

Abstract
Background:
Pulmonary infections remain the most common cause of Acute Respiratory Distress Syndrome (ARDS), a pulmonary inflammatory disease with high mortality, for which no targeted therapy currently exists. We have previously demonstrated an ameliorated syndrome with early, broad spectrum Histone Deacetylase (HDAC) inhibition in a murine model of gram-negative pneumonia-induced Acute Lung Injury (ALI), the underlying pulmonary pathologic phenotype leading to ARDS. With the current project we aim to determine if selective inhibition of a specific HDAC leads to a similar pro-survival phenotype, potentially pointing to a future therapeutic target.
Methods:
C57Bl/6 mice underwent endotracheal instillation of 30×106 Escherichia coli (strain 19138) vs saline (n=24). Half the infected mice were administered Trichostatin A (TSA) 30 minutes later. All animals were sacrificed 6h later for tissue sampling and HDAC quantification, while another set of animals (n=24) was followed to determine survival. Experiments were repeated with selective siRNA inhibition of the HDAC demonstrating the greatest inhibition vs. scrambled siRNA (n=24).
Results:
TSA significantly ameliorated the inflammatory phenotype and improved survival in infected-ALI mice, and HDAC7 was the HDAC with the greatest transcription and protein translation suppression. Similar results were obtained with selective HDAC7 siRNA inhibition compared to scrambled siRNA.
Conclusion:
HDAC7 appears to play a key role in the inflammatory response that leads to ALI after gram-negative pneumonia in mice.
Keywords: Acute Lung Injury, Acute Respiratory Distress Syndrome, Histone Deacetylase Inhibition, Trichostatin-A, Epigenetic Modification, siRNA
Introduction
ARDS represents an inflammatory pulmonary disease, manifesting clinically with severe hypoxia. 1 The syndrome develops as the lungs mount an exaggerated immune response to severe local or systemic inflammatory stimuli.2 More than 200,000 cases of ARDS are diagnosed annually in the United States alone, that are associated with 3.6 million hospital days.3 Its incidence remains high, with an estimated 10.4% of all Intensive Care Unit (ICU) admissions meeting diagnostic criteria for ARDS, and an even higher 23.4% of all mechanically ventilated patients. 4 Despite advances in critical care, mortality remains similarly very high, approximating 40% across all patients with ARDS, and even exceeding 57% in the most severe cases 5 rendering ARDS more lethal than ischemic heart disease and embolic stroke. 5,6
ARDS is characterized by an out-of-proportion release of proinflammatory mediators and diffuse infiltration of the pulmonary parenchyma with inflammatory cells, not dissimilar to sepsis. 7 Despite progress made in critical care, our incomplete understanding of the complex immunological processes taking place in ARDS, has so far limited the development of effective treatment strategies. Broad spectrum anti-inflammatory therapies, including steroidal and non-steroidal anti-inflammatory agents, not only aimed at treating, 8–10 but also preventing the development of ARDS, 11 have so far failed to improve clinically relevant outcomes. The above underline the importance of identifying the key molecular pathways activated in ARDS and targeting them in a timely fashion. 12
We have previously demonstrated an ameliorated clinical syndrome and enhanced survival in a clinically relevant model of E. coli pneumonia-induced murine model of ALI, 13 the underlying pulmonary pathologic phenotype that leads to ARDS, with timely,14 broad-spectrum HDAC inhibition. (ARDS is a clinical syndrome that is very difficult to model in small animals, as it takes into account for its diagnosis the ratio of partial arterial pressure of O2 over inspired fraction of O2, findings on serial chest radiographs, evidence of a preserved cardiac ejection function, and absence of cardiogenic pulmonary edema. The above are challenging to obtain in small animals. As a result, the nearly universal approach to ARDS modeling in small animals is to use models of Acute Lung Injury (ALI). The latter is a histopathologic diagnosis present in the lungs of patients with ARDS, that involves the widespread migration of neutrophils into the lungs, hyaline deposition in the pulmonary interstitial space, thickening of the basal membrane and degradation of the blood-alveolar barrier and presence of inflammatory non-cardiogenic pulmonary edema.)
Lipopolysaccharide (LPS) is a key component of the gram-negative bacterial cell membrane, and is commonly secreted after such infections. LPS is known to bind to the Toll-like Receptor 4 (TLR4) complex on antigen-presenting cells, including B-cells, dendritic cells and macrophages. This interaction initiates several downstream pathways, including the phosphorylation of Heat Shock Protein-90 (HSP90) (via induction of pp60Src-dependent Y300), potentiating its chaperone function,15 and activation of the NF-kB pathway, a well-described transcription factor, eventually translocating into the nucleus and leading to upregulation of proinflammatory proteins IL-1b, IL-6, TNFa.16,17 Both these pathways have been linked to widespread migration of neutrophils to the pulmonary parenchyma, and breakdown of the alveolar-endothelial barrier, leading to non-hydrostatic pulmonary edema, both hallmarks of Acute Lung Injury (ALI). It has been shown that inhibition of the HDAC complex leads to hyperacetylation of HSP-90, limiting its chaperone action,18,19 and breaking down MyDD87 and IRAK-1, both NF-kB pathway intermediary proteins.20 These changes have led to an ameliorated ALI and enhanced survival with broad spectrum HDAC inhibition by both our group and others.13,14,16,19,21–23 What is unknown, however, is whether a specific HDAC target is chiefly responsible for these proinflammatory changes taking place in the lungs in vivo, and whether its specific inhibition will lead to a similar anti-inflammatory, pro-survival phenotype. With this project, we aim to determine whether one or more specific HDACs can be targeted for a similar result. In line with our previous experiments, where ALI is induced in mice after a severe gram negative (E. coli) pneumonia, and broad spectrum HDAC inhibition conferred an ameliorated anti-inflammatory profile and improved survival, we elected to determine whether Trichostatin A (TSA), a narrower spectrum, Class II HDAC inhibitor, would have similar effects. We elected to use TSA towards identifying one or more specific HDACs that may be key to the upregulation of the pulmonary inflammatory response that leads to ALI, as it has been previously demonstrated to be effective in ALI of septic etiology.24,25
Methods
Mice
Male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were acclimated for >5 days and maintained in pathogen-free conditions until experiments, at 6–8 weeks of age. All mice were allowed food and water ab libitum, and all procedures were approved by the Institution’s Animal Care and Use Committee and were conducted per the ARRIVE guidelines.
Escherichia coli
Escherichia coli (serotype 06:K2:H1, strain 19138 -ATCC, Manassas, VA) was cultured to the desired concentration in Luria broth and quantified by optical density.
Endotracheal Instillations and Acute Lung Injury
After isofluorane anesthesia, mice underwent direct endotracheal instillation using the tongue pull technique. Fifty microliters of sterile normal saline (n=6) or 30×106 of E. coli in the same amount of saline (n=24) was instilled endotracheally. Of the 24 mice that were challenged with E. coli, half were randomly selected and administered 1mg/kg of Trichostatin A (TSA) (Sigma-Aldrich, St.Louis, MO) in phosphate-buffer saline (PBS) containing 1% DMSO, and the other half with an equal volume (100µl) of vehicle (PBS containing 1% DMSO) intraperitoneally (IP) 30 minutes post endotracheal instillation. The 30 minute timepoint was selected as we have previously demonstrated that delayed HDAC inhibition loses its anti-inflammatory effects.14
The animals were humanely sacrificed 6 hours later and underwent thoracotomy (after isofluorane anesthesia, followed by cervical dislocation and cardiac puncture). Blood specimens were collected via cardiac puncture, while their left hilum was ligated for left lung preservation and histologic examination. A tracheostomy with a 22G angiocath was performed and bronchoalveolar lavage (BAL) of the right lung was obtained with two flushes of 2 cc HBSS/EDTA to a 1 cc volume of BAL fluid (BALF).
Histology and Confirmation of Acute Lung Injury
The left lung was fixed in 4% formalin, embedded in paraffin, sectioned, and mounted on microscopy slides. The samples were processed with xylene, stained with hematoxylin and eosin, and imaged with an Olympus Upright microscope (Olympus Corporation, Tokyo, Japan) by Histology core personnel unaware of the treatment assignments.
BALF Cell Count and Differential
Cell counts and differentials were obtained in the BALF by Cytospin (Thermo Fisher Scientific, Waltham, MA). Cell counts and viability were determined with a hemacytometer and trypan blue dye (Sigma-Aldrich, St.Louis, MO).
Bacterial Burden
Serial dilutions of BALF in sterile water were plated on 5% sheep blood-agar and Colony Forming Units (CFU) were counted after incubation at 37°C for 24h.
Cytokine Expression
BALF was spun at 1200 g for 15 min, and supernatant aliquots were stored at −80°C. Cytokine levels were quantified as previously described. 13 The pro-inflammatory cytokines tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and the CXC chemokines Keratinocyte Chemoattractant (KC) and Macrophage Inflammatory Protein-2 (MIP-2) levels were determined in samples by ELISA. 26 These cytokines and chemokines were selected, having been previously implicated in the pathophysiology of both ALI and ARDS. 27–30
Gene Expression Analysis
RNA was extracted from lung tissues with Trizol treatment (Thermo Fisher Scientific, Waltham, MA) per the manufacturer’s instructions. Reverse transcripts were prepared with Reverse Transcription First Strand Kit System (Qiagen, Germantown, MD). Gene-specific primers were designed and obtained from Integrated DNA Technology (IDT, Coralville, IA). For the forward (F) and reverse (R) please refer to online supplement.
Qualitative Real Time Polymerase Chain Reaction (qRT-PCR) reactions were carried out in a 20µl final volume containing 450ng of copy DNA, 2µl of primers (0.2µM each) and 10ul of iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA). Reactions were run on Step-one PCR systems (Applied Biosystems, Foster City, CA), running each sample in duplicate. Relative gene expression quantification was performed using the comparative threshold cycle real-time PCR method. Data were normalized to the RNA Polymerase 2A (PolR2A) gene expression.
The HDAC mRNA that was noted to undergo the largest change between the mice with the full-blown ALI syndrome and those treated with TSA was selected for further study and selective inhibition, as described below. (This was HDAC7, as described in the Results section).
Protein Expression Analysis
Pulmonary tissue was homogenized in RIPA buffer with protease/phosphatase inhibitors (Thermo Fisher Scientific, Waltham, MA). After lysis, samples were incubated on ice for 10 min and centrifuged at 10000 rpm for 20 min at 4°C. The concentration of HDAC7 (as described in the previous paragraph) was determined with the Bradford Protein Assay kit (BioRad, Hercules, CA), and Western Blots were performed with anti-HDAC7 antibodies (Sigma-Aldrich, St.Louis, MO). A total of 30µg of each protein sample was loaded and separated by gel electrophoresis and incubated overnight at 4°C with primary antibodies against HDAC7 (Li-Cor, Lincoln, NE). Fluorescent secondary antibodies at 1:15000 were then used and images captured with ImageJ (NIH, Bethesda, MD). The HDAC7 levels were normalized to beta-actin.
siRNA in vivo inhibition
Non-specific, control-siRNA (scrambled) and mouse HDAC7 specific (HDAC7-siRNA), High-Performance Liquid Chromatography (HPLC) purified siRNAs were purchased from Sigma (HDAC7 was selected due to its selective inhibition by TSA in our current experiments). HDAC7-siRNA 21nt was designed by Rosetta algorithm prediction, targeting the HDAC7 gene 789 nucleotides downstream from the Refseq sequence.
A total of 100µg/100µl of control-siRNA or HDAC7-siRNA was instilled endotracheally in mice, and HDAC7 protein expression was measured at 24, 48 and 72 hours. The time point of at least 40% inhibition was selected for our proof-of-concept subsequent experiments. With >40% HDAC7 inhibition (3 days after HDAC7 siRNA instillation), 30×106 CFU E. coli was instilled in both groups. After 6 hours, the animals were sacrificed and blood, tissue samples and BALF were obtained, as previously described.
Survival Analysis
A separate set of animals underwent 30×106 CFU E. coli endotracheal instillation and intraperitoneal injection of 1mg/kg of TSA or buffer as described earlier (n=12/group). Survival was monitored for 7 days. In a separate set of animals with HDAC7-siRNA (n=12) or scramble control-siRNA (n=12), 30×106 CFU of E. coli was instilled endotracheally after 72h, and survival was monitored again for 7 days.
Statistics
Statistical analyses were performed with Prism (GraphPad, La Jolla, CA). Data are presented as mean±standard errors. Comparisons across groups were performed with Student’s t-test or one-way Analysis of Variance (ANOVA) as indicated. Kaplan-Meier curves and log-rank test were used to assess differences in survival. Statistical significance was declared at p<0.05.
Results
Histopathology & Neutrophil Recruitment
While sections from sham animals demonstrated normal parenchymal architecture as expected (Figure 1A), histopathological findings consistent with ALI were observed in the E. coli animals: Numerous neutrophils in the alveolar compartment and in the interstitium. 13,28 Alveolar wall thickening and the expansion of the interstitial compartments were also noted, consistent with pulmonary edema. Loss of architectural integrity and red cell extravasation were also noted, suggestive of vascular endothelial disruption. E. coli + TSA animals demonstrated improved histopathology, with fewer neutrophils, less pulmonary edema, alveolar wall thickening, and red cell decompartmentalization. Parenchymal architectural integrity was also preserved.
Figure 1.
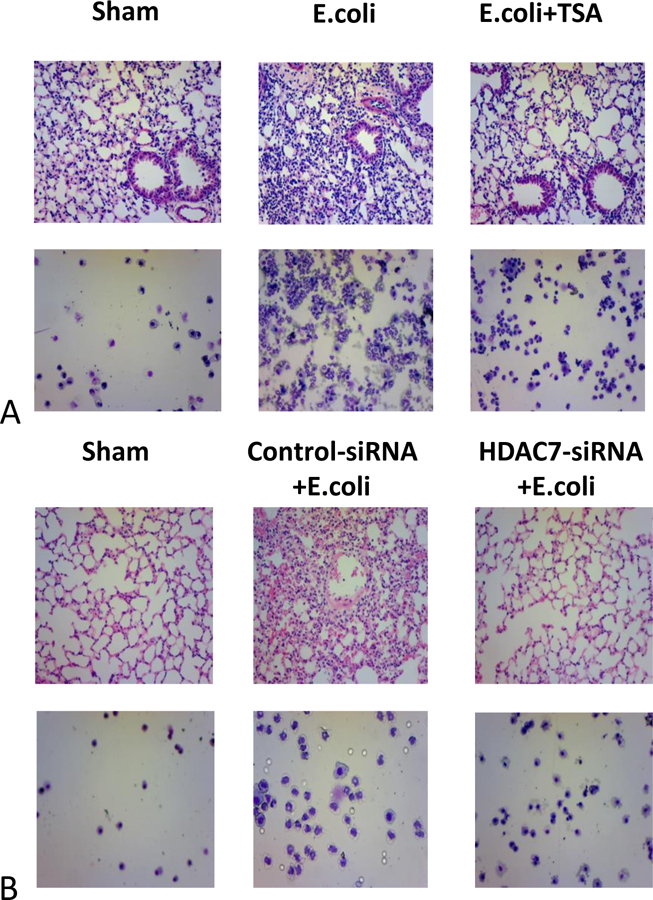
A. Histopathology sections from lung specimens and Cytospin images of sham, E. coli-infected plus intraperitoneal vehicle, and E. coli-infected plus intraperitoneal TSA. B. Histopathology sections from lung specimens and Cytospin images of sham, E. coli-infected plus endotracheal control siRNA, and E. coli-infected plus endotracheal HDAC7-siRNA.
BALF cytology demonstrated a small number of alveolar resident macrophages in the sham animals, with many more neutrophils recruited in the infected mice. The macrophages seen in the E. coli mice was similar to that of the sham animals. The number of macrophages was unchanged in the E. coli + TSA mice, although the chemoattracted neutrophils were significantly reduced. Numeric assessment of these in the BALF confirmed this observation (Figure 2A). Similar observations were noted in the E. coli + HDAC7-siRNA mice, while control siRNA did not appear to make a significant difference in either histopathology, nor neutrophil recruitment (Figure 1B, 2B).
Figure 2.
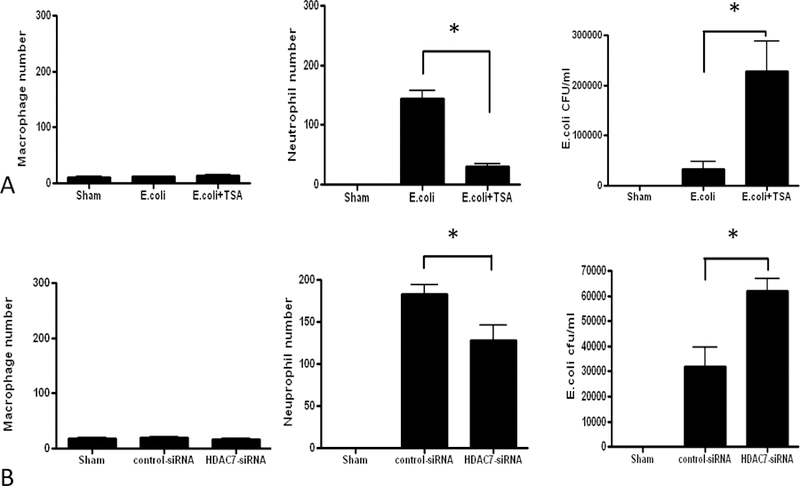
A. Macrophage and neutrophil counts and E. coli CFUs in BALF from sham, E. coli-infected plus intraperitoneal vehicle, and E. coli-infected plus intraperitoneal TSA. B. Macrophage and neutrophil counts and E. coli CFUs in BALF from sham, E. coli-infected plus endotracheal control siRNA, and E. coli-infected plus endotracheal HDAC7-siRNA. (* denotes p<0.05)
Pulmonary Bacterial Burden
No bacteria were found in the sham animals, a higher count in the infected animals, and an even higher one in the E. coli + TSA animals (Figure 2A). Bacterial counts in the BALF of control-and E. coli + HDAC7-siRNA were noted to parallel those of sham and E. coli + TSA mice (Fig 2B).
Cytokine Expression
The pro-inflammatory cytokines TNF-α and IL-6 were noted to be significantly elevated in the BALF of the E. Coli mice compared to the sham animals, and both were found to be decreased in the E. coli + TSA mice (Figure 3A), an observation that suggests a mitigated pulmonary immune response due to HDAC inhibition. BALF chemokines MIP-2 and KC were increased in the E. coli animals compared to shams, and E. coli + TSA appeared to affect only the former, consistent with prior findings. 13 Only TNF-a was noted to be decreased significantly in BALF with HDAC7-siRNA inhibition, while IL-6, MIP-2 and KC were essentially unchanged between the E. coli + HDAC7-siRNA and E. coli + control-siRNA animals (Figure 3B).
Figure 3.
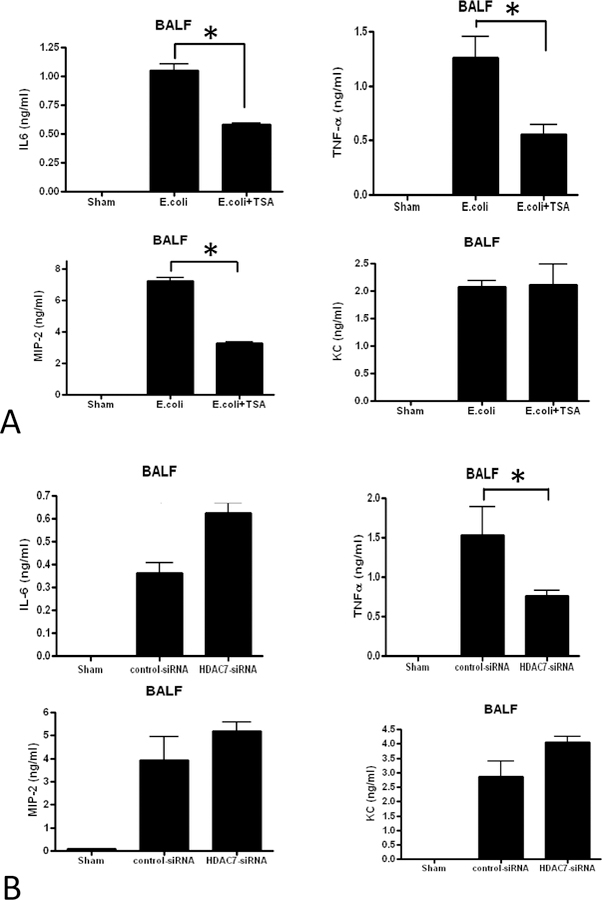
A. Cytokine IL-6, TNFa, MIP2 and KC levels in BALF from sham, E. coli-infected plus intraperitoneal vehicle, and E. coli-infected plus intraperitoneal TSA. B. Cytokine IL-6, TNFa, MIP2 and KC levels in BALF from sham, E. coli-infected plus endotracheal control siRNA, and E. coli-infected plus endotracheal HDAC7-siRNA. (* denotes p<0.05)
HDAC Gene transcription analysis
Having determined the baseline gene expression of all well-described HDACs (HDAC1–11 & Sirt1) in the lungs of sham animals relative to endogenous RNA PolR2A, we noted that mRNA transcription of HDAC2, HDAC7 and HDAC8 decreased significantly in the E. coli animals. The only HDAC whose transcription decreased further in a statistically significant fashion with TSA administration compared to the E. coli animals was HDAC7 (Figure 4A), suggestive that TSA is likely a selective HDAC7 inhibitor in the lungs, at least in our murine model of gram-negative pneumonia induced ALI. The presence of infection led to a decrease in HDAC2, HDAC7 and HDAC8 mRNA transcription, and an increase in HDAC4 and HDAC10 in the E. coli + control-siRNA animals, while an increase in HDAC8 was noted in the E. coli + HDAC7-siRNA mice. The results of this analysis are summarized in Figure 4B.
Figure 4.
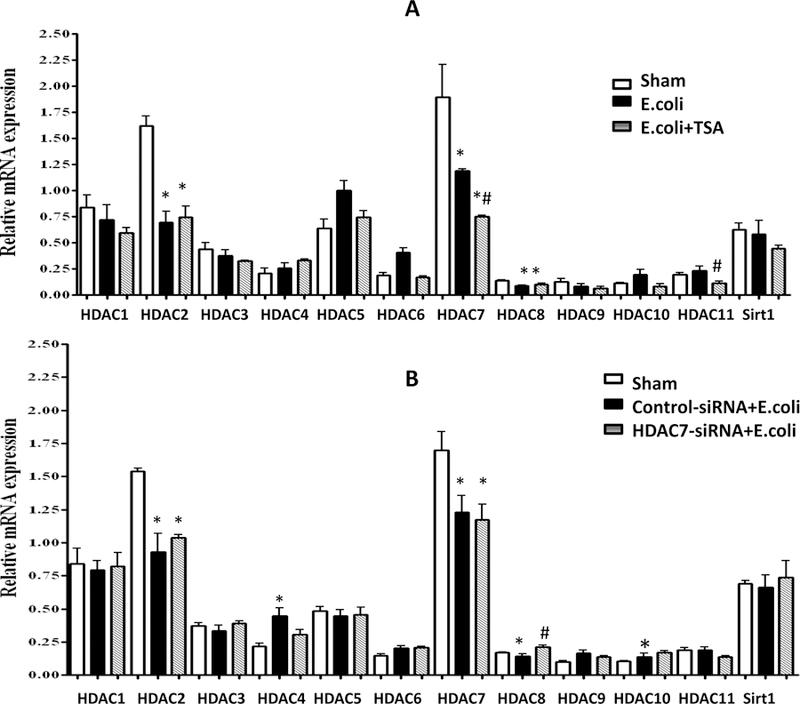
HDAC gene transcription analysis in sham, E. coli-infected and intraperitoneal vehicle, and E. coli-infected plus intraperitoneal TSA (A), and sham, E. coli-infected plus endotracheal control-siRNA, and E. coli-infected plus endotracheal HDAC7-siRNA (B). (* denotes statistical significance p<0.05 compared to sham; # denotes statistical significance p<0.05 between E. coli and E. coli+TSA or between E. coli+control-siRNA and E. coli+HDAC7-siRNA respectively).
Protein expression analysis
Although HDAC7 gene transcription is high at baseline in sham animals and decreases in the E. coli animals, and more so in the E. coli + TSA mice, HDAC7 protein levels do not correspond to these mRNA levels, being low at baseline, increasing significantly in the E. coli animals, and decreasing again in the E. coli + TSA mice (Figure 5A).
Figure 5.
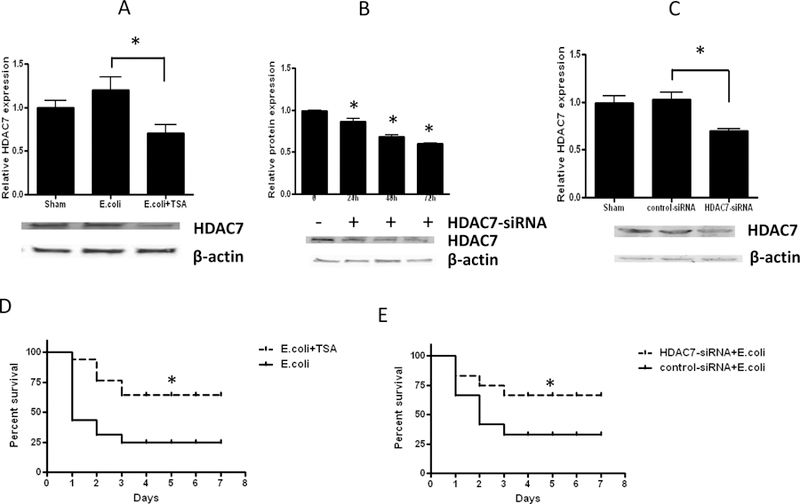
Protein expression analysis. A: Immunoblot analysis of HDAC7 protein in homogenized lung parenchyma in sham, E. coli-infected plus intraperitoneal vehicle, and E. coli-infected plus intraperitoneal TSA animals. B: HDAC7 protein inhibition in animals at varied time points with endotracheal instillation of HDAC7-siRNA. C: Inhibition of HDAC7 protein synthesis with 72h endotracheally pre-administered HDAC7-siRNA. D. Survival analysis in E. coli-infected plus intraperitoneal vehicle versus E. coli-infected plus intraperitoneal TSA. E. Survival analysis in E. coli-infected plus endotracheal control-siRNA versus E. coli-infected plus endotracheal HDAC7-siRNA (B). (* denotes statistical significance at p<0.05)
Prior to E. coli infection, we tested the ability to selectively inhibit HDAC7 in the mouse lungs by a priori endotracheal instillation of HDAC7 siRNA. We demonstrated >40% inhibition of quantifiable HDAC7 protein in the lungs with use of HDAC7-siRNA 72h ahead of the application of the infectious stimulus (Figure 5B). This siRNA corresponds to a non-biased approach to HDAC7 inhibition, as at the time of the experiment the cellular source of HDAC7 was unclear (we aim to address this issue in an upcoming project). Mice treated with non-silencing scrambled control-siRNA did not effectively change HDAC7 protein expression (Figure 5C), suggesting satisfactory use of our HDAC7 siRNA methodology.
Survival
Survival was improved in the E. coli + TSA animals, improving from approximately 25% in the E. coli mice to 75% (Figure 5D). The E. coli + HDAC7-siRNA mice, just like the E. coli + TSA animals, demonstrated a similarly significant improvement in survival (Figure 5E).
Discussion
Pulmonary infections remain by far the most common cause of ARDS, 4 and a key mechanism by which bacterial infections activate the host immune response is through activation of the Toll-Like Receptor (TLR) -NF-kB pathway: Pathogen Associated Molecular Patterns (PAMPs) bind to plasma membrane TLRs to stimulate nuclear translocation of NF-kB, driving transcription of proinflammatory cytokines. 31,32 These kB-regulated proteins including KC, MIP-2, TNF-α, IL-6 and IL-1b, have been shown to play a pivotal role in the pathogenesis of lung injury in ARDS. 30,33–35 HDAC inhibition is postulated to decrease intermediary Myeloid Differentiation Factor 88 (MyD88) and Interleukin-1 Receptor Kinase (IRAK1) protein expression in sepsis, seemingly by enhancing their degradation through Heat Shock Protein 90 (HSP90) hyper-acetylation. 19,36 Little is known, however, about the role of HDAC inhibition in ALI.
We have previously demonstrated an anti-inflammatory and pro-survival phenotype with broad HDAC inhibition with Valproic acid, 13 when administered within a short therapeutic window. 14 However, the therapeutic index of Valproic Acid in both mice and humans is fairly narrow, 37 and it has been reported to inhibit essentially all zinc-dependent HDAC enzymes. 38 In addition, the specific role of the various HDACs in the pulmonary inflammatory response is poorly understood. With this project we sought to determine whether a more specific target for anti-inflammatory regulation exists among the pulmonary HDACs.
TSA, a fungal toxin, is a well-described broad HDAC inhibitor in vitro, 39 while it appears to be a more selective HDAC inhibitor in vivo. 40 We selected to use TSA in our experiments, as it has been described to confer a protective advantage against endothelial barrier dysfunction in both Lipopolysaccharide (LPS)-19 and streptococcal-induced models of severe pneumonia/ALI in mice, 41 as well as minimize ventilator-induced lung injury 25 and ALI in polymicrobial sepsis. 23
With early TSA administration, we demonstrate an anti-inflammatory pro-survival phenotype, not dissimilar to that achieved with much broader HDAC inhibition. 13 TSA also decreased HDAC7 gene expression and protein synthesis, a finding consistent with the work published by Li et al. 40 Based on these findings, we elected to selectively inhibit HDAC7, to determine whether similar observations could be made. With HDAC7 playing a pivotal role in the normal cardiopulmonary embryogenesis 42–44 and HDAC7 nullizygous mice not surviving past the embryonic level, 45 it would not be possible to investigate the effects of knocking out the HDAC7 gene in our mice, so we elected to selectively inhibit it with direct endotracheal instillation of HDAC7-siRNA. A benefit of this post-transcriptional inhibition of HDAC7 is that its decreased (as opposed to near zero) levels may be more akin to the effects of pharmacologic inhibition of the enzyme, and hence potentially more clinically relevant. We sought to confirm that HDAC7 protein expression is reduced in the pulmonary parenchyma of our mice and demonstrated in our proof-of-concept experiment that endotracheal pre-instillation of HDAC7-siRNA affords >40% HDAC7 protein synthesis suppression. This epigenetic manipulation confers similar pathology, cytology, inflammatory and survival benefits with TSA, although IL-6 and MIP-2 do not change significantly. This finding may correspond to the fact that siRNA may act as a Damage Associated Molecular Patterns and may in its own right partly activate TLR-dependent pathways, as these elevations were similar with both the control and HDAC7 siRNA. 46
More interestingly, HDAC7 mRNA is not statistically different from control-siRNA-treated animals, suggesting that a negative feedback loop must be in place, attempting to restore HDAC7 levels. Another interesting finding is that HDAC8 mRNA is increased in the HDAC7-siRNA-treated mice, suggesting that cross talk may exist at least between the two enzymes.
The above observations suggest that HDAC7 may play an important role in the regulation of the pulmonary response that leads to the lung injury of ARDS after gram-negative pneumonia. This finding is corroborated by the work of Joshi et al who also demonstrated ameliorated endothelial barrier dysfunction and limitation of the lung injury in cultured LPS-activated human lung microvascular endothelial cells with TSA. 19 Shakespear et al 20 also demonstrated that HDAC7 promotes TLR4-dependent proinflammatory gene expression in macrophages, a finding consistent with ours.
We also demonstrated that baseline HDAC7 gene transcription is high at baseline in the sham animals and decreases during gram-negative infection. This transcription is inhibited further with both TSA and highly selective HDAC7-siRNA inhibition. However, HDAC7 protein expression is low at baseline, increases during E. coli infection and improves with both TSA and HDAC7-siRNA administration. This finding demonstrates that HDAC7 mRNA transcription does not correlate with protein translation in healthy animals, suggesting that post-transcriptional modifications take place at baseline that either leads to breakdown of HDAC7 mRNA through a poorly understood mechanism; limit the amount of HDAC7 mRNA that undergoes effective protein translation; or to high translated HDAC7 protein breakdown and turnover. This observation is consistent with findings from Shakespear et al,20 who demonstrated that only one of two alternatively spliced HDAC7 mRNA molecules leads to the proinflammatory version of the protein, while the other is inert. While the Shakespear study utilized thioglycolate-elicited peritoneal macrophages, and we can only make an inference by association, we assume that it is the alveolar macrophages that are the chief HDAC7 producers in the stimulated murine lung, and aim to confirm this hypothesis in the aforementioned upcoming project.
There are a several limitations in the current project: We did not determine the effect of HDAC7 inhibition to other HDACs. It appears that important cross-talk may exist across the different classes of HDACs, as selective inhibition of HDAC7 affects the transcription of many other HDACs. This is a topic we aim to examine further in the near future. Also, we did not examine the effect of apoptosis to the overall anti-inflammatory and pro-survival effect of HDAC7 inhibition. In addition, we have not assessed the effect of HDAC inhibition in non-gram-negative infectious etiology ARDS models, where different pro-inflammatory pathways may become activated, which will be the topic of future studies. Lastly, we did not examine possible toxicity selective HDAC7 inhibition may have on the animals, although we estimate that to be likely less significant than that of other steroidal anti-inflammatories and broad-spectrum HDAC inhibitors. Further studies will be required to look at the toxicity HDAC7 inhibition exerts, as well as the specific pathways it may be involved in toward minimizing the ALI of ARDS. Notably, the present study is descriptive in nature, and its intended purpose is to shed light on potential specific HDACs that may be chiefly responsible for regulating the local immune response that leads to ALI/ARDS after a gram-negative pneumonia in mice, as a proof of concept. In a subsequent project, work on which has already begun, we plan to determine the cellular source of HDAC7 (likely the alveolar macrophage), the stimuli that trigger its protein synthesis, its downstream cellular targets, and the functionally active versus inert versions of HDAC7 (HDAC7-u and HDAC7-s), as described by other investigators.
Conclusion
In conclusion, we demonstrate that highly selective inhibition of HDAC7 limits the pro-inflammatory effects of severe gram-negative pneumonia that typically progresses to ALI and ARDS, and significantly improves survival. Our study provides new insights into the pathophysiology of the syndrome, at least in cases of gram-negative infections, paving the way towards identification of potential therapeutic targets in the management of gram-negative infection-induced ARDS.
Supplementary Material
Acknowledgements
The project was funded partly by the NIH R01 GM117519 and R21 AI112887.
Footnotes
Conflicts of Interest: None
References
- 1.Rezoagli E, Fumagalli R, Bellani G. Definition and epidemiology of acute respiratory distress syndrome. Ann Transl Med; 5:282, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. The Journal of clinical investigation; 122:2731–40, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. The New England journal of medicine; 353:1685–93, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley DF, et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. Jama;315:788–800, 2016. [DOI] [PubMed] [Google Scholar]
- 5.Madotto F, Pham T, Bellani G, Bos LD, Simonis FD, Fan E, Artigas A, Brochard L, Schultz MJ, Laffey JG, et al. Resolved versus confirmed ARDS after 24 h: insights from the LUNG SAFE study. Intensive care medicine 2018. [DOI] [PubMed]
- 6.Phua J, Badia JR, Adhikari NK, Friedrich JO, Fowler RA, Singh JM, Scales DC, Stather DR, Li A, Jones A, et al. Has mortality from acute respiratory distress syndrome decreased over time?: A systematic review. American journal of respiratory and critical care medicine;179:220–7, 2009. [DOI] [PubMed] [Google Scholar]
- 7.ARDS Definition Task Force, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS. Acute respiratory distress syndrome: the Berlin Definition. Jama; 307:2526–33, 2012. [DOI] [PubMed] [Google Scholar]
- 8.Bernard GR, Luce JM, Sprung CL, Rinaldo JE, Tate RM, Sibbald WJ, Kariman K, Higgins S, Bradley R, Metz CA, et al. High-dose corticosteroids in patients with the adult respiratory distress syndrome. The New England journal of medicine; 317:1565–70, 1987. [DOI] [PubMed] [Google Scholar]
- 9.McAuley DF, Laffey JG, O’Kane CM, Perkins GD, Mullan B, Trinder TJ, Johnston P, Hopkins PA, Johnston AJ, McDowell C, et al. Simvastatin in the acute respiratory distress syndrome. The New England journal of medicine; 371:1695–703, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Steinberg KP, Hudson LD, Goodman RB, Hough CL, Lanken PN, Hyzy R, Thompson BT, Ancukiewicz M, National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. The New England journal of medicine; 354:1671–84, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Kor DJ, Carter RE, Park PK, Festic E, Banner-Goodspeed VM, Hinds R, Talmor D, Gajic O, Ware LB, Gong MN, et al. Effect of Aspirin on Development of ARDS in At-Risk Patients Presenting to the Emergency Department: The LIPS-A Randomized Clinical Trial. Jama; 315:2406–14, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubenfeld GD. Who cares about preventing acute respiratory distress syndrome? American journal of respiratory and critical care medicine; 191:255–60, 2015. [DOI] [PubMed] [Google Scholar]
- 13.Kasotakis G, Galvan M, King E, Sarkar B, Stucchi A, Mizgerd JP, Burke PA, Remick D. Valproic acid mitigates the inflammatory response and prevents acute respiratory distress syndrome in a murine model of Escherichia coli pneumonia at the expense of bacterial clearance. J Trauma Acute Care Surg; 82:758–65, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasotakis G, Galvan MD, Osathanugrah P, Dharia N, Bufe L, Breed Z, Mizgerd JP, Remick DG. Timing of valproic acid in acute lung injury: prevention is the best therapy? The Journal of surgical research; 220:206–12, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barabutis N, Handa V, Dimitropoulou C, Rafikov R, Snead C, Kumar S, Joshi A, Thangjam G, Fulton D, Black SM, et al. LPS induces pp60c-src-mediated tyrosine phosphorylation of Hsp90 in lung vascular endothelial cells and mouse lung. American journal of physiology Lung cellular and molecular physiology; 304:L883–93, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Chang G, Huang J, Wang Y, Ma N, Roy AC, Shen X. Sodium Butyrate Inhibits the Inflammation of Lipopolysaccharide-Induced Acute Lung Injury in Mice by Regulating the Toll-Like Receptor 4/Nuclear Factor kappaB Signaling Pathway. J Agric Food Chem; 67:1674–82, 2019. [DOI] [PubMed] [Google Scholar]
- 17.Liu YL, Liu YJ, Liu Y, Li XS, Liu SH, Pan YG, Zhang J, Liu Q, Hao YY. Hydroxysafflor yellow A ameliorates lipopolysaccharide-induced acute lung injury in mice via modulating toll-like receptor 4 signaling pathways. Int Immunopharmacol; 23:649–57, 2014. [DOI] [PubMed] [Google Scholar]
- 18.Chatterjee A, Dimitropoulou C, Drakopanayiotakis F, Antonova G, Snead C, Cannon J, Venema RC, Catravas JD. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. American journal of respiratory and critical care medicine; 176:667–75, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joshi AD, Barabutis N, Birmpas C, Dimitropoulou C, Thangjam G, Cherian-Shaw M, Dennison J, Catravas JD. Histone deacetylase inhibitors prevent pulmonary endothelial hyperpermeability and acute lung injury by regulating heat shock protein 90 function. American journal of physiology Lung cellular and molecular physiology; 309:L1410–9, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shakespear MR, Hohenhaus DM, Kelly GM, Kamal NA, Gupta P, Labzin LI, Schroder K, Garceau V, Barbero S, Iyer A, et al. Histone deacetylase 7 promotes Toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J Biol Chem; 288:25362–74, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li LF, Lee CS, Lin CW, Chen NH, Chuang LP, Hung CY, Liu YY. Trichostatin A attenuates ventilation-augmented epithelial-mesenchymal transition in mice with bleomycin-induced acute lung injury by suppressing the Akt pathway. PLoS One; 12:e0172571, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni YF, Wang J, Yan XL, Tian F, Zhao JB, Wang YJ, Jiang T. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respiratory research; 11:33, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Jin S, Wang C, Jiang R, Wan J. Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture-induced polymicrobial sepsis. World journal of surgery; 34:1676–83, 2010. [DOI] [PubMed] [Google Scholar]
- 24.Thangavel J, Samanta S, Rajasingh S, Barani B, Xuan YT, Dawn B, Rajasingh J. Epigenetic modifiers reduce inflammation and modulate macrophage phenotype during endotoxemia-induced acute lung injury. J Cell Sci; 128:3094–105, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen HY, Li L, Fu ZJ. Histone deacetylase inhibitors trichostatin A and suberoylanilide hydroxamic acid attenuate ventilator-induced lung injury. Pharmazie; 69:55–9, 2014. [PubMed] [Google Scholar]
- 26.Osuchowski MF, Siddiqui J, Copeland S, Remick DG. Sequential ELISA to profile multiple cytokines from small volumes. J Immunol Methods; 302:172–81, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, Phillips RJ, Strieter RM. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. The Journal of clinical investigation; 110:1703–16, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizgerd JP, Lupa MM, Kogan MS, Warren HB, Kobzik L, Topulos GP. Nuclear factor-kappaB p50 limits inflammation and prevents lung injury during Escherichia coli pneumonia. American journal of respiratory and critical care medicine; 168:810–7, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Mizgerd JP, Spieker MR, Doerschuk CM. Early response cytokines and innate immunity: essential roles for TNF receptor 1 and type I IL-1 receptor during Escherichia coli pneumonia in mice. Journal of immunology; 166:4042–8, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Critical care medicine; 27:304–12, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene; 18:6853–66, 1999. [DOI] [PubMed] [Google Scholar]
- 32.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature; 472:476–80, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. The Journal of clinical investigation; 107:1529–36, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li XY, Donaldson K, Brown D, MacNee W. The role of tumor necrosis factor in increased airspace epithelial permeability in acute lung inflammation. American journal of respiratory cell and molecular biology; 13:185–95, 1995. [DOI] [PubMed] [Google Scholar]
- 35.Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F 2nd, Park DR, Pugin J, Skerrett SJ, Hudson LD, Martin TR. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. American journal of respiratory and critical care medicine; 164:1896–903, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Chong W, Li Y, Liu B, Zhao T, Fukudome EY, Liu Z, Smith WM, Velmahos GC, deMoya MA, Alam HB. Histone deacetylase inhibitor suberoylanilide hydroxamic acid attenuates Toll-like receptor 4 signaling in lipopolysaccharide-stimulated mouse macrophages. The Journal of surgical research; 178:851–9, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sztajnkrycer MD. Valproic acid toxicity: overview and management. J Toxicol Clin Toxicol; 40:789–801, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J; 20:6969–78, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bieliauskas AV, Pflum MK. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev; 37:1402–13, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S, Wang B, Xu Y, Zhang J. Autotaxin is induced by TSA through HDAC3 and HDAC7 inhibition and antagonizes the TSA-induced cell apoptosis. Mol Cancer; 10:18, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yagi K, Ishii M, Namkoong H, Fujii H, Asami T, Suzuki S, Asakura T, Mizoguchi K, Kamo T, Tasaka S, et al. Histone Deacetylase Inhibition Protects Mice Against Lethal Postinfluenza Pneumococcal Infection. Critical care medicine; 44:e980–7, 2016. [DOI] [PubMed] [Google Scholar]
- 42.Margariti A, Zampetaki A, Xiao Q, Zhou B, Karamariti E, Martin D, Yin X, Mayr M, Li H, Zhang Z, et al. Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circ Res;106:1202–11, 2010. [DOI] [PubMed] [Google Scholar]
- 43.Yang J, Margariti A, Zeng L. Analysis of Histone Deacetylase 7 (HDAC7) Alternative Splicing and Its Role in Embryonic Stem Cell Differentiation Toward Smooth Muscle Lineage. Methods Mol Biol;1436:95–108, 2016 [DOI] [PubMed] [Google Scholar]
- 44.Kretsovali A, Hadjimichael C, Charmpilas N. Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells Int:184154, 2012. [DOI] [PMC free article] [PubMed]
- 45.Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell;126:321–34, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Mansoori B, Mohammadi A, Shir Jang S, Baradaran B. Mechanisms of immune system activation in mammalians by small interfering RNA (siRNA). Artif Cells Nanomed Biotechnol;44:1589–96, 2016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.