. Mwaskom/Seaborn: V0.8.1 (September 2017), 2017, DOI: 10.5281/zenodo.883859. [DOI] [Google Scholar]
- (29).Goloborodko AA, Levitsky LI, Ivanov MV, Gorshkov MV Pyteomics-a Python Framework for Exploratory Data Analysis and Rapid Software Prototyping in Proteomics. Journal of The American Society for Mass Spectrometry 2013, 24, 301–304, DOI: 10.1007/s13361-012-0516-6. [DOI] [PubMed] [Google Scholar]
- (30).Bittremieux W, Tabb DL, Impens F, Staes A,et al. Quality Control in Mass Spectrometry-Based Proteomics. Mass Spectrometry Reviews 2018, 37, 697–711, DOI: 10.1002/mas.21544. [DOI] [PubMed] [Google Scholar]
- (31).Bandeira N, Tsur D, Frank A, Pevzner PA Protein Identification by Spectral Networks Analysis. Proceedings of the National Academy of Sciences 2007, 104, 6140–6145, DOI: 10.1073/pnas.0701130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bittremieux W, Laukens K, Noble WS Extremely Fast and Accurate Open Modification Spectral Library Searching of High-Resolution Mass Spectra Using Feature Hashing and Graphics Processing Units. bioRxiv 2019, DOI: 10.1101/627497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Creasy DM, Cottrell JS Unimod: Protein Modifications for Mass Spectrometry. PROTEOMICS 2004, 4, 1534–1536, DOI: 10.1002/pmic.200300744. [DOI] [PubMed] [Google Scholar]
- (34).Noble WS Mass Spectrometrists Should Search Only for Peptides They Care About. Nature Methods 2015, 12, 605–608, DOI: 10.1038/nmeth.3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tsiatsiani L, Heck AJR Proteomics beyond Trypsin. FEBS Journal 2015, 282, 2612–2626, DOI: 10.1111/febs.13287. [DOI] [PubMed] [Google Scholar]
- (36).Savidor A, Barzilay R, Elinger D, Yarden Y,et al. Database-Independent Protein Sequencing (DiPS) Enables Full-Length de Novo Protein and Antibody Sequence Determination. Molecular & Cellular Proteomics 2017, 16, 1151–1161, DOI: 10.1074/mcp.O116.065417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Blank-Landeshammer B, Kollipara L, Biß K, Pfenninger M,et al. Combining de Novo Peptide Sequencing Algorithms, a Synergistic Approach to Boost Both Identifications and Confidence in Bottom- up Proteomics. Journal of Proteome Research 2017, 16, 3209–3218, DOI: 10.1021/acs.jproteome.7b00198. [DOI] [PubMed] [Google Scholar]
- (38).Griss J, Stanek F, Hudecz O, Dürnberger G,et al. Spectral Clustering Improves Label-Free Quantification of Low-Abundant Proteins. Journal of Proteome Research 2019, 18, 1477–1485, DOI: 10.1021/acs.jproteome.8b00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wang M, Carver JJ, Phelan VV, Sanchez LM, et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nature Biotechnology 2016, 34, 828–837, DOI: 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cafferty BJ, Ten AS, Fink MJ, Morey S,et al. Storage of Information Using Small Organic Molecules. ACS Central Science 2019, DOI: 10.1021/acscentsci.9b00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary section 1: Simulated digestion of English dictionary. Supplementary figure S1: Simulated peptide length.