

Abstract
Anti‐programmed death‐1 (PD‐1)/programmed death‐ligand 1 (PD‐L1) therapy, which is one of the most promising cancer therapies, is licensed for treating various tumors. Programmed death‐ligand 1, which is expressed on the surface of cancer cells, leads to the inhibition of T lymphocyte activation and immune evasion if it binds to the receptor PD‐1 on CTLs. Anti‐PD‐1/PD‐L1 Abs inhibit interactions between PD‐1 and PD‐L1 to restore antitumor immunity. Although certain patients achieve effective responses to anti‐PD‐1/PD‐L1 therapy, the efficacy of treatment is highly variable. Clinical trials of anti‐PD‐1/PD‐L1 therapy combined with radiotherapy/chemotherapy are underway with suggestive evidence of favorable outcome; however, the molecular mechanism is largely unknown. Among several molecular targets that can influence the efficacy of anti‐PD‐1/PD‐L1 therapy, PD‐L1 expression in tumors is considered to be a critical biomarker because there is a positive correlation between the efficacy of combined treatment protocols and PD‐L1 expression levels. Therefore, understanding the mechanisms underlying the regulation of PD‐L1 expression in cancer cells, particularly the mechanism of PD‐L1 expression following DNA damage, is important. In this review, we consider recent findings on the regulation of PD‐L1 expression in response to DNA damage signaling in cancer cells.
Keywords: clinical protocol, combined modality therapy, cytotoxic T lymphocyte, DNA damage, signal transduction
Abbreviations
- Ab
antibody
- ATM
ataxia‐telangiectasia mutated
- ATR
ataxia telangiectasia and Rad3 related
- BER
base excision repair
- BRCA
breast cancer susceptibility protein
- cGAS
cyclic GMP‐AMP synthase
- Chk
checkpoint kinase
- CTL
cytotoxic T lymphocyte
- DAMP
damage‐associated molecular pattern
- DC
dendritic cell
- DNA‐PKcs
DNA‐dependent protein kinase catalytic subunit
- DSB
double‐strand break
- HLA
human leukocyte antigen
- HMGB1
high mobility group box‐1
- HR
homologous recombination
- IFN
interferon
- IFNAR
interferon alpha/beta receptor
- IFNGR
interferon gamma receptor
- IR
ionizing radiation
- IRF1
interferon regulatory factor 1
- JAK
Janus kinase
- MMR
mismatch repair
- MRE11
meiotic recombination 11
- MSI
microsatellite instability
- MyD88
myeloid differentiation primary response 88
- NHEJ
non‐homologous end joining
- ORF
open reading frame
- PARP
poly‐(ADP‐ribose) polymerase
- PARPi
poly‐(ADP‐ribose) polymerase inhibitor
- PD‐1
programmed cell death 1
- PD‐L1
programmed death‐ligand 1
- RPA
replication protein A
- SSB
single‐strand break
- ssDNA
single‐stranded DNA
- STAT
signal transducer and activator of transcription
- STING
stimulator of interferon genes
- TLR4
toll‐like receptor 4
- TMB
tumor mutational burden
- TRIF
TIR‐domain‐containing adapter‐inducing interferon‐β
- XRCC4
X‐ray repair cross‐complementing protein 4
1. REGULATION OF PD‐L1 EXPRESSION IN RESPONSE TO DNA DAMAGE SIGNALING IN CANCER CELLS
Cancer therapies such as radiotherapy and chemotherapy cause cell death through DNA damage. Recent studies have suggested that the DNA damage response is an important factor influencing the efficacy of cancer immunotherapy.1, 2 DNA damage induced by IR, etoposide, camptothecin, cisplatin, mitomycin C, and alkylating agents upregulates PD‐L1 expression in cancer cells.3 In this section, we describe the current state of knowledge of the molecular mechanisms underlying the regulation of PD‐L1 expression in response to DSB induction and the influence of DSB repair and signaling.
A DSB is a critical form of DNA damage, and the capability to repair DSBs significantly influences cell fate. Among the available cancer treatments, IR, etoposide, and camptothecin therapy kills cancer cells by inducing DSBs. In human cells, DSBs are repaired by NHEJ or HR (Figure 1).4, 5 Immediately following the introduction of DSBs, cells emit a warning signal that arrests the cell cycle by activating a cell cycle checkpoint signal.6 Double‐strand break repair and cell cycle checkpoint arrest cooperate to facilitate the recovery of cells after DSBs. Following the induction of DSBs, ATM, which is the central DNA damage signal transducer, is rapidly and transiently activated at the DSB site.7 Activation of ATM occurs predominantly at unresected DSB ends, including those undergoing NHEJ (Figure 1).8 In contrast, ATR, the other central DNA damage signal transducer, is activated at DSB ends undergoing HR (Figure 1).8, 9 In the axis of cell cycle checkpoint activation, ATM activates Chk2 and p53, whereas ATR activates Chk1 (Figure 1). The ATM‐ATR/Chk1‐dependent phosphorylation signaling cascade activates further signal transduction, leading to cell cycle arrest.6 P53 is frequently inactivated in cancer cells; therefore, the G1/S checkpoint is rarely effectively activated in cancer cells. In contrast, the G2/M checkpoint is activated with relative efficiency in cancer cells, which explains why such cells exposed to DNA‐damaging agents frequently accumulate in G2 phase. The ATR/Chk1 pathway mainly contributes to the arrest of cells in G2 phase.10 During HR, DSB ends are processed by DNA nucleases to generate 3′‐overhangs (called “DSB end resection”). The process of DSB end resection is triggered by CtIP/MRE11‐dependent endonuclease incision, which nicks the 5′ strand at DSB ends. Subsequently, the exonuclease activities of MRE11, exonuclease 1, and DNA2 expand resection by digesting DNA bidirectionally to produce a sufficient length of ssDNA.5, 11 Following resection, the generated ssDNA is required for DNA strand invasion and D‐loop formation. Additionally, ATR is activated by ssDNA coated with RPA, and the activation of the ATR/Chk1 pathway is, therefore, highly associated with the magnitude of DSB end resection (Figure 1).9, 12 As the ssDNA serves as a scaffold for ATR‐mediated Chk1 activation, ATR/Chk1 is also activated at ssDNA gaps when DNA replication is stalled.13
Figure 1.
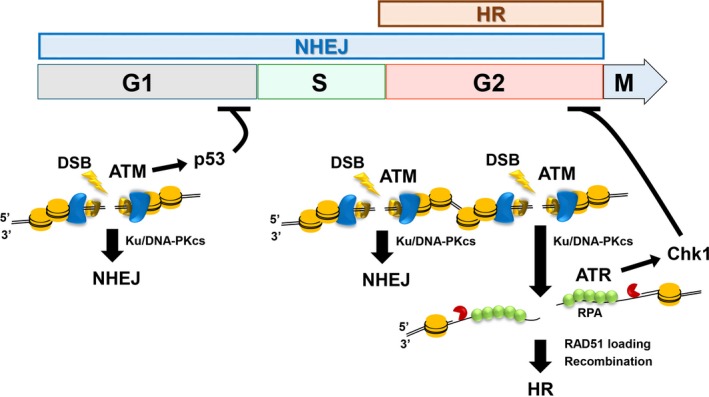
Orchestration of double‐strand break (DSB) repair and its associated signaling activity. DSBs are repaired by non‐homologous end joining (NHEJ) or homologous recombination (HR). NHEJ repairs DSBs throughout the cell cycle except for M phase in mammalian cells, whereas HR functions only in S/G2 phase following DNA replication. DSB ends undergoing NHEJ, which are not resected, activate ataxia‐telangiectasia mutated (ATM). In contrast, DSB ends that undergo resection by DNA nucleases promote HR. The Ku70/80 (Ku) and DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs) complex binds to most DSB ends to protect them from DNA nucleases and thereby promote NHEJ. In G1 phase, an ATM/p53‐dependent pathway activates G1/S checkpoint arrest, whereas in G2 phase, ataxia telangiectasia and Rad3 related (ATR)/checkpoint kinase (Chk1) activates G2/M checkpoint arrest. As ATM is required for resection, ATM contributes to G2/M checkpoint arrest. RPA, replication protein A
Several recent studies reported that DNA damage induces the expression of PD‐L1 mRNA, which results in the increase in the cell surface expression of PD‐L1.3, 14, 15, 16 This process depends on the activity of the ATM‐ATR/Chk1 signal transduction, suggesting that the expression of PD‐L1 is controlled by DNA damage signaling. Thus, the activation of the ATM‐ATR/Chk1 signal during the repair process above is a critical step leading to the upregulation of PD‐L1 after exogenous genotoxic stress. In the next paragraph, we introduce the concept that there is greater upregulation of DSB‐induced PD‐L1 in a repair defective background.
In our recent study, we found that depletion of Ku70/80 or BRCA2 significantly enhances the upregulation of PD‐L1 expression after IR.3 Ku70/80 and DNA‐PKcs bind to most DNA break ends immediately after the induction of DSBs (Figure 1).11, 17 Among the multiple roles of DNA‐PKcs in NHEJ, it aids the recruitment of NHEJ repair factors following its autophosphorylation. In addition to a role for Ku in recruiting DNA‐PKcs and facilitating NHEJ, the role of immediate binding of Ku70/80 to the DSB ends has been considered to protect DSB ends from inappropriate DNA digestion by DNA nucleases.18, 19 Consistent with this notion, depletion of Ku70/80 complexes enhances DSB end resection, which has been ascribed to the failure of DSB end protection, followed by increased ATR/Chk1 activation compared with that of control cells. Consistent with the increased activation of ATR/Chk1 signaling, depletion of Ku70/80 enhances further upregulation of the expression of DNA damage‐dependent PD‐L1.3 Additionally, BRCA2 depletion also induces upregulation of PD‐L1 expression after DSB formation. BRCA2 is required for HR by functioning to promote the switch from RPA to RAD51 on regions of ssDNA (Figure 1). Therefore, BRCA2 depletion impairs the ability to switch from RPA to RAD51 and consequently RPA accumulates at DSB ends, which is associated with continuous activation of ATR/Chk1 signaling. Thus, increased upregulation of PD‐L1 expression in BRCA2‐depleted cells is considered to be caused by the continuous activation of ATR/Chk1 signaling. Consistent with this idea, increased upregulation of PD‐L1 expression in BRCA2‐depleted cells is significantly suppressed by inhibition of ATR/Chk1 signaling.3 These results suggest that ATR/Chk1 serves as a central relay point, promoting the upregulation of PD‐L1 expression in response to exogenous DNA damage.
Moreover, we recently found that oxidative DNA damage upregulates cell surface PD‐L1 expression in cancer cells.14 Oxidative stress causes SSB and base damage, which are repaired by SSB repair and BER, respectively. Furthermore, depletion of NTH1, a central component of BER, increases the upregulation of PD‐L1 expression in response to oxidative stress, supporting the notion that DNA damage signaling induced by oxidative stress upregulates PD‐L1.14 Similar to the events at DSBs, ATR/Chk1 signaling is required for the upregulation of PD‐L1 expression after oxidative DNA damage. However, because oxidative DNA damage does not directly introduce DSBs, we hypothesize that ATR/Chk1 signaling is activated after oxidative DNA damage through replication‐associated DNA damage in S phase.14 As ATR/Chk1 can be activated at single‐strand gaps during the stalling of DNA replication, replication stress induced by oxidative stress could also be involved in the upregulation of PD‐L1 irrespective of direct DSB induction.
As a downstream component of ATR/Chk1 signaling, STAT1/3‐IRF1 play an important role in generating the signal that activates the transcription of PD‐L1 mRNA.3 Generally, in the context of the immune response, PD‐L1 expression is controlled by STAT1/3 phosphorylation and IRF1 expression following the stimulation of IFNγ.20, 21 Interferon regulatory factor 1 binds to the promoter region of PD‐L1 to upregulate PD‐L1 transcription.21 Interestingly, we found that phosphorylation of STAT1/3 as well as IRF1 expression are induced by DNA damage.3 Furthermore, the increase in IRF1 expression by DSBs is suppressed by a specific ATM inhibitor, suggesting that the ATM‐ATR/Chk1 pathway is required for STAT1/3‐IRF1‐dependent PD‐L1 expression (Figure 2).
Figure 2.
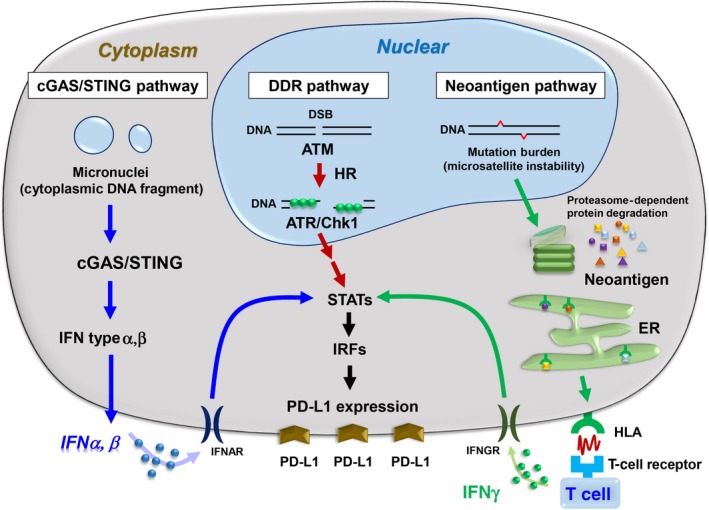
Regulation of programmed death‐ligand 1 (PD‐L1) expression in the context of DNA damage‐induced signaling in cancer cells. PD‐L1 expression is differentially regulated by neoantigens, cyclic GMP‐AMP synthase (cGAS)/stimulator of interferon genes (STING), ataxia telangiectasia and Rad3 related (ATR)/checkpoint kinase (Chk1), and the damage‐associated molecular pattern pathways in cancer cells. ATM, ataxia‐telangiectasia mutated; DSB, double‐strand break; ER, endoplasmic reticulum; HR, homologous recombination; IFN, interferon; IFNAR, interferon alpha/beta receptor; IFNGR, interferon gamma receptor; IRF, interferon regulatory factor
However, surprisingly, in contrast to the upregulation of PD‐L1 in cancer cells after DNA damage, the DNA damage‐dependent upregulation of PD‐L1 expression does not occur in primary normal human dermal fibroblasts.22 Cultured primary fibroblasts usually have a greater G1 population compared with cancer cells. As activation of the ATR/Chk1 pathway requires ssDNA‐RPA formation, which specifically occurs in S/G2 during the progression of HR, the activity of ATR/Chk1 signaling after DNA damage might be insufficient to promote the expression of PD‐L1 in primary fibroblasts. Alternatively, signaling through the STAT1/3‐IRF1 pathway might not be effectively activated because of negative epistatic regulation or through posttranslational modifications in the signal cascade.
In summary, ATM‐ATR/Chk1‐dependent upregulation of PD‐L1 expression represents an early immune response in cancer cells that survive after DNA damage. Following DSB end resection during repair in G2 cells, the signal generated by ATR/Chk1 is transmitted approximately 1‐2 hours after IR, continuing for 24‐48 hours. For example, 10 Gy X‐rays activate ATR/Chk1 signaling to cause G2/M checkpoint arrest, which is maintained for 24‐48 hours. Because the output of the G1/S checkpoint machinery is generally downregulated in cancer cells (see above), the cells that accumulate in G2 activate ATR/Chk1 signaling in the process of HR. Consistent with the timing of the accumulation of G2 phase, the levels of PD‐L1 mRNA increase 16 hours after IR.3 Thus, DNA damage signal‐dependent upregulation of PD‐L1 expression on the cell surface occurs during an early time of the immune response against surviving cancer cells that are arrested at the G2/M cell cycle checkpoint.
2. CYTOSOLIC DNA FRAGMENTS ACTIVATE THE cGAS/STING‐MEDIATED IFN PATHWAY AFTER DNA DAMAGE
In response to DSBs, cell cycle checkpoint arrest is induced by ATM‐ and ATR/Chk1‐dependent signaling. Damaged cells are arrested at each phase of the cell cycle through the checkpoint machinery such as the G1/S, intra‐S, and G2/M checkpoints. For example, the G2/M checkpoint plays a critical role in preventing genome instability by suppressing the transition of the cell cycle toward M phase.6 However, the G2/M checkpoint cannot sensitively monitor DSBs; that is, more than 10‐20 DSBs are required to activate G2/M checkpoint arrest.8, 23 Such insensitive checkpoint monitoring can occur in normal human cells, and generally, the sensitivity decreases in cancer cells, that is, DNA‐damaged cancer cells progress into M phase with a greater number of DSBs compared to normal cells.10 If cells with DSBs enter mitosis, the DNA fragments generated during cell segregation form micronuclei in the subsequent G1 phase (Figure 3). Thus, micronuclei are formed through mitosis when cells do not complete DSB repair in G2 phase, and the frequency of micronuclei formation is enhanced when the G2/M checkpoint is impaired. For example, defective ATM or ATR/Chk1 signaling impairs G2/M checkpoint arrest, which enhances the formation of micronuclei early after DNA damage (Figure 3).
Figure 3.
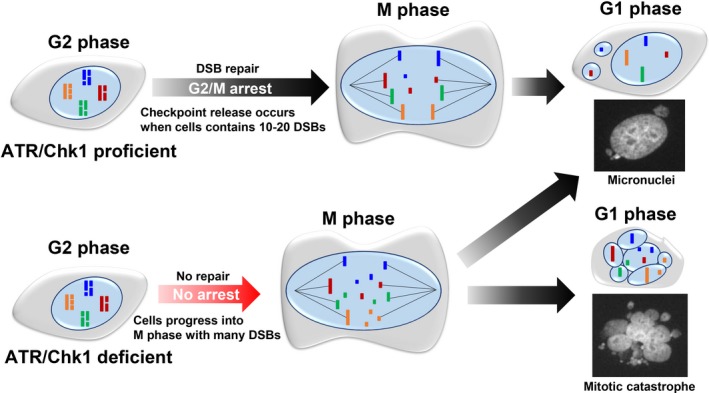
Release of G2/M checkpoint arrest with unrepaired double‐strand breaks (DSBs) causes micronuclei or mitotic catastrophe in the next G1 phase. Cells with intact G2/M checkpoint machinery are able to arrest the cell cycle phase until most DSBs are repaired; however, as G2/M checkpoint arrest in human cells is insensitive, particularly in cancer cells, G2 cells commence progression into M phase with 10‐20 DSBs, resulting in the formation of micronuclei. In contrast, cells with ataxia telangiectasia and Rad3 related (ATR)/checkpoint kinase (Chk1) deficiency progress to M phase without checkpoint arrest and insufficient time for optimal repair. The failure of G2/M checkpoint arrest causes severe DNA fragmentation during M phase, which result in multiple micronuclei. Such cells harboring multiple nuclear fragmentations are categorized as undergoing mitotic catastrophe
The cGAS/STING pathway, previously known as the cytoplasmic DNA sensing factor, detects viral or other exogenous DNAs to exclude them from cells.24 Similarly, micronuclei formed by DNA damage is recognized as cytoplasmic DNA, thus activating the cGAS/STING pathway, which leads to upregulation of IRF3‐IFN type‐I signal transduction and nuclear factor‐kappa B‐dependent release of inflammatory cytokines, followed by an overall immune response (Figure 2).25, 26 The notion that the cGAS/STING pathway is involved in the immune response within the tumor environment is supported by the finding that a specific Chk1 inhibitor used in clinical trials upregulates cGAS/STING signaling subsequent to the formation of micronuclei, possibly due to defective G2/M checkpoint arrest.27 Micronuclei‐induced cGAS/STING signaling under Chk1 inhibition stimulates the immune response, including PD‐L1 expression.27 Poly (ADP‐ribose) polymerase promotes SSB repair and BER by recruiting the required repair proteins.28 Poly (ADP‐ribose) polymerase inhibitor traps PARP1/2 on damaged DNA, which causes DNA replication stress in S phase. The presence of such lesions involving trapped PARP1/2 places a reliance on HR and such lesions are enhanced in HR‐defective cancer cells. The treatment of PARPi upregulates PD‐L1 expression in breast cancer cell lines.29 One of the mechanisms upregulating PD‐L1 following PARPi treatment is thought to be dependent on the inactivation of glycogen synthase kinase 3β, which results in PD‐L1 stabilization.29, 30 In addition to the mechanisms above, recent studies reported that clinical PARPi potentiates an antitumor effect due to the upregulation of PD‐L1 expression and the enhancement of cytotoxic T cell infiltration.31, 32 In these reports, the authors suggest that PARPi‐induced PD‐L1 upregulation is considered to be due to the cGAS/STING‐dependent immune activation following the increase in the formation of micronuclei. The DNA damage‐induced micronuclei are formed when mitotic cells contain unrepaired DNA or chromosomal translocations, and particularly the formation of micronuclei is enhanced in cells treated with PARPi or in cells defective in HR, such as BRCA1‐deficient cancer cells.31, 32 Such DNA damage‐induced micronuclei could effectively elicit the cGAS/STING‐dependent immune activation, followed by the upregulation of PD‐L1 in cancer cells. In contrast, another study identified an alternative pathway suggesting that ATM activates STING signaling in nuclei independent of cGAS after DNA damage.33 Thus, several distinct signaling pathways required for immune activation following DNA damage have been intensively investigated. In addition, from the clinical point of view, cGAS/STING‐dependent immune activation is considered to be involved in promoting the abscopal effect (a systemic antitumor response distant from the X‐ray‐irradiated tumors).25
The ATR/Chk1‐dependent upregulation of PD‐L1 expression occurs during G2/M checkpoint arrest, that is, at an early time after DNA damage. However, we propose that the cGAS/STING‐dependent immune response is induced at the mid‐phase of the immune response after DNA damage because micronuclei are generated following the release of cells from G2/M checkpoint arrest.
3. AN IMMUNE RESPONSE MEDIATED BY CELL DEATH SIGNAL AFTER DNA DAMAGE
In the tumor environment, signals generated by dying cells stimulate the immune response to the surrounding viable cancer cells after DNA damage. After radiotherapy and chemotherapy, when cancer cells incur excessive DNA damage, cells undergo apoptosis or mitotic catastrophe‐mediated cell death. The signals from dying or dead cells are generated by DAMP molecules that upregulate immune signaling in the tumor environment. High mobility group box‐1, which binds to histones to modulate chromatin structure, is released during cell death. Released HMGB1 from dying cells activates the TLR4 pathway to increase MyD88/TRIF signaling.34 Thereafter, the HMGB1‐mediated TLR4/MyD88/TRIF pathway stimulates immune activity. Such cell death releasing DAMPs is called immunogenic cell death.35 While DAMPs stimulate immune activity, leading to further cancer cell‐killing, HMGB1‐mediated TLR4/MyD88/TRIF signaling upregulates PD‐L1 expression in surrounding viable cancer cells.36 Consistent with this finding, there is a positive correlation between TLR4 and PD‐L1 expression in tumors.37 In addition, elevated TLR4 expression is associated with poor survival of cancer patients.37 Thus, DAMP‐mediated upregulation of PD‐L1 expression could contribute to the downregulation of immune activity during radiotherapy and DNA damage‐dependent chemotherapy. After radiotherapy and chemotherapy, ATP, calreticulin, and heat shock proteins are released and function as DAMPs.35 Thus, several signals from dying and dead cells can affect the immune response associated with immune activation and suppression, possibly through the upregulation of PD‐L1 expression in tumor cells.
Among the types of DNA damage induced by radiotherapy or chemotherapy, DSBs critically influence cell fate.4 In human cells, most DSBs are repaired by NHEJ or HR. Particularly after IR, because of the significant contribution of NHEJ to DSB repair, NHEJ‐defective cells show substantial radiosensitivity. Because NHEJ is an essential repair pathway required for cell survival, NHEJ‐null tumors are rarely observed. Interestingly, however, tumors expressing low levels of Ku70 or XRCC4 (a core component of NHEJ) show better therapeutic outcomes after radiotherapy that might promote the release of DAMPs subsequent to radiotherapy.38 In contrast, it is well known that the activity of the HR pathway is downregulated in certain tumors.39 For example, BRCA1/2, which play central roles in HR, cause hereditary breast and ovarian cancers, and HR activity is impaired in tumors in which BRCA1/2 are mutated. DNA replication‐associated DSBs induced by chemotherapeutic agents, such as PARPi, are mainly repaired by HR.39 Hence, if NHEJ or HR activity is downregulated in tumors, such tumors should show greater sensitivity to radiotherapy and chemotherapy, and NHEJ‐ or HR‐defective tumors likely undergo cell death, compared with NHEJ‐ or HR‐proficient tumors. Thus, NHEJ‐ or HR‐defective tumors after radiotherapy or chemotherapy could release greater DAMPs into the tumor environment. Such DAMPs could cause further immune‐mediated cell death in the surrounding tumors due to DSB repair deficiency. Hence, we also propose that patients with insufficient DSB repair activity should be considered candidates to receive anti‐PD‐1/PD‐L1 therapy, particularly combined with radiotherapy, chemotherapy, or both.40
4. REGULATION OF PD‐L1 EXPRESSION IN TUMORS IN THE CONTEXT OF MUTATIONAL BURDEN, MICROSATELLITE INSTABILITY, AND DNA REPAIR
Cancer cells evade the immune system through the acquisition of immunoresistance.41 During tumorigenesis, the upregulation of PD‐L1 expression in tumor cells could contribute to immunoresistance. Expression of PD‐L1 in tumor cells is regulated by IFNs, which are mainly classified as type I (IFNα and IFNβ) and type II (IFNγ). Particularly, the signaling stimulated by IFNγ induces the upregulation of PD‐L1 expression, which is stronger and more continuous compared with the stimuli generated by IFNα and IFNβ.20 Activated T cells, in response to IFN stimuli, upregulate JAK1/2‐STAT1/3 signaling.20 Phosphorylated STAT1/3, generated through IFN‐dependent signaling, induces upregulation of IRF1 expression.21 The transcriptional activator IRF1 binds to the promoter region of PD‐L1 to induce transcription.
The total mutational level (also known as TMB) is considered an effective biomarker for anti‐PD‐1/PD‐L1 therapy, because high TMB should confer abnormal protein production caused by mutational changes such as deletion/insertion‐dependent frameshifts in the ORF of a gene. Such abnormal proteins are recognized and degraded by proteasomes, generating peptides. Some such peptides (neoantigens) are presented on the cell surface as an HLA‐neoantigen complex (Figure 2). The HLA‐neoantigen complex is recognized by the T‐cell receptor, which activates T cells. Activated T cells release IFNs into the tumor microenvironment. The released IFNs are internalized by other tumor cells through the IFN receptors and activate signaling through the STAT1/3‐IRF1 pathway (as described above), leading to immune stimulation, including the upregulation of PD‐L1 expression in the tumor cells (Figure 2).21 Thus, mutated proteins are supposed to mediate the upregulation of PD‐L1 expression and could contribute to the establishment of an immunosuppressive environment. Therefore, a model for neoantigen‐dependent T cell activation proposes a relationship between TMB and the efficacy of anti‐PD‐1/PD‐L1 therapy.42 Interestingly, a recent study showed that, despite tumor heterogeneity, clonal neoantigens preferentially elicit T cell immunoreactivity.43 Although the local TMB could cause local neoantigen production, current knowledge provides a notion that clonal neoantigen effectively promotes neoantigen‐reactive T cells. Thus, neoantigen heterogeneity could influence the efficacy of immune checkpoint therapy.
Alternatively, accumulating reports have shown that the rate of MSI correlates strongly with the efficacy of anti‐PD‐1/PD‐L1 therapy.44 Therefore, MSI is considered a more promising marker to assess the effectiveness of anti‐PD‐1/PD‐L1 therapy. A microsatellite is a tract of a few base pairs of a DNA sequence repeated 5‐50 times. Although the number of microsatellite sequences can change during DNA replication, MMR is able to correct not only mispaired bases but also insertions and deletions; therefore, lack of MMR activity causes aberrations in microsatellite repeats, resulting in an increase in genome‐wide MSI. Interestingly, tumors with mutations in BER genes also show a high frequency of MSI, and BER‐deficient tumors express higher levels of neoantigens and PD‐L1.14 If BER deficiency fails to remove an alkylated or an oxidized base, it could cause mispairing throughout the genome, including microsatellite sequences.
Accumulating evidence shows that tumor cells with defective MMR show greater antitumor activity following anti‐PD‐1/PD‐L1 treatments, which improves long‐term survival.45, 46 Therefore, MSI is considered a better therapeutic marker for anti‐PD‐1/PD‐L1 cancer therapy than TMB. Although the choice between MSI and TMB as a biomarker for anti‐PD/PD‐L1 therapy has not been elucidated at a molecular level, MSI has an advantage in terms of cost and convenience for examination. Currently, single‐base microsatellite sequences are used as a marker to assess the efficacy of anti‐PD‐1/PD‐L1 therapy. Such single‐base microsatellite sequences could be distributed throughout the human genome, possibly in gene bodies, although the number of repeats is less than those of other representative microsatellite sequences.
In summary, accumulating evidence shows that tumors with a high mutational burden and MSI, which confers constitutive upregulation of PD‐L1 expression, are predicted to display sensitivity to anti‐PD‐1/PD‐L1 therapy. Thus, at present, the use of MSI is a reasonable choice as the most effective marker predicting the response to PD‐1/PD‐L1 blockade. However, as microsatellite repeats might not always correlate with the production of neoantigens, further studies are required to precisely identify the links between MSI, neoantigens, and the therapeutic effect of the PD‐1/PD‐L1 blockade, to further optimize MSI as a marker.
5. SUMMARY AND PERSPECTIVES
The highly encouraging results published in 2015 of anti‐PD‐1 Ab therapy given to patients with advanced melanoma and non‐small cell lung carcinoma, were followed by reports of successful PD‐1/PD‐L1 therapy of patients with other cancers.47, 48, 49 Furthermore, clinical and preclinical studies indicate that the combination of anti‐PD‐1/PD‐L1 Ab with conventional cancer therapies, such as radiotherapy and chemotherapy, could be, at least in some cases, even more effective.50, 51 However, our knowledge of the underlying molecular mechanism is insufficient and hinders development of the optimal treatment to achieve precision medicine.
If the chronology of the regulation of PD‐L1 expression caused by DNA damage is considered, 4 steps can be proposed (Figure 4, phases I‐IV). In the absence of exogenous sources of DNA damage, such as radiotherapy and chemotherapy, PD‐L1 expression is controlled by a neoantigen‐IFN‐STAT‐IRF‐dependent pathway induced by the degradation of abnormal proteins elicited by mutations in their ORFs. Such mutations could be caused by defective MMR and DNA replication, and endogenous oxidative DNA damage in the tumor environment (Figure 4, phase I). After radiotherapy, chemotherapy, or both, DSBs activate DNA damage signaling, for example, through ATM‐ATR/Chk1, resulting in the upregulation of PD‐L1 expression in surviving tumor cells (Figure 4, phase II). After long‐term G2 arrest, G2 cells with DSBs progress into M phase due to insensitive G2/M checkpoint arrest. Cell cycle progression from G2 to M phase in cells with DSBs causes DNA fragmentation. The DNA fragments are visualized as micronuclei in the next G1 phase, which are then recognized as cytoplasmic DNAs and activate the cGAS/STING pathway. The cGAS/STING pathway stimulates immune responses, including the upregulation of PD‐L1 expression (Figure 4, phase III). Cancer cells die if they accumulate excessive unrepairable DNA damage, or if DNA damage causes a functionally critical mutation generated by chromosomal translocations or rearrangements. The dead cells act as immunogens, triggering the release of DAMPs that ultimately stimulate immune signaling, including upregulation of PD‐L1 expression (Figure 4, phase IV). Thus, from the introduction of DNA damage to the determination of cell fate, PD‐L1 expression in the tumor environment is differentially regulated at each stage after DNA damage. For the development of an effective strategy combining anti‐PD‐1/PD‐L1 Ab treatment with chemo/radiotherapy, further investigation of the mechanism underlying PD‐L1 expression and its impact on the immune response will require intensive collaborative efforts between basic, translational, and clinical researchers.
Figure 4.
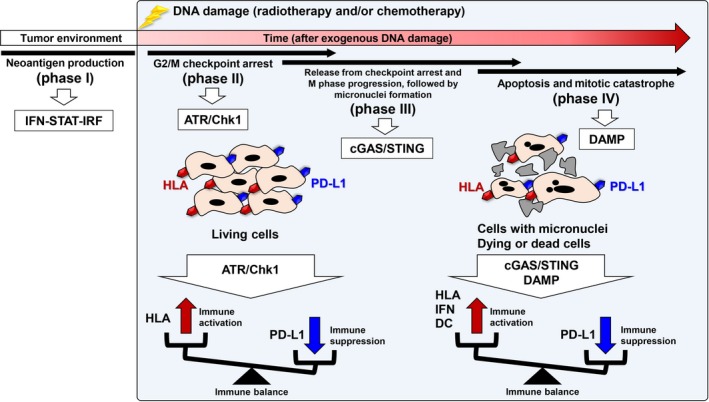
Chronology of the regulation of immune reactions induced by DNA damage‐dependent cellular responses. After DNA damage, cell cycle progression is arrested at the G2/M checkpoint. For example, 48‐72 h after exposure to 10‐Gy X‐rays, G2/M checkpoint arrest is released and G2 cells progress into M phase with double‐strand breaks, followed by the formation of micronuclei in the next G1. Finally, cancer cells receive a lethal dose of DNA damage. The upregulation of programmed death‐ligand 1 (PD‐L1) expression is induced in each process, although through distinct molecular mechanisms. Thus, anti‐PD‐1/PD‐L1 therapy could be given when the upregulation of PD‐L1 expression is induced under conditions of symmetrically stimulated immune activation. ATR, ataxia telangiectasia and Rad3 related; cGAS, cyclic GMP‐AMP synthase; Chk1, checkpoint kinase 1; DAMP, damage‐associated molecular pattern; DC, dendritic cell; HLA, human leukocyte antigen; IFN, interferon; IRF, interferon regulatory factor; STING, stimulator of interferon genes
DISCLOSURE
The authors have no conflict of interest.
ACKNOWLEDGEMENTS
The work was supported by the Japan Society for the Promotion of Science KAKENHI Grants provided to AS (grant no. JP17H04713) and HS (grant no. JP17K16420). Support was extended by the Cell Science Research Foundation and the Daiichi Sankyo Foundation of Life Science to AS, by the Yasuda Medical Foundation to AS and HS, and by the Takeda Science Foundation to AS and HS. This work was supported by the Program of the Network‐type Joint Usage/Research Center for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University, and Fukushima Medical University.
Sato H, Jeggo PA, Shibata A. Regulation of programmed death‐ligand 1 expression in response to DNA damage in cancer cells: Implications for precision medicine. Cancer Sci. 2019;110:3415–3423. 10.1111/cas.14197
REFERENCES
- 1. Mouw KW, D'Andrea AD. DNA repair deficiency and immunotherapy response. J Clin Oncol. 2018;36:1710‐1713. [DOI] [PubMed] [Google Scholar]
- 2. Mouw KW, Konstantinopoulos PA. From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD‐L1 immune checkpoint activation. Br J Cancer. 2018;118:933‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sato H, Niimi A, Yasuhara T, et al. DNA double‐strand break repair pathway regulates PD‐L1 expression in cancer cells. Nat Commun. 2017;8:1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shibata A, Jeggo P. A historical reflection on our understanding of radiation‐induced DNA double strand break repair in somatic mammalian cells; interfacing the past with the present. Int J Radiat Biol. 2019;95:945‐956. [DOI] [PubMed] [Google Scholar]
- 5. Shibata A. Regulation of repair pathway choice at two‐ended DNA double‐strand breaks. Mutat Res. 2017;803–805:51‐55. [DOI] [PubMed] [Google Scholar]
- 6. Shibata A, Jeggo PA. DNA double‐strand break repair in a cellular context. Clin Oncol (R Coll Radiol). 2014;26:243‐249. [DOI] [PubMed] [Google Scholar]
- 7. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197‐210. [PubMed] [Google Scholar]
- 8. Shibata A, Barton O, Noon AT, et al. Role of ATM and the damage response mediator proteins 53BP1 and MDC1 in the maintenance of G(2)/M checkpoint arrest. Mol Cell Biol. 2010;30:3371‐3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rhind N. Changing of the guard: how ATM hands off DNA double‐strand break signaling to ATR. Mol Cell. 2009;33:672‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deckbar D, Jeggo PA, Lobrich M. Understanding the limitations of radiation‐induced cell cycle checkpoints. Crit Rev Biochem Mol Biol. 2011;46:271‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shibata A, Jeggo P, Lobrich M. The pendulum of the Ku‐Ku clock. DNA Repair. 2018;71:164‐171. [DOI] [PubMed] [Google Scholar]
- 12. Jazayeri A, Falck J, Lukas C, et al. ATM‐ and cell cycle‐dependent regulation of ATR in response to DNA double‐strand breaks. Nat Cell Biol. 2006;8:37‐45. [DOI] [PubMed] [Google Scholar]
- 13. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Permata TBM, Hagiwara Y, Sato H, et al. Base excision repair regulates PD‐L1 expression in cancer cells. Oncogene. 2019;38:4452‐4466. [DOI] [PubMed] [Google Scholar]
- 15. Sun LL, Yang RY, Li CW, et al. Inhibition of ATR downregulates PD‐L1 and sensitizes tumor cells to T cell‐mediated killing. Am J Cancer Res. 2018;8:1307‐1316. [PMC free article] [PubMed] [Google Scholar]
- 16. Vendetti FP, Karukonda P, Clump DA, et al. ATR kinase inhibitor AZD6738 potentiates CD8 + T cell‐dependent antitumor activity following radiation. J Clin Investig. 2018;128:3926‐3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shibata A, Conrad S, Birraux J, et al. Factors determining DNA double‐strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mimitou EP, Symington LS. Ku prevents Exo1 and Sgs1‐dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010;29:3358‐3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun J, Lee KJ, Davis AJ, Chen DJ. Human Ku70/80 protein blocks exonuclease 1‐mediated DNA resection in the presence of human Mre11 or Mre11/Rad50 protein complex. J Biol Chem. 2012;287:4936‐4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shin DS, Zaretsky JM, Escuin‐Ordinas H, et al. Primary resistance to PD‐1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garcia‐Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD‐L1 and PD‐L2 expression. Cell Rep. 2017;19:1189‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hagiwara Y, Sato H, Permata TBM, et al. Analysis of programmed death‐ligand 1 expression in primary normal human dermal fibroblasts after DNA damage. Hum Immunol. 2018;79:627‐631. [DOI] [PubMed] [Google Scholar]
- 23. Deckbar D, Birraux J, Krempler A, et al. Chromosome breakage after G2 checkpoint release. J Cell Biol. 2007;176:749‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ablasser A, Goldeck M, Cavlar T, et al. cGAS produces a 2’‐5’‐linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mackenzie KJ, Carroll P, Martin CA, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sen T, Rodriguez BL, Chen L, et al. Targeting DNA damage response promotes antitumor immunity through STING‐mediated T‐cell activation in small cell lung cancer. Cancer Discov. 2019;9:646‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caldecott KW. DNA single‐strand break repair. Exp Cell Res. 2014;329:2‐8. [DOI] [PubMed] [Google Scholar]
- 29. Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD‐L1 expression and enhances cancer‐associated immunosuppression. Clin Cancer Res. 2017;23:3711‐3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li CW, Lim SO, Xia W, et al. Glycosylation and stabilization of programmed death ligand‐1 suppresses T‐cell activity. Nat Commun. 2016;7:12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pantelidou C, Sonzogni O, De Oliveria Taveira M, et al. PARP inhibitor efficacy depends on CD8(+) T‐cell recruitment via intratumoral STING pathway activation in BRCA‐deficient models of triple‐negative breast cancer. Cancer Discov. 2019;9:722‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ding L, Kim HJ, Wang Q, et al. PARP inhibition elicits STING‐dependent antitumor immunity in Brca1‐deficient ovarian cancer. Cell Rep. 2018;25:2972‐2980 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dunphy G, Flannery SM, Almine JF, et al. Non‐canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF‐kappaB signaling after nuclear DNA damage. Mol Cell. 2018;71:745‐760 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll‐like receptor 4‐dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050‐1059. [DOI] [PubMed] [Google Scholar]
- 35. Rodriguez‐Ruiz ME, Rodriguez I, Leaman O, et al. Immune mechanisms mediating abscopal effects in radioimmunotherapy. Pharmacol Ther. 2019;196:195‐203. [DOI] [PubMed] [Google Scholar]
- 36. Liu J, Hamrouni A, Wolowiec D, et al. Plasma cells from multiple myeloma patients express B7‐H1 (PD‐L1) and increase expression after stimulation with IFN‐γ and TLR ligands via a MyD88‐, TRAF6‐, and MEK‐dependent pathway. Blood. 2007;110:296‐304. [DOI] [PubMed] [Google Scholar]
- 37. Wang K, Wang J, Wei F, Zhao N, Yang F, Ren X. Expression of TLR4 in non‐small cell lung cancer is associated with PD‐L1 and poor prognosis in patients receiving pulmonectomy. Front Immunol. 2017;8:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takada Y, Someya M, Matsumoto Y, et al. Influence of Ku86 and XRCC4 expression in uterine cervical cancer on the response to preoperative radiotherapy. Med Mol Morphol. 2016;49:210‐216. [DOI] [PubMed] [Google Scholar]
- 39. Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31:955‐960. [DOI] [PubMed] [Google Scholar]
- 40. Parkes EE, Walker SM, Taggart LE, et al. Activation of STING‐dependent innate immune signaling by S‐phase‐specific DNA damage in breast cancer. J Natl Cancer Inst. 2017;109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991‐998. [DOI] [PubMed] [Google Scholar]
- 42. Xu‐Monette ZY, Zhang M, Li J, Young KH. PD‐1/PD‐L1 blockade: have we found the key to unleash the antitumor immune response? Front Immunol. 2017;8:1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter‐inhibitory checkpoints. Cancer Discov. 2015;5:43‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372:2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science. 2017;357:409‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320‐330. [DOI] [PubMed] [Google Scholar]
- 48. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med. 2015;373:123‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Borghaei H, Paz‐Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med. 2015;373:1627‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015;16:e498‐e509. [DOI] [PubMed] [Google Scholar]