

Abstract
Bone morphogenetic protein (BMP) signaling plays important roles in glioblastoma multiforme (GBM), a lethal form of brain tumor. BMP reduces GBM tumorigenicity through its differentiation‐ and apoptosis‐inducing effects on glioma‐initiating cells (GIC). However, some GIC do not respond to the tumor suppressive effects of BMP. Using a phosphoreceptor tyrosine kinase array, we found that EPHA6 (erythropoietin‐producing hepatocellular carcinoma receptor A6) phosphorylation was regulated by BMP‐2 signaling in some GIC. Analysis of The Cancer Genome Atlas showed that EPHA6 expression was lower in patients with GBM than in the normal brain, and that high EPHA6 expression was correlated with better prognosis. EPHA6 receptor increased the susceptibility of both sensitive and resistant GIC to BMP‐2‐induced apoptosis. The cooperative effect on apoptosis induction depended on the kinase activity of BMP type I receptor but was independent of EPHA6 kinase function. Overexpression of the EPHA6 receptor in GIC resulted in the formation of a protein complex of EPHA6 receptor and the BMP type I receptor ALK‐2, which was associated with BMP‐induced apoptosis in GIC. Intracranial injection of GIC into nude mice showed that gain‐of‐function of EPHA6 together with BMP‐2 pretreatment slowed GBM tumor progression in the mouse brain and promoted mouse survival. In summary, EPHA6 together with BMP‐2 signaling led to apoptotic cell death in GIC, and thus is a putative tumor suppressor in GBM.
Keywords: apoptosis, bone morphogenetic protein‐2, cancer stem cell, EPHA6, glioblastoma
Abbreviations
- BMP
bone morphogenetic protein
- EPHA6
erythropoietin‐producing hepatocellular carcinoma receptor A6
- GBM
glioblastoma multiforme
- GIC
glioma‐initiating cell
- GSEA
gene set enrichment analysis
- RTK
receptor tyrosine kinase
- TCGA
The Cancer Genome Atlas
1. INTRODUCTION
Glioblastoma multiforme is an aggressive form of brain tumor with a median survival of 14 months and 5‐year survival expectancy of 5%.1 GBM cells are heterogeneous and highly infiltrative. Stem‐like cells, such as GIC, remaining after surgical removal of the tumor bulk are often resistant to radiation and anticancer drugs, including temozolomide, leading to recurrence of GBM.2 GBM tumorigenesis is highly regulated by RTK. Genomic studies cataloged in TCGA identified that somatic alterations in the RTK/Ras/PI3K‐Akt pathway occur in 88% of GBM cases, including amplification and/or mutations in the EGFR gene (encoding epidermal growth factor receptor) and in the PDGFRA gene (encoding platelet‐derived growth factor receptor‐α).3, 4 Numerous therapies targeting RTK signaling have been developed and tested in clinical trials and have shown varying levels of success.5
The BMP family of growth factors has been proposed as potential non‐cytotoxic therapeutic agents for inhibiting the growth of GIC by inducing differentiation6 and sensitization to temozolomide.7 BMP signaling promotes the differentiation of GIC by BMP type I receptors and the intracellular signaling pathway.8, 9 Moreover, our previous study showed that BMP‐4 and BMP‐7 induce apoptosis by activating the BMP type I receptor ALK‐2.9 Although the BMP signaling pathway can be activated in GIC, some cells are resistant to BMP‐induced differentiation or growth inhibition.8, 10 Similarly, some GIC are refractory to BMP‐induced apoptosis. We hypothesized that resistance to BMP‐ALK‐2‐induced apoptosis in GIC is related to RTK activity given their major role in causing the resistance of cancer cells to cell death and growth inhibition.11
Erythropoietin‐producing hepatocellular carcinoma receptor A6 belongs to the Eph receptor family, which constitutes the largest family among RTK and is subdivided into EphA and EphB receptors.12 Eph receptors form large signaling clusters, which is facilitated by binding to Eph receptor‐interacting protein (ephrin) ligands on neighboring cells, thus activating both forward and reverse signaling. Eph signaling regulates cell adhesion, repulsion, differentiation, cytokinesis, cell survival, and apoptosis during development and tissue homeostasis.12, 13 Eph receptors can also signal independently of ephrin binding and kinase activity. In GBM, EPHA2 and EPHA3 were reported to ligand‐independently increase stemness, proliferation, and radiation resistance.14, 15 In a ligand‐dependent method, EPHA2 receptor is downregulated and dephosphorylated at Ser897 by ephrin‐A1‐Fc, thus forming a less invasive GBM tumor with growth inhibition.14, 16 Targeting antibodies against EPHA2 and EPHA3 also blocked oncogenic effects of EPHA2 and EPHA3, and suppressed tumorigenesis.17, 18 Additional studies implicate EPHA4,19 EPHA5,20 and EPHA721 as glioma promoters. However, little is known about the role of EPHA6 in GBM.
To evaluate whether BMP‐ALK‐2 modulates RTK activity in GIC apoptosis, we carried out a phospho‐RTK screening array. We found that tyrosine phosphorylation of EPHA6 was upregulated after BMP‐2 treatment in GIC expressing endogenous ALK‐2. EPHA6 gain‐of‐function together with BMP‐2 stimulation resulted in apoptosis in GIC that were resistant to BMP‐2‐induced cell death. Mechanistically, EPHA6 physically interacted with the ALK‐2 receptor, whereas EPHA6 kinase activity was dispensable. Our data show that the cooperation of EPHA6 with BMP‐2 signaling inhibits the tumorigenicity of GBM, which could serve as a potential therapeutic target or biomarker.
2. MATERIALS AND METHODS
2.1. Cell culture and cell viability
TGS‐01, TGS‐03, TGS‐04, and TGS‐05 cells are grade IV glioblastoma cells derived from surgically resected GBM tumors.22 The cells were maintained under neurosphere culture conditions as described previously.9, 22, 23 Briefly, the medium consisted of DMEM/F12 supplemented with Glutamax, B27 supplement, 15 μg/mL human recombinant insulin (all from Gibco, Thermo Fisher Scientific), 6 mg/mL D‐(+)‐glucose (Sigma‐Aldrich, Merck), 20 ng/mL epidermal growth factor, and 20 ng/mL basic fibroblast growth factor (both from Peprotech). Cell viability assay was evaluated using a WST‐8 kit (Nacalai Tesque).
2.2. Ligand and recombinant proteins
Recombinant human BMP‐2 was a gift from Bioventus LLC. BMP‐2 was given at 100 ng/mL in the experiments unless otherwise indicated. Recombinant human EPHA6‐Fc and IgG1‐Fc were procured from R&D Systems (Bio‐Techne).
2.3. Tyrosine phosphorylation screening
TGS‐01, TGS‐04, and TGS‐05 cells were incubated with 100 ng/mL BMP‐2 for 24 hours prior to cell lysis. Phosphorylated proteins were assessed using a human phospho‐RTK array kit (ary001b and ary003b; R&D Systems). Samples were processed according to the manufacturer's protocol.
2.4. Plasmid construction and adenovirus production
Adenovirus for LacZ (ad‐LacZ) and constitutively active (CA) kinase ALK‐2 QD (mutation at the Gly‐Ser domain, Q207D) (ad‐ALK‐2‐CA) vectors were described previously.24 Human EPHA6 (accession NP_001073917.2) was amplified by PCR. C‐terminally FLAG‐tagged full‐length human EPHA6 cDNA was cloned into an adenoviral vector (ad‐EPHA6‐WT) as previously described.9 The EPHA6 K757R mutant was constructed using a mutagenesis primer 5′‐GTTGCCATTAGAACTTTGAAA‐3′ and cloned into the adenoviral vector (ad‐EPHA6‐KD). The mutation site was based on the corresponding kinase‐dead mutant of EPHA8 K666M25 and EPHA3 K653R.26 All sequences were confirmed by Sanger sequencing analysis. For in vitro experiments, ad‐ALK‐2‐CA was infected at multiplicity of infection (MOI) 30. Ad‐EPHA6‐WT and ad‐EPHA6‐KD were infected at MOI 120 in TGS‐03 and TGS‐04 cells, and at MOI 60 in TGS‐01 cells. The corresponding MOI for ad‐LacZ was used as a control.
2.5. Immunoblotting and coimmunoprecipitation
The lysis buffer consisted of 1% Nonidet P‐40, 150 mmol/L NaCl, 20 mmol/L Tris‐HCl (pH 7.5), cOmplete EDTA‐free protease inhibitor cocktail (Roche Life Science) and phosphatase inhibitor cocktail (Nacalai Tesque). For coimmunoprecipitation, 0.5 mL cell lysate was incubated with 25 or 30 μL precoated Dynabeads protein A or M280 sheep antimouse IgG (both from Invitrogen, Thermo Fisher Scientific), respectively, overnight or for 6 hours at 4°C with gentle rotation. Dynabeads were precoated with 2 μg primary antibody per sample. Samples were washed four times with lysis buffer and eluted in SDS‐PAGE loading buffer at 95°C for 3 minutes. Immunoblotting was carried out as previously described.9
2.6. Flow cytometry
Single cell suspensions were stained with Annexin‐V‐APC and propidium iodide (eBioscience, Thermo Fischer Scientific) for 15 minutes at room temperature before detection using a Gallios (Beckman Coulter) flow cytometer. Results were analyzed with Flow Jo software (TreeStar).
2.7. Tumor orthotopic transplantation assay and in vivo bioluminescence imaging
TGS‐01 cells expressing luciferase were transduced with adenovirus to express LacZ (ad‐LacZ), EPHA6 WT (ad‐EPHA6‐WT), or EPHA6 kinase‐dead mutant (ad‐EPHA6‐KD) (MOI 60) together with pretreatment with 200 ng/mL BMP‐2 for 2 days prior to intracranial injection in 6‐week‐old female BALB/c nude mice (Sankyo Labo Service). A total of 75 000 cells in 3 μL medium was stereotactically injected over a 1‐minute period. Injection coordinates, imaging, and killing methods were conducted as previously described.9, 27 Log‐rank test was used for statistical analysis.
2.8. Phylogenetic tree of EphA kinase domain
Amino acid sequences of the kinase domains of human EPHA1‐8 were obtained from the NCBI protein database (www.ncbi.nlm.nih.gov). The sequences were aligned with Clustal Omega (www.clustal.org/omega)28 and the phylogenetic tree was illustrated using FigTree v1.4.4 (tree.bio.ed.ac.uk/software/figtree).
2.9. Patient survival and EPHA6 expression analysis
For EPHA6 expression analysis, microarray data from the REpository for Molecular BRAin Neoplasia DaTa (REMBRANDT) and TCGA were extracted from Project Betastasis (http://www.betastasis.com). Significant differences were analyzed by Tukey's honestly significant difference test corrected for multiple comparisons. Statistical analysis was carried out in R software (http://www.R-project.org) (*P < .05, **P < .01, ***P < .001).
To analyze patient datasets from TCGA Pan‐Cancer clinical data,29 Z‐scored expression values of mRNA were obtained from cBioPortal30, 31 in April 2019. Patients were divided into tertiles based on their mRNA expression levels. Differences between groups were evaluated by the log‐rank test using the R package cmprsk as described previously.9, 32
2.10. Statistical analysis
Analysis of variance followed by Tukey‐Kramer post‐hoc test using GraphPad Prism 6 (GraphPad, Inc.) was carried out to determine the P‐values and significance is shown in the figures.
2.11. Study approval
All animal studies were approved by the Animal Experiment Committee of the Graduate School of Medicine, The University of Tokyo. Use of patient‐derived glioblastoma cells was approved by the Research Ethics Committee of the Graduate School of Medicine, The University of Tokyo.
A list of inhibitors and antibodies, as well as detailed methods for RNA sequencing and analysis, are available in the Supplementary Information.
2.12. Accession numbers
Raw RNA‐seq data of patient‐derived glioma cells are available at the DDBJ Japanese Genotype‐phenotype Archive for genetic and phenotypic human data33 with accession no. JGAS00000000077.
Detailed methods are available in Doc S1.
3. RESULTS
3.1. EPHA6 is a putative GBM tumor suppressor that sensitizes GIC to BMP‐induced apoptosis
To explore the potential mechanisms of BMP‐ALK‐2‐induced apoptosis, we assessed phosphorylation of RTK affected by BMP type I receptor activity. We used three GIC cells: TGS‐01 and TGS‐04 cells expressing higher relative expression of ACVR1 (encoding ALK‐2 protein),9 and TGS‐05 cells expressing lower levels of ACVR1 (Figure S1A). These cells were then subjected to a protein array assay to detect phosphorylation changes in 49 RTK before and after BMP‐2 stimulation (Figure S1B). Phosphorylation was downregulated by BMP‐2 in the majority of the screened RTK (Table S1). EPHA6 was the only RTK in the array where phosphorylation was upregulated by BMP‐2 in both TGS‐01 and TGS‐04 cells but not in TGS‐05 cells (Figure 1A and Table S1), indicating that EPHA6 is potentially activated by BMP‐2 signaling in ALK‐2‐expressing cells.
Figure 1.
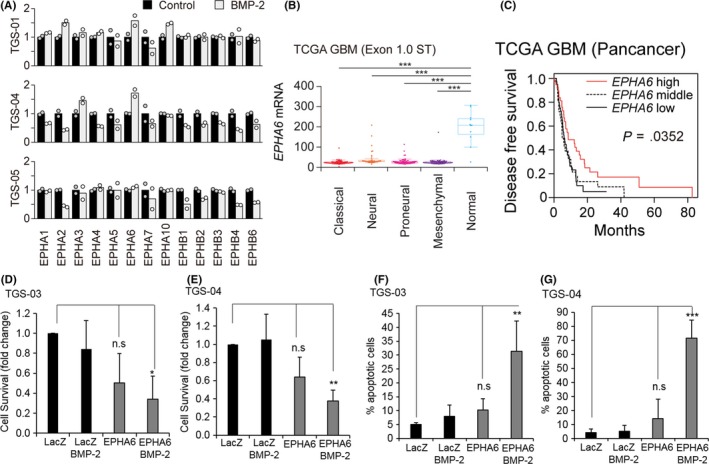
Erythropoietin‐producing hepatocellular carcinoma receptor A6 (EPHA6) is a glioblastoma tumor suppressor, which functions with bone morphogenetic protein 2 (BMP‐2) to induce apoptosis. A, EPHA and EPHB phosphorylation in patient‐derived glioma‐initiating cells (GIC) in response to BMP‐2 treatment evaluated by receptor tyrosine kinase (RTK) phospho‐array analysis. TGS‐01, TGS‐04, and TGS‐05 were untreated (control, black bars) or treated with BMP‐2 (gray bars) for 24 h prior to analysis. Phosphorylation of the control conditions was normalized to 1 in each RTK. B, EPHA6 expression in normal brain or glioblastoma tissues in The Cancer Genome Atlas (TCGA) datasets. ***P < .001. Glioblastoma was classified into four subtypes according to Verhaak et al.34 C, Kaplan‐Meier analysis of disease‐free survival of patients with glioblastoma from TCGA dataset (n = 112). Survival analysis was carried out using a log‐rank test. D, E, WST cell survival assay in TGS‐03 and TGS‐04 cells, respectively, after adenoviral transduction of LacZ control (ad‐LacZ) or wild‐type EPHA6 (ad‐EPHA6‐WT) in the presence or absence of BMP‐2 for 6 d. Data represent mean ± standard deviation (SD) of n = 3 independent experiments. *P < .05, **P < .01 and n.s. (not significant). F, G, Apoptosis assays by FACS analysis in TGS‐03 and TGS‐04, respectively. The same experimental setting as (D, E) was used, and cells were labeled with Annexin‐V and propidium iodide. Data represent mean ± SD of n = 4 (TGS‐03) and n = 3 (TGS‐04) independent experiments
We next evaluated whether EPHA6 is relevant in a larger population of glioma. Database analysis indicated that compared to the normal brain, EPHA6 mRNA expression is significantly lower in all four major GBM subtypes (classical, neural, proneural, and mesenchymal34) (Figure 1B) and in other types of glioma such as astrocytoma and oligodendroglioma (Figure S1C). Moreover, high EPHA6 expression was significantly correlated with longer disease‐free survival of patients with GBM (Figure 1C). These results indicate that EPHA6 may be a putative tumor suppressor and its activity could be modulated by BMP‐2.
As a result of low levels of EPHA6 expressed in GBM and to investigate the potential role of EPHA6 as a BMP‐2‐induced tumor suppressor, we examined the function of this gene by increasing its expression through adenoviral transduction in TGS‐03 and TGS‐04 cells. These cells responded to BMP‐induced phosphorylation of SMAD1/59 (data not shown; see Figure 2A); however, they are relatively resistant to both BMP‐2‐induced cytostasis and apoptosis (Figure S1D). BMP induced apoptosis in TGS‐04 cells at early passages,9 but, after multiple passages, the cells became resistant to cytostasis and apoptosis induction by BMP‐2. BMP‐2 stimulation or EPHA6 overexpression alone did not significantly reduce cell survival. However, EPHA6 expression together with BMP‐2 stimulation significantly decreased TGS‐03 and TGS‐04 cell survival (Figure 1D,E). Similarly, BMP‐2 treatment or EPHA6 overexpression alone induced only modest levels of apoptosis, whereas the combination of BMP‐2 treatment and EPHA6 overexpression significantly enhanced the apoptosis of TGS‐03 and TGS‐04 cells (Figures 1F,G and S1E, F). These findings suggest that EPHA6 inhibits the survival of some GIC by promoting BMP‐2‐induced apoptosis.
Figure 2.
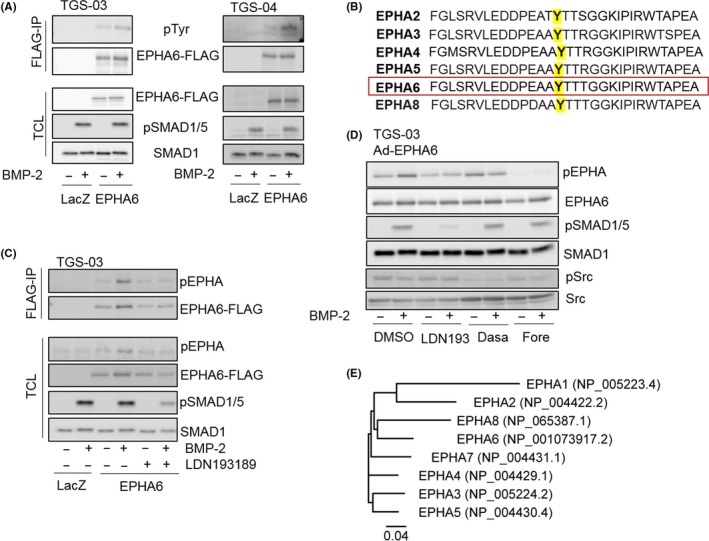
Bone morphogenetic protein 2 (BMP‐2) induces erythropoietin‐producing hepatocellular carcinoma receptor A6 (EPHA6) tyrosine phosphorylation. A, Immunoblot analysis of TGS‐03 cells (left) and TGS‐04 cells (right) infected with ad‐LacZ or wild‐type EPHA6 (ad‐EPHA6‐WT) with or without BMP‐2 stimulation for 24 h. Cell lysates were subjected to immunoprecipitation (IP) with anti‐FLAG antibody for EPHA6, followed by phospho‐tyrosine or FLAG immunoblotting. pTyr, phospho‐tyrosine; pSMAD1/5, phospho‐SMAD1/5; TCL, total cell lysate. B, Amino acid sequence in the conserved activation loop region of EPHA2, EPHA3, EPHA4, EPHA5, EPHA6, and EPHA8. The tyrosine residue recognized by phospho‐EPHA (pEPHA) antibody when phosphorylated is highlighted in yellow (Y779 for EPHA3 and Y925 for EPHA6). C, Immunoblots of TGS‐03 cells infected with ad‐LacZ or ad‐EPHA6‐WT and treated or untreated with BMP‐2 and 0.5 μmol/L LDN193189 for 24 h. Cell lysates were subjected to IP with FLAG antibody, followed by phospho‐EPHA or FLAG immunoblotting. D, Immunoblots of TGS‐03 cells infected with ad‐EPHA6‐WT, and treated with or without BMP‐2, DMSO control or 1 μmol/L inhibitors for 24 h as indicated. LDN193, LDN193189; Dasa, dasatinib; Fore, foretinib; pSrc, phospho‐Src. E, Phylogenetic tree of the kinase domain of the EPHA receptors
3.2. Bone morphogenetic protein signaling regulates EPHA6 tyrosine phosphorylation
Consistent with the phospho‐array results, EPHA6 overexpression and BMP‐2 stimulation increased tyrosine phosphorylation of EPHA6 in TGS‐03 and TGS‐04 cells (Figure 2A). To detect EPHA6 tyrosine phosphorylation, we used a phospho‐EPHA antibody that recognizes tyrosine‐779 phosphorylation of EPHA3.26 Because of sequence homology, the antibody cross‐reacts with other EPHA at the corresponding sites including EPHA6 (Y925) (Figure 2B).
BMP‐2‐induced phosphorylation of EPHA6 protein and associated SMAD1/5 phosphorylation were prevented by a BMP type I receptor kinase inhibitor LDN193189 in TGS‐03 cells, suggesting that BMP receptor activity contributes to EPHA6 phosphorylation (Figure 2C,D). Similar results were obtained using TGS‐01 GIC (Figure S1G), in which BMP‐2 induced cytostasis and apoptosis (Figure S1D). To further characterize EPHA6‐specific phosphorylation among other EPHA proteins expressed in GBM, such as EPHA2 and EPHA3,14, 15 we used small molecule inhibitors of RTK. The c‐Met inhibitor foretinib (also known as EXEL‐2880/GSK‐1363089) was reported to inhibit EPHA6 with a dissociation constant (K d) of 1.1 nmol/L, whereas dasatinib (Src and EPHA2/3 inhibitor) does not (K d = 2100 nmol/L).35 Consistently, foretinib diminished EPHA6 phosphorylation in TGS‐03 and TGS‐01 cells overexpressing EPHA6, whereas dasatinib did not (Figures 2D and S1G). These results further confirm that the phospho‐EPHA signal detected represented EPHA6 tyrosine phosphorylation rather than phosphorylation of EPHA2 or EPHA3.
Interestingly, the EPHA6 kinase domain has the closest homology to that of EPHA8 (Figure 2E). EPHA8 induces neuronal‐like differentiation by sustained MAPK activation in murine glioma‐neuroblastoma hybrid cells without the requirement for ephrin binding or kinase activity.25 Thus, we investigated whether EPHA6 kinase activity is required for BMP‐2‐induced apoptosis.
3.3. EPHA6 and BMP receptor kinase activities are differentially required for apoptosis
To assess the role of EPHA6 kinase activity in GIC apoptosis, we introduced a kinase dead (KD) mutant of EPHA6 (K757R). In both TGS‐03 and TGS‐04 cells, EPHA6 tyrosine phosphorylation was abolished when the KD mutant of EPHA6 was expressed, clearly contrasting the results for WT EPHA6 (Figure 3A). Apoptosis assays were carried out to compare EPHA6‐WT and EPHA6‐KD. Both WT and the KD mutant of EPHA6 induced apoptosis, which was enhanced in the presence of BMP‐2 treatment (Figure 3B,C). This result suggests that EPHA6 promotes apoptosis independently of its kinase activity.
Figure 3.
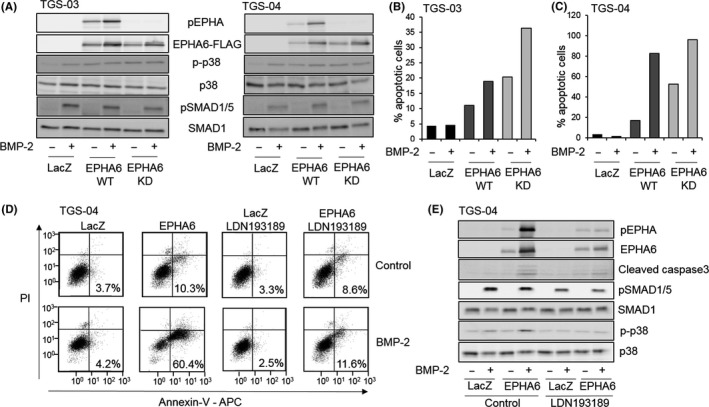
Kinase activities of bone morphogenetic protein (BMP) type I receptor and erythropoietin‐producing hepatocellular carcinoma receptor A6 (EPHA6) are differentially required in apoptosis induction. A, Immunoblot analysis of TGS‐03 and TGS‐04 cells infected with ad‐LacZ, ad‐EPHA6‐WT or adenovirus with kinase‐dead EPHA6 K757R (ad‐EPHA6‐KD) and stimulated with or without BMP‐2 for 2 d as indicated. pEPHA, phospho‐EPHA; p‐p38, phospho‐p38; pSMAD1/5, phospho‐SMAD1/5. B, C, Quantification of apoptosis of TGS‐03 (B) and TGS‐04 (C) cells infected with ad‐LacZ, ad‐EPHA6‐WT and ad‐EPHA6‐KD, stimulated with or without BMP‐2 for 5 d with FACS analysis. Graphs represent mean values from two independent experiments. D, FACS diagrams of TGS‐04 cells infected with ad‐LacZ or ad‐EPHA6‐WT with or without 1 μmol/L LDN193189 and BMP‐2 for 6 d. Percentages represent apoptotic cells. Y‐axis plots propidium iodide signal and X‐axis plots Annexin‐V signal. E, Immunoblots of TGS‐04 cells with the same experimental conditions as in (D) for 4 d
Consistent with previous results, inhibition of BMP type I receptor kinase by LDN193189 blocked apoptosis induced by the cooperation of EPHA6 overexpression and exogenous BMP‐2 (Figure 3D,E), showing the importance of BMP type I receptor kinase activity in apoptosis induction. Results of the immunoblots further support that EPHA6 and BMP‐2 upregulated apoptosis, as indicated by expression of the apoptosis marker cleaved caspase 3 with a concomitant increase in phosphorylated p38 MAPK (Figure 3E), similar to previous observations of ALK‐2‐induced apoptosis.9 Both effects were suppressed by LDN193189 (compare lane 4 to lane 8, Figure 3E).
3.4. EPHA6 physically interacts with BMP type I receptor ALK‐2
To further characterize the mechanism of EPHA6 cooperation with ALK‐2, we investigated whether these receptors interact with each other. Coimmunoprecipitation experiments by pulling down FLAG‐tagged EPHA6 or HA‐tagged ALK‐2 in TGS‐03 and TGS‐04 cells showed that overexpressed EPHA6 protein physically interacted with the ALK‐2‐CA receptor (Figure 4A,B). Furthermore, binding of EPHA6 to the ALK‐2‐CA receptor was reduced in the presence of LDN193189 (Figure 4C), implying that ALK‐2 kinase activity supports binding to EPHA6 and that this interaction is correlated with their cooperative effects in causing apoptosis.
Figure 4.
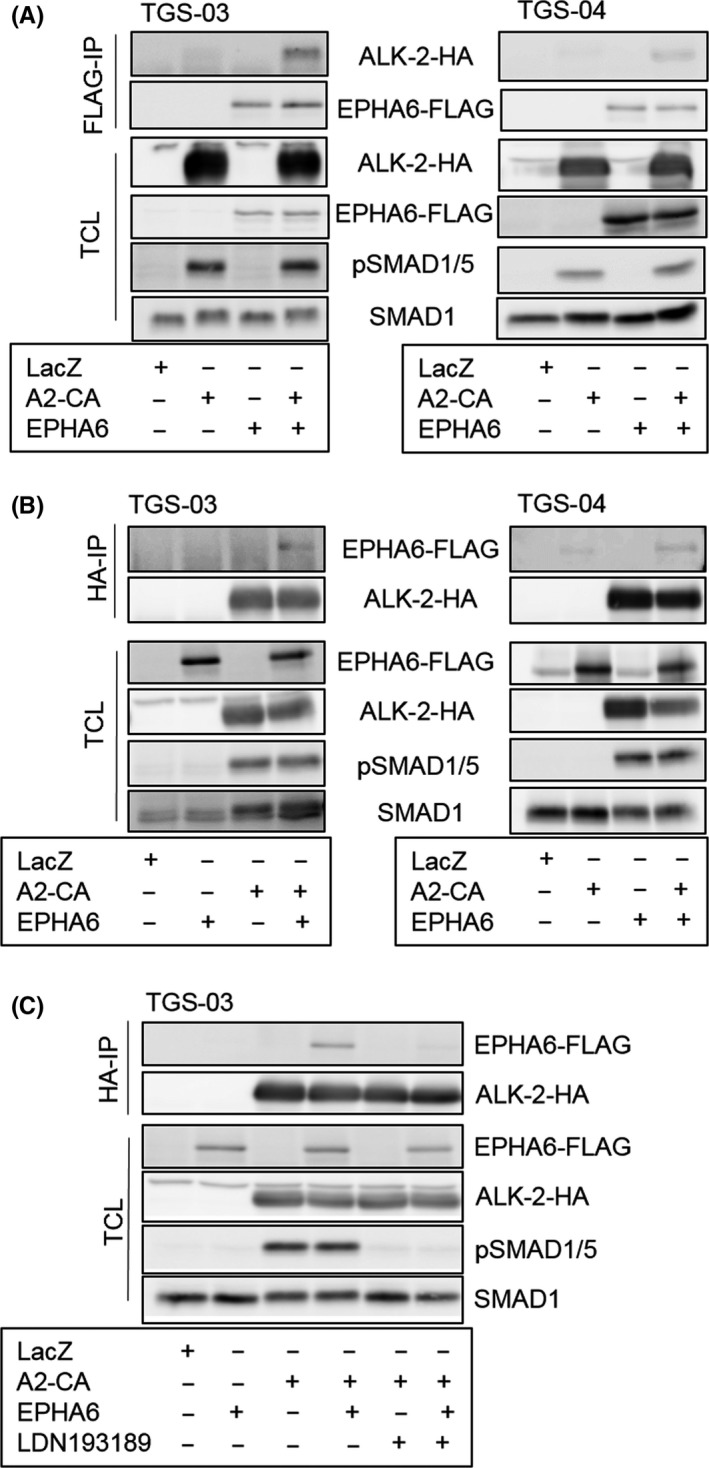
Erythropoietin‐producing hepatocellular carcinoma receptor A6 (EPHA6) physically interacts with bone morphogenetic protein (BMP) type I receptor ALK‐2. A, Immunoblots showing coimmunoprecipitation of ALK‐2 with EPHA6. TGS‐03 and TGS‐04 cells overexpressing LacZ, wild‐type EPHA6, and constitutively active ALK‐2 (A2‐CA) were immunoprecipitated (IP) with anti‐FLAG antibody 1 day after adenovirus transduction, followed by HA and FLAG immunoblotting for ALK‐2 and EPHA6, respectively. TCL, total cell lysate; pSMAD1/5, phospho‐SMAD1/5. B, Immunoblots of TGS‐03 and TGS‐04 cells after immunoprecipitation of ALK‐2‐CA protein using anti‐HA antibody and the corresponding TCL with the same experimental setting as in (A). C, Immunoblot analysis of TGS‐03 cells showing coimmunoprecipitation of EPHA6 with HA‐tagged ALK‐2‐CA treated with or without 1 μmol/L LDN193189 for 1 d. LacZ overexpression serves as a negative control for all experiments in Figure 4
3.5. EPHA6 promotes apoptosis in BMP‐2‐sensitive TGS‐01 cells
TGS‐01 cells were used because of their tumor‐forming ability in the brain in immunocompromised mice9 and sensitivity to BMP‐2‐induced apoptosis (Figure S1D). BMP‐2 signaling alone induced apoptosis in TGS‐01 cells infected with ad‐LacZ and this effect was augmented by overexpression of EPHA6‐WT or EPHA6‐KD (Figure 5A), confirming the results observed in TGS‐03 and TGS‐04 cells. Additionally, we carried out RNA‐sequencing to compare the transcriptome of TGS‐01 cells infected with ad‐LacZ control, ad‐EPHA6‐WT, or ad‐EPHA6‐KD. Gene enrichment analysis showed significant increases in apoptosis‐related genes in response to BMP‐2 treatment under the LacZ control condition (Figure 5B), which were further increased by overexpression of EPHA6‐WT and EPHA6‐KD compared to that of LacZ in the presence of BMP‐2 signaling (Figure 5C,D, respectively). In the absence of BMP‐2, the WT or KD mutant of EPHA6 alone also showed significant upregulation of apoptosis‐related genes compared to the LacZ control (Figure S2A,B, respectively).
Figure 5.
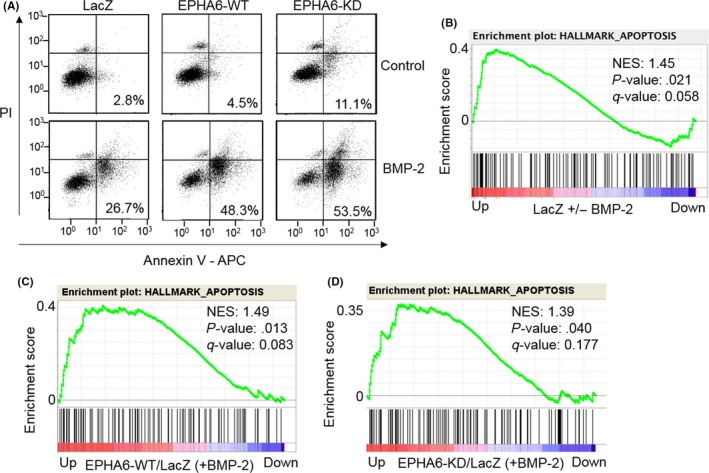
Wild‐type and kinase‐dead erythropoietin‐producing hepatocellular carcinoma receptor A6 (EPHA6) function similarly to promote bone morphogenetic protein (BMP)‐2‐induced apoptosis in TGS‐01 cells. A, FACS analysis of TGS‐01 cells infected with ad‐LacZ, ad‐EPHA6‐WT and ad‐EPHA6‐KD treated or untreated with BMP‐2 for 7 d. Percentages represent apoptotic cells. Propidium iodide (PI) signal is plotted on Y‐axis and Annexin‐V signal on X‐axis. B‐D, TGS‐01 cells were infected with ad‐LacZ, ad‐EPHA6‐WT, and ad‐EPHA6‐KD in the presence or absence of BMP‐2 for 24 h and RNA‐sequencing analysis was carried out. Genes with fragments per kilobase of exon per million mapped fragments (FPKM) values equal or higher than 3 are selected for gene set enrichment analysis. Plots show enrichment of apoptotic genes in LacZ cells treated with vs without BMP‐2 treatment (B), EPHA6‐WT vs LacZ‐expressing cells (both treated with BMP‐2) (C), and EPHA6‐KD vs LacZ‐expressing cells (both treated with BMP‐2) (D). NES, normalized enrichment score
Because EPHA6 kinase activity was not shown to be a prerequisite for apoptosis, we further examined the function of EPHA6 binding to ephrin‐A ligands by using EPHA6‐Fc consisting of the N‐terminal ephrin‐binding domain conjugated to IgG1. EPHA6 could bind to all ephrin‐A ligands (A1‐A5)36 and EPHA6‐Fc has been used to block endogenous ephrin‐A ligands in the mouse brain.37 In TGS‐01 cells, EPHA6‐Fc treatment minimally affected BMP‐2‐induced apoptosis compared to the IgG‐Fc control (Figure S2C,D). This result suggests a ligand‐independent mechanism in addition to the EPHA6 kinase‐independent mechanism for the augmented EPHA6/BMP‐2 apoptosis activity in GIC.
3.6. EPHA6 gain‐of‐function suppresses GBM growth in vivo
Tumorigenic abilities of TGS‐01 cells overexpressing LacZ control, EPHA6‐WT or EPHA6‐KD were examined by orthotopic inoculation into nude mice in the presence or absence of BMP‐2 pretreatment (Figure 6). BMP‐2 pretreatment alone did not delay tumor progression of GIC expressing LacZ; however, EPHA6‐KD mutant expression inhibited tumor growth, resulting in extended survival of mice compared to LacZ control (Figure 6A,B). This tumor‐suppressive effect was enhanced by BMP‐2 pretreatment as observed in the survival curve (Figure 6A), in agreement with the data obtained in cell culture experiments. TGS‐01 cells overexpressing EPHA6‐WT with BMP‐2 pretreatment also showed smaller tumors and longer survival of mice than the LacZ control with BMP‐2 pretreatment (Figure 6C,D). Nevertheless, TGS‐01 cells overexpressing EPHA6‐KD with BMP‐2 pretreatment exerted the strongest tumor‐suppressive effect compared to both EPHA6‐WT and LacZ control (Figure 6C,D).
Figure 6.
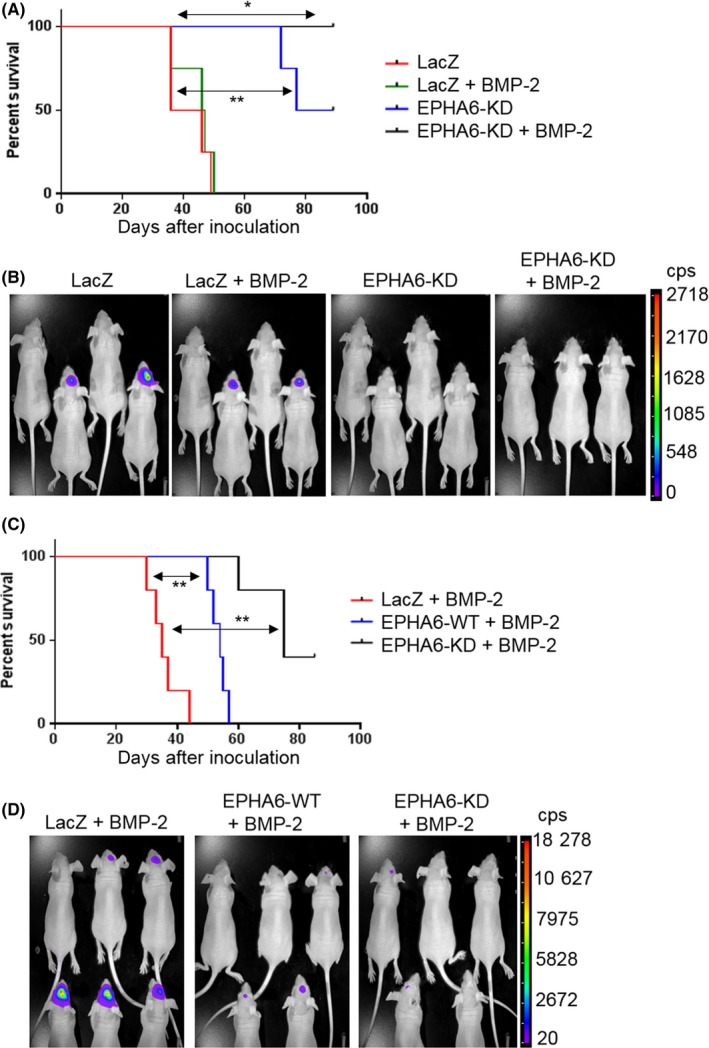
Erythropoietin‐producing hepatocellular carcinoma receptor A6 (EPHA6) gain‐of‐function impedes glioblastoma multiforme tumor growth in a mouse orthotopic model. A, Survival curve of mice inoculated with TGS‐01‐luc (luciferase) cells infected with ad‐LacZ or ad‐EPHA6‐KD and pretreated or not pretreated with 200 ng/mL BMP‐2 for 2 d. n = 4 for all groups except the EPHA6‐KD group treated with BMP‐2 (n = 3). LacZ + BMP‐2 vs EPHA6‐KD + BMP‐2 (*P < .05); LacZ vs EPHA6‐KD (**P < .01). B, Tumor formation shown by luciferase signals in the groups of mice as in experiment (A) at 3 wk‐time point. cps, count per second. C, Survival curve of mice inoculated with TGS‐01‐luc cells infected with ad‐LacZ, ad‐EPHA6‐WT, or ad‐EPHA6‐KD pretreated with 200 ng/mL BMP‐2 for 2 d. n = 5 for all groups. LacZ + BMP‐2 vs EPHA6‐WT + BMP‐2 (**P < .01); LacZ + BMP‐2 vs EPHA6‐KD + BMP‐2 (**P < .01). (D) Tumor formation shown by luciferase signals in the groups of mice as in experiment (C) at the 3‐wk time point. One mouse from LacZ + BMP‐2 group was omitted in the survival analysis (C) due to premature death during anesthesia.
4. DISCUSSION
The present study showed that EPHA6 is a pro‐apoptotic receptor that interacts with the BMP type I receptor to sensitize cells to BMP‐2‐induced cell death in apoptosis‐responsive or apoptosis‐resistant GIC (Figures 1 and 5). BMP bind to type II and type I receptors. Among the three different BMP type I receptors expressed in glioblastoma cells, we reported that only ALK‐2, but not ALK‐3 or ALK‐6, induced GIC apoptosis, and that knockdown of ALK‐2 abolished the apoptosis‐inducing activity of BMP‐4 and BMP‐7, indicating that ALK‐2 is responsible for inducing apoptosis in GIC.9 Although BMP‐2 does not directly bind to ALK‐2,38 it may activate ALK‐2 by indirectly binding through ALK‐339 or other coreceptors. BMP also bind to three different type II receptors, namely BMPRII, ActRII/ACVR2A, and ActRIIB/ACVR2B.40 Additional studies are needed to determine which type II receptors are involved in inducing cytostasis and apoptosis in human GIC.
Bone morphogenetic proteins induce differentiation, cytostasis, and apoptosis in human GIC.6, 8, 9, 10 However, some GIC do not respond to BMP signaling with regard to cytostasis8, 10 and apoptosis induction (Figures 1D‐G and S1D). Some BMP‐resistant GIC may not express BMP receptors,8 whereas other GIC may acquire resistance to BMP‐mediated cytostasis/apoptosis through alterations in their intracellular signaling pathways. The mechanism(s) of BMP‐2‐induced apoptosis resistance in TGS‐03 and TGS‐04 are unknown, although the results of this study indicate that EPHA6 overexpression could reverse this resistance. TGS‐04 cells used in the present study became resistant to cytostasis/apoptosis induction by BMP‐2 after multiple passages of the cells, suggesting that certain signaling pathways were perturbed in these cells. Interestingly, BMPRII was shown to inhibit ALK‐2‐induced cell death in multiple myeloma cells by blocking ALK‐2 oligomerization with type II activin receptors ActRII and ActRIIB, thus reducing ALK‐2 activity.41 Selective BMPRII versus ActRII/IIB binding may also play a role in inducing cytostasis and apoptosis in human GIC. EPHA6 association with ALK‐2 might alter its interaction with type II receptors, thus influencing signaling directions and sensitivity to BMP‐induced apoptosis.
Bone morphogenetic proteins transduce intracellular signals through SMAD and non‐SMAD pathways.40 For EPHA6 to function as a pro‐apoptotic factor in GIC, ALK‐2 kinase activity was required for optimum induction of cell death,9 and p38 MAPK phosphorylation was increased by both BMP‐2 and EPHA6 during apoptosis induction (Figure 3). Future study may show whether EPHA6 binding to ALK‐2 promotes p38 phosphorylation. Additionally, how BMP‐2 signaling promotes EPHA6 phosphorylation on tyrosine residue(s) (Figure 2) and whether transphosphorylation by BMP receptors is involved remain unclear. Members of the transforming growth factor beta (TGF‐β) family receptors, such as TGF‐β type I receptor ALK‐5, exert both serine/threonine kinase and tyrosine kinase activities;42, 43 however, tyrosine kinase activity has not been shown for ALK‐2. Autophosphorylation may also be facilitated by clustering of the EPHA6 receptor. In agreement with this possibility, EPHA6 was not phosphorylated at tyrosine‐925 when EPHA6‐KD was transduced into TGS‐03 and TGS‐04 cells (Figure 3A). BMP‐regulated decrease of EPHA2 protein levels in GIC10 may also lead to increased EPHA6 mRNA transcription which contributes to EPHA6 phosphorylation. Accordingly, increase in EPHA6 mRNA transcription could be observed in TGS‐04 cells (Figure S3). In the mouse heart, EphA2 deficiency was associated with higher EphA6 expression and reduced cardiomyocyte survival after infarction.44, 45 In addition to EPHA6, other RTK, including ROR2, MuSK, and TrkC, have been reported to associate with BMP receptors.46, 47, 48 ROR2 RTK and ALK‐6 form a protein complex, leading to transphosphorylation of ROR2 and activation of non‐SMAD pathways,46 whereas MuSK RTK binds to ALK‐3 or ALK‐6 and stimulates BMP‐SMAD signaling independently of its tyrosine kinase activity.47
EPHA6 kinase activity was not necessary for apoptosis induction (Figure 3), and for impeding tumor growth (Figure 6). Effects of EPHA6 on apoptosis are likely to be independent of ephrin ligand binding (Figure S2). Although many Eph receptors are linked to oncogenic functions in GBM,49 some can cause apoptosis independently of ligand binding or kinase activity. For example, EPHA4 induces cell death in the absence of ephrin‐B3 ligand in subventricular zone neuroblasts during adult neurogenesis50 and in glioblastoma.51 Furthermore, the intrinsically kinase‐inactive EPHB6 promotes breast cancer cell death by anoikis mediated by EPHA2 signaling inhibition52 and apoptosis through mitochondrial fragmentation, rendering cells susceptible to death receptor signaling.53 c‐Kit RTK, which binds to BMPRII,54 was similarly reported to induce apoptosis in the absence of its ligand and its apoptotic activity was enhanced by the loss of its kinase function.55
Collectively, our data imply that EPHA6 expression is beneficial for GBM inhibition, particularly in combination with activation of BMP‐2 signaling. How this interaction works in vivo remains an unanswered question. These results suggest that EPHA6 expression or protein levels could be used as biomarkers for identification of subsets of GBM patients who might benefit from BMP treatment. Although the levels of EPHA6 expression are lower in brain tumors than in normal brain, BMP‐2 induced the expression of EPHA6 mRNA in TGS‐04 cells (Figure S3), suggesting that certain signals can induce the expression of EPHA6 in GIC. It is also interesting to investigate whether EPHA6, BMP‐2, or ALK‐2‐CA can be delivered using oncolytic viruses for future GBM treatment.56
DISCLOSURE
Howard J. Seeherman was employed by Bioventus LLC. The other authors have no conflicts of interest to declare.
Supporting information
ACKNOWLEDGMENTS
We thank A. Moustakas for critical reading of the manuscript, S. I. Kubota for RNA sequencing support and Y. Kakiuchi for animal care. This work was supported by KAKENHI (Grant‐in‐Aid for Early Career Scientists 19K16735, E.R.) from the Japan Society for the Promotion of Science (JSPS), and KAKENHI (Grants‐in‐Aid for Scientific Research on Innovative Area) on Integrated Analysis and Regulation of Cellular Diversity (Grant number 17H06326, K.M.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT).
Raja E, Morikawa M, Nishida J, et al. Tyrosine kinase Eph receptor A6 sensitizes glioma‐initiating cells towards bone morphogenetic protein‐induced apoptosis. Cancer Sci. 2019;110:3486–3496. 10.1111/cas.14187
REFERENCES
- 1. Delgado‐López PD, Corrales‐García EM. Survival in glioblastoma: a review on the impact of treatment modalities. Clin Transl Oncol. 2016;18:1062‐1071. [DOI] [PubMed] [Google Scholar]
- 2. Fung NH, Grima CA, Widodo SS, et al. Understanding and exploiting cell signalling convergence nodes and pathway cross‐talk in malignant brain cancer. Cell Signal. 2019;57:2‐9. [DOI] [PubMed] [Google Scholar]
- 3. Cancer Genome Atlas Research Network . Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pearson JRD, Regad T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct Target Ther. 2017;2:e17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour‐initiating cells. Nature. 2006;444:761‐765. [DOI] [PubMed] [Google Scholar]
- 7. Tso JL, Yang S, Menjivar JC, et al. Bone morphogenetic protein 7 sensitizes O6‐methylguanine methyltransferase expressing‐glioblastoma stem cells to clinically relevant dose of temozolomide. Mol Cancer. 2015;14:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee J, Son MJ, Woolard K, et al. Epigenetic‐mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma‐initiating cells. Cancer Cell. 2008;13:69‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raja E, Komuro A, Tanabe R, et al. Bone morphogenetic protein signaling mediated by ALK‐2 and DLX2 regulates apoptosis in glioma‐initiating cells. Oncogene. 2017;36:4963‐4974. [DOI] [PubMed] [Google Scholar]
- 10. Darmanis S, Gallant CJ, Marinescu VD, et al. Simultaneous multiplexed measurement of RNA and proteins in single cells. Cell Rep. 2016;14:380‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu AM, Huang PH. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010;70:3857‐3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol. 2013;5:a009159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jungas T, Perchey RT, Fawal M, et al. Eph‐mediated tyrosine phosphorylation of citron kinase controls abscission. J Cell Biol. 2016;214:555‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Binda E, Visioli A, Giani F, et al. The EphA2 receptor drives self‐renewal and tumorigenicity in stem‐like tumor‐propagating cells from human glioblastomas. Cancer Cell. 2012;22:765‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Day BW, Stringer BW, Al‐Ejeh F, et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell. 2013;23:238‐248. [DOI] [PubMed] [Google Scholar]
- 16. Miao H, Li DQ, Mukherjee A, et al. EphA2 mediates ligand‐dependent inhibition and ligand‐independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferluga S, Tomé CM, Herpai DM, D'Agostino R, Debinski W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget. 2016;7:59860‐59876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qazi MA, Vora P, Venugopal C, et al. Cotargeting ephrin receptor tyrosine kinases A2 and A3 in cancer stem cells reduces growth of recurrent glioblastoma. Cancer Res. 2018;78:5023‐5037. [DOI] [PubMed] [Google Scholar]
- 19. Fukai J, Yokote H, Yamanaka R, Arao T, Nishio K, Itakura T. EphA4 promotes cell proliferation and migration through a novel EphA4‐FGFR1 signaling pathway in the human glioma U251 cell line. Mol Cancer Ther. 2008;7:2768‐2778. [DOI] [PubMed] [Google Scholar]
- 20. Almog N, Ma L, Raychowdhury R, et al. Transcriptional switch of dormant tumors to fast‐growing angiogenic phenotype. Cancer Res. 2009;69:836‐844. [DOI] [PubMed] [Google Scholar]
- 21. Wang LF, Fokas E, Juricko J, et al. Increased expression of EphA7 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. BMC Cancer. 2008;8:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF‐β signaling maintains tumorigenicity of glioma‐initiating cells through Sry‐related HMG‐box factors. Cell Stem Cell. 2009;5:504‐514. [DOI] [PubMed] [Google Scholar]
- 23. Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum‐cultured cell lines. Cancer Cell. 2006;9:391‐403. [DOI] [PubMed] [Google Scholar]
- 24. Fujii M, Takeda K, Imamura T, et al. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol Biol Cell. 1999;10:3801‐3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gu C, Shim S, Shin J, et al. The EphA8 receptor induces sustained MAP kinase activation to promote neurite outgrowth in neuronal cells. Oncogene. 2005;24:4243‐4256. [DOI] [PubMed] [Google Scholar]
- 26. Shi G, Yue G, Zhou R. EphA3 functions are regulated by collaborating phosphotyrosine residues. Cell Res. 2010;20:1263‐1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakurai T, Isogaya K, Sakai S, et al. RNA‐binding motif protein 47 inhibits Nrf2 activity to suppress tumor growth in lung adenocarcinoma. Oncogene. 2016;35:5000‐5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu J, Lichtenberg T, Hoadley KA, et al. An integrated TCGA pan‐cancer clinical data resource to drive high‐quality survival outcome analytics. Cell. 2018;173:400‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harada M, Morikawa M, Ozawa T, et al. Palbociclib enhances activin‐SMAD‐induced cytostasis in estrogen receptor‐positive breast cancer. Cancer Sci. 2019;110:209‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kodama Y, Mashima J, Kosuge T, et al. The DDBJ Japanese genotype‐phenotype archive for genetic and phenotypic human data. Nucleic Acids Res. 2015;43:18‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046‐1051. [DOI] [PubMed] [Google Scholar]
- 36. Charmsaz S, Scott AM, Boyd AW. Targeted therapies in hematological malignancies using therapeutic monoclonal antibodies against Eph family receptors. Exp Hematol. 2017;54:31‐39. [DOI] [PubMed] [Google Scholar]
- 37. Rudolph J, Zimmer G, Steinecke A, Barchmann S, Bolz J. Ephrins guide migrating cortical interneurons in the basal telencephalon. Cell Adh Migr. 2010;4:400‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seeherman HJ, Berasi SP, Brown CT, et al. A BMP/activin A chimera is superior to native BMPs and induces bone repair in nonhuman primates when delivered in a composite matrix. Sci Transl Med. 2019;11: pii:eaar4953. [DOI] [PubMed] [Google Scholar]
- 39. Lavery K, Swain P, Falb D, et al. BMP‐2/4 and BMP‐6/7 differentially utilize cell surface receptors to induce osteoblastic differentiation of human bone marrow‐derived mesenchymal stem cells. J Biol Chem. 2008;283:20948‐20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35‐51. [DOI] [PubMed] [Google Scholar]
- 41. Olsen OE, Sankar M, Elsaadi S, et al. BMPR2 inhibits activin and BMP signaling via wild‐type ALK2. J Cell Sci. 2018;131:pii: jcs213512. [DOI] [PubMed] [Google Scholar]
- 42. Derynck R, Budi EH. Specificity, versatility, and control of TGF‐β family signaling. Sci Signal. 2019;12:pii:eaav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Heldin C‐H, Moustakas A. Signaling receptors for TGF‐β family members. Cold Spring Harb Perspect Biol. 2016;8:a022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O'Neal WT, Griffin WF, Kent SD, et al. Deletion of the EphA2 receptor exacerbates myocardial injury and the progression of ischemic cardiomyopathy. Front Physiol. 2014;5:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DuSablon A, Kent S, Coburn A, Virag J. EphA2‐receptor deficiency exacerbates myocardial infarction and reduces survival in hyperglycemic mice. Cardiovasc Diabetol. 2014;13:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sammar M, Stricker S, Schwabe GC, et al. Modulation of GDF5/BRI‐b signalling through interaction with the tyrosine kinase receptor Ror2. Genes Cells. 2004;9:1227‐1238. [DOI] [PubMed] [Google Scholar]
- 47. Yilmaz A, Kattamuri C, Ozdeslik RN, et al. MuSK is a BMP co‐receptor that shapes BMP responses and calcium signaling in muscle cells. Sci Signal. 2016;9:ra87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jin W, Yun C, Kim HS, Kim SJ. TrkC binds to the bone morphogenetic protein type II receptor to suppress bone morphogenetic protein signaling. Cancer Res. 2007;67:9869‐9877. [DOI] [PubMed] [Google Scholar]
- 49. Day BW, Stringer BW, Boyd AW. Eph receptors as therapeutic targets in glioblastoma. Br J Cancer. 2014;111:1255‐1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Furne C, Ricard J, Cabrera JR, et al. EphrinB3 is an anti‐apoptotic ligand that inhibits the dependence receptor functions of EphA4 receptors during adult neurogenesis. Biochim Biophys Acta. 2009;1793:231‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Royet A, Broutier L, Coissieux MM, et al. Ephrin‐B3 supports glioblastoma growth by inhibiting apoptosis induced by the dependence receptor EphA4. Oncotarget. 2017;8:23750‐23759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Akada M, Harada K, Negishi M, Katoh H. EphB6 promotes anoikis by modulating EphA2 signaling. Cell Signal. 2014;26:2879‐2884. [DOI] [PubMed] [Google Scholar]
- 53. El Zawily AM, Toosi BM, Freywald T, et al. The intrinsically kinase‐inactive EPHB6 receptor predisposes cancer cells to DR5‐induced apoptosis by promoting mitochondrial fragmentation. Oncotarget. 2016;7:77865‐77877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hassel S, Yakymovych M, Hellman U, Rönnstrand L, Knaus P, Souchelnytskyi S. Interaction and functional cooperation between the serine/threonine kinase bone morphogenetic protein type II receptor with the tyrosine kinase stem cell factor receptor. J Cell Physiol. 2006;206:457‐467. [DOI] [PubMed] [Google Scholar]
- 55. Wang H, Boussouar A, Mazelin L, et al. The proto‐oncogene c‐Kit inhibits tumor growth by behaving as a dependence receptor. Mol Cell. 2018;72:413‐425. [DOI] [PubMed] [Google Scholar]
- 56. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials