

Abstract
Targeting the function of membrane transporters in cancer stemlike cells is a potential new therapeutic approach. Cystine‐glutamate antiporter xCT expressed in CD44 variant (CD44v)‐expressing cancer cells contributes to the resistance to oxidative stress as well as cancer therapy through promoting glutathione (GSH)‐mediated antioxidant defense. Amino acid transport by xCT might, thus, be a promising target for cancer treatment, whereas the determination factors for cancer cell sensitivity to xCT‐targeted therapy remain unclear. Here, we demonstrate that high expression of xCT and glutamine transporter ASCT2 is correlated with undifferentiated status and diminished along with cell differentiation in head and neck squamous cell carcinoma (HNSCC). The cytotoxicity of the xCT inhibitor sulfasalazine relies on ASCT2‐dependent glutamine uptake and glutamate dehydrogenase (GLUD)‐mediated α‐ketoglutarate (α‐KG) production. Metabolome analysis revealed that sulfasalazine treatment triggers the increase of glutamate‐derived tricarboxylic acid cycle intermediate α‐KG, in addition to the decrease of cysteine and GSH content. Furthermore, ablation of GLUD markedly reduced the sulfasalazine cytotoxicity in CD44v‐expressing stemlike HNSCC cells. Thus, xCT inhibition by sulfasalazine leads to the impairment of GSH synthesis and enhancement of mitochondrial metabolism, leading to reactive oxygen species (ROS) generation and, thereby, triggers oxidative damage. Our findings establish a rationale for the use of glutamine metabolism (glutaminolysis)‐related genes, including ASCT2 and GLUD, as biomarkers to predict the efficacy of xCT‐targeted therapy for heterogeneous HNSCC tumors.
Keywords: ASCT2, CD44 variant, glutamate dehydrogenase, head and neck cancer, xCT
1. INTRODUCTION
Cancer cells reprogram their energy metabolism to favor aerobic glycolysis or oxidative phosphorylation and, thus, manifest metabolic features that differ from those of the corresponding normal cells.1, 2 The metabolic changes also often result in attenuation of reactive oxygen species (ROS) accumulation through either suppression of mitochondrial respiration or promotion of antioxidant defense through synthesis of the major antioxidant glutathione (GSH), and they thereby contribute to maintenance of intracellular redox homeostasis and resistance to anticancer treatment.2, 3, 4, 5 Several types of tumors are hierarchically organized, and are sustained by a distinct subpopulation of cancer stemlike cells that possess an enhanced antioxidant defense system compared with non‐stemlike cancer cells.6, 7 In contrast to differentiated non‐stemlike cancer cells that rely solely on glycolysis,8 cancer stemlike cells tend to be highly glycolytic or reliant on oxidative phosphorylation, depending on the cancer type, suggesting that the antioxidant defense system adapts to the metabolic reprogramming in the stemlike cells.
Our investigations into the mechanism underlying the regulation of intracellular redox homeostasis in CD44 variant (CD44v)‐expressing cancer stemlike cells recently revealed that CD44v interacts with the xCT subunit of system xc(−) and thereby stabilizes its localization at the cell surface.9 System xc(−) is a cystine‐glutamate antiporter that is composed of both the light‐chain subunit xCT (SLC7A11) and a heavy‐chain subunit (CD98hc, SLC3A2) and which imports cystine into cells in exchange for intracellular glutamate.10, 11 The molecular interaction between CD44v and xCT enhances cystine uptake and thereby promotes GSH synthesis from cysteine. It thus potentiates antioxidant defense and confers treatment resistance in CD44v‐expressing cancer stemlike cells. However, the relevance of metabolic reprogramming to xCT activity in cancer cells has remained unclear.
Sulfasalazine is administered clinically for the treatment of inflammatory bowel disease and rheumatoid arthritis.12 It has also recently been found to act as a specific inhibitor of xCT‐dependent cystine transport.11, 13, 14 Inhibitors of xCT, including sulfasalazine and erastin, have been shown to induce an iron‐dependent oxidative cell death, or ferroptosis, in cancer cells.15, 16 Given that cancer cells often manifest an increased intracellular iron concentration due to a high level of expression of transferrin receptor 1 (which mediates cellular iron uptake) and low abundance of ferroportin (which contributes to iron efflux),17 xCT‐targeted therapy is expected to effectively induce cell death in cancer cells without affecting normal tissue. We previously found that among the hierarchically organized cell population of head and neck squamous cell carcinoma (HNSCC) tumors, treatment with sulfasalazine‐induced oxidative cell death in and thereby reduced the number of stemlike tumor cells that express CD44v at a high level (CD44vhigh), without affecting cells that express CD44v at a low or undetectable level (CD44vlow‐neg).18, 19 Furthermore, a recent phase I study of sulfasalazine treatment in patients with advanced gastric cancer revealed a reduction in the size of the CD44v‐expressing tumor cell subpopulation in posttreatment biopsy tissue from 4 of 8 patients.20 These observations indicate that CD44vhigh stemlike tumor cells tend to rely on the CD44v‐xCT system for their survival to a greater extent than do CD44vlow‐neg tumor cells. However, the factors determining the sensitivity to sulfasalazine in heterogenous cancer cells remain unclear.
We have now found that induction of oxidative stress by sulfasalazine treatment requires ASCT2‐mediated glutamine uptake and the production of α‐ketoglutarate (α‐KG) mediated by glutamate dehydrogenase (GLUD) in CD44vhigh stemlike HNSCC cells.
2. MATERIALS AND METHODS
2.1. Cell lines
OSC19 cells were obtained as described previously18; HSC‐2, and HSC‐4 cells were from RIKEN Cell Bank. The tongue squamous cell carcinoma patient‐derived primary cancer YH cells were established at Kumamoto University Hospital after informed consent was obtained and following the guidelines of the Ethics Committee of Kumamoto University. HSC‐4 cells stably expressing xCT or harboring the empty vector were established by retroviral infection with the pMXs‐IP plasmid containing human xCT cDNA or with pMXs‐IP alone.21 HSC‐2 cells stably expressing mCherry were established by retroviral infection with the pMXs‐IRES‐mCherry plasmid, which is constructed by replacement of the puromycin resistant gene in pMXs‐IP to mCherry. After infection of pMXs‐IRES‐mCherry retrovirus, mCherry positive cells were purified by cell sorting. All cells were cultured in DMEM supplemented with 10% FBS and maintained under 5% CO2 at 37°C.
2.2. Antibodies
For immunoblot analysis, CD44 was detected with rabbit polyclonal antibodies as previously described,22 xCT with rabbit monoclonal antibodies (#12691, Cell Signaling Technology; 1:1000), involucrin with mouse monoclonal antibodies (ab68, Abcam; 1:200), ASCT2 with rabbit monoclonal antibodies (#8057, Cell Signaling Technology; 1:1000 dilution), Myc with rabbit monoclonal antibodies (ab32072, Abcam; 1:1000), and β‐actin with mouse monoclonal antibodies (sc‐47778, Santa Cruz Biotechnology; 1:200). For flow cytometry, CD44v was detected with rat monoclonal antibodies specific for human CD44v (1 μg/mL) as previously described.7 ASCT2 was detected with newly generated rat monoclonal antibodies (10 μg/mL). Mouse CD31 (102410, Biolegend; 1:200), CD45 (MABF319, Millipore; 1:200) and TER119 (116212, Biolegend; 1:200) were each stained with allophycocyanin‐conjugated rat monoclonal antibodies. For immunofluorescence and immunohistochemical analysis, human ASCT2 was stained with rabbit polyclonal antibodies (HPA035240, Sigma; 1:200), CD44v with the rat monoclonal antibodies described above (1 μg/mL), involucrin with mouse monoclonal antibodies (ab68, Abcam; 1:200) or rabbit polyclonal antibodies (HPA055211, Atlas Antibodies; 1:25), and mCherry with rabbit polyclonal antibodies (ab167453, abcam; 3.33 μg/mL).
2.3. In vivo drug treatment
HSC‐2 cells (2 × 106) were implanted subcutaneously in the flank of athymic nude mice. The mice were then injected intraperitoneally with physiological saline, sulfasalazine (350 mg/kg per day) or cisplatin (2 mg/kg every 3 days) for 35 days. All animal experiments were performed in accordance with protocols approved by the Ethics Committee of Keio University.
2.4. Metabolome analysis
Metabolome analysis of HSC‐2 and control or GLUD siRNA‐transfected OSC19 cells was performed with the use of the C‐SCOPE package of Human Metabolome Technologies (Yamagata, Japan) and either capillary electrophoresis and time‐of‐flight mass spectrometry (CE‐TOFMS) for cation analysis or capillary electrophoresis and tandem mass spectrometry (CE‐MS/MS) for anion analysis based on methods described previously.23, 24
2.5. Measurement of oxidative consumption and extracellular acidification
Oxidative consumption (OCR) and extracellular acidification (ECAR) were determined with the use of a Seahorse XF Extracellular Flux Analyzer (Seahorse Bioscience). Cells were seeded in 24‐well plates (1.5 × 104 cells per well) and cultured overnight, after which the culture medium was replaced with extracellular flux (XF) assay medium (Seahorse Bioscience) containing 25 mM glucose and 10 mM glutamine, and the cells were cultured for 1 hour in a CO2‐free incubator before measurement of OCR and ECAR.
2.6. Statistical analysis
Data are presented as means ± SD and were analyzed with the unpaired Student's t test or log‐rank test with the use of Excel 2013 (Microsoft) or IBM SPSS statistics version 23 (IBM), respectively. A P value of <0.05 was considered statistically significant.
2.7. Data availability
Microarray data are available in the GEO database under the accession number GSE97569.
2.8. Other methods
Additional methodology is included in Appendix S1.
3. RESULTS
3.1. ASCT2‐mediated glutamine transport is essential for xCT inhibitor sensitivity in head and neck squamous cell carcinoma cells
To examine whether the CD44v‐xCT‐dependent antioxidant system is selectively activated in stemlike undifferentiated cells, we adopted an adhesion‐restricted culture system that induces cellular differentiation of HNSCC cells.18, 25 Consistent with our previous observations,18 the limited adhesion converted the undifferentiated HSC‐2 (HSC‐2‐Undiff) human HNSCC cells into the keratinocyte differentiation marker involucrin‐expressing (involucrin+) differentiated HSC‐2 (HSC‐2‐Diff) cells in vitro. (Figure 1A). Furthermore, the abundance of xCT, whose expression and activity at the cell surface are regulated by CD44v in HNSCC cells,18 was also decreased in HSC‐2‐Diff cells (Figure 1A). These results thus suggested that the CD44v‐xCT‐dependent antioxidant system is selectively activated in HSC‐2‐Undiff cells but not in HSC‐2‐Diff cells.
Figure 1.
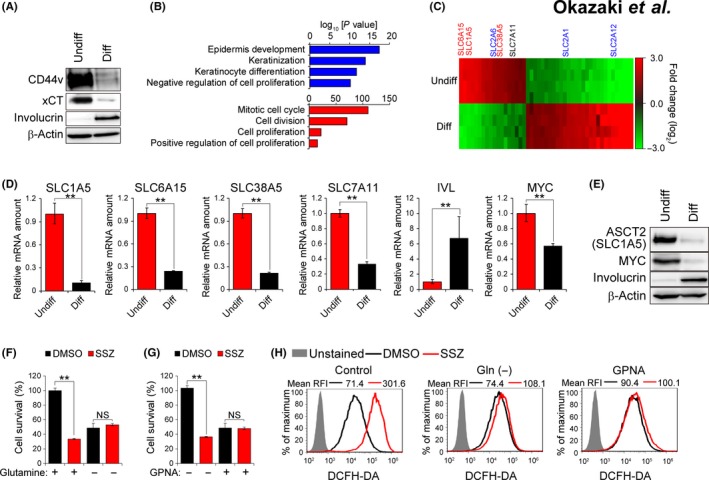
Sulfasalazine‐induced oxidative stress requires glutamine uptake mediated by ASCT2. A, Immunoblot analysis of CD44v, xCT, involucrin and β‐actin (loading control) in HSC‐2 cells cultured under normal (Undiff) or adhesion‐restricted conditions for 96 h (Diff). B, Gene ontology (GO) analysis of genes whose expression was upregulated (blue) or downregulated (red) in HSC‐2 cells cultured under the adhesion‐restricted condition. C, Heat map for SLC family genes whose expression was upregulated (red) or downregulated (green) with an absolute fold change value of >2.5 and a P value of <0.01 as revealed by microarray analysis of HSC‐2 cells cultured under normal (Undiff) or adhesion‐restricted conditions for 72 h (Diff). The gene names of glutamine transporter are shown in red, and those of glucose transporter in blue. D, Quantitative RT‐PCR analysis of SLC1A5, SLC6A15, SLC38A5, SLC7A11, involucrin (IVL) and MYC mRNA in HSC‐2 cells cultured under normal (Undiff) or adhesion‐restricted conditions for 72 h (Diff). Data were normalized by the amount of RPS17 mRNA and are means ± SD from 3 independent experiments. **P < 0.01 (Student's t test). E, Immunoblot analysis of ASCT2, MYC and involucrin in HSC‐2 cells cultured under normal (Undiff) or adhesion‐restricted conditions for 72 h (Diff). F and G, Survival of HSC‐2 cells cultured under the normal condition with sulfasalazine (400 μM) for 48 h in the absence or presence of 4 mM glutamine (F) or of 2 mM GPNA (G). Data are expressed relative to the corresponding value for cells not treated with sulfasalazine and are means ± SD from 3 independent experiments. **P < 0.01 (Student's t test). H, HSC‐2 cells cultured under the normal condition with sulfasalazine (400 μM) or DMSO vehicle for 24 h in the absence of glutamine or in the presence of GPNA (2 mM) were stained (or not) with dichloro‐dihydro‐fluorescein diacetate (DCFH‐DA) and subjected to flow cytometric analysis for measurement of intracellular reactive oxygen species. RFI, relative fluorescence intensity
To further examine the impact of cellular differentiation on the CD44v‐xCT‐dependent antioxidant system, we performed microarray analysis of HSC‐2‐Undiff cells and HSC‐2‐Diff cells (Figure S1A). Adhesion restriction increased the expression of genes related to epidermis development (GO: 0008544), keratinization (GO: 0031424), keratinocyte differentiation (GO: 0030216) and negative regulation of cell proliferation (GO: 0008285; Figure 1B), confirming that adhesion‐restricted culture effectively induced the differentiation of HSC‐2 cells in vitro. We next examined the gene expression of SLC7A11 (xCT) and found that the abundance of xCT mRNA was downregulated in association with cellular differentiation (Figure 1C,D), indicating that xCT expression is regulated at both protein and mRNA levels in HNSCC cells. Such comprehensive analysis as well as reverse transcription (RT) and real‐time PCR analysis revealed that the genes for the major glutamine transporters, including SLC1A5 (ASCT2), SLC6A15 [B(0)AT2] and SLC38A5 (SNAT5 or SN2), were selectively expressed in CD44v‐expressing HSC‐2‐Undiff cells compared with involucrin+ HSC‐2‐Diff cells (Figure 1C,D, Table S1). In contrast, expression of genes for the SLC2 facilitative GLUT transporter family, including SLC2A6 (GLUT6), SLC2A1 (GLUT1) and SLC2A12 (GLUT12), was not associated with the differentiation status of HSC‐2 cells (Figure 1C). Together, these results suggested that glutamine metabolism might be selectively activated in CD44v‐expressing undifferentiated HNSCC cells. Given that MYC was previously shown to upregulate the transcription of SLC1A5 and SLC38A5 genes,26 we examined the expression of MYC in these cells. The abundance of MYC mRNA and protein was substantially higher in HSC‐2‐Undiff cells than in HSC‐2‐Diff cells (Figure 1D,E), suggesting that MYC might play a role in the metabolic reprogramming in CD44v‐expressing undifferentiated HNSCC cells.
ASCT2 is a major cell surface transporter that mediates the uptake of neutral amino acids, including glutamine,27 and increased ASCT2 expression has been shown to be associated with disease progression and poor prognosis in HNSCC, including oral cancer.28 Immunoblot analysis revealed that the abundance of ASCT2 protein was markedly increased in HSC‐2‐Undiff cells compared with the corresponding HSC‐2‐Diff cells (Figure 1E), suggesting that cellular differentiation status affects both glutamine uptake and glutamate efflux at the level of ASCT2 and xCT expression, respectively, in HNSCC cells.
To determine whether enhanced glutamine metabolism (glutaminolysis) might confer sensitivity to xCT‐targeted therapy in CD44v‐expressing HNSCC cells, we examined the effect of extracellular glutamine depletion or ASCT2 inhibitor treatment on the induction of oxidative stress by sulfasalazine. Removal of glutamine or addition of the ASCT2 inhibitor l‐γ‐glutamyl‐p‐nitroanilide (GPNA)29 markedly attenuated the inhibitory effect of sulfasalazine on cell survival (Figure 1F,G) as well as the sulfasalazine‐induced intracellular accumulation of ROS (Figure 1H) in HSC‐2‐Undiff cells. We next examined the relevance of ASCT2‐mediated glutamine uptake to the sulfasalazine sensitivity in primary HNSCC cells. Treatment with ASCT2 inhibitor GPNA markedly reduced the anti–proliferative effect of sulfasalazine in YH cells, the HNSCC patient‐derived primary cancer cells (Figure S1B), suggesting that ASCT2‐mediated glutamine uptake enhances SSZ sensitivity in HNSCC cells. Together, these results thus indicated that the xCT‐dependent antioxidant system might counteract the oxidative stress resulting from enhanced glutaminolysis in CD44v‐expressing HNSCC cells.
3.2. ASCT2+/CD44vhigh head and neck squamous cell carcinoma tumor cells are more sensitive to sulfasalazine compared with ASCT2 tumor cells
We next examined whether the xCT inhibitor sulfasalazine might selectively impair the survival and proliferation of ASCT2‐expressing (ASCT2+) CD44vhigh tumor cells in a mouse xenograft model based on HSC‐2 cells, which also possess the ability to undergo differentiation and to give rise to differentiated‐type HNSCC tumors in vivo. Almost all HSC‐2 cells cultured under the normal condition express CD44v and ASCT2 at a high level (Figure 2A), but these cells form differentiated‐type HNSCC tumors consisting of CD44vhigh undifferentiated tumor cells and CD44vlow‐neg/involucrin+ differentiated tumor cells in vivo (Figure S2A), as we previously described.18 ASCT2+ tumor cells were detected only in the basal portion of the CD44v+ area adjacent to stromal tissue (Figure 2B and Figure S2B), suggesting that ASCT2‐promoted glutaminolysis is operative in a subpopulation of CD44v‐expressing stemlike tumor cells.
Figure 2.
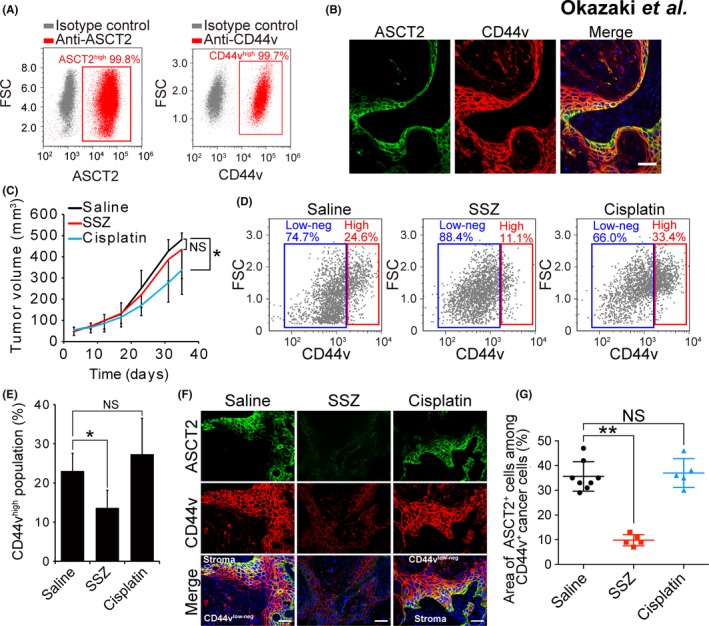
ASCT2+/CD44vhigh head and neck squamous cell carcinoma (HNSCC) cells are highly sensitive to xCT inhibition. A, Flow cytometric analysis of ASCT2 and CD44v expression in HSC‐2 cells. FSC, forward scatter. B, Immunohistofluorescence staining of ASCT2 (green) and CD44v (red) in tumors formed in athymic mice injected with HSC‐2 cells. Nuclei in the merged image were stained with 4ʹ,6‐diamidino‐2‐phenylindole (DAPI, blue). Scale bar, 50 μm. C, Volume of tumors formed by HSC‐2 cells in nude mice treated with saline (control), sulfasalazine (350 mg/kg per day) or cisplatin (2 mg/kg every 3 days). Data are means ± SD for 5 mice per group. *P < 0.05 (Student's t test); NS, not significant. D, Flow cytometric analysis of CD44v expression in lineage marker‐negative cells isolated from tumors formed by HSC‐2 cells in nude mice treated with saline, sulfasalazine or cisplatin as in (C). E, Quantification of CD44vhigh tumor cells as in (D). Data are means ± SD for 3 mice per group. *P < 0.05 (Student's t test). F, Immunohistofluorescence staining for ASCT2 (green) and CD44v (red) in tumors formed by HSC‐2 cells in nude mice treated as in (C). Nuclei in the merged images were stained with DAPI (blue). Scale bars, 50 μm. G, Quantification of the area occupied by ASCT2+ cells in the CD44v‐expressing region of tumors formed by HSC‐2 cells in nude mice as in (F). Each data point corresponds to an individual field of view, and bars indicate mean ± SD values. **P < 0.01 (Student's t test)
We then examined the effects of administration of the anticancer drug cisplatin or sulfasalazine on these heterogeneous HSC‐2 tumors. Treatment with cisplatin resulted in a significant reduction in the volume of tumors formed by HSC‐2 cells in the xenograft model, whereas that with sulfasalazine had no such effect (Figure 2C). However, flow cytometric analysis of lineage marker‐negative tumor cells revealed that the administration of sulfasalazine, but not that of cisplatin, markedly reduced the proportion of CD44vhigh undifferentiated tumor cells (Figure 2D,E), indicating that sulfasalazine was selectively cytotoxic for these cells. To examine the relevance of ASCT2 expression to sulfasalazine sensitivity in stemlike tumor cells, we performed immunohistofluorescence analysis of ASCT2 and CD44v. Sulfasalazine administration markedly reduced the relative area occupied by ASCT2+/CD44vhigh tumor cells, whereas treatment with cisplatin had no such effect (Figure 2F,G). Collectively, these results suggested that xCT‐targeted therapy selectively depleted ASCT2+/CD44vhigh undifferentiated stemlike tumor cells in HNSCC tumors.
3.3. Glutaminolysis‐related genes expression is associated with the stemlike gene signature in head and neck squamous cell carcinoma
Given that HNSCC stemlike and non‐stemlike cells are distinguished by the expression of involucrin and CD44,30 we studied RNA‐sequencing (RNA‐seq) data for HNSCC patients obtained from The Cancer Genome Atlas (TCGA) database to examine the relation between CD44v and involucrin expression and disease progression. An increased abundance of CD44v9 mRNA in tumors was significantly associated with a poor prognosis in HNSCC patients (Figure 3A). Furthermore, a CD44v9high/involucrinlow gene signature, which characterizes stemlike tumor cells, was associated with a shorter overall survival compared with a CD44v9low/involucrinhigh signature, characteristic of non‐stemlike tumor cells (Figure 3A). Expansion of CD44vhigh stemlike tumor cells thus appeared to be related to disease progression in HNSCC.
Figure 3.
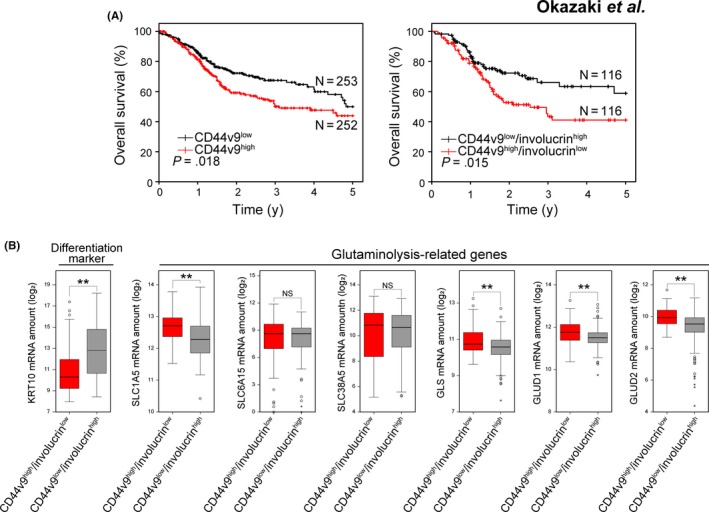
A stemlike gene signature is associated with poor patient survival and glutaminolysis‐related gene expression in head and neck squamous cell carcinoma (HNSCC). A, Kaplan‐Meier plots of overall survival for patients with HNSCC tumors characterized by CD44v9high or CD44v9low (left) or CD44v9high/involucrinlow or CD44v9low/involucrinhigh (right) gene expression signatures. Data are from the TCGA cohort. P values were determined with the log‐rank test. B, Box‐whisker plot of the expression of KRT10 (differentiation marker) and glutaminolysis‐related (SLC1A5, SLC6A15, SLC38A5, GLS, GLUD1 and GLUD2) genes on the CD44v9high/involucrinlow and CD44v9low/involucrinhigh gene signatures for head and neck squamous cell carcinoma (HNSCC) tumors in the TCGA cohort. **P < 0.01 (Student's t test); NS, not significant
To investigate whether glutaminolysis might be upregulated in tumors with a CD44v9high/involucrinlow gene signature, we next examined the relation between this signature and the expression level of genes that contribute to glutaminolysis. The expression of glutaminolysis‐related genes including those for SLC1A5 (ASCT2), GLUD1, GLUD2 and glutaminase (GLS), was significantly higher in HNSCC tumors with the CD44v9high/involucrinlow stemlike gene signature, whereas that of the genes for SLC6A15 and SLC38A5 was similar in HNSCC tumors with CD44v9high/involucrinlow or CD44v9low/involucrinhigh gene signatures (Figure 3B), suggesting that the upregulation of glutaminolysis in CD44vhigh stemlike tumor cells might be associated with the disease progression in HNSCC.
3.4. xCT inhibition not only suppresses glutathione synthesis but enhances mitochondrial metabolism in head and neck squamous cell carcinoma cells
Extracellular glutamine is imported into cells by glutamine transporters and then converted to glutamate by GLS, and the resulting glutamate is converted by GLUD to α‐KG in mitochondria.31, 32 To examine the impact of xCT inhibition on cellular metabolism in ASCT2+/CD44vhigh HNSCC cells, we treated HSC‐2 cells and OSC19 cells which manifest high expression of CD44v and ASCT2 with sulfasalazine and then subjected them to metabolome analysis. Sulfasalazine treatment significantly reduced the amounts of cysteine and GSH, whereas it increased those of tricarboxylic acid (TCA) cycle intermediates, including α‐KG, succinate, fumarate, malate and citrate, in both cell lines (Figure 4A), suggesting that inhibition of glutamate release by sulfasalazine results in the accumulation of glutamate, its conversion to α‐KG, and increased activity of the TCA cycle.
Figure 4.
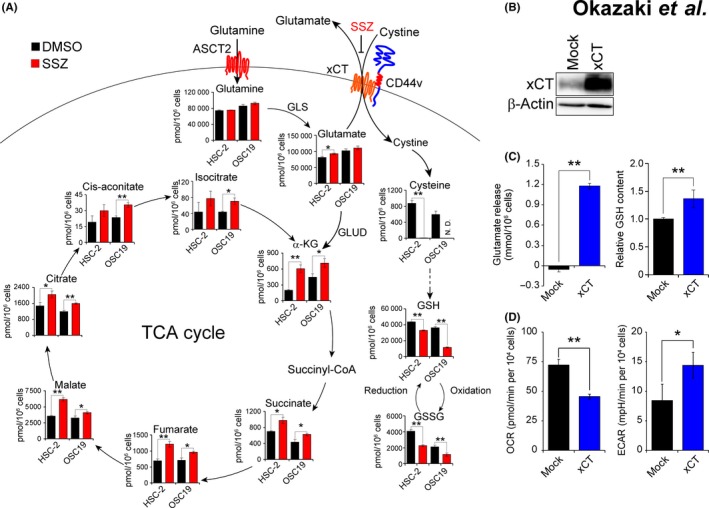
Inhibition of xCT promotes mitochondrial metabolism in CD44vhigh head and neck squamous cell carcinoma (HNSCC) cells. A, The content of metabolites related to the GSH synthesis pathway (GSSG, oxidized glutathione) or the TCA cycle was determined for HSC‐2 and OSC19 cells cultured in the presence of sulfasalazine (SSZ, 400 μM) or DMSO vehicle for 6 h. Data are means ± SD from 3 independent experiments. *P < 0.05, **P < 0.01 (Student's t test). N.D., not detected. B, Immunoblot analysis of xCT in HSC‐4 cells stably infected with a retrovirus encoding xCT or with the corresponding empty virus (Mock). C, Glutamate release over 24 h and glutathione (GSH) content for cells as in (B). Data are means ± SD from 3 independent experiments. **P < 0.01 (Student's t test). D, Oxidative consumption (OCR) and extracellular acidification (ECAR) for cells as in (B). Data are means ± SD from 3 independent experiments. *P < 0.05, **P < 0.01 (Student's t test)
To investigate the effect of enhanced xCT‐dependent glutamate release on cellular metabolism in HNSCC cells, we stably infected HSC‐4 cells (in which endogenous xCT is expressed at a low level) with a retrovirus encoding human xCT or with the corresponding empty virus (Figure 4B). Forced expression of xCT in HSC‐4 cells resulted in a significant increase in glutamate release as well as in the intracellular abundance of GSH (Figure 4C), suggesting that the exogenous xCT enhanced cystine‐glutamate antiporter activity and thereby promoted GSH synthesis in these cells. With the use of these cells, we next measured the rates of OCR and ECAR, which are indicators of mitochondrial respiration and glycolysis, respectively. Forced expression of xCT significantly reduced OCR (Figure 4D), suggesting that increased amino acid transport by xCT suppressed mitochondrial metabolism. In contrast, ECAR was significantly increased in the cells expressing exogenous xCT (Figure 4D), suggesting that the glycolytic pathway was activated in these cells in a compensatory manner. Together, these results indicated that activation of xCT‐dependent amino acid transport contributes to a shift in cellular metabolism from mitochondrial respiration toward aerobic glycolysis in HNSCC cells.
3.5. Competition between dependent glutamine uptake and glutamate dehydrogenase and xCT for glutamate in head and neck squamous cell carcinoma cells
Glutamate can be used both for xCT‐mediated cystine uptake and for GLUD‐mediated α‐KG generation in mitochondria.31 To examine the relevance of GLUD expression to xCT‐dependent glutamate transport and GSH metabolism in cancer cells, we determined the effects of GLUD knockdown in sulfasalazine‐sensitive ASCT2+/CD44vhigh OSC19 cells on the amounts of glutamine, glutamate, α‐KG, cysteine and GSH. Given that GLUD1 and GLUD2 share 97% amino acid sequence identity, we generated siRNA that target both isoforms of the enzyme (Figure 5A). Metabolome analysis revealed that GLUD knockdown resulted in a significant increase in the amount of glutamine and a significant decrease in that of α‐KG (Figure 5B, Figure S3A), suggesting that GLUD plays an essential role in glutaminolysis leading to α‐KG generation in ASCT2+/CD44vhigh HNSCC cells. It should be noted that GLUD knockdown did not affect the glutamate levels, probably because production of glutamate‐derived amino acids, including proline and citrulline, was increased (Figure S3A). Furthermore, GLUD depletion attenuated the sulfasalazine‐induced increase in the abundance of α‐KG (Figure 5B), suggesting that xCT‐dependent glutamate export negatively affects GLUD‐dependent α‐KG generation. GLUD knockdown also significantly increased cysteine, GSH and total glutathione (GSH + GSSG) levels, as well as attenuated the sulfasalazine‐induced depletion of these compounds in OSC19 cells (Figure 5C). Collectively, these results suggested that xCT‐dependent amino acid transport was activated by GLUD depletion in HNSCC cells as a result of the competition between xCT and GLUD for glutamate.
Figure 5.
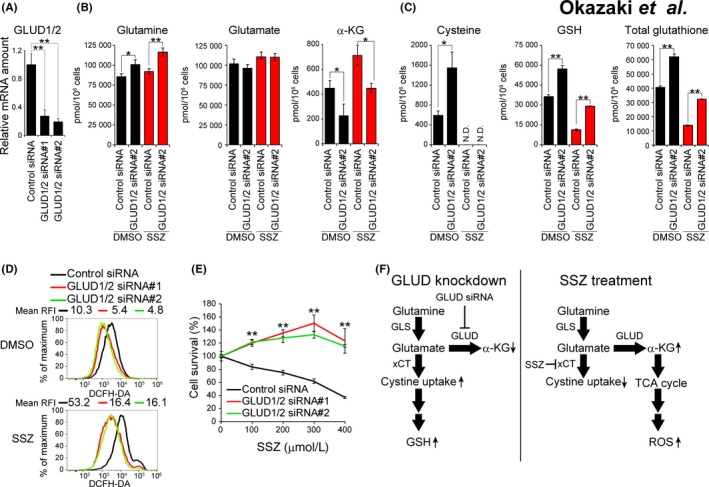
Role of α‐KG production in oxidative stress induced by xCT inhibition. A, Quantitative RT‐PCR analysis of GLUD1 and GLUD2 mRNA abundance in OSC19 cells transfected with control or GLUD1/2 (#1 or #2) siRNAs. Data were normalized by the amount of GAPDH mRNA and are means ± SD from 3 independent experiments. **P < 0.01 (Student's t test). B and C, OSC19 cells transfected with control or GLUD1/2 #2 siRNAs were cultured with sulfasalazine (400 μM) or DMSO vehicle for 6 h and then subjected to metabolome analysis for determination of the amounts of metabolites related to glutaminolysis (B) or to the glutathione (GSH) antioxidant system (C). Data are means ± SD from 3 independent experiments. *P < 0.05, **P < 0.01 (Student's t test). N.D., not detected. D, OSC19 cells transfected with control or GLUD1/2 (#1 or #2) siRNA were cultured with sulfasalazine (400 μM) or DMSO vehicle for 24 h and were then stained with DCFH‐DA for determination of ROS abundance by flow cytometry. E, OSC19 cells transfected with control or GLUD1/2 (#1 or #2) siRNA were cultured with the indicated concentrations of sulfasalazine for 48 h and then assayed for cell viability. Data are means ± SD from 3 independent experiments. **P < 0.01 vs the corresponding value for cells transfected with the control siRNA (Student's t test). F, Competition between xCT‐mediated cystine uptake and GLUD‐mediated α‐KG generation for the utilization of glutamate in HNSCC cells either depleted of GLUD (left) or treated with the xCT inhibitor sulfasalazine (right)
We then examined the relevance of GLUD expression to intracellular ROS accumulation in HNSCC cells. Knockdown of GLUD resulted in a marked reduction in the intracellular ROS level in ASCT2+/CD44vhigh OSC19 cells (Figure 5D), suggesting that GLUD‐mediated α‐KG production promotes ROS generation. Depletion of GLUD also markedly attenuated the ROS accumulation induced by sulfasalazine treatment in these cells (Figure 5D). Furthermore, GLUD knockdown prevented the negative effect of sulfasalazine on the survival of OSC19 cells and YH cells (Figure 5E, Figure S3B,C). Together, these observations suggested that xCT‐dependent glutamate release suppresses GLUD‐mediated α‐KG production and subsequent ROS generation. The competition between xCT and GLUD for glutamate utilization might, thus, be expected to sensitize ASCT2+/CD44vhigh HNSCC cells to xCT inhibitors (Figure 5F).
4. DISCUSSION
The 52 families of SLC transporters comprise 395 membrane‐bound proteins that include ion‐coupled transporters, exchangers and passive transporters and which play key roles in the movement of various substrates across biological membranes.33, 34 The expression of several types of SLC transporter has been found to be upregulated in cancer cells, with these proteins mediating the uptake of nutrients essential for tumor growth and survival.35 We have now shown that CD44vhigh undifferentiated HNSCC cells express glutamine transporters at high levels. Glutamine transporters belong to the SLC1, SLC1‐6, SLC1‐7 and SLC1‐38 families of SLC transporters,33, 36 and the expression of high‐affinity glutamine transporters such as ASCT2 and SNAT5 (SN2) in cancer cells has been shown to be regulated by oncogenic MYC.26 We also observed that the expression of MYC was markedly increased in CD44vhigh HSC‐2‐Undiff cells compared with the corresponding involucrin+ HSC‐2‐Diff cells, suggesting that the MYC‐regulated transcriptional program might play an important role in glutaminolysis in CD44v‐expressing undifferentiated HNSCC cells.
The plasma membrane proteins CD44v, ASCT2 and xCT are widely expressed in several types of human cancer, including HNSCC, with upregulation of their expression having been associated with poor prognosis.28, 37 We found that in the differentiated‐type of HNSCC tumors formed by HSC‐2 cells, ASCT2 was expressed at a high level in a subpopulation of CD44v‐expressing undifferentiated tumor cells located adjacent to stromal tissue. Thus, ASCT2 might be responsible for glutamine uptake from tumor stroma in CD44v‐expressing tumor cells. Treatment with sulfasalazine, but not that with cisplatin, markedly depleted CD44v‐expressing undifferentiated tumor cells, especially those also positive for ASCT2, in tumors formed by HSC‐2 cells in nude mice, suggesting that xCT‐targeted therapy is effective for elimination of ASCT2‐expressing stemlike HNSCC tumor cells and that the ASCT2+/CD44v9high/involucrinlow gene signature might predict the efficacy of such therapy in HNSCC.
Triple‐negative breast cancer cells that are glutamine auxotrophs were shown to be highly sensitive to xCT inhibition by sulfasalazine,38 suggesting that glutaminolysis is associated with xCT dependency. In the present study, metabolome analysis revealed that sulfasalazine treatment not only reduced the intracellular abundance of cysteine and GSH but also increased that of α‐KG in CD44vhigh HNSCC cells. In contrast, forced expression of xCT in xCTlow HSC‐4 cells markedly attenuated OCR and enhanced ECAR, suggesting that an increase in the extent of xCT‐dependent glutamate release resulted in suppression of mitochondrial respiration and activation of glycolysis. The expression of xCT may, thus, shift the metabolic phenotype of HNSCC cells toward aerobic glycolysis and thereby suppress endogenous ROS production.
Glutamate dehydrogenase catalyzes the conversion of glutamate to α‐KG and ammonia and is the primary driver of mitochondrial glutamate metabolism leading to TCA progression.39 We found that knockdown of GLUD markedly attenuated the increase in α‐KG content and the induction of oxidative stress in sulfasalazine‐treated HNSCC cells, suggesting that GLUD‐dependent α‐KG production might be related to the oxidative stress induced by sulfasalazine in CD44v‐expressing HNSCC cells. In contrast, depletion of GLUD increased the intracellular abundance of cysteine and GSH, suggesting that xCT competes with GLUD for the utilization of glutamate. High GLUD activity thus limits the glutamate availability for xCT leading to the suppression of cystine transport in stemlike HNSCC cells and thereby sensitizes them to xCT‐targeted therapy.
Our findings establish a rationale for the use of not only CD44v but glutaminolysis‐related genes including ASCT2 and GLUD as biomarkers to predict the efficacy of xCT‐targeted therapy for heterogeneous HNSCC tumors.
DISCLOSURE
The authors declare no potential conflicts of interest.
Supporting information
ACKNOWLEDGMENTS
We thank I. Ishimatsu, M. Ishikawa and S. Hayashi for technical assistance, M. Sato and M. Kobori for help with preparation of the manuscript, M. Fujiwara (Core Instrumentation Facility, Keio University School of Medicine) for assistance with microarray analysis, and S. Suzuki (Department of Physiology, Keio University School of Medicine) for assistance with flow cytometric analysis. This work was supported by a grant from the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) of the Japan Agency for Medical Research and Development (AMED) (to O.N.).
Okazaki S, Umene K, Yamasaki J, et al. Glutaminolysis‐related genes determine sensitivity to xCT‐targeted therapy in head and neck squamous cell carcinoma. Cancer Sci. 2019;110:3453–3463. 10.1111/cas.14182
Okazaki and Umene contributed equally to this work.
REFERENCES
- 1. Chen J‐Q, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim Biophys Acta. 2012;1826:370‐384. [DOI] [PubMed] [Google Scholar]
- 2. Levine AJ, Puzio‐Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340‐1344. [DOI] [PubMed] [Google Scholar]
- 3. Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic Biol Med. 2012;53:421‐436. [DOI] [PubMed] [Google Scholar]
- 4. Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS‐regulated metabolic pathway. Nature. 2013;496:101‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364‐373. [DOI] [PubMed] [Google Scholar]
- 6. Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nagano O, Okazaki S, Saya H. Redox regulation in stem‐like cancer cells by CD44 variant isoforms. Oncogene. 2013;32:5191‐5198. [DOI] [PubMed] [Google Scholar]
- 8. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016;114:1305‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(‐) and thereby promotes tumor growth. Cancer Cell. 2011;19:387‐400. [DOI] [PubMed] [Google Scholar]
- 10. Sato H, Shiiya A, Kimata M, et al. Redox imbalance in cystine/glutamate transporter‐deficient mice. J Biol Chem. 2005;280:37423‐37429. [DOI] [PubMed] [Google Scholar]
- 11. Lo M, Wang Y‐Z, Gout PW. The x(c)‐ cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008;215:593‐602. [DOI] [PubMed] [Google Scholar]
- 12. Klotz U, Maier K, Fischer C, Heinkel K. Therapeutic efficacy of sulfasalazine and its metabolites in patients with ulcerative colitis and Crohn's disease. N Engl J Med. 1980;303:1499‐1502. [DOI] [PubMed] [Google Scholar]
- 13. Chen R‐S, Song Y‐M, Zhou Z‐Y, et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin‐1/beta‐catenin pathway. Oncogene. 2009;28:599‐609. [DOI] [PubMed] [Google Scholar]
- 14. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)‐ cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633‐1640. [DOI] [PubMed] [Google Scholar]
- 15. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13:342‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoshikawa M, Tsuchihashi K, Ishimoto T, et al. xCT inhibition depletes CD44v‐expressing tumor cells that are resistant to EGFR‐targeted therapy in head and neck squamous cell carcinoma. Cancer Res. 2013;73:1855‐1866. [DOI] [PubMed] [Google Scholar]
- 19. Okazaki S, Shintani S, Hirata Y, et al. Synthetic lethality of the ALDH3A1 inhibitor dyclonine and xCT inhibitors in glutathione deficiency‐resistant cancer cells. Oncotarget. 2018;9:33832‐33843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shitara K, Doi T, Nagano O, et al. Dose‐escalation study for the targeting of CD44v+ cancer stem cells by sulfasalazine in patients with advanced gastric cancer (EPOC1205). Gastric Cancer. 2016;20:341‐349. [DOI] [PubMed] [Google Scholar]
- 21. Tsuchihashi K, Okazaki S, Ohmura M, et al. The EGF receptor promotes the malignant potential of glioma by regulating amino acid transport system xc(‐). Cancer Res. 2016;76:2954‐2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okamoto I, Kawano Y, Tsuiki H, et al. CD44 cleavage induced by a membrane‐associated metalloprotease plays a critical role in tumor cell migration. Oncogene. 1999;18:1435‐1446. [DOI] [PubMed] [Google Scholar]
- 23. Ohashi Y, Hirayama A, Ishikawa T, et al. Depiction of metabolome changes in histidine‐starved Escherichia coli by CE‐TOFMS. Mol BioSyst. 2008;4:135‐147. [DOI] [PubMed] [Google Scholar]
- 24. Ooga T, Sato H, Nagashima A, et al. Metabolomic anatomy of an animal model revealing homeostatic imbalances in dyslipidaemia. Mol BioSyst. 2011;7:1217‐1223. [DOI] [PubMed] [Google Scholar]
- 25. Connelly JT, Gautrot JE, Trappmann B, et al. Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat Cell Biol. 2010;12:711‐718. [DOI] [PubMed] [Google Scholar]
- 26. Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105:18782‐18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanai Y, Hediger MA. The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 2004;447:469‐479. [DOI] [PubMed] [Google Scholar]
- 28. Toyoda M, Kaira K, Ohshima Y, et al. Prognostic significance of amino‐acid transporter expression (LAT1, ASCT2, and xCT) in surgically resected tongue cancer. Br J Cancer. 2014;110:2506‐2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Esslinger CS, Cybulski KA, Rhoderick JF. Ngamma‐aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg Med Chem. 2005;13:1111‐1118. [DOI] [PubMed] [Google Scholar]
- 30. Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci. 2007;104:973‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hensley CT, Wasti AT, Deberardinis RJ. Review series glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678‐3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kanai Y, Clémençon B, Simonin A, et al. The SLC1 high‐affinity glutamate and neutral amino acid transporter family. Mol Aspects Med. 2013;34:108‐120. [DOI] [PubMed] [Google Scholar]
- 34. Lin L, Yee SW, Kim RB, Giacomini KM. SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov. 2015;14:543‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang Y, Sadée W. Membrane transporters and channels in chemoresistance and ‐sensitivity of tumor cells. Cancer Lett. 2006;239:168‐182. [DOI] [PubMed] [Google Scholar]
- 36. Pochini L, Scalise M, Galluccio M, Indiveri C. Membrane transporters for the special amino acid glutamine: structure/function relationships and relevance to human health. Front Chem. 2014;2:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Herold‐Mende C, Seiter S, Born AI, et al. Expression of CD44 splice variants in squamous epithelia and squamous cell carcinomas of the head and neck. J Pathol. 1996;179:66‐73. [DOI] [PubMed] [Google Scholar]
- 38. Timmerman LA, Holton T, Yuneva M, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple‐negative breast tumor therapeutic target. Cancer Cell. 2013;24:450‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frigerio F, Casimir M, Carobbio S, Maechler P. Tissue specificity of mitochondrial glutamate pathways and the control of metabolic homeostasis. Biochim Biophys Acta. 2008;1777:965‐972. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data are available in the GEO database under the accession number GSE97569.