

Abstract
Coxsackievirus and adenovirus receptor (CAR) is a single‐pass transmembrane protein that is associated with adenoviral infection. CAR is involved in the formation of epithelial tight junctions and promotes tumor growth in some cancers. Previously, we developed mouse monoclonal antibodies against human CAR and found that one, mu6G10A, significantly inhibited tumor growth in xenografts of human cancer cells. Herein, we generated and characterized a mouse‐human chimeric anti‐CAR antibody (ch6G10A) from mu6G10A. ch6G10A had binding activity, inducing antibody‐dependent cellular cytotoxicity and complement‐dependent cytotoxicity, and in vivo anti‐tumor activity against CAR‐expressing prostate cancer DU‐145 cells. In addition, cancer tissue array analysis confirmed that CAR is highly expressed in neuroendocrine lung cancers including small cell lung cancer, and treatment with ch6G10A effectively inhibited in vivo subcutaneous tumor growth of NCI‐H69 small cell lung cancer cells in nude mice. Moreover, treatment with mu6G10A effectively inhibited both in vivo orthotopic tumor growth and distant metastatic formation in mouse xenograft models of a highly metastatic subline of human small cell lung cancer DMS273 cells. These results suggest that targeting therapy to CAR with a therapeutic antibody might be effective against several cancer types including small cell lung cancer.
Keywords: CAR, mouse‐human chimeric antibody, orthotopic transplantation model, prostate cancer, small cell lung cancer
Abbreviations
- ADCC
antibody‐dependent cellular cytotoxicity
- CAR
coxsackievirus and adenovirus receptor
- CDC
complement‐dependent cytotoxicity
- CTA
cancer tissue array
- FcγRIIIA
Fcγ receptor IIIA
- NK
natural killer
- SCLC
small cell lung cancer
1. INTRODUCTION
Coxsackievirus and adenovirus receptor (CAR) protein that encoded by CXADR gene, is a single‐pass transmembrane protein and is involved in the formation and/or maintenance of epithelial tight junctions.1 CAR has an essential role in the development of the heart and lymphatic system in mice.2, 3 Because CAR is the primary cellular receptor for adenoviruses,1 its expression level is considered an important factor for adenovirus infection and adenoviral vector‐mediated treatment.
It has been reported that high CAR expression levels occur in various human cancers.4, 5, 6, 7, 8, 9, 10 Moreover, CAR promotes tumor growth in some cancer types. CXADR silencing in lung cancer with high CAR expression suppressed tumor formation ability in xenografts,11 whereas CXADR knockdown in oral squamous cell carcinoma resulted in the inhibition of both anchorage‐independent growth and metastatic tumor formation in vivo.12 These previous studies suggest that CAR might be an appropriate target molecule for cancer therapy.
Previously, we found that LNCaP‐CR cells, a highly tumorigenic subline of the LNCaP human prostate cancer cell line,13 express higher levels of CAR than their parental cells.14 We also developed mouse mAbs against human CAR and found that one of these antibodies (clone 6G10A) significantly inhibited tumor growth in xenografts of human prostate, pancreatic, and colorectal cancer.14 Based on these findings, we proposed that an anti‐CAR antibody might be a feasible candidate for cancer immunotherapy.14
Because the immunogenicity of antibodies needs to be reduced for their therapeutic use in humans, mouse‐human chimerization of mouse mAbs is an important step in the development of therapeutic antibodies.15 In the current study, we generated a mouse‐human chimeric anti‐CAR antibody (ch6G10A) from 6G10A mouse anti‐CAR antibody (mu6G10A), and characterized ch6G10A by flow cytometry, western blotting, ADCC/CDC analyses, and in vivo anti‐tumor activity against a prostate cancer cell line. In addition, we carried out a CTA analysis to investigate CAR expression levels in lung, prostate, and brain tumors, and examined the anti‐tumor activities of anti‐CAR antibodies against SCLC using mouse xenograft models.
2. MATERIALS AND METHODS
2.1. Cell lines and reagents
KHYG‐1/FcγRIIIA cells, a human NK cell line stably expressing Fcγ receptor IIIA (FcγRIIIA), were previously established16 and were maintained in RPMI‐1640 medium (Nissui Pharmaceutical) supplemented with 10% FBS (PAN Biotech GmbH) and 100 units of human interleukin (IL)‐2 (FUJIFILM Wako Pure Chemical Corporation). Human SCLC cell lines NCI‐H69, DMS53, and DMS114 were maintained in RPMI‐1640 medium containing 10% FBS. Human prostate cancer cell line DU‐145, human SCLC cell line DMS273, GFP‐labeled subline DMS273‐GFP, highly metastatic subline G3H,17 and C5B (S. Sakamoto, H. Inoue & M. Kawada, unpubl. data, manuscript in preparation) were maintained in DMEM (Nissui) containing 10% FBS.
2.2. Generation of mouse‐human chimeric anti‐human CAR
A mouse anti‐human CAR mAb, mu6G10A, was developed as described previously.14 Generation of mouse‐human chimeric anti‐human CAR (ch6G10A) was previously described.18 Briefly, the appropriate V H and V L cDNAs of mu6G10A and C H and C L of human IgG1 were subcloned into pcDNA3.3/Neo or pcDNA3.1/Zeo vectors (Thermo Fisher Scientific Inc.), respectively. Antibody expression vectors were transfected into CHO‐S cells (Thermo Fisher Scientific Inc.) using Lipofectamine 2000 reagent (Thermo Fisher Scientific Inc.). Stable transfectants of CHO‐S/ch6G10A were selected by culturing the transfectants in medium containing 1 mg/mL G418 (Nacalai Tesque, Inc.) and 0.5 mg/mL zeocin (InvivoGen). Stable transfectants were cultured for 14 days in CHO‐S‐SFM II medium (Thermo Fisher Scientific Inc.), and then ch6G10A immunoglobulin was purified from the culture supernatant using Protein G Sepharose (GE Healthcare United Kingdom, Ltd). Purity of ch6G10A was evaluated by SDS‐PAGE and Coomassie Brilliant Blue staining.
2.3. Flow cytometry
Cells were fixed in 4% neutral buffered formalin for 10 minutes and suspended in PBS containing 0.1% NaN3. Cells were resuspended in PBS containing 0.5% BSA and incubated with anti‐CAR antibodies for 1 hour at 4°C and then with PE‐labeled secondary antibodies for 1 hour at room temperature. After washing with PBS containing 0.1% NaN3, cells were analyzed by FACSCalibur (BD Biosciences).
2.4. Western blot analysis
Western blot analysis was carried out as described previously.19 Briefly, cultured cell pellets were lysed with lysis buffer (20 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 1% Triton X‐100, 10% glycerol, 1 mmol/L EDTA, 50 mmol/L NaF, 50 mmol/L β‐glycerophosphate, 1 mmol/L Na3VO4, 1× cOmplete Protease Inhibitor Cocktail; Roche). Lysates were centrifuged at 13 000 g for 15 minutes at 4°C, and the supernatants were boiled in SDS sample buffer containing 0.5 mol/L β‐mercaptoethanol. These samples were separated by 12.5% SDS‐PAGE and transferred to PVDF membranes (Merck KGaA). Protein levels were detected using the following antibodies: rabbit polyclonal antibodies specific for CAR (H‐300/sc‐15405; Santa Cruz Biochemical) and rabbit polyclonal antibodies specific for HSP90 (H‐114/sc‐7947; Santa Cruz Biochemical).
2.5. Immunohistochemical analysis of cancer tissue arrays
Cancer tissue arrays (BC041115c for lung cancer, BC19013 for prostate cancer, and GL803b for brain cancer) were purchased from US Biomax. The arrays were deparaffinized and rehydrated. Then, antigens were unmasked using citrate buffer for 10 minutes at 96°C. Prior to antibody reactions, endogenous peroxidase was inactivated with 3% hydrogen peroxidase for 10 minutes at room temperature followed by rinsing with distilled water and TBST. The arrays were blocked with 10% horse serum for 1 hour at 37°C and incubated with primary antibody against CAR (H‐300/sc‐15405, 1:750; Santa Cruz Biochemical) overnight at 4°C. After washing three times with TBST, the arrays were incubated with secondary antibody peroxidase‐conjugated anti‐rabbit IgG (ImmPRESS Reagent kit; Vector Laboratories) for 30 minutes at room temperature. The arrays were then washed three times with TBST and developed using ImmPACT DAB (Vector Laboratories) for 1 minute, and then briefly immersed in hematoxylin for counterstaining and evaluation under light microscopy. Estimated visual intensity of CAR immunostaining was graded on an arbitrary three‐point scale: negative (0), positive (1), strongly positive (2).
2.6. Antibody‐dependent cellular cytotoxicity
Antibody‐dependent cellular cytotoxicity was measured with a calcein AM release assay as described previously.14 Briefly, for ADCC activity, target cells were labeled with 10 μg/mL calcein AM (Dojindo) for 30 minutes at 37°C, washed three times with complete medium, and further incubated for 1 hour at 37°C. Labeled target cells (5 × 104 cells) and effector cells were then cocultured in 96‐well plates and incubated for 4 hour at 37°C. After centrifugation, fluorescence intensity (F) of cell‐free supernatants was measured (excitation 485 nm, emission 538 nm). Cytolytic activity was calculated using the following formula:
Assays were carried out in triplicate at least.
2.7. Complement‐dependent cytotoxicity
For CDC activity, target cells (2 × 104 cells) were inoculated into 96‐well plates and incubated with 5% rabbit complement (Cedarlane) and ch6G10A, mu6G10A, control human IgG, or control mouse IgG for 4 hours at 37°C. Cell viability was determined using MTT assay. Assays were done in triplicate at least.
2.8. In vivo anti‐tumor activity
Animal experiments were approved by the Institute Committee for Animal Experiments at the Institute of Microbial Chemistry and carried out according to the ethics guidelines of our institute. Female BALB/c nude mice and female SCID‐beige mice were obtained from Charles River Japan (Kanagawa, Japan) and were maintained in a specific pathogen‐free barrier facility inhouse. Mice aged 8 weeks were used for the in vivo anti‐tumor assay. To prepare the subcutaneous xenograft model, 1 × 107 DU‐145 cells or 1 × 106 NCI‐H69 cells in 0.1 mL of 62.5% growth factor‐reduced Matrigel (BD Biosciences) were injected s.c. into the left flanks of the SCID‐beige mice. When the tumor size had reached approximately 100 mm3, i.v. injection of the antibodies was started. To prepare the orthotopic transplanted xenograft model of the highly metastatic subline of DMS273 human SCLC cells, 2.5 × 105 C5B cells were injected into the left lungs of nude mice as described previously.17 An Olympus OV110 Small Animal Imaging System (Olympus Corp.) was used for imaging orthotopic and metastatic tumor formation in the orthotopic transplanted xenograft model. Length (L) and width (W) of the subcutaneous and orthotopic tumors were measured by calipers and tumor volume (TV) was calculated using the formula: TV = (L × W 2)/2. All the in vivo experiments were carried out at least twice with similar results.
2.9. Statistical analysis
Results are expressed as mean ± SD. Statistical analysis was carried out by Student's t test or Mann‐Whitney U test as described in the figure legends. P < .05 was considered statistically significant.
3. RESULTS
3.1. Generation and characterization of a mouse‐human chimeric anti‐CAR antibody (ch6G10A)
Previously, we developed a mouse mAb against human CAR (mu6G10A) that recognized an extra‐membranous region of CAR and showed anti‐tumor activities against CAR‐expressing cancer cells in xenograft models.14 To reduce the immunogenicity of antibodies in humans, we attempted to generate a mouse‐human chimeric antibody (ch6G10A) by fusing the V H and V L regions of mu6G10A with the C H and C L regions of human IgG1, respectively. As shown in Figure 1A, ch6G10A reacted with the human prostate cancer cell line DU‐145, which highly expresses human CAR, as strongly as mu6G10A by flow cytometry. Because mu6G10A can induce both ADCC and CDC,14 we next examined whether ch6G10A can also induce both ADCC and CDC. When a human NK cell line, KHYG‐1/FcγRIIIA, which stably expresses FcγRIIIA was used as effector cells,16 ADCC by ch6G10A was observed against DU‐145 cells (Figure 1B). ch6G10A also significantly induced CDC activity against DU‐145 cells when using rabbit complement (Figure 1C). Then, we evaluated the anti‐tumor activity of ch6G10A against DU‐145 cells in vivo using a subcutaneous xenograft model in nude mice. ch6G10A, control human IgG (500 μg/mice) or saline was injected i.v. at days 0 and 7, and human NK cells KHYG‐1/FcγRIIIA were also injected around the tumors at days 0 and 7. ch6G10A significantly inhibited the growth of xenograft DU‐145 tumors in vivo (43% inhibition compared with control human IgG, P < .01) without significant body weight loss in the host mice (Figure 2, data not shown). These results showed that ch6G10A had ADCC‐/CDC‐inducing activities and in vivo anti‐tumor activity against CAR‐expressing prostate cancer cells and that we successfully generated a mouse‐human chimeric anti‐CAR antibody with potent anti‐tumor activity.
Figure 1.
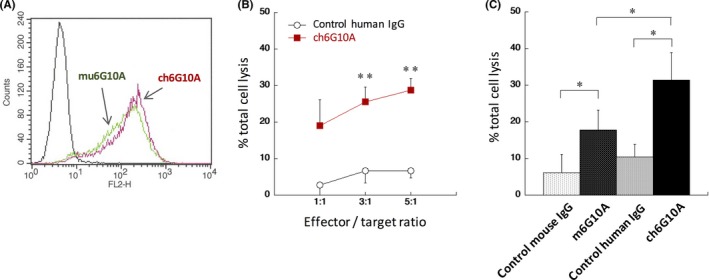
Generation of a mouse‐human chimeric anti‐coxsackievirus and adenovirus receptor (CAR) mAb, ch6G10A. A, Flow cytometric analysis. DU‐145 human prostate cancer cells were incubated with 10 μg/mL ch6G10A, a mouse‐human chimeric anti‐CAR mAb and subjected to flow cytometric analysis. B, Assessment of antibody‐dependent cellular cytotoxicity activity. DU‐145 cells were incubated with human natural killer cell line KHYG‐1/Fcγ receptor IIIA in the presence of the indicated antibodies at 100 μg/mL for 4 h. Results are expressed as means ± SD of three independent experiments carried out in triplicate. Statistical analysis was done by Student's t test (**P < .01 vs control). C, Assessment of complement‐dependent cytotoxicity activity. DU‐145 cells were incubated with 5% rabbit complement in the presence of the indicated antibodies at 10 μg/mL for 4 h. Results are expressed as means ± SD of three independent experiments carried out in triplicate. Statistical analysis was done by Student's t test (*P < .05 vs control)
Figure 2.
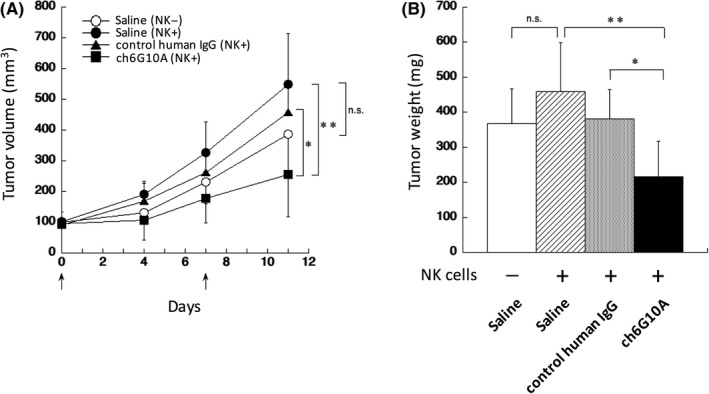
In vivo anti‐tumor effect of chimeric anti‐coxsackievirus and adenovirus receptor mAbs on a subcutaneous transplanted xenograft model of DU‐145 human prostate cancer cells. Effect of treatment with ch6G10A on the in vivo growth of DU‐145 cells. DU‐145 cells (1 × 107 cells) were implanted s.c. into BALB/c nude mice. Treatment started when average tumor volume had reached 100 mm3 (as day 0). A total of 500 μg of ch6G10A, control human IgG or saline was injected i.v. at days 0 and 7 (indicated by arrows). Human natural killer (NK) cells KHYG‐1/Fcγ receptor IIIA (4 × 105 cells) were injected around the tumors at days 0 and 7 (indicated by arrows). A, Tumor volume. B, Tumor weight at 11 days. Results are expressed as means ± SD (IgG control; n = 7, ch6G10A; n = 5). Statistical analysis was carried out by Student's t test (*P < .05, **P < .01, n.s., not significant)
3.2. Expression of CAR in human lung, prostate, and brain tumors
Our previous results suggest that CAR is highly expressed in tumor tissues such as prostate, lung, and brain.14 Because we used only small numbers of tissue sections in the previous study, we further assessed the expression of CAR in these tumors through immunohistochemical staining of CTA using commercially available antibody against CAR (H‐300) (Figure 3). CTA analysis showed that CAR was considerably expressed in lung squamous cell carcinoma (19/40; 48%), lung adenocarcinoma (14/45; 31%), lung neuroendocrine cancer (including small cell lung cancer) (7/12; 58%), prostate adenocarcinoma (45/56; 80%), and glioblastoma (23/39; 59%). Importantly, expression of CAR was significantly higher in all lung cancer types than in normal lung tissue (lung squamous cell carcinoma and lung neuroendocrine cancer, P < .01; lung adenocarcinoma, P < .05), and was significantly higher in glioblastoma than in normal brain tissue (P < .05). Thus, our results show that CAR was highly expressed in these human tumor tissues.
Figure 3.
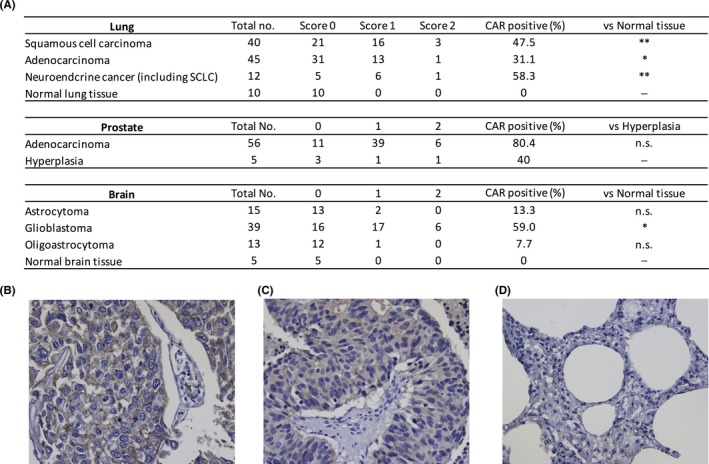
Coxsackievirus and adenovirus receptor (CAR) expression in various human tumor tissues. A, CAR expression in human lung, prostate, and brain tumors. Immunohistochemical staining was carried out on cancer tissue arrays using CAR antibody H‐300. Estimated visual intensity of CAR immunostaining was graded on an arbitrary three‐point scale: negative (0), positive (1), strongly positive (2). CAR, coxsackievirus and adenovirus receptor; SCLC, small cell lung cancer. Statistical analysis was done by Mann‐Whitney U test (*P < .05, **P < .01, n.s., not significant). B‐D, Representative CAR staining in the lung cancer tissue array. B, SCLC (score 2). C, SCLC (score 1). D, Normal lung tissue (score 0). All data were documented under a magnification of ×400
3.3. In vivo anti‐tumor activity of ch6G10A antibody against NCI‐H69 small cell lung cancer cells
In addition to our CTA results, other studies also showed that CAR was highly expressed in SCLC patients.5, 8, 10 Therefore, we investigated CAR protein levels in four human SCLC cell lines (DMS114, DMS53, DMS273, and NCI‐H69) by western blot analysis, and found that CAR was expressed in three of the four lines and was particularly overexpressed in NCI‐H69 cells (Figure 4A). These expression data of CAR prompted us to evaluate the in vivo anti‐tumor activity of ch6G10A against NCI‐H69 using a subcutaneous xenograft model in nude mice. ch6G10A or control human IgG (500 μg/mice) was injected i.v. at days 0 and 7, and human NK cells KHYG‐1/FcγRIIIA were injected around the tumors at days 0 and 7. As shown in Figure 4B,C, ch6G10A significantly inhibited the growth of xenograft NCI‐H69 tumors in vivo (37% inhibition compared with control human IgG, P < .01) without significant body weight loss in the host mice (data not shown). These results showed that ch6G10A had anti‐tumor activity against CAR‐expressing SCLC cells and suggested that SCLC might be a candidate disease for targeting therapy to CAR with therapeutic antibody.
Figure 4.
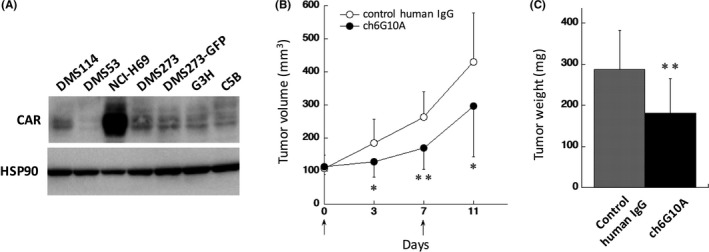
In vivo anti‐tumor effect of chimeric anti‐coxsackievirus and adenovirus receptor (CAR) mAb on a subcutaneous transplanted xenograft model of NCI‐H69 human small cell lung cancer (SCLC) cells. A, CAR expression in human SCLC cell lines. Whole‐cell lysates (20 μg) of indicated cell lines were separated by 12.5% SDS‐PAGE and analyzed by western blotting using CAR antibody H‐300. HSP90 was used as a loading control. CAR, coxsackievirus and adenovirus receptor; B‐C, Effect of treatment with ch6G10A on the in vivo growth of NCI‐H69 cells in a subcutaneous transplanted xenograft model. NCI‐169 cells (1 × 106 cells) were transplanted s.c. into BALB/c nude mice. Treatment started when average tumor volume had reached 100 mm3 (as day 0). Total of 500 μg of ch6G10A or control human IgG was injected i.v. at days 0 and 7 (indicated by arrows). Human natural killer cells KHYG‐1/Fcγ receptor IIIA (4 × 105 cells) were injected around the tumors at days 0 and 7 (indicated by arrows). B, Tumor volume. C, Tumor weight at 11 days. Results are expressed as means ± SD (IgG control; n = 14, ch6G10A; n = 10). Statistical analysis was carried out by Student's t test (*P < .05, **P < .01)
3.4. In vivo anti‐tumor activity of anti‐CAR antibody against an orthotopic transplantation model of SCLC
We previously developed an orthotopic transplantation model of SCLC using GFP‐labeled human SCLC cell line DMS273 (DMS273‐GFP) and two highly metastatic sublines (G3H, C5B); this model has significant metastatic activity and comparable metastatic tropism with that of SCLC patients.17 Because CAR expression was clearly observed in these cells (Figure 4A), we attempted to elucidate whether anti‐CAR antibody could suppress orthotopic tumor growth and distant metastasis formation in the model using C5B cells. Owing to the difficulty of giving systemic human NK cells to xenograft mice, which is essential for the anti‐tumor activity of ch6G10A, we used mouse monoclonal CAR antibody mu6G10A in this experiment. Intravenous injection with mu6G10A or control mouse IgG (250 μg/mice) was started a day after orthotopic transplantation of C5B cells. The injection was carried out once a week for 5 weeks. Orthotopic tumor growth was prominently inhibited by mu6G10A (78% inhibition compared with control mouse IgG, P < .005) without significant body weight loss in the host mice (Figure 5, data not shown). Incidence of distant metastases formation was 67% in the control mouse IgG‐treated group and 33% in the mu6G10A‐treated group, and significantly fewer metastasis‐positive organs were observed in the mu6G10A‐treated group compared with the control mouse IgG‐treated group (P < .05; Figure 5B). Thus, treatment with anti‐CAR antibody efficiently inhibited tumor growth in the lung as well as metastasis of SCLC. These results support the notion that SCLC might be a potential disease for targeting therapy to CAR with therapeutic antibody.
Figure 5.
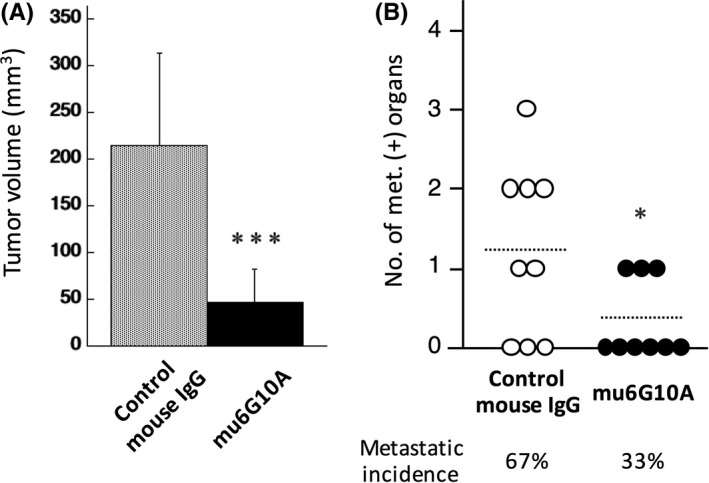
In vivo anti‐tumor effect of mouse coxsackievirus and adenovirus receptor (CAR) mAb on an orthotopic transplanted xenograft model of the highly metastatic subline of DMS273 human small cell lung cancer (SCLC) cells. A‐B, Effect of treatment with mu6G10A on the in vivo growth of C5B cells, the highly metastatic subline of DMS273, in an orthotopic transplanted xenograft model. C5B cells (2.5 × 105 cells) were orthotopically transplanted into the lungs of BALB/c nude mice (day 0). Total of 250 μg of mu6G10A or control mouse IgG was injected i.v. once a week from day 1 for 5 weeks. Mice were killed and analyzed on days 30‐39. A, Orthotopic tumor volume at the time of death. B, Distant metastatic formation. Dotted lines show the means of the number of metastasis‐positive organs of each group. Percentages show distant metastasis incidence. Results are expressed as means ± SD (n = 9). Statistical analysis was carried out by Student's t test (*P < .05, ***P < .005 vs control)
4. DISCUSSION
In the present study, we successfully generated a mouse‐human chimeric anti‐CAR monoclonal antibody, ch6G10A, which has ADCC‐ and CDC‐inducing activities and in vivo anti‐tumor activity against CAR‐expressing cancer cells. In addition, CTA analysis showed that CAR is highly expressed in neuroendocrine lung cancer including SCLC, and treatment with anti‐CAR antibody inhibited both subcutaneous and orthotopic tumor growth in mouse xenograft models of human SCLC cell lines. These results suggest that targeting therapy to CAR with therapeutic antibodies might be effective against several cancers including SCLC.
Our CTA analysis showed significantly higher expression of CAR in neuroendocrine lung cancers including SCLC compared with normal lung tissues (Figure 3), consistent with the findings of previous studies.5, 8, 10 Furthermore, treatment with anti‐CAR antibody effectively inhibited both in vivo tumor growth (subcutaneous and orthotopic) and distant metastasis formation in mouse xenograft models of human SCLC cell lines (Figures 4 and 5). SCLC accounts for 10%‐20% of all cases of lung cancer worldwide and shows aggressive growth and early metastatic spread. The current main treatment against SCLC is chemotherapy but the high chance of disease relapse after chemotherapy results in poor median survival.20 Therefore, novel therapeutic strategies against SCLC are urgently needed. CAR expression data in SCLC and our current results with anti‐CAR antibody collectively suggested that therapeutic antibodies against CAR might be an alternative strategy for SCLC treatment. Our CTA analysis also showed significantly higher expression of CAR in lung squamous cell carcinoma, lung adenocarcinoma, and glioblastoma than in matched adjacent normal tissues. Because there are many cases of these diseases, validation of the anti‐tumor activity of our anti‐CAR antibody will be meaningful for future investigations.
Our previous study showed that mu6G10A exerts anti‐tumor activity primarily through both ADCC and CDC activities.14 In this study, we generated ch6G10A with the human IgG1 constant region because human IgG1 exerts higher ADCC/CDC activities among the IgG1 subclass. Therefore, it is presumed that ch6G10A also exerts its anti‐tumor activity primarily through both ADCC and CDC. If this is the case, the anti‐tumor activity of ch6G10A could be enhanced by ADCC‐enhancing technologies such as afucosylation of the Fc domain.21 Thus, ch6G10A could be a potent lead antibody for cancer immunotherapy.
We showed that the treatment with mu6G10A efficiently inhibited tumor growth in the lung as well as metastasis in the SCLC metastatic model (Figure 5). Although it is very important as to whether ch6G10A also inhibits SCLC metastasis, there are no suitable metastatic models for the evaluation of chimeric mAb at this point. However, there is an interesting humanized mouse strain, NOG‐IL‐2 Tg, which can induce human NK cells from hematopoietic stem cells (HSC).22 The report suggested that chimeric anti‐human CCR4 mAb (Poteligeo, Kyowa‐Kirin, Tokyo, Japa) suppressed tumor growth of human lymphoma cells in human HSC‐transferred NOG‐IL‐2 Tg mice through ADCC. If SCLC metastatic models can be generated using mice, evaluation of the effects of ch6G10A in SCLC metastasis might be possible.
Another important point about the result of Figure 5 is the mechanism by which anti‐CAR mAb inhibited SCLC metastasis. It is still unclear whether anti‐CAR mAb directly reacts with tumor cells in blood/lymph vessels and metastatic sites. However, as a result of technical limitations of our model, it is very difficult to answer this question. Because we use fluorescence imaging to chase the metastatic tumor in our model, killing and dissection of mice are needed for quantitative detection of metastatic tumors. This makes it difficult to start treatment with antibody after confirming formation of metastatic tumors in the model. In addition, enucleation of the orthotopic tumor in lung will result in death of the model mouse. This also makes it difficult to examine the direct effect of anti‐CAR mAb on tumor cells in metastatic sites of the model.
One of the major points for the clinical safety of therapeutic antibodies is reactivity with normal tissues. We previously showed that mu6G10A reacts with normal skin, prostate, and kidney tissue.14 At this point, we could not avoid the possibility that the reactivity of 6G10A to normal tissues could cause some adverse events when the 6G10A‐based antibody is given to humans. Thus, further safety evaluations of the 6G10A‐based antibody using animal models such as primates or human CAR knock‐in mice are required. If the reactivity of 6G10A to normal tissues causes severe problems, cancer specificity of the antibody should be improved.
In conclusion, our studies and other reports collectively suggest that CAR should be considered a candidate target for cancer immune therapy against several cancers including SCLC.
DISCLOSURE
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
We thank T. Ohishi, A. Harakawa, and Y. Kohda (BIKAKEN) for helpful discussions and their technical assistance. This work was supported by grants from the Japan Society for the Promotion of Science (26460481 and 17K08777) (S.S.) and from the Kobayashi Foundation for Cancer Research (M.K.). This work was also supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research [BINDS]) from AMED under Grant Number JP18am0101078 (Y.K.). We also thank H. Nikki March, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Sakamoto S, Inoue H, Kaneko MK, et al. Generation and evaluation of a chimeric antibody against coxsackievirus and adenovirus receptor for cancer therapy. Cancer Sci. 2019;110:3595–3602. 10.1111/cas.14196
Sakamoto and Inoue contributed equally to this work.
REFERENCES
- 1. Coyne CB, Bergelson JM. CAR: a virus receptor within the tight junction. Adv Drug Deliv Rev. 2005;57:869‐882. [DOI] [PubMed] [Google Scholar]
- 2. Dorner AA, Wegmann F, Butz S, et al. Coxsackievirus‐adenovirus receptor (CAR) is essential for early embryonic cardiac development. J Cell Sci. 2005;118:3509‐3521. [DOI] [PubMed] [Google Scholar]
- 3. Mizra M, Pang M‐P, Amr Zaini M, et al. Essential role of the coxsackie‐ and adenovirus receptor (CAR) in development of the lymphatic system in mice. PLoS ONE. 2012;7:e37523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brüning A, Stickeler E, Diederich D, et al. Coxsackie and adenovirus receptor promotes adenocarcinoma cell survival and is expressionally activated after transition from preneoplastic precursor lesions to invasive adenocarcinomas. Clin Cancer Res. 2005;11:4316‐4320. [DOI] [PubMed] [Google Scholar]
- 5. Wang Y, Wang S, Bao Y, et al. Coxsackievirus and adenovirus receptor expression in non‐malignant lung tissues and clinical lung cancer. J Mol Histol. 2006;37:153‐160. [DOI] [PubMed] [Google Scholar]
- 6. Persson A, Fan X, Salford LG, Widegren B, Englund E. Neuroblastomas and medulloblastomas exhibit more coxsackie adenovirus receptor expression than gliomas and other brain tumors. Neuropathology. 2007;27:233‐236. [DOI] [PubMed] [Google Scholar]
- 7. Giaginis C, Zarros A, Papaefthymiou M, Papadopouli A, Sfiniadakis I, Theocharis S. Coxsackievirus and adenovirus receptor expression in human endometrial adenocarcinoma: possible clinical implications. World J. Surg. Oncol. 2008;6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bao Y, Wang Y, Ma L, et al. Expression of coxsackie and adenovirus receptor in small cell lung cancer. Chin Ger J Clin Oncol. 2009;8:504‐505. [Google Scholar]
- 9. Giaginis C, Zarros A, Alexandrou P, Klijanienko J, Delladetsima I, Theocharis S. Evaluation of coxsackievirus and adenovirus receptor expression in human benign and malignant thyroid lesions. APMIS. 2010;118:210‐221. [DOI] [PubMed] [Google Scholar]
- 10. Wunder T, Schmid K, Wicklein D, et al. Expression of the coxsackie adenovirus receptor in neuroendocrine lung cancers and its implications for oncolytic adenoviral infection. Cancer Gene Ther. 2013;20:25‐32. [DOI] [PubMed] [Google Scholar]
- 11. Veena MS, Qin M, Andersson A, Sharma S, Batra RK. CAR mediates efficient tumor engraftment of mesenchymal type lung cancer cells. Lab Invest. 2009;89:875‐886. [DOI] [PubMed] [Google Scholar]
- 12. Saito K, Sakaguchi M, Iioka H, et al. Coxsackie and adenovirus receptor is a critical regulator for the survival and growth of oral squamous carcinoma cells. Oncogene. 2014;33:1274‐1286. [DOI] [PubMed] [Google Scholar]
- 13. Kawada M, Inoue H, Usami I, et al. Establishment of a highly tumorigenic LNCaP cell line having inflammatory cytokine resistance. Cancer Lett. 2006;242:46‐52. [DOI] [PubMed] [Google Scholar]
- 14. Kawada M, Inoue H, Kajikawa M, et al. A novel monoclonal antibody targeting coxsackie virus and adenovirus receptor inhibits tumor growth in vivo. Sci Rep. 2017;7:40400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berger M, Shankar V, Vafai A. Therapeutic applications of monoclonal antibodies. Am J Med Sci. 2002;324:14‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kobayashi E, Motoi S, Sugiura M, et al. Antibody‐dependent cellular cytotoxicity and cytokine/chemokine secretion by KHYG‐1 cells stably expressing FcγRIIIA. Immunol Lett. 2014;161:59‐64. [DOI] [PubMed] [Google Scholar]
- 17. Sakamoto S, Inoue H, Ohba S, et al. New metastatic model of human small‐cell lung cancer by orthotopic transplantation in mice. Cancer Sci. 2015;106:367‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaneko MK, Kunita A, Abe S, et al. Chimeric anti‐podoplanin antibody suppresses tumor metastasis through neutralization and antibody‐dependent cellular cytotoxicity. Cancer Sci. 2012;103:1913‐1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sakamoto S, Kojima F, Momose I, Kawada M, Adachi H, Nishimura Y. Decalpenic acid induces early osteoblastic markers in pluripotent mesenchymal cells via activation of retinoic acid receptor γ. Biochem Biophys Res Commun. 2012;422:751‐757. [DOI] [PubMed] [Google Scholar]
- 20. Oronsky B, Reid TR, Oronsky A, Carter CA. What's new in SCLC? A review. Neoplasia. 2017;19:842‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zahavi D, AlDeghaither D, O'Connell A, Weiner L. Enhancing antibody‐dependent cell‐mediated cytotoxicity: a strategy for improving antibody‐based immunotherapy. Antib Ther. 2018;1:7‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katano I, Takahashi T, Ito R, et al. Predominant development of mature and functional human NK cells in a novel human IL‐2–producing transgenic NOG mouse. J Immunol. 2015;194:3513‐3525. [DOI] [PubMed] [Google Scholar]