

Abstract
Poly ADP‐ribose polymerase inhibitors (PARPi) have shown promising therapeutic efficacy in triple‐negative breast cancer (TNBC) patients. However, resistance ultimately develops, preventing a curative effect from being attained. Extensive investigations have indicated the diversity in the mechanisms underlying the PARPi sensitivity of breast cancer. In this study, we found that DNA damage binding protein 2 (DDB2), a DNA damage‐recognition factor, could protect TNBC cells from PARPi by regulating DNA double‐strand break repair through the homologous recombination pathway, whereas the depletion of DDB2 sensitizes TNBC cells to PARPi. Furthermore, we found that DDB2 was able to stabilize Rad51 by physical association and disrupting its ubiquitination pathway‐induced proteasomal degradation. These findings highlight an essential role of DDB2 in modulating homologous recombination pathway activity and suggest a promising therapeutic target for TNBC.
Keywords: DDB2, homologous recombination, PARPi sensitivity, Rad51, triple‐negative breast cancer
1. INTRODUCTION
Poly ADP‐ribose polymerase (PARP), which detects and binds DNA at the site of single‐strand breaks, recruits DNA repair molecules by generating PAR chains and plays an essential role in DNA damage response when cancer cells defend themselves against the pernicious effects of genotoxic agents.1 Poly ADP‐ribose polymerase inhibitors (PARPi) trap PARP1 at the DNA and prevent its dissociation, thereby creating barriers for replication forks,2 which results in the accumulation of unrevised genetic errors. DNA breaks are then recognized and repaired by the DNA double‐strand break (DSB) pathways, such as nonhomologous end‐joining3 and homologous recombination (HR). The concept of synthetic lethality, whereby a fault in either one of two genes has little influence on cell viability but a combination of defects in both genes results in death, was proposed nearly a century ago.4 The first clinically approved drug designed to exploit the concept, PARPi, impedes PARP‐mediated DNA lesion repair created by radiotherapy or chemotherapy, and has been extensively developed in cancer therapy with promising clinical results.
Triple‐negative breast cancer (TNBC) is an aggressive subtype that accounts for 10%‐17% of all mammary malignancies and is clinically characterized by a high rate of recurrence, early metastasis, and poor prognosis.5 Due to expression defects of estrogen receptors, progesterone receptors, and human epidermal growth factor receptor 2, which leads to a lack of approaches to targeted agents, treatment of TNBC patients has been extremely challenging. Olaparib, a credible inhibitor of PARP1/2 that has been reported to strengthen the effects of DNA‐damaging agents,6 has shown considerable efficacy in BRCA‐deficient breast tumor patients.7, 8, 9, 10, 11 However, the therapeutic effect of PARPi in metastatic breast cancer is not as efficient as that in ovarian cancer,7, 8, 9, 10, 11, 12 moreover, as with other targeted therapies, resistance to PARPi in TNBC cells ultimately develops, and thus the potential methods to sensitize TNBC cells to PARPi urgently need to be developed. Currently, there are mainly 2 approaches moving forward to improve the therapeutic efficacy of PARPi. One is to develop methods confirming biomarkers predictive of the sensitivity to PARP inhibition. The other is to impede DNA repair pharmacologically and consequently sensitize cancer cells to PARP inhibition.
DNA damage binding protein 2 (DDB2) was first reported as a damage sensor that detects cyclobutane pyrimidine dimers in chromatin induced by UV light and that facilitates the nucleotide excision repair (NER) pathway13; DDB2 was later identified to mediate cisplatin sensitivity of ovarian cancer cells.14, 15 Recently, DDB2 was reported to regulate the HR pathway in non‐small cell lung cancer by facilitating the phosphorylation of checkpoint kinase 1.16
In this research, we sought to determine whether DDB2 impacts the HR pathway and PARPi sensitivity in TNBC and to provide mechanistic insights into the functions of DDB2 in DNA repair and potential ways to maximize the clinical efficacy of PARPi in this aggressive subtype of mammary malignancies.
2. MATERIALS AND METHODS
2.1. Cell lines and reagents
Two TNBC lines (SUM159 and MDA‐MB‐231) and 2 non‐TNBC lines (T47D and MCF7) were chosen for this study. SUM159 cells (kindly provided by Dr Stephen Ethier, Karmanos Cancer Institute) were incubated in Ham’s F‐12 (Invitrogen) with 5% FBS (HyClone), 10 mg/mL gentamicin (Life Technologies), and 1 mg/mL hydrocortisone (Sigma‐Aldrich). MDA‐MB‐231 was obtained from the ATCC and grown in RPMI‐1640 medium (Invitrogen) with 10% FBS (HyClone), 1% antibiotic‐antimycotic, and 10 mg/mL gentamicin (Life Technologies). T47D and MCF7 were also obtained from ATCC and cultured in DMEM (Invitrogen) with 10% FBS (HyClone) and 1% antibiotic‐antimycotic. All cells were cultured at 37°C and 5% CO2.
2.2. Plasmid construction, transfection, and stable cell line establishment
The whole coding sequences of DDB2 and Rad51 were acquired by RT‐PCR from HUVEC mRNA. The DDB2 and Rad51 cDNAs were cloned into a pEGFP or pcDNA3.1 vectors to generate the pEGFP‐DDB2 or pcDNA3.1‐Rad51 recombinant plasmids. The DDB2 shRNAs were purchased from Santa Cruz Biotechnology. A nonspecific shRNA sequence (Dharmacon) was used as a control. For transient transfection, 50 nmol/L shRNA was blend with 4 µL Lipofectamine RNAimax reagent (Invitrogen), cultured at room temperature for 20 minutes, and added to the cells.
2.3. Constructs and viral infection
For stable transfection, TNBC cells were transfected with DDB2, shDDB2, Rad51, or shRad51 (Santa Cruz Biotechnology) lentiviral particles following the manufacturer’s protocol. Scrambled lentiviral particles were used as a control. Puromycin (0.375‐1 μg/mL) was used for selection 48 hours after infection. All oligonucleotides were transfected using Lipofectamine 2000 (Invitrogen).
2.4. Colony formation assay
Cells were incubated as described previously in 60‐mm tissue culture plates with olaparib (dissolved in DMSO) for 24 hours. The culture media was replaced with media without olaparib. Ten to 14 days later, the colonies were fixed with 100% methanol and stained with 0.1% crystal violet in 20% methanol for 15 minutes. Survival fraction was shown as the count of visible colonies/plated cells × 100%.
2.5. Western blot analysis
Cell lysates were dissolved in SDS‐PAGE (10%‐15%) and were transferred electrophoretically to PVDF membranes. The membranes were blocked in PBS‐Tween 20 with 5% nonfat milk at room temperature for 1 hour followed by incubation with primary Ab at 4°C overnight. Then an HRP‐conjugated secondary Ab (Santa Cruz Biotechnology) was used to bind with primary Ab on the membrane. Subsequently, the bound Abs were detected by the ECL method (Amersham Biosciences). Antibodies against DDB2 (Abcam), Rad51 (Abcam), γ‐H2AX (Novus Biologicals), BRCA1 (Abcam), cleaved caspase‐3 (Cell Signaling Technology), ATM (Abcam), and β‐actin (Bethyl Laboratories) were applied for western blot assays.
2.6. Flow cytometric analysis
Cells were treated with PARPi following ectopic genetic modulation as detailed below. Both detached and adherent cells were collected for apoptosis assays. An annexin V‐FITC apoptosis detection kit (BD Pharmingen) were used for annexin V‐positive cell detection. Cells were labeled with FITC‐conjugated annexin V and propidium iodide without permeabilization followed by analyzing with a FACScan flow cytometer with CellQuest software. The proportion of apoptotic cells was calculated based on the staining of annexin V‐phycoerythrin/7‐AAD.
2.7. Homologous recombination DNA repair assay
The system was invented and provided as a gift by Dr Maria Jasin of the Memorial Sloan‐Kettering Cancer Center. Homologous recombination DNA repair assay was carried out as in our previous study.17 Briefly, the DR‐GFP HR repair substrate included 2 different GFP genes with specific mutations. I‐SceI endonuclease ectopic expression resulted in DSBs in one of the 2 differentially mutated GFP genes. Then the DSB was restored by gene conversion, which will generate a functional GFP gene. The DR‐GFP plasmid was then transfected into SUM‐159 or MDA‐MB‐231 cells, and positive clones that had integrated a copy of the reporter gene were detected. To obtain stable DR‐GFP TNBC cells, hygromycin was used for selection. DR‐GFP cells were then cotransfected with shRNAs and the pCBA‐SceI plasmid to evaluate HR repair of DSBs. Flow cytometric analysis was then applied to detect the proportion of GFP+ cells caused by DSB‐induced HR repair.
2.8. In vivo ubiquitination assay
Cells were cotransfected with HA‐Rad51 and Myc‐Ub. Twenty‐four hours later, the cells were treated with 150 μmol/L PARPi for 24 hours. An in vivo ubiquitination assay was carried out as described in a previous study.18 The complex that was immunoprecipitated with an anti‐HA Ab was subjected to western blot analysis using Ab against Myc. The immunoprecipitations were carried out under denatured conditions.
2.9. Glutathione S‐transferase pulldown assay
Expression of GST‐DDB2 in Escherichia coli BL21 cells was induced with 0.8 mmol/L isopropyl‐β‐D‐thiogalactopyranoside at 16°C for 6 hours and purified with GST beads (GE Healthcare Life Sciences). In the GST pulldown assay, bacterial‐expressed GST‐DDB2 or GST bound to glutathione‐Sepharose beads was incubated overnight with cell lysates at 4°C after 3 washes with GST‐binding buffer. The bound proteins were rinsed with PBS with Triton X‐100 and then subjected to immunoblotting with the indicated Abs and Coomassie blue staining.
2.10. Immunofluorescence
The cells were seeded at 1 × 104 cells/well in 6 chamber slides (Corning) and cultured for 5 days at 37℃ before confluence. The cells were rinsed 3 times with PBS and fixed (1% formaldehyde and 0.1% Triton X‐100) for 20 minutes at room temperature. After blocking with blocking solution (1% BSA and 0.1% Triton X‐100) for 1 hour, the cells were then incubated with the primary polyclonal Abs anti‐DDB2 and anti‐Rad51 (Abcam), diluted at 1:100 in blocking solution, and cultured at 4℃ overnight. The next day, the cells were rinsed with PBS/0.1% Triton X‐100 followed by 2 washes with PBS. After incubation with the secondary Ab (Invitrogen) at room temperature for 2 hours, the slides were incubated with Hoechst 33342 for 5 minutes. Finally, the slides were rinsed in PBS and then mounted with glycerol. Images of cellular immunofluorescence were obtained using an LSM800 confocal microscope (Carl Zeiss) driven by ZEN 2.3 software (Carl Zeiss).
2.11. Statistical analysis
All tests were repeated in triplicate. Results were analyzed using Student’s t test for pairwise mean comparisons. Differences with P < .05 were regarded as statistically significant.
3. RESULTS
3.1. Depletion of DDB2 confers sensitivity to PARPi in TNBC cells
First, we analyzed the expression of DDB2 in 2 TNBC cell lines (SUM159 and MDA‐MB‐231) and 2 non‐TNBC cell lines (T47D and MCF7), compared with that in a normal human mammary epithelial cell line (HMEC), by Western blot analysis. Among these 5 cell lines, HMEC and MDA‐MB‐231 had the lowest expression of DDB2 protein, SUM159 cells had slightly higher expression, and T47D and MCF7 had the highest expression (Figure S1A). Next, to determine the role of DDB2 in TNBC cells in response to PARPi, we overexpressed DDB2 in MDA‐MB‐231 cells, which had a lower expression of DDB2 than SUM159 cells, as described previously (Figure 1A). MDA‐MB‐231 cells that overexpressed DDB2 formed significantly more colonies following PARPi treatment than control cells, indicating that the overexpression of DDB2 reversed the reduction of colony‐forming capacity caused by PARPi (Figure 1B). As the gain of DDB2 could cause PARPi resistance in TNBC cells, we asked whether DDB2 depletion could confer sensitivity to PARPi. We next silenced DDB2 in SUM159 cells (Figure 1C), which have a relatively higher expression of DDB2 than MDA‐MB‐231 cells. We noted that DDB2 silencing conferred PARPi sensitivity to SUM159 cells, which was reflected by a decreased colony‐forming ability following PARPi treatment compared with that in SUM159 cells expressing the control vector (Figure 1D). In addition, we assessed the impact of DDB2 on non‐TNBC cells in response to PARPi. Following DDB2 knockdown in T47D and MCF7 cell lines, both formed fewer colonies following PARPi treatment than the controls; however, the differences were not significant, indicating that the impact of DDB2 on the colony‐forming capacity in non‐TNBC lines after PARPi therapy is not as significant as that in TNBC lines (Figure S1B‐E). We also assessed the apoptosis rate in the TNBC lines in response to PARPi. As shown in Figure 1E,F, compared to the controls, a significant increase in the count of apoptotic cells (annexin V‐FITC and propidium iodide) with a concomitant increase in the cleavage of caspase‐3 (Figure 1I) was detected in DDB2‐knockout SUM159 cells, whereas an obvious reduction in the number of apoptotic cells (Figure 1G,H) with a simultaneous decrease in cleaved caspase‐3 (Figure 1I), was observed in DDB2 upregulated MDA‐MB‐231 cells. Conversely, DDB2 depletion in non‐TNBC lines did not cause as significant apoptosis in response to PARPi as that in TNBC lines (Figure S1F‐J). Together, these findings indicate that DDB2 depletion confers sensitivity to PARPi exclusively in TNBC cells.
Figure 1.
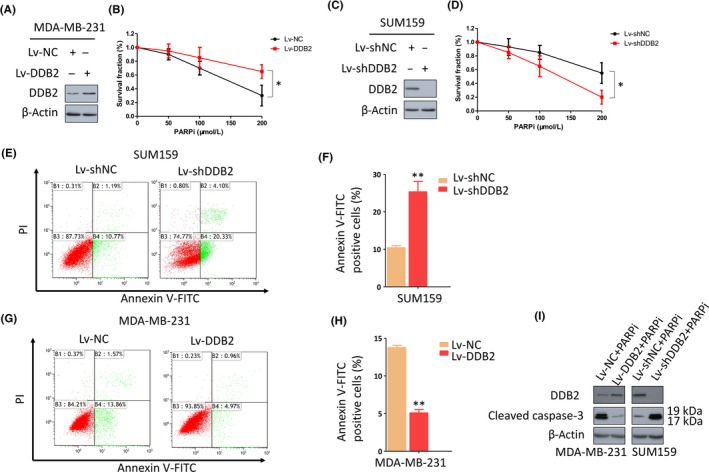
DNA damage binding protein 2 (DDB2) depletion confers sensitivity to poly ADP‐ribose polymerase inhibitors (PARPi) in triple‐negative breast cancer cells. A, MDA‐MB‐231 cells that expressed low levels of DDB2 were transfected with either a control lentivirus (Lv‐NC) or a DDB2 lentivirus (Lv‐DDB2). Immunoblotting was undertaken to evaluate the upregulation efficiency of DDB2 by Lv‐DDB2. β‐Actin was detected as a loading control. B, Clonogenic formation assay to evaluate the sensitivity of these cells to a PARPi. *P < .05. C, SUM159 cells that expressed high levels of DDB2 were transfected with either a control shRNA (Lv‐shNC) or DDB2 shRNA (Lv‐shDDB2). Immunoblotting was used to evaluate the knockdown efficiency of shDDB2. β‐Actin was detected as a loading control. D, Clonogenic formation assay to evaluate the sensitivity of these cells to a PARPi. *P < .05. E,F, DDB2‐depleted SUM159 cells were exposed to 150 μmol/L PARPi for 24 h, and apoptosis was measured by flow cytometry. Data are shown as the mean of 3 independent experiments ± SEM. **P < .01. G‐I, DDB2‐overexpressed MDA‐MB‐231 cells were exposed to 150 μmol/L PARPi for 24 h, and apoptosis was measured by flow cytometry. Data are shown as the mean of 3 independent experiments ± SEM. **P < .01. I, Western blot analysis of cleaved caspase‐3 expression in DDB2‐depleted SUM159 cells or DDB2‐overexpressing MDA‐MB‐231 cells in the presence of PARPi. β‐Actin served as the loading control
3.2. Depletion of DDB2 induces a delay in HR‐mediated DNA repair
Given the phenomenon that DDB2 depletion sensitized TNBC cells to PARPi therapy, we further explored the changing pattern of DDB2 expression after PARPi treatment in TNBC lines. We noted that DDB2 foci formed in nuclei after PARPi treatment (Figure 2A) and correspondingly elevated the expression of DDB2 in both TNBC lines (Figure 2B). More experiments were applied to assess DDB2 protein after other DNA‐damaging treatments; similarly, DDB2 protein levels rose dramatically in both TNBC lines after cisplatin (Figure S2A‐C) and irradiation (Figure S2A,B,D) treatment. These data indicated that the DDB2 foci formation and elevation of DDB2 is universal after DNA‐damaging treatment. For further investigation into the effects of DDB2 on PARPi‐induced DNA damage and repair, we analyzed the impacts of DDB2 depletion on γH2AX, a reliable marker of DSBs, through assessing γH2AX expression with western blotting after PARPi treatment. Compared to the controls, DDB2 overexpression in MDA‐MB‐231 cells obviously accelerated DNA damage repair after PARPi treatment, which was reflected by the immediate clearance of γH2AX (Figure 2C), whereas DDB2 depletion in SUM159 cells led to the persistence of high levels of γH2AX after PARPi treatment, indicating a delay in DNA damage repair (Figure 2D). In addition, DDB2 overexpression accelerated, whereas DDB2 depletion slowed, γH2AX clearance in TNBC lines after either cisplatin (Figure S2E,F) or irradiation (Figure S2G,H) treatment, which indicated that DDB2 silence induces a delay in HR pathway DNA damage repair under various kinds of genotoxic treatment, in additon to PARPi.
Figure 2.
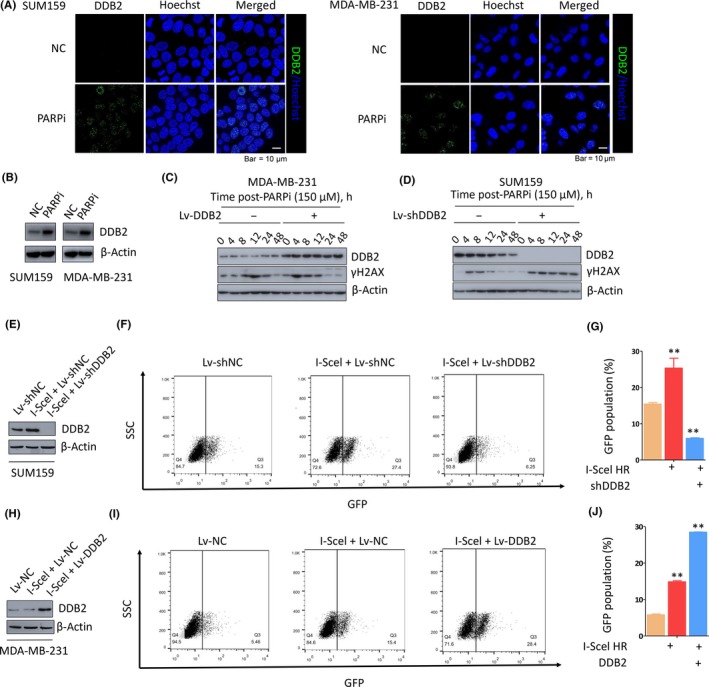
DNA damage binding protein 2 (DDB2) depletion induces a delay in homologous recombination (HR)‐mediated DNA repair. A, Immunofluorescence assay showing formation of DDB2 foci in SUM159 and MDA‐MB‐231 cells, which increased significantly after poly ADP‐ribose polymerase inhibitor (PARPi) therapy. B, Western blot analysis of DDB2 expression after PARPi treatment in SUM159 and MDA‐MB‐231 cells. β‐Actin served as the loading control (NC). C, Western blot analysis of DDB2 and γH2AX expression in MDA‐MB‐231 cells transfected with DDB2 shRNA or control shRNA at different time points after PARPi withdrawal. D, Western blot analysis of DDB2 and γH2AX expression in SUM159 cells transfected with DDB2 shRNA or control shRNA at different time points after PARPi withdrawal. E‐G, SUM159 cells carrying the recombination substrate (DR‐GFP) were transfected with expression vectors for DDB2 shRNA (Lv‐shDDB2) or control shRNA (Lv‐shNC). **P < .01. H‐J, MDA‐MB‐231 cells carrying DR‐GFP were transfected with expression vectors for DDB2 (Lv‐DDB2) or control RNA (Lv‐NC). The I‐SceI expression plasmid was transiently transfected, and the GFP‐positive cells were analyzed 48 h later by flow cytometry. **P < .01
On the basis of the above evidence that DDB2 is required for DSB repair, we investigated whether DDB2 has impacts on HR repair. We assessed the HR repair efficacy for DSB induced by I‐SceI in SUM159 cells using the HR DNA repair assay. A significant 77% decrease was observed in the count of GFP+ cells after DDB2 silencing (Figure 2E‐G) in comparison to that in the controls, indicating that the specific defect in HR repair is, at least in part, due to the compromised DDB2 function. In contrast, a significant 2‐fold increase in the number of GFP+ cells was observed in MDA‐MB‐231 cells after DDB2 overexpression compared to that in the control cells (Figure 2H‐J). Collectively, these results suggest that DDB2 is essential for HR pathway DSB repair.
3.3. Depletion of DDB2 downregulates Rad51, leading to defective HR repair
To further investigate the mechanism underlying DDB2‐mediated HR repair and PARPi resistance, we analyzed the protein expression of major HR‐related genes, including BRCA1, ATM, and Rad51, in TNBC cell lines. We noted that, compared to the controls, the level of Rad51 was downregulated after DDB2 depletion (Figure 3A) and the apoptotic rate significantly rose under PARP inhibition, which was reversed by ectopically expressed Rad51 in SUM159 cells (Figure 3B,C). In MDA‐MB‐231 cells, Rad51 was upregulated after DDB2 overexpression (Figure 3D) and the apoptotic rate fell after PARPi therapy, which was reversed by Rad51 silencing (Figure 3E,F). In contrast, the expression of the other HR pathway‐related proteins was not significantly changed after DDB2 downregulation or upregulation (Figure S3A,B). Furthermore, we assessed the regulation of DDB2 on Rad51 in immunofluorescence assays. The results showed that DDB2 depletion reduced the number of Rad51 foci in the nucleus of SUM159 cells under PARP inhibition, which was reversed by Rad51 upregulation (Figures 3G and S3C). Conversely, DDB2 overexpression raised the number of Rad51 foci in the nucleus of MDA‐MB‐231 cells under PARP inhibition, which was reversed by Rad51 silence (Figures 3H and S3D). Interestingly, we found in these assays that DDB2 colocalized with Rad51 in the nucleus of TNBC cells under PARPi treatment. We further assayed for the HR‐mediated repair of I‐SceI‐induced DSBs in SUM159 cells after DDB2 and Rad51 modulation. The significant 78% decrease in the count of GFP+ cells that was observed after DDB2 silencing compared to that in the controls (Figure 3I,J) was rescued by Rad51 overexpression. In contrast, the significant 2‐fold increase in the count of GFP+ cells that was observed after DDB2 overexpression, in comparison to that in the controls, was diminished after Rad51 silencing in MDA‐MB‐231 cells (Figure 3K,L), indicating that DDB2 mediates HR activity by regulating the level of Rad51. Together, these data suggest that DDB2 depletion leads to decreased HR pathway activity by downregulating Rad51.
Figure 3.
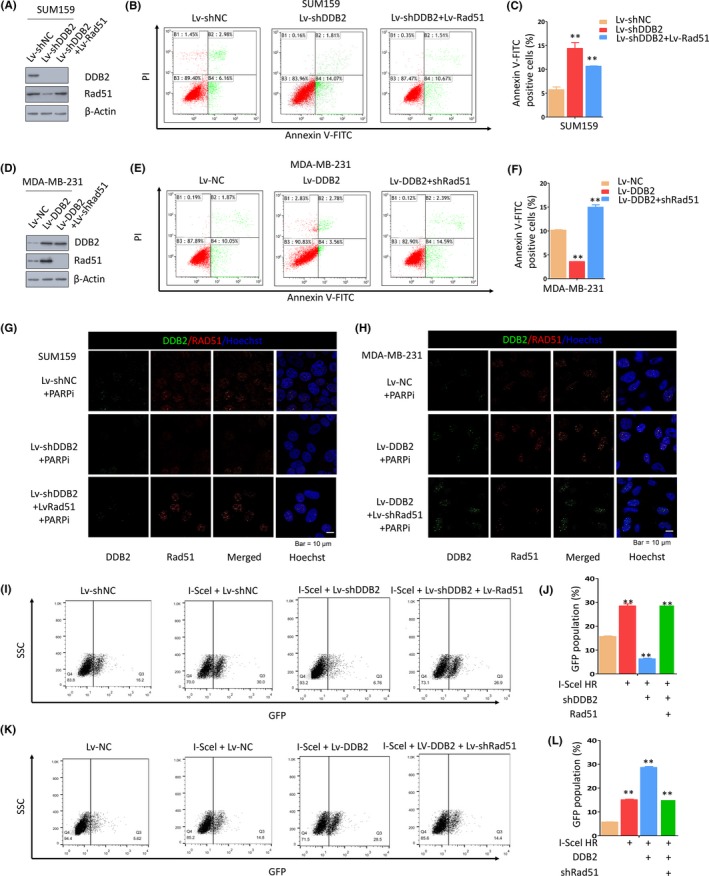
DNA damage binding protein 2 (DDB2) depletion downregulates Rad51, leading to defective homologous recombination (HR) repair. A‐C, DDB2‐depleted SUM159 cells with or without Rad51 upregulation were exposed to 150 μmol/L poly ADP‐ribose polymerase inhibitor (PARPi) for 24 h, and apoptosis was measured by flow cytometry. Data are shown as the mean of 3 independent experiments ± SEM. **P < .01. D‐F, DDB2‐overexpressing MDA‐MB‐231 cells with or without Rad51 silencing were exposed to 150 μmol/L PARPi for 24 h, and apoptosis was measured by flow cytometry. Data are shown as the mean of 3 independent experiments ± SEM. **P < .01. G, Immunofluorescent staining of DDB2 and Rad51 in SUM159 cells with DDB2 depletion alone or both DDB2 depletion and PARPi treatment. H, Immunofluorescent staining of DDB2 and Rad51 in MDA‐MB‐231 with DDB2 upregulation alone or both DDB2 upregulation and PARPi treatment. I,J, SUM‐159 cells carrying the recombination substrate (DR‐GFP) were transfected with expression vectors for DDB2 shRNA or both DDB2 shRNA and Rad51 shRNA. The I‐SceI expression plasmid was transiently transfected, and the GFP‐positive cells were analyzed 48 h later by flow cytometry. K,L, MDA‐MB‐231 cells carrying DR‐GFP were transfected with expression vectors for DDB2 or both DDB2 RNA and Rad51 shRNA
3.4. Depletion of DDB2 increases Rad51 polyubiquitination and proteasomal degradation, leading to defective HR repair and sensitivity to PARPi
We next determined how DDB2 affected Rad51 levels. Quantitative PCR was carried out at first to assess the mRNA level of Rad51 in control SUM159 cells and in DDB2‐knockdown SUM159 cells. However, no statistically significant difference in the Rad51 mRNA level was detected after DDB2 silencing compared to that in controls (Figure S4A,B), in this way excluding the possibility that Rad51 expression was regulated at the transcriptional level by DDB2. Next, we further examined whether DDB2 posttranslationally regulates Rad51 protein stability. Compared to the controls, DDB2 knockdown significantly shortened the half‐life of Rad51, from more than 8 hours to less than 2 hours after PARPi treatment (Figure 4A), whereas DDB2 upregulation prolonged the half‐life of Rad51 (Figure 4B). Moreover, we treated SUM159 DDB2 silenced or control cells with MG132, a proteasome inhibitor. As shown in Figure 4C, MG132 restored the expression levels of Rad51 in cells with DDB2 depletion after PARPi therapy; in contrast, MG132 further increased the expression levels of Rad51 in cells with DDB2 upregulation compared to that in cells treated with the vehicle (Figure 4D). These data suggested that DDB2 regulated Rad51 stability. To determine whether this phenomenon was dependent on PARPi treatment, we undertook experiments without PARPi treatment. Consistent with previous results, DDB2 knockdown shortened (Figure S4C), whereas DDB2 upregulation prolonged (Figure S4D), the half‐life of Rad51 significantly in TNBC lines without PARPi treatment. Similarly, MG132 restored the expression of Rad51 in cells with DDB2 depletion (Figure S4E) and increased the expression of Rad51 in cells with DDB2 upregulation (Figure S4F) without PARPi treatment. This evidence suggests that DDB2 could manipulate the protein stability of Rad51 through a proteasome‐dependent pathway with or without PARPi treatment, as the ubiquitin‐proteasome system (UPS) executed the protein degradation through the proteasomal pathway. Then we examined whether DDB2 mediated Rad51 proteolysis through the UPS. SUM159 DDB2 silenced or control cells were transiently transfected with plasmids encoding Myc‐tagged ubiquitin and HA‐tagged Rad51. The depletion of DDB2 in SUM159 cells significantly reduced Rad51 protein levels and caused a drastic increase in polyubiquitination levels compared to control cells 2 hours after PARPi withdrawal (Figure 4E). Parallel experiments were applied in MDA‐MB‐231 cells. We found that the upregulation of DDB2 in MDA‐MB‐231 cells led to a marked decrease in the ubiquitination level of Rad51 compared to control cells (Figure 4F). More experiments confirmed that, in the absence of PARPi treatment, DDB2 manipulated the ubiquitination level of Rad51 following the same pattern as that in the presence of PARPi treatment in TNBC cells (Figure S4G,H). As shown in Figure 3G,H, DDB2 colocalized with Rad51 in the nucleus, therefore we supposed that DDB2 might manipulate Rad51 by physical association. Hence we undertook GST pulldown assays and found the interaction between DDB2 and Rad51, suggesting that DDB2 stabilizes Rad51 by inhibiting its ubiquitination (Figure 4G). In previous studies, RING1,19 UCHL3,20 UAF1,21 FBH122, 23 UBL1,24, 25 UBC9,26 and BLM27 were reported to manipulate the ubiquitination of Rad51 by direct interaction. Therefore, using immunoprecipitation assays, we checked whether any of these proteins was involved in this process. However, none was found to interact with either DDB2 or Rad51 to play a role in DDB2‐modulated Rad51 ubiquitination. Collectively, these data suggest that the inhibition of DDB2 promotes Rad51 polyubiquitination and proteasomal degradation, resulting in defective HR repair and sensitivity to PARPi in TNBC cells. The schematic diagram indicating the mechanism by which DDB2 regulates HR pathway DSB repair through stabilizing Rad51 is shown in Figure 4H.
Figure 4.
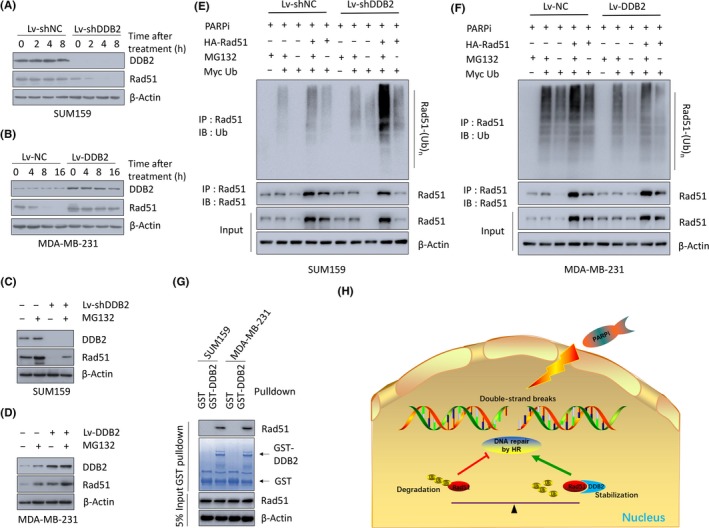
DNA damage binding protein 2 (DDB2) depletion increases Rad51 polyubiquitination and proteasomal degradation, leading to defective homologous recombination (HR) repair and sensitivity to poly ADP‐ribose polymerase inhibitors (PARPi). A, SUM159 DDB2‐depleted or control cells were pretreated for 15 min with 20 mmol/L cycloheximide followed by 150 μmol/L PARPi. DDB2 depletion decreased the apparent half‐life of the Rad51 protein in the presence of cycloheximide. B, MDA‐MB‐231 DDB2‐overexpression or control cells were pretreated for 15 min with 20 mmol/L cycloheximide followed by 150 μmol/L PARPi. DDB2 overexpression increased the apparent half‐life of the Rad51 protein in the presence of cycloheximide. C, Western blot analysis of DDB2, Rad51, and β‐actin in SUM159 cells transduced with DDB2 shRNA (Lv‐shDDB2) or control shRNA after treatment with 150 μmol/L PARPi in the absence or presence of 10 mmol/L MG132. D, Western blot analysis of DDB2, Rad51, and β‐actin in MDA‐MB‐231 cells transduced with DDB2 RNA (Lv‐DDB2) or control RNA after treatment with 150 μmol/L PARPi in the absence or presence of 10 mmol/L MG132. E, Level of Rad51 ubiquitination was detected in SUM159 DDB2‐depleted or control cells 2 h after PARPi removal. F, Level of Rad51 ubiquitination was detected in MDA‐MB‐231 DDB2‐overexpressing or control cells 2 h after PARPi removal. G, Cell lysates of SUM159 and MDA‐MB‐231 cells were incubated with bead‐bound GST or GST‐DDB2. Proteins retained on Sepharose were then subjected to immunoblotting with anti‐Rad51 Abs. H, Schematic model of the function of DDB2 on Rad51, HR repair, and the sensitivity to PARPi. IB, immunoblotting agent; IP, immunoprecipitant
4. DISCUSSION
DNA damage binding protein 2 has been previously revealed to be a potential regulator of the radiosensitivity of non‐small cell lung cancer cells.16 A previous study reported that DDB2 could enhance HR pathway activity, which plays an essential role in DNA repair in response to DSBs, by facilitating the ionizing radiation‐induced Chk1 phosphorylation, which is essential in cell cycle arrest and DNA repair in response to ionizing radiation‐induced DNA DSBs. In this study, the impact of DDB2 on the PARPi sensitivity of TNBC was first confirmed. We determined that, compared to the controls, the silencing of DDB2 in TNBC cell lines promoted PARPi sensitivity, whereas the overexpression of DDB2 inhibited PARPi sensitivity based on clonogenic survival tests, annexin‐V‐FITC apoptosis tests, and cleaved caspase‐3 expression assays. In comparison, DDB2 depletion did not sensitize non‐TNBC cells significantly. Furthermore, we showed that, compared to the controls, depletion of DDB2 dampened the activation of the HR pathway, whereas the ectopic expression of DDB2 activated the HR pathway of DNA DSB repair based on γH2AX expression tests and an HR DNA repair assay. Knockdown of DDB2 also sensitized TNBC cells to cisplatin and irradiation, suggesting that DDB2 depletion enhanced the effect of various kinds of genotoxic therapy on TNBC cells. Moreover, we screened HR‐related proteins and verified that Rad51 is a target of DDB2 that modulates HR pathway activity. The ectopic expression of Rad51 in SUM159 cells reversed the PARPi sensitivity that was induced by DDB2 knockdown, whereas silencing Rad51 expression enhanced the MDA‐MB‐231 cells PARPi sensitivity that was induced by DDB2 overexpression. These data were further supported by findings that showed that, compared to the controls, DDB2 overexpression stabilized Rad51 expression, whereas DDB2 depletion destabilized Rad51 expression. Importantly, we found that DDB2 is critical in mediating Rad51 stabilization through UPS‐mediated protein degradation. Consistently, the function of DDB2 in regulating Rad51 stability was the same under conditions without PARP inhibition. In recent years, besides the role of DDB2 in DNA repair, new functions of DDB2 have been discovered, for example, enhancing apoptosis by downregulating Bcl‐214, 15 and p21,28 inhibiting metastasis of colon cancer by blocking epithelial‐mesenchymal transition,29 promoting cancer cell growth,30 and impeding the activity and invasiveness of breast cancer cells by manipulating the activity of the nuclear factor‐κB pathway.31 It is also widely accepted that DDB2 is a tumor suppressor, based on the phenomenon that DDB2−/− mice were susceptible to carcinogenesis induced by UV irradiation and developed spontaneous malignant tumors with high frequency.32, 33 It was also confirmed that DDB2 was induced after irradiation in radioresistant non‐small cell lung cancer cells, suggesting that DDB2 might be critical in the cellular response to irradiation‐induced DNA damage.34 Taken together, these results indicate that the contribution of DDB2 to genotoxic sensitivity could be crucial not only to chemo‐ and radiosensitivity but also to PARPi sensitivity in breast, lung, and other types of cancers.
As the most important function of DDB2 is regulating the NER pathway DNA, we asked whether HR deficiency in DDB2‐depleted cells could be in part due to lack of NER function. To address this question, we first determined whether NER had an impact on Rad51 and reviewed published works regarding NER and Rad51. In previous reports, Rad51, the essential regulator in the HR pathway, was neither established as a regulator in the NER pathway nor impacted by the NER pathway. These studies supported that lack of NER will not exacerbate HR deficiency caused by DDB2 depletion. Furthermore, we asked whether inactivation of NER might be partly responsible for HR deficiency, and we reviewed published works regarding NER and HR. Homologous recombination is a major DSB repair mechanism that activates during the S and G2 phases. By comparison, NER is an important pathway for DNA bulky adducts repair unrelated to replication.35 However, there is scant evidence supporting the possibility of an interplay between NER and other DNA repair pathways. In conclusion, there has been no evidence up to now supporting that HR deficiency in DDB2‐depleted cells could be in part due to lack of NER function.
Although the systemic delivery of DDB2 inhibitors to breast cancer cells is not yet implemented, the impact of DDB2 depletion on the enhancement of PARPi sensitivity strongly suggests that more work toward the exploration of anti‐DDB2 therapeutics is fully warranted.
Collectively, we showed that DDB2 is critical for the modulation of HR repair and PARPi sensitivity in TNBC cells by regulating Rad51 stability, which could provide new insight into the development of treatment strategies for breast cancer.
DISCLOSURE
The authors declare no conflicts of interest.
Supporting information
ACKNOWLEDGEMENTS
Yan Zhuang designed this research. Jian‐Wei Lu provided the technical support. Lin Zhao, Yue Yu, and Cheng‐shuai Si performed the experiments. This work was funded by grants from Jiangsu Cancer Hospital’s Natural Science Foundation (ZM201705), the National Natural Science Foundation of China (No. 81902453), and the Natural Science Foundation of Jiangsu Province (BK20181083).
Zhao L, Si C‐S, Yu Y, Lu J‐W, Zhuang Y. Depletion of DNA damage binding protein 2 sensitizes triple‐negative breast cancer cells to poly ADP‐ribose polymerase inhibition by destabilizing Rad51. Cancer Sci. 2019;110:3543–3552. 10.1111/cas.14201
Zhao, Si, and Yu contributed equally to this work.
REFERENCES
- 1. Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP‐ribose) and PARP‐1. Genes Dev. 2012;26:417‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Can Res. 2012;72:5588‐5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lieber MR. The mechanism of double‐strand DNA break repair by the nonhomologous DNA end‐joining pathway. Annu Rev Biochem. 2010;79:181‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA. New strategies for triple‐negative breast cancer–deciphering the heterogeneity. Clin Cancer Res. 2014;20:782‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Menear KA, Adcock C, Boulter R, et al. 4‐[3‐(4‐cyclopropanecarbonylpiperazine‐1‐carbonyl)‐4‐fluorobenzyl]‐2H‐phthalazin‐ 1‐one: a novel bioavailable inhibitor of poly(ADP‐ribose) polymerase‐1. J Med Chem. 2008;51:6581‐6591. [DOI] [PubMed] [Google Scholar]
- 7. Kortmann U, McAlpine JN, Xue H, et al. Tumor growth inhibition by olaparib in BRCA2 germline‐mutated patient‐derived ovarian cancer tissue xenografts. Clin Cancer Res. 2011;17:783‐791. [DOI] [PubMed] [Google Scholar]
- 8. Evers B, Drost R, Schut E, et al. Selective inhibition of BRCA2‐deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res. 2008;14:3916‐3925. [DOI] [PubMed] [Google Scholar]
- 9. Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105:17079‐17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof‐of‐concept trial. Lancet. 2010;376:245‐251. [DOI] [PubMed] [Google Scholar]
- 11. Tutt A, Robson M, Garber JE, et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof‐of‐concept trial. Lancets. 2010;376:235‐244. [DOI] [PubMed] [Google Scholar]
- 12. Zimmer AS, Gillard M, Lipkowitz S, Lee JM. Update on PARP inhibitors in breast cancer. Curr Treat Options Oncol. 2018;19:21.s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dualan R, Brody T, Keeney S, Nichols AF, Admon A, Linn S. Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage‐specific DNA binding protein. Genomics. 1995;29:62‐69. [DOI] [PubMed] [Google Scholar]
- 14. Barakat BM, Wang QE, Han C, et al. Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int J Cancer. 2010;127:977‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao R, Han C, Eisenhauer E, et al. DNA damage‐binding complex recruits HDAC1 to repress Bcl‐2 transcription in human ovarian cancer cells. Mol Cancer Res. 2014;12:370‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou N, Xie G, Cui T, et al. DDB2 increases radioresistance of NSCLC cells by enhancing DNA damage responses. Tumour Biol. 2016;37:14183‐14191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peng C, Chen Z, Wang S, et al. The Error‐Prone DNA Polymerase kappa Promotes Temozolomide Resistance in Glioblastoma through Rad17‐Dependent Activation of ATR‐Chk1 Signaling. Can Res. 2016;76:2340‐2353. [DOI] [PubMed] [Google Scholar]
- 18. Liu W, Wu G, Li W, Lobur D, Wan Y. Cdh1‐anaphase‐promoting complex targets Skp2 for destruction in transforming growth factor beta‐induced growth inhibition. Mol Cell Biol. 2007;27:2967‐2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ahmed KM, Pandita RK, Singh DK, Hunt CR, Pandita TK. beta1‐integrin impacts Rad51 stability and DNA double‐strand break repair by homologous recombination. Mol Cell Biol. 2018;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo K, Li L, Li Y, et al. A phosphorylation‐deubiquitination cascade regulates the BRCA2‐RAD51 axis in homologous recombination. Genes Dev. 2016;30:2581‐2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang F, Longerich S, Miller AS, et al. Promotion of RAD51‐mediated homologous DNA pairing by the RAD51AP1‐UAF1 complex. Cell Rep. 2016;15:2118‐2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chu WK, Payne MJ, Beli P, Hanada K, Choudhary C, Hickson ID. FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51. Nat Commun. 2015;6:5931. [DOI] [PubMed] [Google Scholar]
- 23. Tsutsui Y, Kurokawa Y, Ito K, et al. Multiple regulation of Rad51‐mediated homologous recombination by fission yeast Fbh1. PLoS Genet. 2014;10:e1004542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li W, Hesabi B, Babbo A, et al. Regulation of double‐strand break‐induced mammalian homologous recombination by UBL1, a RAD51‐interacting protein. Nucleic Acids Res. 2000;28:1145‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shen Z, Pardington‐Purtymun PE, Comeaux JC, Moyzis RK, Chen DJ. UBL1, a human ubiquitin‐like protein associating with human RAD51/RAD52 proteins. Genomics. 1996;36:271‐279. [DOI] [PubMed] [Google Scholar]
- 26. Kovalenko OV, Plug AW, Haaf T, et al. Mammalian ubiquitin‐conjugating enzyme Ubc9 interacts with Rad51 recombination protein and localizes in synaptonemal complexes. Proc Natl Acad Sci USA. 1996;93:2958‐2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ouyang KJ, Woo LL, Zhu J, Huo D, Matunis MJ, Ellis NA. SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol. 2009;7:e1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stoyanova T, Roy N, Kopanja D, Bagchi S, Raychaudhuri P. DDB2 decides cell fate following DNA damage. Proc Natl Acad Sci USA. 2009;106:10690‐10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roy N, Bommi PV, Bhat UG, et al. DDB2 suppresses epithelial‐to‐mesenchymal transition in colon cancer. Can Res. 2013;73:3771‐3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kattan Z, Marchal S, Brunner E, et al. Damaged DNA binding protein 2 plays a role in breast cancer cell growth. PLoS One. 2008;3:e2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ennen M, Klotz R, Touche N, et al. DDB2: a novel regulator of NF‐kappaB and breast tumor invasion. Can Res. 2013;73:5040‐5052. [DOI] [PubMed] [Google Scholar]
- 32. Yoon T, Chakrabortty A, Franks R, Valli T, Kiyokawa H, Raychaudhuri P. Tumor‐prone phenotype of the DDB2‐deficient mice. Oncogene. 2005;24:469‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Itoh T, Iwashita S, Cohen MB, Meyerholz DK, Linn S. Ddb2 is a haploinsufficient tumor suppressor and controls spontaneous germ cell apoptosis. Hum Mol Genet. 2007;16:1578‐1586. [DOI] [PubMed] [Google Scholar]
- 34. Yang HJ, Kim N, Seong KM, Youn H, Youn B. Investigation of radiation‐induced transcriptome profile of radioresistant non‐small cell lung cancer A549 cells using RNA‐seq. PLoS ONE. 2013;8:e59319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moriel‐Carretero M, Aguilera A. A postincision‐deficient TFIIH causes replication fork breakage and uncovers alternative Rad51‐ or Pol32‐mediated restart mechanisms. Mol Cell. 2010;37:690‐701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials