

Abstract
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI) have been used as the first‐line treatment of non‐small cell lung cancers (NSCLC) harboring EGFR‐activating mutations, but acquired resistance is ubiquitous and needs to be solved urgently. Here, we introduce an effective approach for overcoming resistance to the EGFR‐TKI, AZD9291, in NSCLC cells using SHR‐A1403, a novel c‐mesenchymal‐epithelial transition factor (c‐Met)‐targeting antibody‐drug conjugate (ADC) consisting of an anti‐c‐Met monoclonal antibody (c‐Met mAb) conjugated to a microtubule inhibitor. Resistant cells were established by exposing HCC827 to increasing concentrations of EGFR‐TKI. c‐Met was found to be overexpressed in most resistant cells. AZD9291 resistance was partially restored by combination of AZD9291 and crizotinib only in resistant cells overexpressing phospho‐c‐Met, which synergistically inhibited c‐Met‐mediated phosphorylation of the downstream targets ERK1/2 and AKT. In resistant cells overexpressing c‐Met, neither crizotinib nor c‐Met mAb was able to overcome AZD9291 resistance. In contrast, SHR‐A1403 strongly inhibited proliferation of AZD9291‐resistant HCC827 overexpressing c‐Met, regardless of the levels of c‐Met phosphorylation. SHR‐A1403 bound to resistant cells overexpressing c‐Met was internalized into cells and released associated microtubule inhibitor, resulting in cell‐killing activity that was dependent on c‐Met expression levels only, irrespective of the involvement of c‐Met or EGFR signaling in AZD9291 resistance. Consistent with its activity in vitro, SHR‐A1403 significantly inhibited the growth of AZD9291‐resistant HCC827 tumors and caused tumor regression in vivo. Thus, our findings show that SHR‐A1403 efficiently overcomes AZD9291 resistance in cells overexpressing c‐Met, and further indicate that c‐Met expression level is a biomarker predictive of SHR‐A1403 efficacy.
Keywords: AZD9291, c‐Met ADC, c‐Met overexpression, resistance, SHR‐A1403
Abbreviations
- ADC
antibody‐drug conjugate
- c‐Met
c‐mesenchymal‐epithelial transition factor
- c‐Met mAb
c‐Met‐targeting monoclonal antibody
- EGFR
epidermal growth factor receptor
- NSCLC
non‐small cell lung cancer
- TKI
tyrosine kinase inhibitor
1. INTRODUCTION
Non‐small cell lung cancer accounts for 80%‐85% of all lung cancers, and represents the leading cause of cancer‐related deaths in men and women worldwide.1, 2 First‐generation EGFR TKI, gefitinib and erlotinib, and the second‐generation EGFR inhibitor afatinib,3 used as first‐line treatments for advanced NSCLC patients with EGFR‐activating mutations, have shown promising clinical results with superior progression‐free survival compared with platinum doublet chemotherapy regimens.4, 5 Third‐generation EGFR inhibitors, such as AZD9291, have been developed with the aim of overcoming resistance induced by the EGFR T790M mutation, which occurs in approximately half of patients on EGFR‐TKI therapy that show disease progression.6, 7 Although most EGFR mutant NSCLC initially respond to EGFR inhibitors, acquired resistance to targeted therapies inevitably occurs in most of these tumors. A number of studies have investigated mechanisms underlying EGFR‐TKI resistance, showing that acquired secondary EGFR mutations emerge in approximately 50% of EGFR‐mutated patients treated with EGFR‐TKI; activation of parallel signaling pathways, histological transformation, and activation of downstream signaling pathways as a result of acquired mutations also contribute to acquired resistance mechanisms.8, 9, 10, 11, 12, 13
In particular, accumulating evidence has shown that aberrant activation of the hepatocyte growth factor (HGF)/c‐Met pathway, caused by MET gene amplification and protein hyperactivation, is the second‐most frequent mechanism of resistance to EGFR‐TKI.14, 15, 16, 17, 18, 19 c‐Met, encoded by the MET proto‐oncogene, is the cell surface receptor for HGF, which is required for embryogenesis, cell proliferation, survival, and motility.14, 20, 21 To date, inhibitors of HGF/c‐Met signaling have been developed as monotherapies or combination therapies with EGFR‐TKI for the treatment of NSCLC (eg cabozantinib, Exelixis;22 crizotinib, Pfizer;23 tivantinib, ArQule;24 onartuzumab, Roche; rilotumumab; Amgen;24, 25, 26 ABBV‐399, AbbVie27). In lung cancer cells with c‐Met pathway‐induced resistance to EGFR inhibitors, combination of a c‐Met inhibitor and EGFR inhibitor has been shown to efficiently overcome such resistance.28, 29
In the present study, we established a novel strategy for overcoming AZD9291 resistance in HCC827 NSCLC cells using SHR‐A1403, a novel ADC consisting of a c‐Met mAb conjugated to a microtubule inhibitor.30, 31 Unlike the c‐Met inhibitor crizotinib, which only overcame AZD9291 resistance caused by high levels of phospho‐c‐Met, SHR‐A1403 more effectively inhibited the proliferation of AZD9291‐resistant, c‐Met‐overexpressing HCC827 cells, an effect that was dependent on c‐Met expression levels only, irrespective of the involvement of c‐Met or EGFR signaling in AZD9291 resistance. Our findings show that the c‐Met‐targeting ADC, SHR‐A1403, in contrast to a small‐molecule c‐Met inhibitor or c‐Met mAb alone, efficiently overcomes AZD9291 resistance in cells overexpressing c‐Met, and further indicate that c‐Met expression level is a biomarker predictive of SHR‐A1403 efficacy.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
AZD9291, gefitinib, afatinib, and crizotinib were purchased from Selleckchem. SHR‐A1403, the naked anti‐c‐Met monoclonal antibody c‐Met mAb and free toxin SHR152852, were provided by Jiangsu Hengrui Medicine Co. Ltd.31 DyLight 488 N‐hydroxysuccinimide (NHS) ester was purchased from Thermo‐Fisher Scientific. Sulforhodamine B was purchased from Sigma‐Aldrich.
Antibodies against EGFR, phospho‐EGFR (Tyr1173), c‐Met, phospho‐c‐Met (Tyr1234/1235), STAT3, phospho‐STAT3 (Tyr705), AKT, phospho‐AKT (Ser473), ERK1/2, phospho‐ERK1/2 (Thr202/Tyr204), and GAPDH were purchased from Cell Signaling. β‐Tubulin antibody was purchased from Sigma‐Aldrich.
2.2. Cell culture and treatment
HCC827 and PC‐9 cells were obtained from the cell bank of the Chinese Academy of Sciences. Cells with acquired resistance were established by exposing parental cells to increasing concentrations of gefitinib or afatinib (10 nmol/L to 5 μmol/L) for 12 months and selecting clones using the limiting dilution method. Four clones with c‐Met overexpression were isolated from the resultant gefitinib‐resistant HCC827 cell line (HG), two clones with different c‐Met levels were isolated from the resultant afatinib‐resistant HCC827 cell line (HA), and two clones with different c‐Met levels were isolated from the resultant afatinib‐resistant PC‐9 cell line (PA). c‐Met‐overexpression is defined as more than two fold c‐Met protein expression over parental HCC827 cells. Cells were cultured in RPMI‐1640 medium supplemented with 10% (vol/vol) FBS at 37°C in a humidified 5% CO2 atmosphere.
2.3. Cell proliferation assay
Cell growth inhibition was determined using a sulforhodamine B assay, as described previously.32 Briefly, approximately 24 hours after plating, cells in culture medium containing 10% FBS were incubated with different concentrations of drugs, alone or in combination as indicated, for 72 hours. At least three independent experiments were carried out, and the results are presented as mean ± SD.
2.4. Western blotting
After drug treatment, cells were washed twice with cold PBS (137 mmol/L NaCl, 2.7 mmol/L KCl, 10 mmol/L Na2HPO4, and 1.8 mmol/L KH2PO4, pH 7.4), lysed in SDS sample buffer, and boiled for 10 minutes. Cell lysates containing equal amounts of protein were separated by SDS‐PAGE and transferred to PVDF membranes (Millipore). After blocking in 5% nonfat milk in TBST (Tris‐buffered saline containing 0.1% Tween‐20, pH 7.6), membranes were incubated with the indicated primary antibodies at 4°C overnight and then exposed to appropriate secondary antibodies for 2 hours at room temperature. Immunoreactive proteins were visualized using the ECL system from Pierce Chemical.
2.5. Polymeric tubulin fraction assay
Drug‐treated cells were extracted by incubating for 3 minutes at 30°C in lysis buffer (80 mmol/L MES KOH, pH 6.8, 1 mmol/L MgC12, 1 mmol/L EGTA, 0.5% Triton X‐100, and 10% glycerol) containing protease inhibitors. Supernatants containing detergent‐soluble tubulin and detergent‐insoluble polymerized cytoskeletons were extracted with SDS sample buffer (50 mmol/L Tris‐HCl, pH 6.8, 100 mmol/L DTT, 2% SDS, 0.1% bromophenol blue, and 10% glycerol), and β‐tubulin expression was analyzed by western blotting.
2.6. Internalization assay
Cells were incubated with DyLight 488 NHS‐ester‐labeled SHR‐A1403 (1 μg/mL) in cold PBS on ice for 1 hour. After washing three times with PBS, cells were incubated in growth medium at 37°C for 0.5, 1, 1.5 and 2 hours, then incubated on ice for 15 minutes with stripping buffer consisting of 0.05 mol/L glycine (pH 2.45) and 0.1 mol/L NaCl. Internalized fluorescence was analyzed immediately on an ACCURI C6 PLUS instrument (BD Biosciences).
SHR‐A1403 internalization was also analyzed using fluorescence microscopy. Cells were incubated with DyLight 488 NHS‐ester‐labeled SHR‐A1403 (1 μg/mL) for 24 hours at 37°C. The lysosome fluorescent probe Lyso‐Tracker Red (50 nmol/L) was added 1 hour prior to fixation. Alternatively, cells were incubated with Lyso‐Tracker Red (50 nmol/L) for 1 hour at 37°C, then incubated with DyLight 488 NHS‐ester‐labeled SHR‐A1403 (1 μg/mL) for 10 minutes at 4°C. Cells were fixed with 4% paraformaldehyde for 15 minutes and imaged with an Olympus FV1000 confocal microscope.
2.7. In vivo study
Female BALB/cA‐nude mice (5‐6 weeks old) were purchased from Shanghai SLAC Laboratory Animal Co. Human tumor xenografts were established by s.c. inoculation of nude mice with HCC827, HA1 or HG3 cells. Tumor‐bearing mice were randomized into groups and treated with vehicle, AZD9291 intragastric administration (i.g.) or SHR‐A1403 intravenous injection (i.v.) when average tumor volume reached approximately 100‐200 mm3. Tumor volume was calculated as (length × width2)/2, and body weight was monitored as an indicator of general health. All animal experiments were carried out in accordance with guidelines of the Institutional Animal Care and Use Committee of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences (Shanghai, China).
2.8. Data analysis
Data were analyzed using GraphPad Prism Version 5 software (GraphPad Software, Inc.). Non‐linear regression analyses were carried out to generate dose‐response curves and to calculate IC50 values. Results of repeated experiments are presented as means ± SD. A two‐tailed Student's t test was used to test for significance where indicated. Differences were considered significant at a P‐value < .05.
3. RESULTS
3.1. Establishment of EGFR inhibitor‐resistant NSCLC cells
HCC827 cells were grown initially in medium containing 10 nmol/L gefitinib or afatinib, and the concentration was gradually increased to 5 μmol/L over the subsequent 12 months. Thereafter, monoclonal cell lines were selected, including four gefitinib‐resistant monoclonal cell lines (HG1, HG2, HG3, HG4) and two afatinib‐resistant monoclonal cell lines (HA1, HA2). As shown in Table 1, all established gefitinib‐ and afatinib‐resistant cell clones, with resistance ratios ranging from 43.6 to 2376.4 for gefitinib, and from 25.6 to 2396.4 for afatinib, were also strongly resistant to AZD9291, with resistance ratios ranging from 48.0 to 1924.0 (Table 1).
Table 1.
IC50 values for EGFR inhibitors (gefitinib, afatinib and AZD9291) against the proliferation of parental and resistant HCC827 cells
| Cell line | IC50 (nmol/L, mean ± SD)a | ||
|---|---|---|---|
| Gefitinib | Afatinib | AZD9291 | |
| HCC827 | 3.5 ± 0.5 | 0.7 ± 0.0 | 2.6 ± 0.3 |
| HA1 | 1372.0 ± 50.9 (292.0)b | 1677.5 ± 108.2 (2396.4)b | 5002.5 ± 509.8 (1924.0)b |
| HA2 | 3693.5 ± 470.2 (1055.3)b | 1093.5 ± 48.8 (1562.1)b | 1086.5 ± 81.3 (417.9)b |
| HG1 | 8317.5 ± 1658.2 (2376.4)b | 1595.5 ± 112.4 (2279.3)b | 119.4 ± 25.9 (45.9)b |
| HG2 | 4032.5 ± 1928.3 (1152.1)b | 392.2 ± 11.7 (560.3)b | 1111.5 ± 26.2 (427.5)b |
| HG3 | 152.7 ± 11.8 (43.6)b | 17.9 ± 5.4 (25.6)b | 124.8 ± 6.3 (48.0)b |
| HG4 | 1215.5 ± 120.9 (347.3)b | 295.1 ± 123.2 (421.6)b | 511.9 ± 2.8 (196.9)b |
Abbreviation: EGFR, epidermal growth factor receptor.
IC50 were determined using sulforhodamine B assays by treating cells with different concentrations of drugs for 72 h. Data are presented as means ± SD of three independent experiments.
Resistance ratio = IC50 (resistant cells)/IC50 (HCC827).
To investigate the mechanism of resistance to these EGFR inhibitors, particularly AZD9291, we compared the levels of EGFR and its downstream signaling pathway proteins, ERK1/2, AKT and STAT3 (signal transducer and activator of transcription 3), with or without AZD9291 treatment, in resistant cells with those in parental HCC827 cells (Figure 1). Results showed that upon AZD9291 treatment, phospho‐ERK1/2 and phospho‐AKT, which were inhibited in HCC827 cells, were not sensitive to AZD9291 in TKI‐resistant HA1 and HA2 cells (Figure 1B), raising the possibility that the failure of AZD9291 to inhibit ERK1/2 and AKT may be the mechanism underlying AZD9291 resistance.
Figure 1.
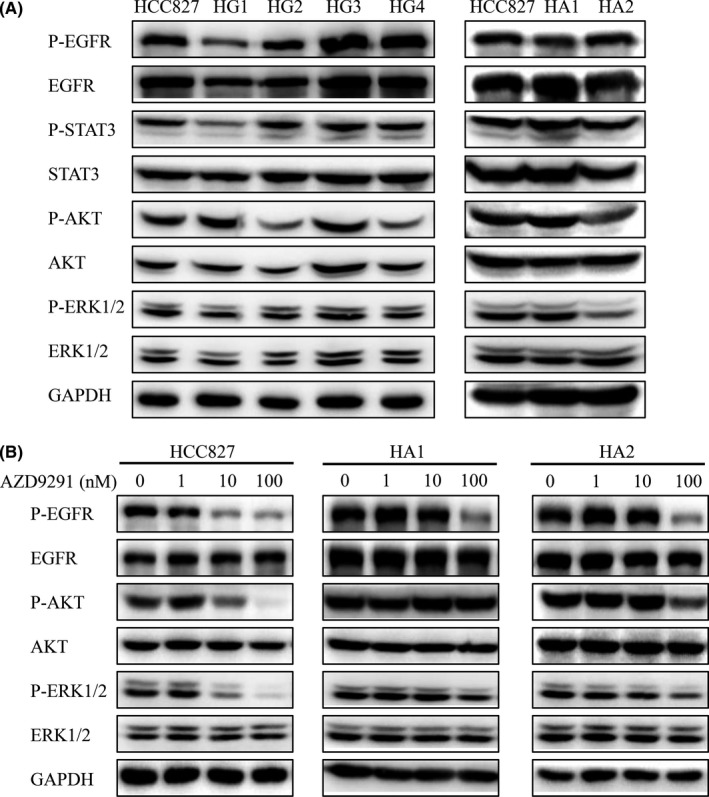
AZD9291 fails to inhibit phosphorylation of the epidermal growth factor receptor (EGFR) downstream AKT and ERK1/2, in EGFR TKI‐resistant HCC827 cells. A, Parental and AZD9291‐resistant HCC827 cells were lysed, and phosphorylation of the EGFR downstream targets AKT and ERK1/2 was detected by western blotting. B, HCC827, HA1, and HA2 cells were treated with AZD9291 for 3 h, and whole‐cell lysates were analyzed by western blotting. Data are representative of three separate experiments
3.2. c‐Met inhibitor crizotinib and c‐Met mAb alone are unable to overcome AZD9291 resistance in cells overexpressing c‐Met
Inability of AZD9291 to inhibit EGFR downstream signals may reflect a “switch” in the kinase dependence of ERK1/2 and AKT activation to other upstream tyrosine kinases, such as c‐Met. Thus, we first determined whether c‐Met was responsible for phosphorylation/activation of PI3K/AKT and ERK1/2. We found that the levels of c‐Met in TKI‐resistant HG1, HG2, HG3, HG4 and HA1 cells, but not HA2 cells, were significantly elevated compared with HCC827 cells (Figure 2A). Phospho‐c‐Met was also greatly increased in HA1 cells, but not other resistant cells, compared with parental HCC827 cells (Figure 2A). To investigate the relationship between downstream ERK1/2 and AKT activation and upstream c‐Met and EGFR, we treated cells with the c‐Met inhibitor crizotinib and c‐Met mAb, alone or in combination with AZD9291. As shown in Figure 2B, crizotinib or c‐Met mAb alone significantly inhibited c‐Met phosphorylation in HCC827 cells, but did not affect AZD9291‐induced ERK1/2 or AKT phosphorylation or inhibition of downstream ERK1/2 and AKT phosphorylation, indicating that ERK1/2 and AKT mainly depend on EGFR, and not on c‐Met, in HCC827 cells. In HA1 cells, containing both high levels of c‐Met and phospho‐c‐Met, crizotinib and c‐Met mAb alone, but not AZD9291, significantly inhibited phosphorylation of c‐Met and its downstream targets ERK1/2 and AKT (Figure 2B), suggesting that, unlike the case in HCC827 cells, ERK1/2 and AKT activation mainly depends on c‐Met in HA1 cells. In addition, in HG1 cells containing high levels of c‐Met, but not phospho‐c‐Met, crizotinib and c‐Met mAb failed to inhibit ERK1/2 and AKT phosphorylation, as they did in HCC827 cells (Figure 2B).
Figure 2.
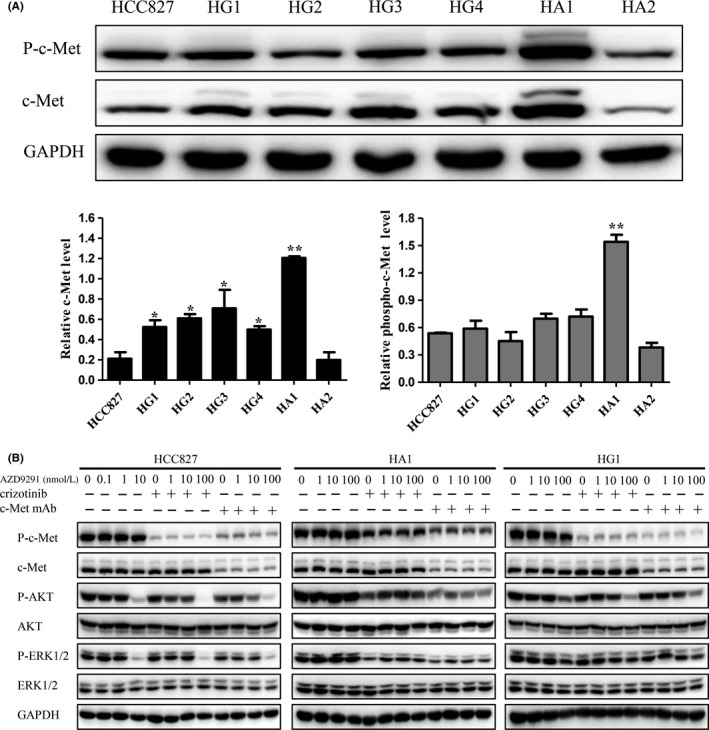
Both the c‐mesenchymal‐epithelial transition factor (c‐Met) inhibitor crizotinib and the c‐Met‐targeting monoclonal antibody (c‐Met mAb) inhibit phosphorylation of the c‐Met downstream targets, AKT and ERK1/2, only in resistant HCC827 cells with elevated phospho‐c‐Met levels. A, Parental and AZD9291‐resistant HCC827 cells were lysed and immunoblotted with anti‐c‐Met and anti‐phospho‐c‐Met antibodies. Relative protein levels of c‐Met and phospho‐c‐Met were normalized to endogenous GAPDH protein for each sample. Data are expressed as mean ± SD of three individual experiments. *P < .05, **P < .01 vs parental HCC827 cells. B, HCC827, HA1, and HG1 cells were treated with AZD9291 plus 100 nmol/L crizotinib or 1 μg/mL c‐Met mAb for 6 h. Whole‐cell lysates were analyzed by western blotting using the indicated antibodies
We next examined the combined effect of crizotinib and c‐Met mAb with AZD9291. Consistent with our finding that ERK1/2 and AKT depended on c‐Met only in HA1 cells containing high levels of phospho‐c‐Met, crizotinib significantly restored sensitivity to AZD9291 only in HA1 cells, and not in other resistant cells containing high levels of c‐Met only (Table 2). Unlike crizotinib, c‐Met mAb did not restore the sensitivity of any resistant cells to AZD9291 (Table 2).
Table 2.
IC50 values for AZD9291, with or without crizotinib or c‐Met mAb, against the proliferation of parental and resistant HCC827 cells
| Cell line | IC50 (mean ± SD)a | ||||
|---|---|---|---|---|---|
| AZD9291 (nmol/L) | Crizotinib (nmol/L) | AZD9291 (nmol/L, + 100 nmol/L crizotinib) | c‐Met mAb (ng/mL) | AZD9291 (nmol/L, + 1 μg/mL c‐Met mAb) | |
| HCC827 | 2.6 ± 0.3 | 3853.0 ± 858.4 | 4.0 ± 1.3 | >30 000.0 | 2.3 ± 0.2 |
| HA1 | 5002.5 ± 509.8 (1924.0)b | 2929.5 ± 105.4 | 111.6 ± 25.8 (27.9)b | >30 000.0 | 2753.5 ± 279.3 (1197.2)b |
| HA2 | 1086.5 ± 81.3 (417.9)b | 3864.0 ± 722.7 | 291.1 ± 86.5 (72.8)b | >30 000.0 | 1163.5 ± 3.5 (505.9)b |
| HG1 | 119.4 ± 25.9 (45.9)b | 2056.5 ± 99.7 | 130.0 ± 50.4 (32.5)b | >30 000.0 | 148.9 ± 19.1 (64.7)b |
| HG2 | 1111.5 ± 26.2 (427.5)b | 3740.0 ± 196.6 | 877.0 ± 138.8 (219.3)b | >30 000.0 | 2059.5 ± 392.4 (895.4)b |
| HG3 | 124.8 ± 6.3 (48.0)b | 2134.0 ± 567.1 | 146.0 ± 22.4 (36.5)b | >30 000.0 | 488.8 ± 71.4 (212.5)b |
| HG4 | 511.9 ± 2.8 (196.9)b | 3217.5 ± 54.5 | 538.8 ± 2.2 (134.7)b | >30 000.0 | 1639.5 ± 150.6 (712.8)b |
Abbreviation: c‐Met mAb, c‐mesenchymal‐epithelial transition factor‐targeting monoclonal antibody.
IC50 were determined using sulforhodamine B assays by treating cells with different concentrations of drugs for 72 h. Data are presented as means ± SD of three independent experiments.
Resistance ratio = IC50 (resistant cells)/IC50 (HCC827).
3.3. SHR‐A1403 overcomes AZD9291 resistance in cells overexpressing c‐Met
Given that the c‐Met inhibitor crizotinib and c‐Met mAb could not restore the sensitivity of c‐Met‐overexpressing resistant cells to AZD9291, we were next interested in examining the ability of the ADC, SHR‐A1403, to overcome AZD9291 resistance. In contrast to crizotinib or c‐Met mAb, which mainly exerted their antitumor activity through inhibition of the c‐Met signaling pathway, SHR‐A1403, consisting of c‐Met mAb and the microtubule‐disrupting agent SHR152852, inhibited the growth of tumor cells not only by inhibiting the c‐Met signaling pathway through induction of c‐Met degradation, but also through microtubule disruption induced by released SHR152852. As shown in Table 3, the inhibitory effect of SHR‐A1403 on the proliferation of c‐Met‐overexpressing resistant cells (HG1, HG2, HG3, HG4, HA1) was much greater than that on HCC827 cells, with sensitivity ratios ranging from 9.2 to 77.9 (Table 3). More importantly, AZD9291‐resistant HA2 cells expressing a low level of c‐Met as compared to parental HCC827 cells were resistant to SHR‐A1403 (Figure 2A, Table 3). These data clearly indicate that SHR‐A1403 is capable of overcoming AZD9291 resistance and this effect is dependent on the expression level of c‐Met. The effect of SHR‐A1403 was confirmed in AZD9291‐resistant PA7 and PA9 cells which were established from PC‐9 cells, another NSCLC cell line, by drug induction. Levels of c‐Met in AZD9291‐resistant PA7 and PA9 cells were significantly elevated compared with PC‐9 cells, but phospho‐c‐Met was greatly increased in PA9 cells only (Figure S1). As expected, SHR‐A1403 effectively overcame AZD9291 resistance in both PA7 and PA9 cells, which was consistent with the results obtained from AZD9291‐resistant HCC827 cells (Table S1).
Table 3.
IC50 values for SHR‐A1403 and SHR152852 against the proliferation of parental and resistant HCC827 cells
| Cell line | IC50 (mean ± SD)a | |
|---|---|---|
| SHR‐A1403 (ng/mL) | SHR152852 (nmol/L) | |
| HCC827 | 918.9 ± 9.6 | 1.6 ± 0.1 |
| HA1 | 11.8 ± 2.9 (77.9)a | 4.9 ± 0.7 (3.1)b |
| HA2 | >30 000.0 | 3.3 ± 1.2 (2.1)b |
| HG1 | 26.6 ± 2.5 (34.5)a | 4.0 ± 2.1 (2.5)b |
| HG2 | 17.8 ± 1.6 (51.6)a | 5.3 ± 0.8 (3.3)b |
| HG3 | 99.4 ± 16.8 (9.2)a | 8.5 ± 3.2 (5.3)b |
| HG4 | 130.8 ± 5.3 (7.0)a | 10.7 ± 0.0 (6.7)b |
IC50 were determined using sulforhodamine B assays by treating cells with different concentrations of drugs for 72 h. Data are presented as means ± SD of three independent experiments.
Sensitivity ratio = IC50 (HCC827)/IC50 (resistant cells).
Resistance ratio = IC50 (resistant cells)/IC50 (HCC827).
We next explored the mechanism underlying the sensitivity of resistant cells to SHR‐A1403. As SHR‐A1403 consists of c‐Met mAb conjugated with the microtubule‐disrupting agent SHR152852, we first tested whether resistant cells were sensitive to c‐Met mAb or SHR152852 alone. As shown in Table 2, resistant cells were not sensitive to c‐Met mAb compared with HCC827 cells, and there were no differences in c‐Met signaling between SHR‐A1403‐treated HCC827 and resistant cells (Figures 3A and S2). These observations suggest that c‐Met mAb activity is not responsible for the sensitivity of resistant cells to SHR‐A1403. Moreover, as shown in Table 3, all resistant cells were insensitive to SHR152852 compared with HCC827 cells. Taken together, these results suggest that the sensitivity of resistant cells to SHR‐A1403 is attributable to the connectedness of c‐Met mAb and SHR152852.
Figure 3.
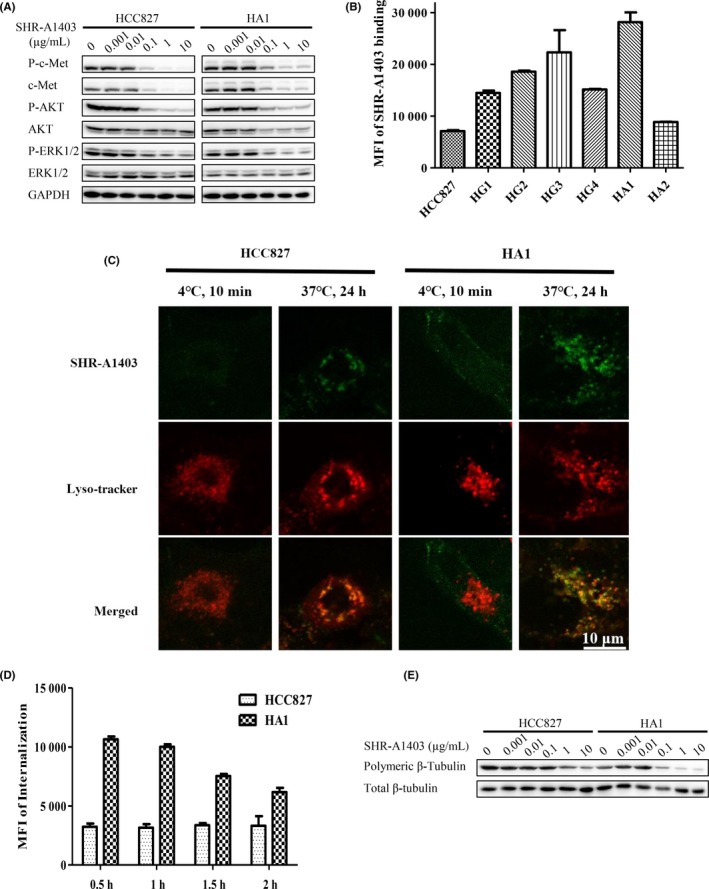
Antitumor activity of SHR‐A1403 is dependent on c‐mesenchymal‐epithelial transition factor (c‐Met) expression levels. A, HCC827 and HA1 cells were treated with different concentrations of SHR‐A1403 for 24 h, and whole‐cell lysates were analyzed by western blotting. B, Binding affinity of DyLight 488 NHS‐ester‐labeled SHR‐A1403 to parental and resistant HCC827 cells was measured by flow cytometry. Representative results are shown. C, Colocalization of SHR‐A1403 (green) with lysosomes (red). Parental and resistant HCC827 cells were incubated with DyLight 488 NHS‐ester‐labeled SHR‐A1403 (1 μg/mL), and lysosomes were labeled with Lyso‐Tracker Red. Samples were analyzed under confocal microscopy. D, Internalization of SHR‐A1403 in HCC827 and HA1 cells. Cells were incubated with DyLight 488 NHS‐ester‐labeled SHR‐A1403 (1 μg/mL) as described in Materials and Methods, and internalized fluorescence was analyzed immediately by flow cytometry. Representative images are shown. E, Effects of SHR‐A1403 on tubulin polymerization in AZD9291‐resistant HCC827 and HA1 cells. Cells were incubated with different concentrations of SHR‐A1403 for 48 h, then intracellular polymeric β‐tubulin and total β‐tubulin were analyzed by western blotting
The main processes involved in SHR‐A1403 activity include binding to cells, internalization within cells and cleavage of SHR‐A1403 to release c‐Met mAb and SHR152852, after which c‐Met mAb and SHR152852 exert their individual actions. To investigate the underlying mechanism of SHR‐A1403 action, we focused on these processes. As shown in Figure 3, SHR‐A1403 bound more extensively to c‐Met‐overexpressing HA1 cells (Figures 3B and S3). Consistent with this, these cells showed greater internalization of SHR‐A1403 (Figure 3C,D) compared with HCC827 cells, raising the possibility that more microtubule‐disrupting agent, SHR152852, is also released. Indeed, microtubules in HA1 cells were much more sensitive to SHR‐A1403 than those in HCC827 cells (Figures 3E and S4). Collectively, these observations confirm greater release of SHR152852 in HA1 cells and establish increased release of SHR152852 as the mechanism by which SHR‐A1403 overcomes AZD9291 resistance.
3.4. SHR‐A1403 inhibits the growth of AZD9291‐resistant, c‐Met‐overexpressing xenografts in vivo
Effects of SHR‐A1403 were further determined in vivo by assessing the growth of HCC827 and HA1 xenograft tumors. Tumor‐bearing mice, established by s.c. inoculation of HCC827 and HA1 cells, were randomized into vehicle, AZD9291 (i.g.) and SHR‐A1403 (i.v.) treatment groups when average tumor volumes reached approximately 100‐200 mm3. As shown in Figure 4, AZD9291 (3 mg/kg) significantly inhibited the growth of HCC827 tumors, but not HA1 tumors (Figure 4A). Consistent with this, AZD9291 inhibited EGFR and downstream AKT phosphorylation in HCC827 tumor tissues, but not in HA1 tumor tissues, 4 hours after injection (Figure 4B), indicating that HA1 tumors are strongly resistant to AZD9291. In contrast, SHR‐A1403 significantly inhibited the growth of HA1 tumors and even induced tumor regression 21 days after injection but did not affect the growth of HCC827 tumors (Figure 4A). Furthermore, although SHR‐A1403 induced c‐Met degradation and inhibited c‐Met phosphorylation in both HCC827 and HA1 tumor tissues, levels of c‐Met and phospho‐c‐Met were much higher in HA1 tumor tissues than in HCC827 tumor tissues (Figure 4C), suggesting that, unlike HCC827 tumors, which depend on EGFR, the growth of HA1 tumors is driven by c‐Met overexpression, and thus is strongly inhibited by SHR‐A1403. The effects of SHR‐A1403 on another c‐Met‐overexpressed, AZD9291‐resistant clone, HG3 xenograft, were also studied in vivo. SHR‐A1403 at the dose of 10 mg/kg significantly inhibited the growth of HG3 xenograft and even induced tumor regression (Figure S5A). Further analysis showed that SHR‐A1403 induced c‐Met degradation and inhibited c‐Met phosphorylation in HG3 tumor tissues (Figure S5B).
Figure 4.
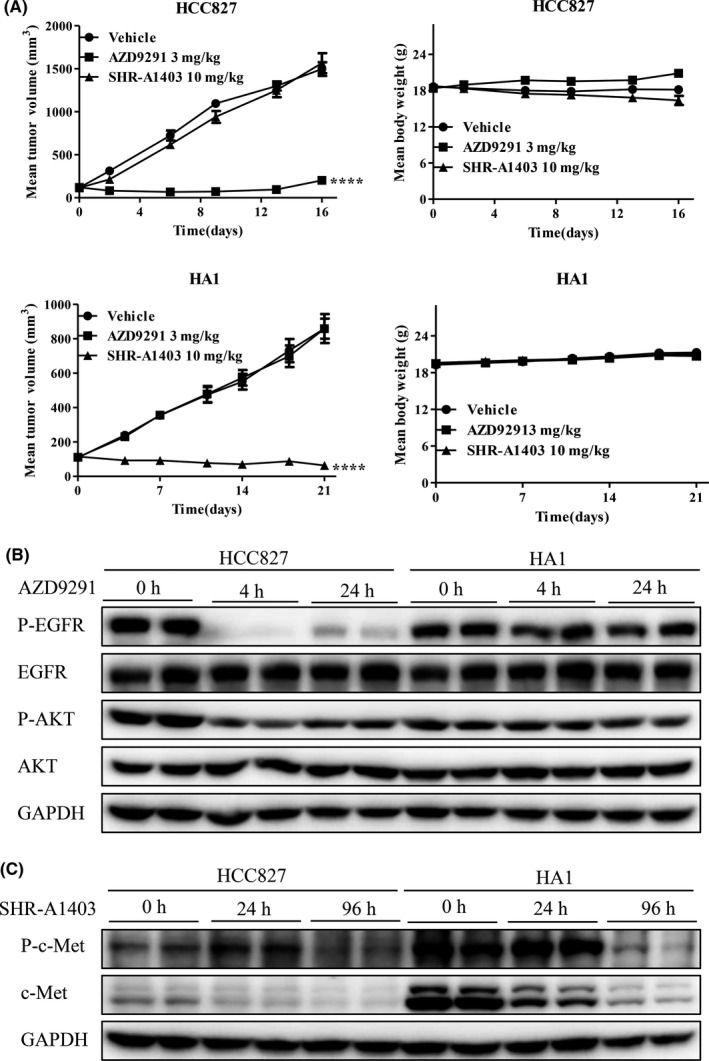
Efficacy of SHR‐A1403 on AZD9291‐resistant HA1 xenografts overexpressing c‐mesenchymal‐epithelial transition factor (c‐Met) in vivo. A, Nude mice bearing HCC827 or HA1 xenografts were treated with vehicle, 10 mg/kg SHR‐A1403 or 3 mg/kg AZD9291, and tumor volume and body weights were measured on the indicated days. B and C, Nude mice bearing HA1 xenografts were i.v. injected with 3 mg/kg AZD9291 or 10 mg/kg SHR‐A1403 once, then tumor tissues were lysed in RIPA buffer, and analyzed by western blotting. Results shown represent means ± SEM (vehicle group, n = 8; SHR‐A1403 group, n = 6). ****P < .0001 vs vehicle
4. DISCUSSION
In recent years, EGFR‐TKI have become the first‐line therapy for NSCLC patients with EGFR‐activating mutations.4 However, drug resistance becomes an obstacle to EGFR‐TKI treatment in the clinic.8, 10, 12, 13 Thus, a variety of reports have focused on the mechanisms of resistance to EGFR‐TKI and have searched for strategies to overcome resistance based on these resistance mechanisms. In the current report, we took a different approach, mainly focusing on the strategy for overcoming resistance to the EGFR‐TKI, AZD9291, instead of first investigating the precise mechanism underlying AZD9291 resistance. Herein, we showed a novel strategy for overcoming AZD9291 resistance, providing the first account of the use of a c‐Met‐targeting ADC rather than a c‐Met‐inhibiting antibody. Based on the distinct antitumor mechanism of the ADC, SHR‐A1403, tumors could be screened for c‐Met expression as a biomarker, and having established c‐Met overexpression, treatment with SHR‐A1403 would be sufficient to overcome drug resistance.27, 33 Thus, we believe that using a c‐Met ADC such as SHR‐A1403 is a more direct and feasible approach for overcoming resistance to EGFR‐TKI and warrants further clinical development.
Unlike the c‐Met inhibitor crizotinib and c‐Met mAb, which exert antitumor activity mainly by inhibiting the c‐Met signaling pathway, the novel c‐Met ADC, SHR‐A1403, inhibits tumor growth through microtubule depolymerization mediated by intracellularly released SHR152852. This different mechanism of action may thus result in clear advantages with respect to overcoming resistance to EGFR‐TKI. As it has been reported that, in addition to EGFR‐dependent resistance mechanisms based on EGFR mutations, EGFR overexpression is responsive to the resistance of AZD9291, aberrant activation of the HGF/c‐Met pathway caused by MET gene overexpression and protein hyperactivation also contribute to the resistance to AZD9291.15, 34 The strategy of combining a c‐Met inhibitor and EGFR‐TKI has been developed for use in AZD9291‐resistant patients in which c‐Met is activated. Because this approach is only effective in cases where the c‐Met signaling pathway is hyperactivated and participates in the resistance to EGFR‐TKI, its limitations are obvious. In cases where only levels of c‐Met, but not phospho‐c‐Met, are high, such as the HG1, HG2, HG3 and HG4 cells reported here (Figure 2A), crizotinib and c‐Met mAb alone were ineffective in overcoming AZD9291 resistance. In this setting, SHR‐A1403 inhibited the proliferation of resistant cells with much greater efficacy than HCC827 cells, and the mechanism was that more SHR‐A1403 bound to c‐Met‐overexpressing resistant cells, leading to greater intracellular release of SHR152852 and subsequent inhibition of cell proliferation. Thus, SHR‐A1403 acts not only by inhibiting the c‐Met signaling pathway, but also by inducing microtubule depolymerization, effectively targeting c‐Met‐overexpressing resistant cells and efficiently overcoming AZD9291 resistance, regardless of whether the c‐Met signaling pathway participates in AZD9291 resistance.
Previous studies have sought to overcome resistance to EGFR inhibitors by combining c‐Met inhibitors with EGFR‐TKI with the goal of causing resistant cells to become as sensitive to EGFR inhibitors as parental cells.35, 36 In the current study, SHR‐A1403 was used as a stand‐alone agent to treat AZD9291‐resistant cells. More importantly, the inhibitory effect of SHR‐A1403 on the proliferation of resistant cells was much greater than that on HCC827 cells both in vitro and in vivo. Thus, our approach using a c‐Met ADC in place of an EGFR inhibitor to overcome resistance is superior to and much more convenient than previous approaches based on a combination strategy.
Another benefit of using the c‐Met ADC, SHR‐A1403, as a strategy for overcoming AZD9291 resistance is that, in assessing whether SHR‐A1403 should be used, screening resistant cells for c‐Met overexpression is sufficient; there is no need to identify the precise resistance mechanism, making the strategy simpler and more direct. Moreover, SHR‐A1403 may also efficiently overcome resistance to additional inhibitors of EGFR or other tyrosine kinases, such as HER2, KDR and PDGFR, among others, in cells expressing elevated c‐Met, possibilities that warrant further investigation.
In summary, we provide a novel strategy for overcoming resistance to the EGFR inhibitor AZD9291 using the c‐Met ADC, SHR‐A1403. c‐Met expression level is the critical biomarker predictive of the efficacy of this strategy, regardless of whether the c‐Met signaling pathway is involved in the resistance mechanism.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This research was supported by grants from the Science and Technology Commission of Shanghai Municipality (no. 18DZ2293200), and the Yunnan Province Sciences and Technology plan (No. 2017ZF010).
Tong M, Gao M, Xu Y, et al. SHR‐A1403, a novel c‐mesenchymal‐epithelial transition factor (c‐Met) antibody‐drug conjugate, overcomes AZD9291 resistance in non‐small cell lung cancer cells overexpressing c‐Met. Cancer Sci. 2019;110:3584–3594. 10.1111/cas.14180
Contributor Information
Haitian Quan, Email: haitianquan@simm.ac.cn.
Liguang Lou, Email: lglou@simm.ac.cn.
REFERENCES
- 1. Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1‐19. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 3. Jain P, Khanal R, Sharma A, Yan F, Sharma N. Afatinib and lung cancer. Expert Rev Anticancer Ther. 2014;14(12):1391‐1406. [DOI] [PubMed] [Google Scholar]
- 4. Nan XL, Xie C, Yu XY, Liu J. EGFR TKI as first‐line treatment for patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer. Oncotarget. 2017;8(43):75712‐75726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee CC, Shiao HY, Wang WC, Hsieh HP. Small‐molecule EGFR tyrosine kinase inhibitors for the treatment of cancer. Expert Opin Investig Drugs. 2014;23(10):1333‐1348. [DOI] [PubMed] [Google Scholar]
- 6. Tang ZH, Cao WX, Su MX, Chen X, Lu JJ. Osimertinib induces autophagy and apoptosis via reactive oxygen species generation in non‐small cell lung cancer cells. Toxicol Appl Pharmacol. 2017;321:18‐26. [DOI] [PubMed] [Google Scholar]
- 7. Soejima K, Yasuda H, Hirano T. Osimertinib for EGFR T790M mutation‐positive non‐small cell lung cancer. Expert Rev Clin Pharmacol. 2017;10(1):31‐38. [DOI] [PubMed] [Google Scholar]
- 8. Wu SG, Shih JY. Management of acquired resistance to EGFR TKI‐targeted therapy in advanced non‐small cell lung cancer. Mol Cancer. 2018;17(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ortiz‐Cuaran S, Scheffler M, Plenker D, et al. Heterogeneous mechanisms of primary and acquired resistance to third‐generation EGFR inhibitors. Clin Cancer Res. 2016;22(19):4837‐4847. [DOI] [PubMed] [Google Scholar]
- 10. Lim SM, Syn NL, Cho BC, Soo RA. Acquired resistance to EGFR targeted therapy in non‐small cell lung cancer: mechanisms and therapeutic strategies. Cancer Treat Rev. 2018;65:1‐10. [DOI] [PubMed] [Google Scholar]
- 11. Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 2015;5(5):390‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eberlein CA, Stetson D, Markovets AA, et al. Acquired resistance to the mutant‐selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. 2015;75(12):2489‐2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11(8):473‐481. [DOI] [PubMed] [Google Scholar]
- 14. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834‐848. [DOI] [PubMed] [Google Scholar]
- 15. Shi P, Oh YT, Zhang G, et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016;380(2):494‐504. [DOI] [PubMed] [Google Scholar]
- 16. Beau‐Faller M, Ruppert AM, Voegeli AC, et al. MET gene copy number in non‐small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol. 2008;3(4):331‐339. [DOI] [PubMed] [Google Scholar]
- 17. Go H, Jeon YK, Park HJ, Sung SW, Seo JW, Chung DH. High MET gene copy number leads to shorter survival in patients with non‐small cell lung cancer. J Thorac Oncol. 2010;5(3):305‐313. [DOI] [PubMed] [Google Scholar]
- 18. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039‐1043. [DOI] [PubMed] [Google Scholar]
- 19. Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signalling pathway in cancer. Eur J Cancer. 2010;46(7):1260‐1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peters S, Adjei AA. MET: a promising anticancer therapeutic target. Nat Rev Clin Oncol. 2012;9(6):314‐326. [DOI] [PubMed] [Google Scholar]
- 21. Mo HN, Liu P. Targeting MET in cancer therapy. Chronic Dis Transl Med. 2017;3(3):148‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298‐2308. [DOI] [PubMed] [Google Scholar]
- 23. Ou SH, Kwak EL, Siwak‐Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal‐epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non‐small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6(5):942‐946. [DOI] [PubMed] [Google Scholar]
- 24. Yap TA, Olmos D, Brunetto AT, et al. Phase I trial of a selective c‐MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol. 2011;29(10):1271‐1279. [DOI] [PubMed] [Google Scholar]
- 25. Jun HT, Sun J, Rex K, et al. AMG 102, a fully human anti‐hepatocyte growth factor/scatter factor neutralizing antibody, enhances the efficacy of temozolomide or docetaxel in U‐87 MG cells and xenografts. Clin Cancer Res. 2007;13(22 Pt 1):6735‐6742. [DOI] [PubMed] [Google Scholar]
- 26. Jin H, Yang R, Zheng Z, et al. MetMAb, the one‐armed 5D5 anti‐c‐Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008;68(11):4360‐4368. [DOI] [PubMed] [Google Scholar]
- 27. Wang J, Anderson MG, Oleksijew A, et al. ABBV‐399, a c‐met antibody‐drug conjugate that targets both MET‐amplified and c‐met‐overexpressing tumors, irrespective of MET pathway dependence. Clin Cancer Res. 2017;23(4):992‐1000. [DOI] [PubMed] [Google Scholar]
- 28. Tong CWS, Wu WKK, Loong HHF, Cho WCS, To KKW. Drug combination approach to overcome resistance to EGFR tyrosine kinase inhibitors in lung cancer. Cancer Lett. 2017;405:100‐110. [DOI] [PubMed] [Google Scholar]
- 29. Neal JW, Dahlberg SE, Wakelee HA, et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second‐line or third‐line treatment of patients with EGFR wild‐type advanced non‐small‐cell lung cancer (ECOG‐ACRIN 1512): a randomised, controlled, open‐label, multicentre, phase 2 trial. Lancet Oncol. 2016;17(12):1661‐1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang C, Zhao X, Sun X, et al. Preclinical pharmacokinetics of a novel anti‐c‐Met antibody‐drug conjugate, SHR‐A1403, in rodents and non‐human primates. Xenobiotica. 2019;49(9):1097‐1105. [DOI] [PubMed] [Google Scholar]
- 31. Yang CY, Wang L, Sun X, et al. SHR‐A1403, a novel c‐Met antibody‐drug conjugate, exerts encouraging anti‐tumor activity in c‐Met‐overexpressing models. Acta Pharmacol Sin. 2019;40:971‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao H, Quan H, Xie C, et al. YHHU0895, a novel synthetic small‐molecule microtubule‐destabilizing agent, effectively overcomes P‐glycoprotein‐mediated tumor multidrug resistance. Cancer Lett. 2012;314(1):54‐62. [DOI] [PubMed] [Google Scholar]
- 33. Liu X, Newton RC, Scherle PA. Developing c‐MET pathway inhibitors for cancer therapy: progress and challenges. Trends Mol Med. 2010;16(1):37‐45. [DOI] [PubMed] [Google Scholar]
- 34. Ou SI, Agarwal N, Ali SM. High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post‐osimertinib progression. Lung Cancer. 2016;98:59‐61. [DOI] [PubMed] [Google Scholar]
- 35. Martinez‐Marti A, Felip E, Matito J, et al. Dual MET and ERBB inhibition overcomes intratumor plasticity in osimertinib‐resistant‐advanced non‐small‐cell lung cancer (NSCLC). Ann Oncol. 2017;28(10):2451‐2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Friese‐Hamim M, Bladt F, Locatelli G, Stammberger U, Blaukat A. The selective c‐Met inhibitor tepotinib can overcome epidermal growth factor receptor inhibitor resistance mediated by aberrant c‐Met activation in NSCLC models. Am J Cancer Res. 2017;7(4):962‐972. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials