

Abstract
There are currently no approved drugs for the treatment of emerging viral infections, such as dengue and Ebola. Adaptor associated kinase 1 (AAK1) is a cellular serine/threonine protein kinase that functions as a key regulator of the clathrin-associated host adaptor proteins and regulates the intracellular trafficking of multiple unrelated RNA viruses. Moreover, AAK1 is overexpressed specifically in dengue virus-infected but not bystander cells. Since AAK1 is a promising antiviral drug target, we have embarked on an optimization campaign of a previously identified 7-azaindole analogue, yielding novel pyrrolo[2,3-b]pyridines with high AAK1 affinity. The optimized compounds demonstrate improved activity against dengue virus both in vitro and in human primary dendritic cells and the unrelated Ebola virus. These findings demonstrate that targeting cellular AAK1 may represent a promising broad-spectrum antiviral strategy.
Graphical abstract
Introduction
Dengue virus (DENV) is an enveloped, positive-sense, single-stranded RNA virus belonging to the Flaviviridae family. DENV is transmitted by the mosquitoes Aedes aegypti and Aedes albopictus, which mainly reside in (sub)tropical climates. Hence, dengue outbreaks are mainly confined to equatorial areas, where more than 100 countries have been declared dengue-endemic.1 The World Health Organization (WHO) estimates that up to 3.9 billion people are at risk for dengue infection at any given time.2 In 2013, the WHO reported 3.2 million cases of severe dengue and more than 9,000 dengue-related deaths worldwide.3 Up to 80% of DENV-infected patients remain asymptomatic. Symptomatic patients usually experience an acute febrile illness, characterized by high fever, muscle and joint pain,, and sometimes rash.1 The likelihood of progression to severe dengue, manifesting by shock, hemorrhage and organ failure, is greater upon secondary infection with a heterologous dengue serotype (of four that circulate) due to antibody-dependent enhancement.4
Ebola virus (EBOV) is a member of the Filoviridae family. Four of the five known EBOV species have been responsible for over twenty outbreaks and over 10,000 deaths since their identification in 1976.6
Current efforts in search for drugs active against DENV focus primarily on viral targets, such as the NS3 helicase, NS2B-NS3 protease, NS4B, NS5 methyltransferase, NS5 polymerase and the viral envelope.7 In search for anti-EBOV drugs, the RNA-dependent RNA polymerase L, the viral surface glycoprotein GP, and viral proteins VP24 and VP35 have been explored as candidate targets.8 However, targeting viral functions is often associated with the rapid emergence of drug resistance and usually provides a ‘one drug, one bug’ approach. DENV and EBOV rely extensively on host factors for their replication and survival. These cellular factors represent attractive candidate targets for antiviral agents, potentially with a higher barrier for resistance. In addition, such host-targeted antivirals are more likely to exhibit broad-spectrum antiviral activity when targeting a host function required for the replication of several unrelated viruses.9,10
Intracellular membrane trafficking is an example of a cellular process that is hijacked by various viruses11. Intracellular membrane trafficking depends on the function of tyrosine and dileucine based signals in host cargo proteins, which are recognized by μ1–5 subunits of the clathrin adaptor protein (AP) complexes AP1–5. Adaptor complexes mediate the sorting of cargo proteins to specific membrane compartments within the cell. While AP2 sorts in the endocytic pathway, AP1 and AP4 sort in the secretory pathway.12
The activity of AP2M1 and AP1M1, the μ subunits of AP2 and AP1, respectively, is regulated by two host cell kinases, adaptor-associated kinase 1 (AAK1) and cyclin G associated kinase (GAK). Phosphorylation of specific threonine residues in AP2M1 and AP1M1 by these kinases is known to stimulate their binding to tyrosine signals in cargo protein and enhance vesicle assembly and internalization. Both AAK1 and GAK regulate clathrin-mediated endocytosis by recruiting clathrin and AP2 to the plasma membrane. AAK1 also regulates clathrin-mediated endocytosis of cellular receptors via alternative sorting adaptors that collaborate with AP-2, e.g. by phosphorylation of NUMB.12 Additionally, AAK1 hasbeen implicated in the regulation of EGFR internalization and recycling to the plasma membrane via its effects on and interactions with alternate endocytic adaptors. We have demonstrated that AAK1 and GAK regulate hepatitis C (HCV) entry and assembly by modulating AP2 activity.12,13 and viral release and cell-to-cell spread via regulation of AP1.9,14 AAK1 and GAK are also required in the life cycles of DENV and EBOV. 9
We have reported that the approved anticancer drugs sunitinib and erlotinib that potently inhibit AAK1 and GAK, respectively, demonstrate broad-spectrum in vitro antiviral activity against different members of the Flaviviridae family (HCV, DENV, Zika virus, West Nile virus), as well as against various unrelated families of RNA viruses. We have also demonstrated that the combination of these two drugs effectively reduces viral load, morbidity and mortality in mice infected with DENV and EBOV.9 These data provide a proof-of-concept that small molecule inhibition of AAK1 and GAK can yield broad-spectrum antiviral agents.9,15 Moreover, using single-cell transcriptomic analysis, AAK1 has been validated as a particularly attractive target since it is overexpressed specifically in DENV-infected and not bystander cells (uninfected cells from the same cell culture), and its expression level increases with cellular virus abundance.15
AAK1 has been studied primarily as a drug target for the treatment of neurological disorders, such as schizophrenia, Parkinson’s disease, neuropathic pain16, bipolar disorders and Alzheimer’s disease.17,18 Consequently, very potent AAK1 inhibitors based on different chemotypes have been disclosed in the patent literature. Figure 1 shows representative examples of these AAK1 inhibitors andtheir enzymatic inhibition data.
Figure 1.

Known AAK1 inhibitors
Despite the fact that AAK1 emerged as a promising antiviral target, none of these compound classes have been evaluated and/or optimized for antiviral activity. The only exception being a series of imidazo[1,2-b]pyridazines, which were originally developed by Lexicon Pharmaceuticals (Figure 2).16 We have previously resynthesized these molecules, confirmed their potent AAK1 affinity and demonstrated their antiviral activity against HCV and DENV.9
Figure 2.

AAK1 inhibitors with documented antiviral activity
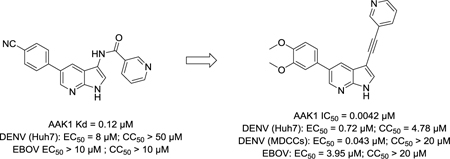
Here, rather than starting from a potent AAK1 inhibitor, we started from a structurally simple compound with reasonable AAK1 activity from which easy structural variation could be introduced. A screening campaign of 577 structurally diverse compounds (representing kinase inhibitor chemical space) across a panel of 203 protein kinases using the DiscoverX binding assay format previously identified a pyrrolo[2,3-b]pyridine or 7-aza-indole derivative (compound 1, Figure 3) as a potent AAK1 inhibitor (KD = 53 nM).19 In this manuscript, we describe our efforts to optimize the AAK1 affinity and anti-DENV activity of compound 1.
Figure 3.

A pyrrolo[2,3-b]pyridine (7-aza-indole) based AAK1 inhibitor
Results and Discussion
Chemistry
Synthesis of 3-substituted-5-aryl pyrrolo[2,3-b]pyridines
A regioselective Sonogashira coupling of trimethylsilylacetylene with commercially available 5-bromo-3-iodo-2-aminopyridine 2 afforded the alkynyl derivative 3 (Scheme 1).20 Compound 3 was then reductively ring closed with a strong base yielding pyrrolo[2,3-b]pyridine 4. Initially, NaH was used as base20, but later on potassium tert-butoxide21 was applied as this gave a cleaner reaction outcome and an improved yield. Initial attempts to nitrate position 3 of the 7-azaindole scaffold employed a mixture of a 65% HNO3 solution and sulfuric acid22. However, the desired product was difficult to isolate from this reaction mixture and therefore, the nitration was performed by treatment of compound 4 with fuming nitric acid.23 The 3-nitro derivative 5 precipitated from the reaction mixture and was conveniently isolated by filtration. Suzuki coupling of compound 5 with a number of arylboronic acids yielded the 3-nitro-5-aryl-pyrrolo[2,3-b]pyridines 6a-h in yields ranging from 65 to 85%.24 Catalytic hydrogenation of the nitro moiety yielded the corresponding amino derivatives 7a-h. Because of the instability of the 3-amino-pyrrolo[2,3-b]pyridines, these were not purified and used as such for further reaction. Coupling with an acid chloride in a mixture of pyridine and dichloromethane25 or alternatively, reaction with a carboxylic acid using (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) as coupling reagent26 yielded a small library of pyrrolo[2,3-b]pyridines 8a-o. A sulfonamide derivative 8p was prepared via reaction of 7g with phenylsulfonyl chloride in pyridine.
Scheme 1.

Synthesis of 3-substituted-5-aryl pyrrolo[2,3-b]pyridines 8a-p
Reagents and conditions. a) TMSA, Pd(PPh3)2Cl2, CuI, Et3N, THF, rt; b) KOtBu, NMP, 80°C; c) HNO3, 0°C to rt; d) Pd(PPh3)4, K2CO3, ArB(OH)2, H2O, dioxane, 105°C; e)H2, Pd/C, THF, rt; f)RCOCl, pyridine, THF, 1M NaOH, rt; g)RCOOH, BOP, Et3N, DMF, rt; h)PhSO2Cl, pyridine, rt.
Synthesis of 3-benzoyl-5-(3,4-dimethoxyphenyl)-pyrrolo[2,3-b]pyridine
Formylation of compound 4 by a Duff reaction27 yielded 3-formyl-5-bromo-azaindole 9 (Scheme 2). The pyrrole nitrogen was protected28 using NaH and tosylchloride yielding compound 10. Suzuki coupling reaction with 3,4-dimethoxyphenylboronic acid furnished compound 11. Nucleophilic addition29 of phenylmagnesium bromide to the aldehyde furnished the secondary alcohol 12. Oxidation30 of the benzylic alcohol using MnO2 afforded ketone 13. Finally, alkaline deprotection31 of the tosyl group yielded the desired compound 14.
Scheme 2.

Synthesis of 3-benzoyl-5-(3,4-dimethoxyphenyl)-pyrrolo[2,3-b]pyridine 14
Reagents and conditions. a) hexamine, H2O, CH3COOH, 120°C ; b) NaH, TsCl, 0°C to rt ; c) 3,4-dimethoxyphenylboronic acid, Pd(PPh3)4, 2M K2CO3, toluene, EtOH, 105°C ; d) 3M PhMgBr, THF, rt; e) MnO2, THF, rt ; f) KOH, EtOH, 80°C.
Synthesis of 3-phenyl-5-(3,4-dimethoxyphenyl)-pyrrolo[2,3-b]pyridine
Iodination of compound 4 with N-iodosuccinimide32 afforded compound 15 (Scheme 3). Reaction of compound 15 with phenylboronic acid led only to recovery of unreacted starting material. Therefore, the pyrrole nitrogen of the 7-azaindole scaffold was protected as a tosyl group,28 affording compound 16. A regioselective Suzuki coupling reaction using phenylboronic acid furnished the 3-phenyl-pyrrolo[2,3-b]pyridine analogue 17. A subsequent Suzuki reaction24 with 3,4-dimethoxyphenylboronic acid yielded compound 18. Finally, alkaline cleavage of the tosyl protecting group afforded the desired target compound 19.31
Scheme 3.

Synthesis of 3-phenyl-5-(3,4-dimethoxyphenyl)-pyrrolo[2,3-b]pyridine 19
Reagents and conditions. a) NIS, acetone, rt ; b) NaH, TsCl, THF, 0°C to rt ; c) PhB(OH)2, Pd(PPh3)4, K2CO3, toluene, EtOH, H2O, 90°C ; d) 3,4-dimethoxyphenylboronic acid, Pd(PPh3)4, K2CO3, toluene, EtOH, H2O, 105°C ; e) KOH, EtOH, 80°C.
Synthesis of 3-alkynyl-5-(3,4-dimethoxyphenyl)-pyrrolo[2,3-b]pyridines
Sonogashira reaction of compound 15 with a number of (hetero)arylacetylenes yielded regioselectively compounds 20a-f in yields varying from 20–70% (Scheme 4).20 In contrast to Suzuki couplings (Scheme 3), protection of the pyrrole nitrogen was not necessary. Subsequent Suzuki coupling24 with 3,4-dimethoxyphenylboronic acid gave access to final compounds 21a-f.
Scheme 4.

Synthesis of 3-alkynyl-5-(3,4-dimethoxyphenyl)-pyrrolo[2,3-b]pyridines 21a-f
Reagents and conditions. a) RC≡CH, Pd(PPh3)2Cl2, CuI, THF, Et3N, rt ; b) 3,4-dimethoxyphenylboronic acid, Pd(PPh3)4, K2CO3, H2O, dioxane, 105 °C.
Synthesis of 1-methyl-1H-pyrrolo[2,3-b]pyridine
Methylation33 of compound 4 using NaH and MeI furnished compound 22 (Scheme 5). Nitration,23 followed by Suzuki coupling24 gave access to compound 24. Finally, catalytic reduction of the nitro group, followed by condensation25 of the exocyclic amino group of compound 25 with nicotinoyl chloride yielded target compound 26.
Scheme 5.

Synthesis of 1-methyl-1H-pyrrolo[2,3-b]pyridine 26.
Reagents and conditions. a) NaH, MeI, THF, 0°C to rt ; b) HNO3, 0°C to rt ; c) 3,4-dimethoxyphenylboronic acid, Pd(PPh3)4, K2CO3, H2O, dioxane, 105°C ; d) H2, THF, rt ; e) nicotinoyl chloride, pyridine, THF, 1M NaOH, rt.
Synthesis of pyrazolo[3,4-b]pyridine
Suzuki coupling24 between commercially available 5-bromo-2-chloronicotinonitrile 27 and 3,4-dimethoxyphenylboronic acid yielded regioselectively compound 28. Nucleophilic displacement of the chlorine by hydrazine, with a concomitant nucleophilic addition at the cyano group34 allowed to construct the pyrazole moiety, yielding compound 29. Finally, amide formation35 using nicotinoyl chloride yielded the final compound 30.
Synthesis of pyrrolo[2,3-b]pyrazine
Treatment of 2-aminopyrazine 31 with N-bromosuccinimide yielded the 3,5-dibromopyrazine intermediate 32 (Scheme 7).36 A regioselective Sonogashira coupling with trimethylsilylacetylene, followed by a reductive ring closure with potassium tert-butoxide furnished pyrrolo[2,3-b]pyrazine 34.37 Iodination38 with N-iodosuccinimide yielded the dihalogenated intermediate 35 that was subsequently treated with 3-ethynylpyridine20 and 3,4-dimethoxyphenylboronic acid24 leading to the desired pyrrolo[2,3-b]pyrazine 37.
Scheme 7.

Synthesis of pyrrolo[2,3-b]pyrazine 37
Reagents and conditions. a) NBS, DMSO, rt ; b) Me3SiC≡CH, Pd(PPh3)2Cl2, CuI, THF, Et3N, rt ; c) KOtBu, NMP, 100°C ; d) NIS, acetone, rt ; e) 3-ethynylpyridine, Pd(PPh3)2Cl2, CuI, THF, Et3N, rt ; f) 3,4-dimethoxyphenylboronic acid, Pd(PPh3)4, K2CO3, H2O, dioxane, 105 °C.
Kinase profiling and X-ray crystallography of compound 1
AAK1 is a serine-threonine kinase that belongs to the NUMB-associated family of protein kinases (NAKs). Other members of this kinase family include BIKE/BMP2K (BMP-2 inducible kinase), GAK (cyclin G associated kinase) and MPSK1 (myristoylated and palmitoylated serine-threonine kinase 1, also known as STK16). As part of an early profiling of hit compound 1, we assessed its selectivity by a binding-displacement assay against each of the four NAK family kinases (Figure 4). Conversion of the experimentally determined IC50 values to Ki values to allow estimation of the selectivity showed that compound 1 was 3-fold more selective for AAK1 over GAK, and 8-fold and 22-fold more selective for AAK1 over BMP2K and STK16, respectively (Table 1).
Figure 4.

Binding displacement assay of compound 1 against the NAK family members.
Table 1.
Selectivity of compound 1 for AAK1 against the NAK family kinases
| Binding Displacement Assay | |||
|---|---|---|---|
| NAK | IC50 (μM) | Ki (μM) | Ki / Ki (AAK1) |
| AAK1 | 1.17 | 0.541 | 1.0 |
| BMP2K | 10.1 | 4.40 | 8.13 |
| GAK | 3.25 | 1.75 | 3.23 |
| STK16 | 20.0 | 11.9 | 22.0 |
To analyse the binding mode of compound 1, the crystal structure of compound 1 bound to AAK1 to 2.0 Å resolution was determined (Figure 5). Data collection and refinement statistics are provided in Supplemental Table 1 (see Supporting Information) and PDB coordinates are deposited under PDB ID 5L4Q. Compound 1 binds in the ATP-binding site of AAK1 in a relatively planar manner (Figure 5), with the pyrrolo[2,3-b]pyridine moiety bound directly between the side-chains of two highly conserved residues: Ala72 from β2 in the kinase N-lobe and Leu183 of the C-lobe, the location where the adenine ring of ATP would bind. The nitrogen atoms of the pyrrolo[2,3-b]pyridine moiety form two hydrogen bonds to the peptide backbone of residues Asp127 and Cys129 at the kinase hinge region (Figure 5). The 4-cyanophenyl moiety is oriented towards the solvent with the phenyl ring directly sandwiched between the backbone of Gly132 of the hinge and Leu52 of β1 in the N-lobe, and the nitrogen of the cyano moiety forming a polar interaction with the side-chain of Asn136. The nicotinamide moiety is bound against the “gatekeeper” residue Met126, and forms a hydrogen bond to the side chain of Lys74, another highly conserved residue that normally links the phosphate of ATP to the αC-helix and required for correct positioning of the N-lobe for efficient catalysis. Compound 1 also interacts with two water molecules, one situated at the back of the ATP pocket that bridges between the oxygen of the amide moiety and the backbone nitrogen of Asp194, and a second at the front of the adenosine binding site directly below Val60 in the β1 strand that interacts with the amide nitrogen and surrounding solvent. The DFG motif (Asp194) is in the conformation expected of active AAK1 (Figure 5), with the activation loop of AAK1 in the same conformation as seen in previous AAK1 and BMP2K crystal structures, including the activation-segment C-terminal helix (ASCH), a structural element that is rare amongst other protein kinases, but conserved across the NAK family.39
Figure 5.

Crystal structure of AAK1 (grey) in complex with compound 1 (purple), PDB ID 5L4Q. A: Overview of the crystal structure of AAK1. Highlighted are areas that are important for kinase function. Compound 1 bound at the hinge is shown. ASCH = Activation Segment C-terminal Helix, a feature unique to kinases of the NAK family. B: 2Fo-Fc Electron density map contoured at 1σ around compound 1, showing the fit of the model to the map. C and D: Detailed views of the interactions of compound 1 in the ATP-binding site from two orientations. Black dotted lines indicate polar interactions, red spheres indicate water molecules. Note that residues 48–63 from β1 and β2 are removed from C for clarity.
Structure-activity relationship (SAR) study
All compounds synthesized in this study were evaluated for AAK1 affinity using two different, commercially available AAK1 binding assays. In the early stage of the program, the proprietary KINOMEscan screening platform of DiscoverX was used. In this assay, compounds that bind the kinase active site prevent kinase binding to an immobilized ligand and reduce the amount of kinase captured on the solid support. Hits are then identified by measuring the amount of kinase captured in test versus control samples via qPCR that detects an associated DNA label.40 Later on in the project, compounds were evaluated by the LanthaScreen™ Eu Kinase Binding Assay (ThermoFisher Scientific), in which binding of an Alexa Fluor™ conjugate or “tracer” to a kinase is detected by addition of a Eu-labeled anti-tag antibody. Binding of the tracer and antibody to a kinase results in a high degree of fluorescence resonance energy transfer (FRET), whereas displacement of the tracer with a kinase inhibitor results in loss of FRET.
The compounds were also assessed for antiviral activity in human hepatoma (Huh7) cells infected with DENV2. Their effect on overall infection was measured at 48 hours postinfection with DENV2 via luciferase assays and the half-maximal effective concentration and the 90% effective concentrations (EC50 and EC90 values, respectively) were calculated. In parallel, the cytotoxicity of the compounds (expressed as the half-maximal cytotoxic concentration or CC50 value) was measured via an AlamarBlue assay in the DENV-infected Huh7 cells.
In all these enzymatic and antiviral assays, sunitinib was included as a positive control (Tables 2, 3, 4, 5 and 6). Sunitinib has potent AAK1 affinity as measured by the KinomeScan format (KD = 11 nM) and LanthaScreen assay (IC50 = 47 nM) anddisplayed potent activity against DENV with an EC50 value of 1.35 μM.
Table 2.
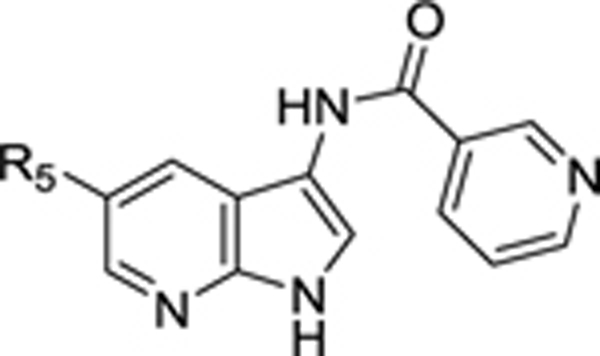
SAR at position 5 of the 7-aza-indole scaffold
 |
| AAK1 enzymatic data | DENV antiviral activity |
Cytotoxicity | ||||
|---|---|---|---|---|---|---|
| Cmpd# | R5 | AAK1 Kd (μM) (DiscoverX) |
AAK1 IC50 (μM) LanthaScreen |
EC50 (μM) |
EC90 (μM) |
CC50 (μM) |
| 1 | 4-cyanophenyl | 0.120 | NDa | 8.37 | 46.8 | 703 |
| 8a | phenyl | 0.0822 | NDa | 5.29 | 87.1 | NEa |
| 8b | 4-F-phenyl | 0.141 | NDa | 6.43 | 25.2 | NEa |
| 8c | 4-CF3-phenyl | NDa | 0.23 | 2.94 | 12.0 | 57.0 |
| 8d | 4-OCH3-phenyl | 0.0194 | NDa | 4.39 | 25.8 | NEa |
| 8e | 4-Me-phenyl | 0.0532 | NDa | 2.60 | 33.6 | 129 |
| 8f | 3-Me-phenyl | NDa | 0.0186 | 4.86 | 10.8 | 16.0 |
| 8g | 3,4-di-OCH3-phenyl | 0.00673 | 0.00432 | 1.64 | 7.46 | 39.7 |
| 8h | 3-thienyl | 0.0161 | NDa | 3.01 | 66.8 | NEa |
| Sunitinib | - | 0.0110 | 0.0474 | 1.35 | 2.71 | 246 |
NE : No effect (no apparent effect on cellular viability up to a concentration of 10 μM).
Table 3.
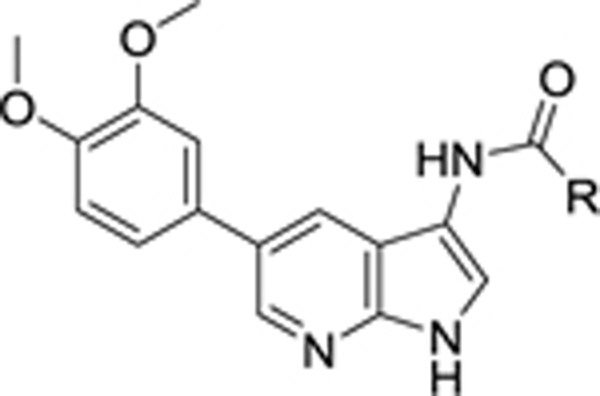
SAR of the N-acyl moiety
 |
| AAK1 enzymatic data |
DENV antiviral activity | Cytotoxicity | |||
|---|---|---|---|---|---|
| Cmpd# | R | AAK1 IC50 (μM) LanthaScreen |
EC50 (μM) | EC90 (μM) | CC50 (μM) |
| 8g | 3-pyridyl | 0.00432 | 1.64 | 7.46 | 39.7 |
| 8i | phenyl | 0.108 | 4.07 | 50.5 | 17.3 |
| 8j | 4-F-phenyl | 0.161 | 8.34 | 30.2 | 15.9 |
| 8k | 4-OCH3-phenyl | 0.0721 | NEa | NEa | 23.6 |
| 8l | 4-pyridyl | 0.381 | 12.6 | 26.8 | NEa |
| 8m | benzyl | 0.612 | 142 | NEa | NEa |
| 8n | cyclopentyl | 0.398 | 3.42 | 8.34 | 15.2 |
| 8o | cyclohexyl | 0.319 | 4.27 | 10.8 | 14.7 |
NE : No effect (no apparent antiviral effect or effect on cellular viability up to a concentration of 10 μM).
Table 4.
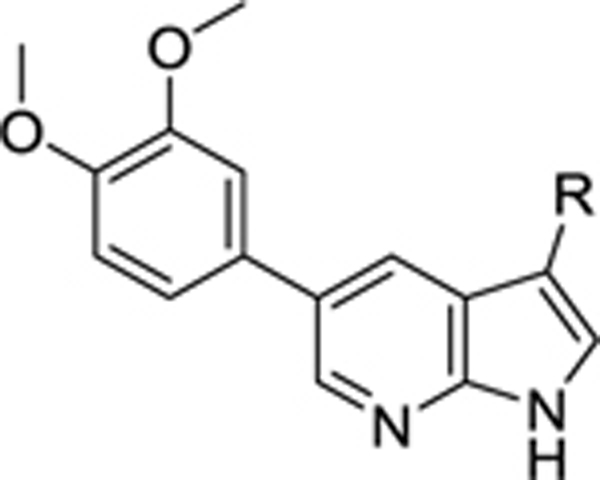
SAR of the linker moiety at position 3
 |
| AAK1 enzymatic data |
DENV antiviral activity | Cytotoxicity | |||
|---|---|---|---|---|---|
| Cmpd# | R | AAK1 IC50 (μM) LanthaScreen |
EC50 (μM) | EC90 (μM) | CC50 (μM) |
| 8i | 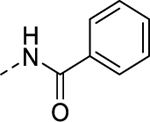 |
0.108 | 4.07 | 50.5 | 17.3 |
| 19 | 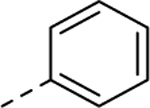 |
0.0236 | 3.02 | 8.34 | 990 |
| 14 | 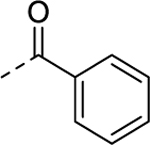 |
0.184 | 1.07 | 3.73 | 35.3 |
| 21a | 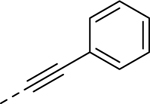 |
0.209 | 5.64 | 10.58 | 20.1 |
| 8p | 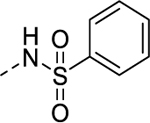 |
4.44 | 8.14 | 106 | NEa |
NE : No effect (no apparent effect on cellular viability up to a concentration of 10 μM).
Table 5.
SAR of phenylacetylene moiety
 |
| AAK1 enzymatic data |
DENV antiviral activity | Cytotoxicity | |||
|---|---|---|---|---|---|
| Cmpd# | R | AAK1 IC50 (μM) LanthaScreen |
EC50 (μM) | EC90 (μM) | CC50 (μM) |
| 21a | phenyl | 0.209 | 5.64 | 10.58 | 20.1 |
| 21b | 3-pyridyl | 0.00402 | 1.00 | 8.12 | 17.0 |
| 21c | 3-OCH3-phenyl | 0.149 | NEa | NEa | 19.6 |
| 21d | 4-CH3-phenyl | 0.420 | 7.45 | 20.5 | NEa |
| 21e | 4-F-phenyl | 0.387 | 4.21 | 8.57 | 19.9 |
| 21f | 3-CN-phenyl | 0.381 | 10.2 | 41.2 | NEa |
NE : No effect (no apparent antiviral effect or effect on cellular viability up to a concentration of 10 μM).
Table 6.
Scaffold modifications
| AAK1 enzymatic data |
DENV antiviral activity | Cytotoxicity | |||
|---|---|---|---|---|---|
| Cmpd# | Structure | AAK1 IC50 (μM) LanthaScreen |
EC50 (μM) | EC90 (μM) | CC50 (μM) |
| 26 | 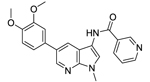 |
3.39 | NEa | NEa | NEa |
| 30 | 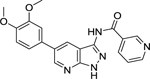 |
0.462 | 2.09 | 19.0 | 16.0 |
| 37 | 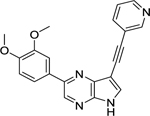 |
0.00927 | 3.28 | 7.68 | 4.81 |
NE : No effect (no apparent antiviral effect or effect on cellular viability up to a concentration of 10 μM).
SAR at position 5 of the 7-aza-indole scaffold
We confirmed the AAK1 binding affinity of hit compound 1, yet in our hands a KD value of 120 nM was measured (vs. the reported KD = 53 nM).19 This hit demonstrated antiviral activity, although rather weak (EC50 = 8.37 μM). Substitution of the 5-(4-cyanophenyl) moiety of compound 1 by phenyl (compound 8a), thienyl (compound 8h) and substituted phenyl rings with electron-withdrawing groups (compound 8c), electron-donating substituents (compounds 8d-g) and a halogen (compound 8b) gave rise to a series of analogues with structural diversity?variety that were at least equipotent to compound 1, suggesting that structural modification at this position is tolerated for AAK1 binding (Table 2). The synthesis of this limited number of analogues allowed us to quickly identify the 5-(3,4-dimethoxyphenyl) congener (compound 8g) with low nM AAK1 binding affinity in both the KinomeScan and LanthaScreen assay, indicating an excellent correlation between the two assays. As compound 8b showed stronger AAK1 affinity than the positive control sunitinib, no additional efforts were done to further explore the SAR at this position of the 7-aza-indole scaffold.
All analogues within this series had an improved antiviral activity against DENV, relative to the original hit 1. The compound with the highest binding affinity for AAK1 (compound 8g) also demonstrated the most effective anti-DENV activity, with EC50 and EC90 values of 1.64 μM and 7.46 μM, respectively (Figure 6).
Figure 6. Compounds 8g and 21b suppress DENV infection more effectively than compound 1.

Dose response of DENV infection (blue) and cell viability (black) to compounds 1, 8g, and 21b measured by luciferase and alamarBlue assays, respectively, 48 hours after infection. Data are plotted relative to vehicle control. Shown are representative experiments from at least two conducted, each with 5 biological replicates; shown are means ± SD.
SAR of the N-acyl moiety
Given the improved AAK1 affinity and antiviral activity of compound 8g, the SAR of this compound was further examined through the replacement of the 3-pyridyl group by a number of (hetero)aromatics and cycloaliphatic groups, while the 3,4-dimethoxyphenyl residue was kept fixed (Table 3). Our findings indicate that the 3-pyridyl moiety is critical for AAK1 binding as all analogues showed a 100-fold decreased AAK1 affinity, when compared to compound 8g, giving rise to AAK1 IC50 values in the 0.1–0.6 μM range. Only the congener with a 4-methoxyphenyl residue (compound 8k) was endowed with an enhanced AAK1 affinity with an IC50 value of 0.072 μM. In correlation with their weaker affinity for AAK1, these compounds had a diminished antiviral activity relative to compound 8g.
SAR of the linker moiety at position 3
To evaluate the importance of the amide linker, a number of surrogates were prepared (Table 4). For synthetic feasibility reasons, compound 8i, having a phenyl residue instead of a 3-pyridyl ring, and endowed with quite potent AAK1 affinity and moderate antiviral activity, was selected as a reference compound. Removing the amide linker furnished the 3-phenyl substituted analogue 19, which was 4-fold more potent as AAK1 ligand than compound 8i, and showed a slightly improved antiviral activity against DENV (EC50 and EC90 values of 3.02 μM and 8.34 μM, respectively). When the amide linker of compound 8i was replaced by a ketone (compound 14) or alkyne (compound 21a) functionality, AAK1 affinity was retained. Compound 14 exhibited an improved antiviral activity in comparison with compound 8i. Finally, replacement of the amide moiety by a sulfonamide linker was not well-tolerated, as compound 8p showed close to 200-fold drop in AAK1 affinity.
SAR of phenylacetylene
The data in Table 4 suggest that the amide moiety is not essential for AAK1 binding and can be replaced. Although the 3-phenyl derivative 19 displayed potent AAK1 affinity, 3,5-diaryl pyrrolo[2,3-b]pyridines are well known in the literature as kinase inhibitors. In contrast, 3-alkynyl-7-aza-indoles are studied to a much less extent. Wetherefore, selected the acetylene derivate 21a to decipher if it was possible to improve AAK1 affinity and antiviral activity by further modifying the substitution pattern. A number of substituted phenylacetylene derivatives was thus prepared (compounds 21b-f). The SAR in this series was quite flat, as all compounds displayed very similar AAK1 affinity with IC50 values in the 0.1–0.4 μM range (Table 5). In contrast, the introduction of a 3-pyridylacetylene (compound 21b) led to a substantial improvement in AAK1 affinity (IC50 = 0.0042 μM) with a concomitant improvement in antiviral activity (Figure 6).
Scaffold modifications
Lastly, our SAR exploration focused on the 7-aza-indole scaffold itself (Table 6). Methylation of the pyrrole nitrogen afforded compound 26, displaying a greatly decreased AAK1 affinity (>800-fold loss in activity relative to compound 8g) and antiviral activity (EC50 > 10 μM). Insertion of an additional nitrogen atom in the pyrrole moiety yielded the pyrazolo[3,4-b]pyridine analogue 30 that is 100-fold less active as an AAK1 ligand relative to the parent compound 8g. When the pyridine moiety of compound 21b was replaced by a pyrazine ring, the pyrrolo[2,3-b]pyrazine analogue 37 was obtained. This compound was endowed with very potent AAK1 affinity (IC50 = 0.00927 μM), comparable to its 7-aza-indole counterpart 21b. Unfortunately, compound 37 demonstrated greater cytotoxicity than 21b in Huh7 cells with CC50’s of4.81 μM vs. 17.0 μM, respectively.
Broad-spectrum antiviral activity
AAK1 has been demonstrated to be important for the regulation of intracellular viral trafficking of multiple unrelated viruses.9 Sunitinib, an approved anticancer drug with potent anti-AAK1 activity, displayed antiviral activity against RNA viruses from six different families9. To evaluate for potential broad-spectrum antiviral coverage of our molecules beyond DENV infection, hit compound 1 and the optimized congeners (compounds 8g and 21b) were tested for their activity against the unrelated EBOV. Huh7 cells were infected with EBOV and treated for 48 hours with each compound (Figure 7). Whereas compound 1 did not show effective activity against EBOV, the more potent AAK1 inhibitors 8g and 21b displayed anti-EBOV activity with EC50 values in the low μM range and CC50 > 10–20 μM.
Figure 7. Compounds 1, 8g and 21b suppress EBOV infection.

Dose response of EBOV infection (blue) and cell viability (black) to compounds 1, 8g and 21b measured by plaque assay (for compound 1) or immunofluorescence assays (for compounds 8g and 21b) and CellTiter-Glo luminescent cell viability assay in Huh7 cells 48 hours after infection. Data are plotted relative to vehicle control. Shown are representative experiments from at least two conducted, each with 3 biological replicates; shown are means ± SD.
Correlation of the antiviral effect of compounds 1, 8g and 21b with functional AAK1 inhibition
To confirm that the observed antiviral activity is correlated with functional inhibition of AAK1 activity, we measured levels of the phosphorylated form of the μ subunit of the AP2 complex, AP2M1, upon treatment with compounds 1, 21b and 8g. Since AP2M1 phosphorylation is transient (due to phosphatase PP2A activity),27 to allow capturing of the phosphorylated state, Huh7 cells were incubated for 30 minutes in the presence of the PP2A inhibitor calyculin A prior to lysis. Treatment with compounds 1, 21b and 8g reduced AP2M1 phosphorylation (Figure 8), indicating modulation of AP2M1 phosphorylation via AAK1 inhibition.
Figure 8. Antiviral effect of compounds 1, 8g and 21b correlate with functional inhibition of AAK1.

Dose response of AP2M1 phosphorylation to treatment with 1 (A), 8g (B), and 21b (C) by Western analysis in lysates derived from Huh7 cells. Representative membranes (from two independent experiments) blotted with anti-phospho-AP2M1 (pAP2M1), anti-AP2M1 (AP2M1), and anti-Actin (Actin) antibodies and quantified data of pAP2M1/actin protein ratio normalized to DMSO controls are shown. ** p < 0.01, *** p < 0.001 by 2-tailed unpaired t test.
Inhibition of DENV infection in human primary monocyte-derived dendritic cells (MDDCs)
To determine the therapeutic potential of the AAK1 inhibitors, their antiviral activity was studied in human primary dendritic cells. Primary cells are a physiologically more relevant model for DENV infection than immortalized cell lines, and are considered an ex vivo model for DENV infection. Compounds 21b and 8g showed a dose-dependent inhibition of DENV infection with EC50 and EC90 values of 0.0428 μM and 1.49 μM and 0.739 μM and 3.32 μM, respectively (Figure 9). This very potent activity in MDDCs associated with minimal cytotoxicity (CC50 > 20 μM), particularly of compound 21b demonstrates the potential of AAK1 inhibitors as antiviral agents.
Figure 9. Ex vivo antiviral activity of 21b and 8g in human primary dendritic cells.

Dose response of DENV infection (blue) and cell viability (black) to compounds 21b (A) and 8g (B) measured by plaque assays and alamarBlue assays, respectively, 72 hours after infection of primary human monocyte-derived dendritic cells (MDDCs). Shown is a representative experiment with cells from a single donor, out of 2 independent experiments conducted with cells derived from 2 donors, each with 6 biological replicates; shown are means ± SD.
Kinase selectivity
To assess the kinase selectivity of the optimized AAK1 inhibitors, compound 21b was screened against 468 kinases via the KINOMEScan assay (DiscoverX) at a single concentration of 10 μM. As can be derived from the kinase interaction map (Figure 10), compound 21b cannot be considered as a selective AAK1 inhibitor. Beyond AAK1, compound 21b targets multiple other kinases (including the other members of the NAK family), which might contribute to its antiviral effect.
Figure 10.

Kinome tree of compound 21b. Kinases that bind compound 21b are marked with red circles. The larger the circle, the stronger the binding affinity.
To quantitatively characterize the selectivity of compound 21b, selectivity scores (S-scores) were calculated41 (Table 7). The S-score is calculated by dividing the number of kinases that a compound binds toby the total number of distinct kinases tested, excluding mutant variants. The S(35) score (calculated by dividing the number of kinases that showed binding at 10% of control or less upon drug treatment by the total number of kinases tested) of 7-aza-indole-based AAK1 inhibitor 21b is 0.526. In comparison, the S(35) score of sunitinib, the approved AAK1 inhibitor that has been often used for measuring antiviral activity, is 0.57 at 3 μM.41 Since the selectivity of compound 21b was measured at a concentration of 10 μM (vs. 3 μM), it can be deduced that, while 21b is not a highly selective AAK1 inhibitor, its selectivity profile is improved relative to that of sunitinib, and it may therefore represent an improved pharmacological tool to probe the role of AAK1 in viral infection.
Table 7.
Selectivity scores (S scores) for compound 21b at 10 μM
| S-score type | Number of Hits | Number of Non-Mutant Kinases |
Selectivity Score |
|---|---|---|---|
| S(35)a | 212 | 403 | 0.526 |
| S(10)b | 138 | 403 | 0.342 |
| S(1)c | 49 | 403 | 0.122 |
S(35) = (number of non-mutant kinases with %Ctrl <35)/(number of non-mutant kinases tested)
S(10) = (number of non-mutant kinases with %Ctrl <10)/(number of non-mutant kinases tested)
S(1) = (number of non-mutant kinases with %Ctrl <1)/(number of non-mutant kinases tested)
Molecular modeling
To rationalize the improved AAK1 affinity of compounds 8g and 21b, when compared to the original hit 1, a docking study was performed using Autodock Vina.42 A control docking experiment with the original inhibitor lkb (compound 1), present in the 5L4Q PDB file, allowed to reproduce the original X-ray position very well. The best Vina docking scores are reported in Table 8. Despite the higher AAK1 affinity of compounds 8g and 21b, their Vina docking scores are worse than reference compound 1. This might be due to the fact that a rigid enzyme in the docking process is used that blocks induced-fit effects.
Table 8.
Vina docking and MM/PBSA results calculated on last 4 ns.
| Compound | Vina score (kcal/mol) |
MMa/GBSAb in kcal/mol (SDd) | MMa/PBSAc in kcal/mol (SDd) |
|---|---|---|---|
| 1 | −10.7 | −31.5 (1.6) | −0.7 (2.3) |
| 8g | −10.3 | −38.6 (2.2) | −3.0 (2.9) |
| 21b | −10.1 | −40.6 (2.2) | −3.1 (2.8) |
MM : molecular mechanics ;
GB: Generalized Born Surface Area;
PBSA: Poisson Boltzmann Surface Area;
SD: standard deviation.
Therefore, for the three docked systems with the best Vina docking score, a molecular dynamics (MD) simulation using the Amber 18 software43 was performed. Figure 11 shows an overlap of representative structures of compounds 1, 8g and 21b, as extracted from the MD simulations. The three inhibitors form a hydrogen bond between the NH of the pyrrole ring and the backbone carbonyl group of D127. The nitrogen at position 3 of the pyridine ring seems to be essential for strong AAK1 affinity. In the X-ray of AAK1 with compound 1, this nitrogen of the nicotinamide moiety makes a hydrogen bond with K74 side chain. However, this hydrogen/ionic bond is not maintained in the molecular dynamics simulations of AAK1 with compound 1, and neither with compounds 8g and 21b. On the other hand, for the three compounds, frequent van der Waals contacts between this nitrogen and the charged nitrogen in the sidechain of K74 (distances < 4 Å) are observed.
Figure 11.

AAK1 snapshot structures with 3 ligands. Compound 1 (green, lightgreen), compound 8g (magenta, rose) and compound 21b (cyan; lightblue), taken extracted from the last 4 ns of the MD trajectory as representative structure from the biggest cluster using the average linkage clustering method available in cpptraj. The AAK1 and lkb X-ray structures are also shown (orange, yellow). Image made by the Chimera software.45
For the last 4 ns of the trajectories, MM/GBSA and MM/PBSA (molecular mechanics energies with a generalized Born surface area continuum solvation model and molecular mechanics energies combined with the Poisson–Boltzmann surface area continuum solvation model, respectively)44 binding affinity calculations were conducted. No entropy contributions were calculated (Table 8). From the data in Table 8, it is clear (more from the General Born model than from the Poisson Boltzmann model) that compounds 8g and 21b are stronger AAK1 binders than reference compound 1. Both methoxy groups of compounds 8g and 21b contribute to the binding energy via van der Waals interactions with specific amino acids of AAK1. Residues L52, L62, F128, C129, R130, G131, G132, Q133, V135 and N136 are involved in binding to compound 21b. The same residues plus E50 make contact with compound 8g. On the other hand, the nitrile function of compound 1 shows clearly less van der Waals contacts with surrounding residues (L52, G132, Q133 and N136, Figure 12).
Figure 12.

AAK1 enzyme with compound 21b (same snapshot as in Figure 11). Amino acids making contact with the two methoxy groups (heavy atom distances < 4 Å) are coloured in yellow and magenta. The magenta colour represents also the residues having Van der Waals contacts with the nitrile group of compound 1. Image made by the Chimera software.45
Conclusions
AAK1 is a promising host target for the development of broad-spectrum antiviral agents. In this manuscript, the first systematic SAR study of AAK1 inhibitors as antiviral agents is presented. Starting from a known ligand with moderate AAK1 binding affinity and anti-DENV activity, a systematic SAR study was carried out. It led to the discovery of AAK1 ligands with low nM AAK1 binding affinity that display improved antiviral activity against DENV. Moreover, the optimized AAK1 inhibitors exhibit very potent activity in DENV-infected primary dendritic cells and anti-EBOV activity, supporting the potential to develop broad-spectrum antiviral agents based on AAK1 inhibition. Kinase profiling revealed that these compounds have an improved selectivity profile relative to sunitinib, yet further research is necessary to improve their kinase selectivity.
Experimental section
All commercial reagents were obtained via Acros Organics, Sigma-Aldrich, AK Scientific and Fluorochem at at least 99% purity unless indicated otherwise. Furthermore, all dry solvents were obtained via Acros Organics with an AcroSeal system and regular solvents were obtained via Fisher Scientific at technical grade. Thin layer chromatography (TLC) of the reactions was performed on silica gel on aluminum foils (60 Å pore diameter) obtained from Sigma-Aldrich and visualized using ultraviolet light. Recording of the NMR spectra was performed using a Bruker 300 MHz, 500 MHz or 600 MHz spectrometer. The chemical shifts are reported in ppm with Me4Si as the reference and coupling constants (J) are reported in hertz. Mass spectra were acquired on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Milford, MA). Samples were infused at 3 μL/min and spectra were obtained in positive or negative ionization mode with a resolution of 15000 (FWHM) using leucine enkephalin as lock mass. Purity of the compounds was analyzed on a Waters 600 HPLC system equipped with a Waters 2487 Dual λ absorbance detector set at 256 nm using a 5μm 4.6×150 mm XBridge Reversed Phase (C18) column. The mobile phase was a gradient over 30 minutes starting from 95% A and 5% B and finishing at 5% A and 95% B with a flow rate of 1 ml per minute (solvent A: MilliQ water; solvent B: acetonitrile). All synthesized final compounds had a purity of at least 95 %. Compound 8o was synthesized according to literature procedure.24
5-Bromo-3-((trimethylsilyl)ethynyl)pyridin-2-amine (3)
To a stirring suspension of 5-bromo-3-iodopyridin-2-amine 2 (520 mg, 1.74 mmol) in degassed triethylamine (10 ml) was added copper iodide (6.63 mg, 0.034 mmol) and Pd(PPh3)2Cl2 (12 mg, 0.017 mmol). The system was flushed with nitrogen and trimethylsilylacetylene (188 mg, 265 μl, 1.91 mmol) was added dropwise over 5 minutes. The reaction was allowed to stir for 3 hours at room temperature. After reaction completion, the solvent was evaporated and water was added. The resulting suspension was extracted three times using ethyl acetate. The combined organic phases were washed with water and brine, dried over MgSO4 and evaporated in vacuo. The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethylacetate (in a ratio of 80:20) as mobile phase, yielding the title compound as a yellow-beige solid (420 mg, 90%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.03 (s, 1H, ArH), 7.69 (s, 1H, ArH), 6.37 (s, 2H, NH2), 0.24 (s, 9H, Si(CH3)3 ).
5-Bromo-1H-pyrrolo[2,3-b]pyridine (4)
To a solution of 5-bromo-3-(trimethylsilyl)ethynyl)pyridin-2-amine 3 (200 mg, 0.743 mmol) in dry NMP (5 ml) was added portionwise KOtBu (100 mg, 0.891 mmol). The reaction mixture was heated to 80°C and stirred for 1 hour. After reaction completion, the mixture was extracted with water and ethyl acetate three times. The combined organic phases were washed twice with water and once with brine, dried over MgSO4 and evaporated in vacuo. The crude residue was then purified using silica gel flash column chromatography (using a mixture of heptane and ethyl acetate in a ratio of 5:1 as mobile phase) yielding the title compound as a white solid (103 mg, 70%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 10.33 (bs, 1H, NH), 8.37 (d, J = 2.1 Hz, 1H, ArH), 8.09 (d, J = 2.1 Hz, 1H, ArH), 7.37 (m, 1H, ArH), 6.47 (m, 1H, ArH).
5-Bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine (5)
5-Bromo-1H-pyrrolo[2,3-b]pyridine 4 (730 mg, 3.7 mmol) was added portionwise to a stirring solution of fuming nitric acid (2 ml) at 0°C over 10 minutes. The reaction was allowed to stir for 30 minutes at 0°C. The mixture was poured into ice water and the formed precipitate was collected via vacuum filtration. The filter cake was washed generously with water and heptane yielding the title compound as a yellow solid (853 mg, 95%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 13.48 (bs, 1H, NH), 8.87 (s, 1H, ArH), 8.51 (s, 2H, ArH). 13C NMR (75 MHz, DMSO-d6): δ (ppm) 145.98, 145.26, 132.13, 129.96, 126.32, 115.17, 114.32.
Synthesis of 3-nitro-5-aryl-pyrrolo[2,3-b]pyridines (6a-h)
General procedure
To a solution of 5-bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine 5 (1 eq) in dioxane (8 ml) was added the appropriate boronic acid (1.2 eq) and 2 ml of a K2CO3 (3 eq) solution. The system was purged three times with argon and heated to 105°C. After stirring for 10 minutes, Pd(PPh3)4 (0.1 eq) was added and the reaction was purged once more with argon. The reaction mixture was stirred at 105°C overnight. After completion, the reaction mixture was cooled to room temperature and filtered through Celite. The filtrate was extracted with water and ethyl acetate. The combined organic phases were washed with brine, dried over MgSO4 and evaporated in vacuo. Purification of the crude residue was achieved by silica gel flash column chromatography using the appropriate solvent mixture.
The following compounds were made according to this procedure
3-Nitro-5-phenyl-1H-pyrrolo[2,3-b]pyridine (6a)
The title compound was synthesized according to the general procedure using 5-bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine 5 (100 mg, 0.413 mmol), phenylboronic acid (61 mg, 0.496 mmol) and K2CO3 (171 mg, 1.24 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 90:10) as the mobile phase, yielded the desired compound as a white solid (83 mg, 84%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 13.39 (bs, 1H, NH), 8.89 (s, 1H, HetH), 8.76 (d, J = 2.1 Hz, 1H, HetH), 8.60 (d, J = 2.2 Hz, 1H, HetH), 7.79 (d, J = 7.2 Hz, 2H, o-PhH), 7.54 (t, J = 7.4 Hz, 2H, m-PhH), 7.45 (t, J = 7.3 Hz, 1H, p-PhH).
5-(4-Fluorophenyl)-3-nitro-1H-pyrrolo[2,3-b]pyridine (6b)
The title compound was synthesized according to the general procedure using 5-bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine 5 (100 mg, 0.413 mmol), 4-fluorophenylboronic acid (69 mg, 0.496 mmol) and K2CO3 (171 mg, 1.24 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 90:10) as the mobile phase yielded the desired compound as a light brown solid (98 mg, 92%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 13.37 (bs, 1H, NH), 8.90 (s, 1H, HetH), 8.82 (d, J = 2.9 Hz, 1H, HetH), 8.68 (d, J = 3.0 Hz, 1H, HetH), 8.01 (m, 4H, PhH).
5-(4-(Trifluoromethyl)phenyl)-3-nitro-1H-pyrrolo[2,3-b]pyridine (6c)
The title compound was synthesized according to the general procedure using 5-bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine 5 (100 mg, 0.413 mmol), 4-trifluoromethylphenylboronic acid (94 mg, 0.496 mmol) and K2CO3 (171 mg, 1.24 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 90:10) as the mobile phase yielded the desired compound as a white solid (110 mg, 87%). 1H NMR (300 MHz, DMSO-d6) δ (ppm) 13.45 (bs, 1H, NH), 8.93 (s, 1H, HetH), 8.84 (d, J = 3.2 Hz, 1H, HetH), 8.70 (d, J = 3.1 Hz, 1H, HetH), 8.45 (m, 1H, PhH), 8.05 (d, J = 8.8 Hz, 2H, PhH), 7.89 (d, J = 8.9 Hz, 1H, PhH).
5-(4-Methoxyphenyl)-3-nitro-1H-pyrrolo[2,3-b]pyridine (6d)
The title compound was synthesized according to the general procedure using 5-bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine 5 (100 mg, 0.413 mmol), 4-methoxyphenylboronic acid (75 mg, 0.496 mmol) and K2CO3 (171 mg, 1.24 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 90:10) as the mobile phase yielded the desired compound as a yellow solid (104 mg, 94%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 13.32 (bs, 1H, NH), 8.86 (s, 1H, HetH), 8.72 (d, J = 2.8 Hz, 1H, HetH), 8.55 (d, J = 3.0 Hz, 1H, HetH), 7.73 (d, J = 8.5 Hz, 2H, PhH), 7.10 (d, J = 8.5 Hz, 2H, PhH), 3.83 (s, 3H, OCH3).
3-Nitro-5-(p-tolyl)-1H-pyrrolo[2,3-b]pyridine (6e)
The title compound was synthesized according to the general procedure using 5-bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine 5 (100 mg, 0.413 mmol), p-tolylboronic acid (67 mg, 0.496 mmol) and K2CO3 (171 mg, 1.24 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 90:10) as the mobile phase yielded the desired compound as a white solid (83 mg, 74%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 13.35 (bs, 1H, NH), 8.88 (s, 1H, HetH), 8.74 (d, J = 3.2 Hz, 1H, HetH), 8.58 (d, J = 3.1 Hz, 1H, HetH), 7.68 (d, J = 7.8 Hz, 2H, PhH), 7.35 (d, J = 7.8 Hz, 2H, PhH), 2.38 (s, 3H, CH3).
3-Nitro-5-(m-tolyl)-1H-pyrrolo[2,3-b]pyridine (6f)
The title compound was synthesized according to the general procedure using 5-bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine 5 (100 mg, 0.413 mmol), m-tolylboronic acid (67 mg, 0.496 mmol) and K2CO3 (171 mg, 1.24 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 90:10) as the mobile phase yielded the desired compound as a white solid (83 mg, 74%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 13.35 (bs, 1H, NH), 8.88 (s, 1H, HetH), 8.74 (d, J = 3.1 Hz, 1H, HetH), 8.59 (d, J = 3.1 Hz, 1H, HetH), 7.58 (t, J = 7.2 Hz, 2H, PhH), 7.42 (t, J = 7.3 Hz, 1H, PhH), 7.26 (d, J = 3.3 Hz, 1H, PhH), 2.42 (s, 3H, CH3).
5-(3,4-Dimethoxyphenyl)-3-nitro-1H-pyrrolo[2,3-b]pyridine (6g)
The title compound was synthesized according to the general procedure using 5-bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine 5 (100 mg, 0.413 mmol), 3,4-dimethoxyphenylboronic acid (63 mg, 0.496 mmol) and K2CO3 (171 mg, 1.24 mmol). The compound precipitated as a yellow solid that was washed twice with dioxane, followed by washing with water yielding the pure title compound (88 mg, 89%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.82 (s, 1H, HetH), 8.72 (d, J = 2.1 Hz, 1H, HetH), 8.54 (d, J = 2.2 Hz, 1H, HetH), 7.30 (m, 2H, PhH), 7.10 (d, J = 8.3 Hz, 1H, PhH), 3.88 (s, 3H, OCH3), 3.82 (s, 3H, OCH3).
3-Nitro-5-(3-thienyl)-1H-pyrrolo[2,3-b]pyridine (6h)
The title compound was synthesized according to the general procedure using 5-bromo-3-nitro-1H-pyrrolo[2,3-b]pyridine 5 (100 mg, 0.413 mmol), 3-thienylboronic acid (69 mg, 0.496 mmol) and K2CO3 (171 mg, 1.24 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 90:10) as the mobile phase yielded the title compound as a brown solid (97 mg, 96%). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 13.41 (bs, 1H, NH), 8.89 (d, J = 3.0 Hz, 1H, HetH), 8.83 (d, J = 3.1 Hz, 1H, HetH), 8.57 (d, J = 3.0 Hz, 1H, HetH), 7.69 (m, 2H, ThH) ppm, 7.22 (t, J = 4.4 Hz, 1H, ThH).
Synthesis of 5-aryl-3-amino- and 5-aryl-3-N-acylamino pyrrolo[2,3-b]pyridines (7a-f and 8a-o)
General procedure
To a solution of a 5-aryl-3-nitro-pyrrolo[2,3-b]pyridine 6a-h (100 mg) in THF (5 ml) was added a slurry of Raney Nickel in water in catalytic amounts. The reaction vessel was flushed three times with hydrogen gas and was stirred under a hydrogen atmosphere for 3 – 4 hours. Upon completion of the reaction, the catalyst was removed and the solvent evaporated in vacuo. The crude residue was used in the next reaction without any purification, due to the rapid decomposition of the 3-amino-pyrrolo[2,3-b]pyridine intermediates 7a-h. To a solution of compounds 7a-h (1 eq) in dry pyridine (3 ml) was added a solution of nicotinoyl chloride hydrochloride (1.2 eq) in CH2Cl2 (2 ml). The reaction was allowed to stir at room temperature for 3 hours. The solvent was evaporated in vacuo. THF (5 ml) was added and the resulting solution was stirred for 5 minutes, followed by the addition of a 1M NaOH solution in water (5 ml). The resulting suspension was stirred for an additional 10 minutes. The precipitate was collected via vacuum filtration and purified using flash column chromatography with the appropriate solvent mixture as mobile phase. Compounds 8a-h were synthesized according to this procedure.
N-(5-Phenyl-1H-pyrrolo[2,3-b]pyridin-3-yl)nicotinamide (8a)
The title compound was synthesized according to the general procedure, using 3-nitro-5-phenyl-1H-pyrrolo[2,3-b]pyridine 6a (84 mg, 0.351 mmol). Purification by silica gel flash column chromatography (eluting with a mixture of dichloromethane and methanol in a gradient gradually ranging from 98:2 to 90:10) afforded the title compound as a dark brown solid (47 mg, 36%). Purity of 99%. 1H NMR (300 MHz, DMSO-d6) δ 11.62 (bs, 1H, NH), 10.58 (bs, 1H, NH), 9.19 (s, 1H, HetH), 8.79 (d, J = 3.0 Hz, 1H, HetH), 8.66 (s, 1H, ArH), 8.58 (s, 1H, ArH), 8.36 (d, J = 6.1 Hz, 1H, ArH), 7.98 (s, 1H, ArH), 7.75 (d, J = 8.8 Hz, 2H, ArH), 7.60 (t, J = 6.0 Hz, 1H, ArH), 7.51 (t, J = 7.3 Hz, 2H, ArH), 7.38 (t, J = 7.4 Hz, 1H, ArH) ppm. 13C NMR (75 MHz, DMSO-d6) δ 163.46, 152.01, 148.87, 145.52, 142.23, 139.16, 135.60, 131.63, 130.52, 129.13, 127.06, 126.93, 125.49, 123.61, 117.52, 114.10, 113.45 ppm. HRMS m/z [M+H]+ calcd for C19H14N4O 315.12403, found 315.1237.
N-[5-(4-Fluorophenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]nicotinamide (8b)
The title compound was synthesized according to the general procedure using 3-nitro-5-(4-fluorophenyl)-1H-pyrrolo[2,3-b]pyridine 6b (98 mg, 0.381 mmol). Purification by silica gel flash column chromatography (using a mixture of dichloromethane/methanol in a ratio gradually ranging from 98:2 to 90:10 as mobile phase) afforded the title compound as a brown solid (44 mg, 34%). Purity of 99%. 1H NMR (300 MHz, DMSO-d6) δ 11.63 (bs, 1H, NH), 10.57 (bs, 1H, NH), 9.17 (s, 1H, HetH), 8.78 (d, J = 5.8 Hz, 1H, ArH), 8.62 (s, 1H, HetH), 8.56 (s, 1H, ArH), 8.36 (d, J = 8.9 Hz, 1H, ArH), 7.98 (d, J = 3.2 Hz, 1H,ArH), 7.77 (t, J = 7.6 Hz, 2H, ArH), 7.61 (d, J = 9.0 Hz, 1H, ArH), 7.35 (t, J = 7.5 Hz, 2H, ArH) ppm. 13C NMR (75 MHz, DMSO-d6) δ 162.24, 161.77 (d, JCF = 242 Hz), 150.23, 148.62, 148.62, 145.58, 142.05, 138.10, 135.66, 128.86 (d, JCF = 8.0 Hz), 126.77, 126.72, 125.74, 122.38, 117.74, 116.00, 115.85 (d, JCF = 20.4), 113.75, 113.47 ppm. HRMS m/z [M+H]+ calcd for C19H13N4OF 333.11460, found 333.1145. HRMS m/z [M+Na]+ calcd for C19H13N4OF 355.09658, found 355.0950.
N-[5-(4-Trifluoromethylphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]nicotinamide (8c)
The title compound was synthesized according to the general procedure using 3-nitro-5-(4-trifluoromethylphenyl)-1H-pyrrolo[2,3-b]pyridine 6c (116 mg, 377.56 μmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and methanol (in a ratio gradually ranging from 98:2 to 90:10) as mobile phase afforded the title compound as a white solid (49 mg, 36%). Purity of 99%. 1H NMR (300 MHz, DMSO-d6) δ 11.73 (bs, 1H, NH), 10.62 (bs, 1H, NH), 9.19 (d, J = 3.2 Hz, 1H, HetH), 8.78 (t, J = 6.1 Hz, 2H, ArH), 8.67 (d, J = 3.1 Hz, 1H, HetH), 8.36 (d, J = 3.0 Hz, 1H, ArH), 7.99 (d, J = 8.9 Hz, 3H, PhH), 7.87 (d, J = 9.0 Hz, 2H, PhH), 7.61 (q, J = 5.9 Hz, 1H, ArH) ppm. 13C NMR (75 MHz, DMSO-d6) δ 163.52, 152.05, 148.85, 145.88, 143.25, 142.35, 135.58, 130.46, 127.48, 126.09, 123.61, 122.80, 117.84, 114.28, 113.52 ppm; missing peaks were observed in an APT spectrum. HRMS m/z [M+H]+ calcd for C20H13N4F3O 383.11141; found 383.1114.
N-[5-(4-Methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]nicotinamide (8d)
The title compound was synthesized according to the general procedure using 3-nitro-5-(4-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridine 6d (105 mg, 0.390 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and methanol (in a ratio gradually ranging from 98:2 to 90:10) as mobile phase afforded the title compound as a pink solid (43 mg, 34%). Purity of 99% 1H NMR (300 MHz, DMSO-d6) δ 11.56 (bs, 1H, NH), 10.56 (bs, 1H, NH), 9.18 (s, 1H, ArH), 8.78 (d, J = 6.1 Hz, 1H, ArH), 8.58 (s, 1H, ArH), 8.52 (s, 1H, ArH), 8.36 (d, J = 9.0 Hz, 1H, PhH), 7.96 (s, 1H, ArH), 7.62 (m, 3H, ArH), 7.08 (d, J = 8.9 Hz, 2H, PhH), 3.82 (s, 3H, OCH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 163.45, 158.74, 151.99, 148.85, 145.22, 141.99, 135.59, 131.54, 130.50, 128.00, 127.55, 124.87, 123.60, 117.43, 114.64, 113.94, 113.45, 55.34 ppm. HRMS m/z [M+H]+ calcd for C20H16N4O2 345.13459, found 345.1343. HRMS m/z [M+Na]+ calcd for C20H16N4O2 367.11656; found 367.1153.
N-(5-(p-Tolyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)nicotinamide (8e)
The title compound was synthesized according to the general procedure using 3-nitro-5-(p-tolyl)-1H-pyrrolo[2,3-b]pyridine 6e (96 mg, 0.379 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and methanol (in a ratio gradually ranging from 98:2 to 90:10) as mobile phase afforded the title compound as a beige solid (48 mg, 37%). Purity of 98%. 1H NMR (300 MHz, DMSO-d6) δ 11.60 (bs, 1H, NH), 10.59 (bs, 1H, NH), 9.19 (s, 1H, ArH), 8.78 (d, J = 2.9 Hz, 1H, ArH), 8.63 (s, 1H, ArH), 8.56 (s, 1H, ArH), 8.36 (d, J = 6.0 Hz, 1H, ArH), 7.99 (s, 1H, ArH), 7.59 (m, 3H, ArH), 7.31 (d, J = 9.3 Hz, 2H, ArH), 2.35 (s, 3H, CH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 163.49, 151.99, 148.85, 145.41, 142.10, 136.28, 136.22, 135.59, 130.51, 129.73, 127.68, 126.73, 125.12, 123.60, 117.50, 114.03, 113.47, 20.77 ppm. HRMS m/z [M+H]+ calcd for C20H16N4O 329.13967; found 329.1398.
N-[5-(m-Tolyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]nicotinamide (8f)
The title compound was synthesized according to the general procedure using 3-nitro-5-(m-tolyl)-1H-pyrrolo[2,3-b]pyridine 6f (77 mg, 0.304 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and methanol (in a ratio gradually ranging from 98:2 to 90:10) as mobile phase afforded the title compound as a beige solid (22 mg, 25% yield).Purity of 98%. 1H NMR (300 MHz, DMSO-d6) δ 11.61 (bs, 1H, NH), 10.59 (bs, 1H, NH), 9.19 (s, 1H, ArH), 8.79 (d, J = 2.8 Hz, 1H, ArH), 8.64 (s, 1H, ArH), 8.57 (s, 1H, ArH), 8.37 (d, J = 9.0 Hz, 1H, ArH), 7.99 (s, 1H, ArH), 7.59 (m, 3H, CH3), 7.39 (t, J = 7.4 Hz, 1H), 7.19 (d, J = 6.1 Hz, 1H), 2.41 (s, 3H) ppm. 13C NMR (75 MHz, DMSO-d6) δ 163.49, 151.99, 148.84, 145.47, 142.25, 139.08, 138.27, 135.59, 130.50, 129.02, 127.84, 127.71, 127.53, 125.39, 124.06, 123.60, 117.47, 114.07, 113.42, 21.28 ppm. HRMS m/z [M+H]+ calcd for C20H16N4O 329.13967; found 329.1397.
N-[5-(3,4-Dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]nicotinamide (8g)
The title compound was synthesized according to the general procedure using 3-nitro-5-(3,4-dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine 6g (80 mg, 0.297 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and methanol (in a ratio gradually ranging from 98:2 to 90:10) as mobile phase afforded the title compound as a beige solid (56 mg, 50%). Purity of 97%. 1H NMR (300 MHz, DMSO-d6) δ 11.57 (bs, 1H, NH), 10.56 (bs, 1H, NH), 9.20 (s, 1H, ArH), 8.78 (d, J = 6.1 Hz, 1H, ArH), 8.58 (d, J = 3.2 Hz, 2H, ArH), 8.37 (d, J = 9.1 Hz, 1H, ArH), 7.97 (s, 1H, ArH), 7.60 (t, J = 6.0 Hz, 1H, ArH), 7.27 (t, J = 7.4 Hz, 2H, ArH), 7.09 (d, J = 9.0 Hz, 1H, ArH), 3.88 (s, 3H, OCH3), 3.81 (s, 3H, OCH3) ppm.13C NMR (75 MHz, DMSO-d6) δ 163.45, 152.00, 149.40, 148.88, 148.43, 145.28, 142.26, 135.61, 132.04, 130.51, 127.84, 124.96, 123.61, 119.23, 117.47, 113.93, 113.39, 112.70, 111.09, 55.92, 55.82 ppm. HRMS m/z [M+H]+ calcd for C21H18N4O3 375.14515, found 375.1447, HRMS m/z [M+Na]+ calcd for C21H18N4O3 397.12713; found 397.1264.
N-[5-(3-Thienyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]nicotinamide (8h)
The title compound was synthesized according to the general procedure using 3-nitro-5-(3-thienyl)-1H-pyrrolo[2,3-b]pyridine 6h (97 mg, 0.395 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and methanol (in a ratio gradually ranging from 98:2 to 90:10) as mobile phase, afforded the title compound as a brown solid (48 mg, 37%). Purity of 99%. 1H NMR (300 MHz, DMSO-d6) δ 11.58 (bs, 1H, NH), 10.54 (bs, 1H, NH), 9.20 (d, J = 2.8 Hz, 1H, ArH), 8.79 (d, J = 2.9 Hz, 1H, ArH), 8.67 (d, J = 2.9 Hz, 1H, ArH), 8.64 (d, J = 3.0 Hz, 1H, ArH), 8.36 (m, 1H, ArH), 7.93 (s, 1H, ArH), 7.84 (s, 1H, ArH), 7.71 (m, 1H, ArH), 7.60 (t, J = 5.9 Hz, 2H, ArH) ppm. 13C NMR (75 MHz, DMSO-d6) δ 163.48, 152.02, 148.86, 145.30, 142.03, 140.21, 135.58, 130.49, 127.30, 126.37, 124.61, 123.60, 123.18, 119.78, 117.63, 113.94, 113.49 ppm. HRMS m/z [M+H]+ calcd for C17H12N4OS 321.08045; found 321.0804.
N-(5-(3,4-Dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzamide (8i)
To a solution of 3-nitro-5-(3,4-dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine 6g (100 mg, 0.334 mmol) in THF (4 ml) was added a catalytic amount of a slurry of Raney Nickel in water. The vessel was flushed three times with hydrogen gas and kept under a hydrogen atmosphere for 4 hours. After complete conversion, the catalyst was removed and the solvent was evaporated in vacuo. The crude product was immediately used for further reaction without purification. To a solution of the crude residue from the previous reaction in pyridine (5 ml) was added a solution of benzoyl chloride (47 μl, 0.401 mmol) in dichloromethane (0.5 ml). The reaction was stirred overnight at room temperature. After reaction completion, the solvent was evaporated in vacuo and the crude residue was extracted three times with water and ethyl acetate. The combined organic layers were dried over MgSO4 and evaporated to dryness. Purification was achieved by silica gel flash column chromatography (using a mixture of dichloromethane and methanol in a ratio of 97:3 as mobile phase), followed by precipitation of the residue from acetone. The precipitate was collected via filtration yielding the title compound as a white solid (27 mg, 22%). Purity of 98%. 1H NMR (300 MHz, DMSO-d6) δ 11.52 (bs, 1H, NH), 10.37 (bs, 1H, NH), 8.56 (d, J = 6.52, 2H, ArH), 8.00 (m, 3H, ArH), 7.56 (d, J = 7.6 Hz, 3H, ArH), 7.25 (m, 2H, ArH), 7.08 (d, J = 8.2 Hz, 1H, ArH), 3.87 (s, 3H, OCH3), 3.80 (s, 3H, OCH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 165.07, 149.30, 148.30, 145.25, 142.12, 134.88, 132.01, 131.42, 128.48, 127.87, 127.70, 125.04, 119.14, 117.27, 114.22, 113.44, 112.52, 110.87, 55.83, 55.74 ppm. HRMS m/z [M+H]+ calcd for C22H19N3O3 374.14990; found 374.1493.
Synthesis of 5-(3,4-dimethoxyphenyl)-3-N-acylamino pyrrolo[2,3-b]pyridines
General procedure
3-Nitro-5-(3,4-dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine 6g (100 mg, 0.334 mmol) was hydrogenated with a catalytic amount of Raney Nickel as a slurry in water (slurry in water? x mg/ml) in THF (5 ml) for 3 hours. The solvents were evaporated in vacuo yielding crude 3-amino-5-(3,4-dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine which was used in the next reaction without further purification (90 mg, 99%). To a solution of 3-amino-5-(3,4-dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine (90 mg, 0.334 mmol) in DMF (3 ml) was added the appropriate carboxylic acid (1 eq.), benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP, 177 mg, 0.401 mmol) and triethylamine (140 μl, 1 mmol). The mixture was stirred overnight. When the reaction reached completion, water was added and the mixture was extracted three times with ethyl acetate. The combined organic layers were washed with brine, dried over Na2SO4 and evaporated in vacuo. The crude residue was purified by silica gel flash chromatography using an appropriate solvent system as mobile phase. An additional purification was performed by preparative TLC using a mixture of dichloromethane and acetone (in a ratio of 8:2) as eluent. The following compounds were prepared according to this procedure.
N-(5-(3,4-Dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-fluorobenzamide (8j)
The title compound was synthesized according to the general procedure using 4-fluorobenzoic acid (47 mg, 0.334 mmol). Purification by silica gel flash column chromatography (using a mixture of dichloromethane and methanol as mobile phase in a ratio of 99:1) yielded the desired compound as a white solid (56 mg, 43%). Purity of 99%. 1H NMR (300 MHz, DMSO-d6) δ 11.53 (bs, 1H, NH), 10.39 (bs, 1H, NH), 8.56 (d, J = 8.5 Hz, 2H, ArH), 8.12 (m, 2H, ArH), 7.93 (s, 1H, ArH), 7.38 (t, J = 8.7 Hz, 2H, ArH), 7.26 (m, 2H, ArH), 7.08 (d, J = 8.3 Hz, 1H, ArH), 3.87 (s, 3H, OCH3), 3.81 (s, 3H, OCH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 164.07 (JCF = 245 Hz), 163.93, 149.30, 148.30, 145.25, 142.12, 131.99, 131.27, 130.57 (JCF = 8.97 Hz), 127.69, 125.05, 119.12, 117.35, 115.39 (JCF = 22 Hz), 114.11, 113.46, 112.53, 110.88, 55.84, 55.75 ppm. HRMS m/z [M+H]+ calcd for C22H18FN3O3 392.14048; found 392.1399.
N-(5-(3,4-Dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-methoxybenzamide (8k)
The title compound was synthesized according to the general procedure using 4-methoxybenzoic acid (51 mg, 0.334 mmol). Purification by silica gel flash column chromatography (using a mixture of dichloromethane and methanol as mobile phase in a ratio of 99:1) yielded the desired compound as a white solid (64 mg, 47%). Purity of 97%. 1H NMR (600 MHz, DMSO-d6) δ 11.46 (s, 1H, NH), 10.16 (s, 1H, NH), 8.55 (d, J = 1.92 Hz, 1H, ArH), 8.53 (d, J = 2.22 Hz, 1H, ArH), 8.01 (m, 2H, ArH), 7.90 (d, J = 2.46 Hz, 1H, ArH), 7.28 (d, J = 2.16 Hz, 1H, ArH), 7.22 (dd, J = 8.22, 2.16 Hz, ArH), 7.08 (m, 3H, ArH), 3.87 (s, 3H, OCH3), 3.85 (s,3H, OCH3), 3.80 (s, 3H, OCH3) ppm. 13C NMR (150 MHz, DMSO-d6) δ 164.43, 161.77, 149.25, 148.24, 145.21, 142.00, 131.99, 129.69, 127.58, 126.92, 124.98, 119.08, 117.14, 114.27, 113.65, 113.48, 112.48, 110.82, 55.78, 55.70, 55.50 ppm. HRMS m/z [M+H]+ calcd for C23H21N3O4 404.16047; found 404.1603.
N-(5-(3,4-Dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-2-phenylacetamide (8m)
The title compound was synthesized according to the general procedure using phenylacetic acid (45 mg, 0.334 mmol). Purification by silica gel flash column chromatography (using a mixture of dichloromethane and methanol as mobile phase in a ratio of 99:1) yielded the desired compound as a white solid (49 mg, 54%). Purity of 99%.1H NMR (300 MHz, DMSO-d6) δ 11.36 (s, 1H, NH), 10.45 (s, 1H, NH), 8.53 (m, 2H, ArH), 7.79 (d, J = 2.3 Hz, 1H, ArH), 7.31 (m, 7H, ArH), 7.09 (d, J = 8.4 Hz, 1H, ArH), 3.88 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 3.76 (s, 2H, CH2) ppm. 13C NMR (75 MHz, DMSO-d6) δ 168.21, 157.37, 149.33, 148.30, 145.01, 142.09, 136.54, 131.94, 129.28, 128.38, 127.55, 126.56, 124.40, 119.00, 115.78, 114.37, 112.78, 112.55, 110.80, 55.83, 55.76, 42.41 ppm. HRMS m/z [M+H]+ calcd for C23H21N3O3 388.16555; found 388.1660.
N-(5-(3,4-Dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)cyclopentanecarboxamide (8n)
To a solution of 3-nitro-5-(3,4-dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine 6g (100 mg, 0.334 mmol) in THF (5 ml) was added a catalytic amount of a slurry of Raney Nickel in water. The vessel was flushed three times with hydrogen gas and kept under a hydrogen atmosphere for 4 hours. After complete conversion, the catalyst was removed and the solvent was evaporated in vacuo. The crude product was immediately used for further reaction without purification. To a solution of the crude residue in pyridine (5 ml) was added a solution of cyclopentylcarbonyl chloride (46 μl, 0.401 mmol) in DCM (2 ml). The reaction was stirred overnight at room temperature. After reaction completion, the solvent was evaporated in vacuo and the crude residue was extracted three times with water and ethyl acetate. The combined organic layers were dried over MgSO4 and evaporated to dryness. Purification by silica gel flash column chromatography (using a mixture of dichloromethane and methanol in a ratio of 97:3 as mobile phase) yielded the title compound as a white solid (39 mg, 32%). Purity of 99% 1H NMR (300 MHz, DMSO-d6) δ 11.33 (bs, 1H, NH), 9.96 (bs, 1H, NH), 8.51 (s, 1H, ArH), 8.44 (s, 1H, ArH), 7.82 (d, J = 2.2 Hz, 1H, ArH), 7.23 (m, 2H, ArH), 7.08 (d, J = 8.3 Hz, 1H, ArH), 3.88 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 2.90 (m, 1H, CH2), 1.77 (m, 8H, CH2) ppm. 13C NMR (75 MHz, DMSO-d6) δ 173.48, 149.31, 148.30, 145.00, 142.05, 132.01, 127.51, 124.19, 119.05, 115.69, 114.50, 112.71, 112.55, 110.80, 55.82, 55.75, 44.55, 30.46, 25.85. HRMS m/z [M+H]+ calcd for C21H23N3O3 366.18120; found 366.1808.
N-(5-(3,4-dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzenesulfonamide (8p)
To a solution of 3-nitro-5-(3,4-dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine (100 mg, 0.334 mmol) in THF (5 ml) was added a catalytic amount of a slurry of Raney Nickel in water. The vessel was flushed with hydrogen gas three times and kept under a hydrogen atmosphere for 4 hours. After complete conversion, the catalyst was removed and the solvent evaporated. The crude was immediately used in the next reaction without further purification because of the rapid decomposition of the amine. To a solution of the crude from the previous reaction in pyridine (5 ml) was added a solution of benzensulfonyl chloride (47 μl, 0.368 mmol) in DCM (1 ml). The reaction was stirred overnight at room temperature. After reaction completion the solvent was evaporated and the crude was extracted with water and ethyl acetate three times. The collected organics were dried with MgSO4 and evaporated to dryness. Flash column chromatography (using a mixture of dichloromethane and methanol as mobile phase in a ratio of 97:3) yielded the desired compound as an off-white solid (34 mg, 25%). Purity of 98%. 1H NMR (600 MHz, DMSO-d6) δ 11.64 (bs, 1H, NH), 9.87 (bs, 1H, NH), 8.42 (d, J = 1.92 Hz, 1H, ArH), 7.73 (s, 1H, ArH), 7.72 (s, 2H, ArH), 7.55 (t, J = 7.5 Hz, 1H, ArH), 7.51 (t, J = 7.74 Hz, 2H, ArH), 7.20 (d, J = 2.52 Hz, 1H, ArH), 7.05 (m, 2H, ArH), 6.99 (d, J = 1.74 Hz, 1H, ArH), 3.85 (s, 3H, OCH3), 3.80 (s, 3H, OCH3) ppm. 13C NMR (150 MHz, DMSO-d6) δ 149.18, 148.27, 145.66, 142.15, 140.05, 132.66, 131.55, 129.03, 128.23, 126.96, 123.76, 121.94, 118.96, 115.65, 113.91, 112.46, 111.06, 110.50, 55.70, 55.68 ppm. HRMS m/z [M+H]+ calcd for C21H19N3O4S 408.10234; found 408.1024.
5-Bromo-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde (9)
To a solution of 5-bromo-1H-pyrrolo[2,3-b]pyridine 4 (500 mg, 2.54 mmol) in a mixture of water and acetic acid (in a ratio of 3:1, 10 ml) was added hexamine (712 mg, 5.08 mmol). The reaction was heated to 120°C and refluxed overnight. The formed precipitate was filtered off, affording the title compound (380 mg, 67%). 1H NMR (300 MHz, DMSO-d6) δ 12.93 (bs, 1H, NH), 9.93 (s, 1H, CHO), 8.55 (s, 1H, HetH), 8.53 (d, J = 2.3 Hz, 1H, HetH), 8.46 (d, J = 2.3 Hz, 1H, HetH) ppm. 13C NMR (75 MHz, DMSO-d6) δ 185.58, 147.92, 145.09, 139.90, 131.01, 118.32, 116.04, 113.86 ppm.
5-Bromo-1-tosyl-pyrrolo[2,3-b]pyridine-3-carbaldehyde (10)
To a suspension of 60% NaH on mineral oil (80 mg, 2 mmol) in dry THF (6 ml) was added 5-bromo-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde 9 (300 mg, 1.33 mmol) at 0°C. After stirring for 20 minutes at room temperature, tosyl chloride (381 mg, 2 mmol) was added and the resulting solution was stirred for another 3 hours at room temperature. After reaction completion, the solvent was evaporated in vacuo and the crude residue was extracted with water and dichloromethane. The combined organic layers were dried over MgSO4 and evaporated in vacuo, affording the title compound (460 mg, 91%). 1H NMR (300 MHz, DMSO-d6) δ 10.03 (s, 1H, CHO), 9.02 (s, 1H, HetH), 8.59 (d, J = 2.2 Hz, 1H, HetH), 8.55 (d, J = 2.2 Hz, 1H, HetH), 8.07 (d, J = 8.4 Hz, 2H, TsH), 7.48 (d, J = 8.2 Hz, 2H, TsH), 2.37 (s, 3H, CH3) ppm.
5-(3,4-Dimethoxyphenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde (11)
To a solution of 5-bromo-1-tosyl-pyrrolo[2,3-b]pyridine-3-carbaldehyde 10 (240 mg, 0.633 mmol) in a mixture of toluene and ethanol (in a ratio of 3:1, 4 ml) was added 3,4-dimethoxyphenylboronic acid (138 mg, 0.759 mmol) and a 2M solution of K2CO3 (950 μl, 1.90 mmol). The system was purged with argon and Pd(PPh3)4 (2mol%) was added. The reaction was heated to 105°C and was stirred for 4 hours. After completion, the solvent was evaporated in vacuo and the residue was extracted with water and ethyl acetate. Purification by silica gel flash chromatography using a mixture of heptane and ethyl acetate (in a ratio of 7:3) as mobile phase afforded the title compound (160 mg, 58%). 1H NMR (300 MHz, DMSO-d6) δ 10.09 (s, 1H, CHO), 9.00 (s, 1H, HetH), 8.76 (s, 1H, HetH), 8.55 (s, 1H, HetH), 8.11 (d, J = 7.7 Hz, 2H, TsH), 7.49 (d, J = 7.4 Hz, 2H, TsH), 7.24 (m, 2H, PhH), 7.07 (d, J = 8.2 Hz, 1H, PhH), 3.85 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 2.37 (s, 3H, CH3) ppm.
(5-(3,4-Dimethoxyphenyl)-1-tosyl-pyrrolo[2,3-b]pyridin-3-yl)(phenyl)methanol (12)
To a solution of 5-(3,4-dimethoxyphenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde 11 (160 mg, 0.367 mmol) in dry THF (5 ml) at 0°C was added a 3M solution of phenylmagnesium bromide in diethylether (159 μl, 0.477 mmol). The reaction mixture was stirred for 1 hour at 0°C. After reaction completion, the mixture was quenched with a saturated NH4Cl solution and extracted with water and ethyl acetate. The combined organic layers were dried over Na2SO4 and evaporated to dryness yielding the title compound (130 mg, 69%). 1H NMR (300 MHz, DMSO-d6) δ 8.62 (s, 1H, ArH), 7.99 (d, J = 8.8 Hz, 3H, ArH), 7.70 (s, 1H, ArH), 7.59 – 6.99 (m, 10H, ArH), 6.13 (d, J = 4.4 Hz, 1H, ArH), 6.02 (s, 1H, ArH), 3.82 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 2.34 (s, 3H, CH3) ppm.
(5-(3,4-Dimethoxyphenyl)-1-tosyl-pyrrolo[2,3-b]pyridin-3-yl)(phenyl)methanone (13)
To a solution of (5-(3,4-dimethoxyphenyl)-1-tosyl-pyrrolo[2,3-b]pyridin-3-yl)(phenyl)methanol 12 (130 mg, 0.253 mmol) in dichloromethane (8 ml) was added manganese dioxide (220 mg, 2.53 mmol) and the reaction was stirred overnight at room temperature. After completion, the mixture was filtered through Celite and the filtrate was evaporated to dryness. The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 6:4) as mobile phase yielding the title compound (64 mg, 49%). 1H NMR (300 MHz, DMSO-d6) δ 8.78 (d, J = 2.1 Hz, 1H, HetH), 8.64 (d, J = 2.1 Hz, 1H, HetH), 8.33 (s, 1H), 8.16 (d, J = 8.3 Hz, 2H, ArH), 7.96 (d, J = 7.2 Hz, 2H, ArH), 7.70 (m, 3H, ArH), 7.47 (d, J = 8.2 Hz, 2H, ArH), 7.25 (m, 2H, ArH), 7.08 (d, J = 8.4 Hz, 1H, ArH), 3.86 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 2.37 (s, 3H, CH3) ppm.
(5-(3,4-Dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)(phenyl)methanone (14)
To a solution of (5-(3,4-dimethoxyphenyl)-1-tosyl-pyrrolo[2,3-b]pyridin-3-yl)(phenyl)methanone 13 (64 mg, 0.124 mmol) in ethanol (10 ml) was added KOH (35 mg, 0.624 mmol) and the mixture was heated to 80°C. The reaction was stirred for 3 hours. After reaction completion, the solvent was evaporated in vacuo and the crude residue was partitioned between water and ethyl acetate. The combined organic layers were dried over MgSO4 and evaporated to dryness, yielding the title compound as a white solid (34 mg, 76%). Purity of 95%. 1H NMR (300 MHz, DMSO-d6) δ 12.74 (bs, 1H, NH), 8.67 (s, 2H, ArH), 8.12 (s, 1H, ArH), 7.84 (d, J = 6.8 Hz, 2H, ArH), 7.61 (m, 3H, ArH), 7.25 (m, 2H, ArH), 7.10 (d, J = 8.3 Hz, 1H, ArH), 3.88 (s, 3H, OCH3), 3.82 (s, 3H, OCH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 190.00, 149.38, 148.64, 148.45, 143.66, 139.77, 136.58, 131.13, 127.30, 119.42, 118.86, 113.88, 112.59, 110.91, 55.77, 55.75 ppm. HRMS m/z [M+H]+ calcd for C22H18N2O3 359.1390; found 359.1393.
5-Bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine (15)
To a solution of 5-bromo-1H-pyrrolo[2,3-b]pyridine 4 (1.00 g, 5.08 mmol) in acetone (16 ml) was added portionwise N-iodosuccinimide (1.26 g, 5.58 mmol). The reaction mixture was stirred for 2 hours at room temperature. The formed precipitate was collected via filtration, yielding the title compound as an off-white solid (1.50 g, 92%). 1H NMR (300 MHz, DMSO-d6) δ 12.36 (s, 1H, NH), 8.32 (d, J = 2.0 Hz, 1H, HetH), 7.87 (d, J = 1.7 Hz, 1H, HetH), 7.81 (d, J = 2.4 Hz, 1H, HetH) ppm.
5-Bromo-3-iodo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (16)
To a solution of 5-bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine 15 (300 mg, 0.929 mmol) in dry THF (10 ml) was added portionwise NaH (41 mg, 1.02 mmol) at 0°C. After stirring for 20 minutes at room temperature, tosyl chloride (230 mg, 1.21 mmol) was added and the reaction was allowed to stir for another 2 hours at room temperature. After reaction completion, the reaction was quenched with water and partitioned between water and ethyl acetate. The combined organic phases were dried over MgSO4 and concentrated in vacuo, yielding the title compound as a white solid (420 mg, 95%). 1H NMR (300 MHz, DMSO-d6) δ 8.51 (d, J = 2.1 Hz, 1H, HetH), 8.22 (s, 1H, HetH), 8.00 (m, 3H, HetH/TsH), 7.43 (d, J = 8.5 Hz, 2H, TsH), 2.34 (s, 3H, CH3) ppm.
5-Bromo-3-phenyl-1-tosyl-1H-pyrrolo[2,3-b]pyridine (17)
To a solution of 5-bromo-3-iodo-1-tosyl-1H-pyrrolo[2,3-b]pyridine 16 (200 mg, 0.419 mmol) in in mixture of toluene and ethanol (in a ratio of 3:1) was added phenylboronic acid (51 mg, 0.419 mmol) and a 2M solution of K2CO3 (420 μl, 0.838 mmol). The reaction was flushed with argon three times, followed by the addition of Pd(PPh3)4 (10 mg, 0.008 mmol). The reaction was stirred at 90°C for 3 hours. After reaction completion, the reaction was filtered through Celite and the filtrate was washed with water and ethyl acetate. The combined organic phases were dried over MgSO4 and evaporated in vacuo. Purification by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 8:2) as mobile phase yielded the title compound as a white solid (150 mg, 84%). 1H NMR (300 MHz, DMSO-d6) δ 8.54 (d, J = 2.1 Hz, 1H, HetH), 8.50 (d, J = 2.1 Hz, 1H, HetH), 8.29 (s, 1H, HetH), 8.04 (d, J = 8.4 Hz, 2H, TsH), 7.79 (d, J = 7.2 Hz, 2H, TsH), 7.45 (m, 5H, PhH), 2.35 (s, 3H, CH3) ppm.
5-(3,4-Dimethoxyphenyl)-3-phenyl-1-tosyl-1H-pyrrolo[2,3-b]pyridine (18)
To a solution of 5-bromo-3-phenyl-1-tosyl-1H-pyrrolo[2,3-b]pyridine (150 mg, 0.351 mmol) in a mixture of toluene and ethanol (in a ratio of 3:1) was added 3,4-dimethoxyphenylboronic acid (77 mg, 0.421 mmol) and a 2M solution of K2CO3 in water (351 μl, 0.702 mmol). The system was flushed with argon and Pd(PPh3)4 (2 mol%) was added. The reaction was stirred for 4 hours at 105°C. Upon reaction completion, the solvents were evaporated in vacuo and the crude residue was extracted with water and ethyl acetate. Purification by silica gel flash chromatography using a mixture of heptane and ethyl acetate (in a ratio of 8:2) as mobile phase yielded the desired compound (100 mg, 59%). 1H NMR (300 MHz, DMSO-d6) δ 8.71 (d, J = 2.0 Hz, 1H, HetH), 8.37 (d, J = 2.0 Hz, 1H, HetH), 8.23 (s, 1H, HetH), 8.08 (d, J = 8.4 Hz, 2H, ArH), 7.85 (d, J = 7.2 Hz, 2H, ArH), 7.46 (m, 5H, ArH), 7.29 (m, 2H, ArH), 7.06 (d, J = 8.3 Hz, 1H, ArH), 3.84 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 2.35 (s, 3H, CH3) ppm.
5-(3,4-Dimethoxyphenyl)-3-phenyl-1H-pyrrolo[2,3-b]pyridine (19)
To a solution of 5-(3,4-dimethoxyphenyl)-3-phenyl-1-tosyl-1H-pyrrolo[2,3-b]pyridine 18 (100 mg, 0.309 mmol) in ethanol (5 ml) was added KOH (87 mg, 1.55 mmol). The reaction was allowed to stir for 2 hours at 80°C. After completion, the solvent was evaporated in vacuo and the crude residue was purified by silica gel flash chromatography using a mixture of heptane and ethyl acetate (in a ratio of 7:3) as mobile phase, affording the title compound (49 mg, 72%). Purity of 97% 1H NMR (300 MHz, DMSO-d6) δ 11.98 (s, 1H, NH), 8.56 (s, 1H, ArH), 8.39 (s, 1H, ArH), 7.90 (d, J = 2.3 Hz, 1H, ArH), 7.79 (d, J = 7.5 Hz, 2H, ArH), 7.45 (t, J = 7.6 Hz, 2H, ArH), 7.28 (m, 3H, ArH), 7.06 (d, J = 8.3 Hz, 1H, ArH), 3.87 (s, 3H, OCH3), 3.80 (s, 3H, OCH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 149.32, 148.56, 148.37, 142.16, 135.18, 132.07, 129.15, 129.06, 126.57, 125.83, 124.63, 119.50, 117.37, 114.73, 112.54, 111.27, 55.84, 55.76 ppm. HRMS m/z [M+H]+ calcd for C21H18N2O2 331.1440; found 331.1437.
Synthesis of 3-alkynyl-5-bromo-pyrrolo[2,3-b]pyridines (20a-f)
General procedure
To a degassed solution of 5-bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine 15 (1 eq) in THF and triethylamine (3 eq) was added CuI (0.02 eq) and Pd(PPh3)2Cl2 (0.01 eq). The resulting mixture was stirred under an inert atmosphere for 10 minutes at room temperature. A solution of the appropriate alkyne (0.95 eq) in THF was added to the mixture. The reaction mixture was stirred for an additional 4 hours at room temperature. After completion, the reaction was filtered through Celite. The filtrate was extracted with water and ethyl acetate, washed with brine and dried over MgSO4. The crude residue was purified by silica gel flash column chromatography with an appropriate mobile phase. Compounds 20a-f were made according to this procedure.
5-Bromo-3-(phenylethynyl)-1H-pyrrolo[2,3-b]pyridine (20a)
The title compound was synthesized according to the general procedure using 5-bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine 15 (200 mg, 0.619 mmol) and phenylacetylene (60 mg, 0.588 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 8:2) as mobile phase yielding the title compound as a white solid (85 mg, 51%). 1H NMR (300 MHz, DMSO-d6) 12.40 (bs, 1H, NH), 8.39 (d, J = 2.2 Hz, 1H, HetH), 8.32 (d, J = 2.1 Hz, 1H, HetH), 8.01 (s, 1H, HetH), 7.60 (m, 2H, PhH), 7.42 (m, 3H, PhH) ppm.5-Bromo-3-(pyridin-3-ylethynyl)-1H-pyrrolo[2,3-b]pyridine (20b)
The title compound was synthesized according to the general procedure using 5-bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine 15 (200 mg, 0.619 mmol) and 3-ethynylpyridine (61 mg, 0.588 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 7:3) as mobile phase yielding the title compound as a white solid (140 mg, 75%). 1H NMR (300 MHz, DMSO-d6) 12.49 (bs, 1H, NH), 8.81 (s, 1H, ArH), 8.56 (d, J = 4.8 Hz, 1H, ArH), 8.40 (s, 2H, ArH), 8.04 (m, 2H, ArH), 7.46 (m, 1H, ArH) ppm.
5-Bromo-3-((3-methoxyphenyl)ethynyl)-1H-pyrrolo[2,3-b]pyridine (20c)
The title compound was synthesized according to the general procedure using 5-bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine 15 (200 mg, 0.619 mmol) and 3-methoxyphenylacetylene (60 mg, 0.588 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 7:3) as mobile phase, yielding the title compound as a white solid (105 mg, 51%). 1H NMR (300 MHz, DMSO-d6) δ 12.41 (bs, 1H, NH), 8.39 (d, J = 2.2 Hz, 1H, HetH), 8.34 (d, J = 2.1 Hz, 1H, HetH), 8.00 (s, 1H, HetH), 7.33 (t, J = 8.1 Hz, 1H, PhH), 7.16 (m, 2H, PhH), 6.96 (m, 1H, PhH), 3.81 (s, 3H, OCH3) ppm.
5-Bromo-3-(p-tolylethynyl)-1H-pyrrolo[2,3-b]pyridine (20d)
The title compound was synthesized according to the general procedure using 5-bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine 15 (200 mg, 0.619 mmol) and p-tolylacetylene (68 mg, 0.588 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 7:3) as mobile phase, yielding the title compound as a white solid (130 mg, 67%). 1H NMR (300 MHz, DMSO-d6) δ 12.36 (bs, 1H, NH), 8.38 (d, J = 2.2 Hz, 1H, HetH), 8.30 (d, J = 2.2 Hz, 1H, HetH), 7.98 (s, 1H, HetH), 7.48 (d, J = 8.1 Hz, 2H, PhH), 7.23 (d, J = 7.9 Hz, 2H, PhH), 2.34 (s, 3H, CH3).
5-Bromo-3-((4-fluorophenyl)ethynyl)-1H-pyrrolo[2,3-b]pyridine (20e)
The title compound was synthesized according to the general procedure using 5-bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine 15 (200 mg, 0.619 mmol) and 4-fluorophenylacetylene (60 mg, 0.588 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 8:2) as mobile phase yielding the title compound as a white solid (110 mg, 56%). 1H NMR (300 MHz, DMSO-d6) δ 12.41 (bs, 1H, NH), 8.38 (d, J = 2.1 Hz, 1H, HetH), 8.34 (d, J = 2.1 Hz, 1H, HetH), 8.00 (s, 1H, HetH), 7.66 (dd, J = 8.7, 5.6 Hz, 2H, PhH), 7.27 (t, J = 8.9 Hz, 2H, PhH) ppm.
3-((5-Bromo-1H-pyrrolo[2,3-b]pyridin-3-yl)ethynyl)benzonitrile (20f)
The title compound was synthesized according to the general procedure using 5-bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine 15 (200 mg, 0.619 mmol) and 3-cyanophenylacetylene (75 mg, 0.588 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 7:3) as mobile phase, yielding the title compound as a white solid (90 mg, 45%). 1H NMR (300 MHz, DMSO-d6) δ 8.45 (d, J = 2.2 Hz, 1H, HetH), 8.40 (d, J = 2.1 Hz, 1H, HetH), 8.14 (s, 1H, HetH), 8.05 (s, 1H, PhH), 7.91 (d, J = 8.0 Hz, 1H, PhH), 7.84 (d, J = 7.5 Hz, 1H, PhH), 7.63 (t, J = 7.9 Hz, 1H, PhH) ppm.
Synthesis of 3-alkynyl-5-(3,4-dimethoxyphenyl)-pyrrolo[2,3-b]pyridines (21a-f)
General procedure
To a solution of a 5-bromo-3-arylethynyl-1H-pyrrolo[2,3-b]pyridine derivatrive (1 eq) in dioxane (4 ml) was added 3,4-dimethoxyphenylboronic acid (1.2 eq) and 1 ml of a K2CO3 (3 eq) solution. The system was purged three times with argon and heated to 105 °C. After stirring for 10 minutes, Pd(PPh3)4 (0.1 eq) was added and the reaction was purged once more with argon. The reaction mixture was stirred at 105°C for 3 hours. After completion, the reaction mixture was cooled to room temperature and filtered through Celite. The filtrate was extracted with water and ethyl acetate. The combined organic layers were washed with brine, dried over MgSO4 and evaporated in vacuo. The crude residue was purified by silica gel flash column chromatography with an appropriate mobile phase. Compounds 21a-f were made according to this procedure.
5-(3,4-Dimethoxyphenyl)-3-(phenylethynyl)-1H-pyrrolo[2,3-b]pyridine (21a)
The title compound was synthesized according to the general procedure using 3-(phenylethynyl)-1H-pyrrolo[2,3-b]pyridine 20a (80 mg, 0.269 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 9:1) as mobile phase yielding the title compound as a brown solid (36 mg, 38%). Purity of 99%. 1H NMR (300 MHz, DMSO-d6) δ 12.21 (bs, 1H, NH), 8.61 (s, 1H, ArH), 8.25 (s, 1H, ArH), 7.96 (d, J = 2.2 Hz, 1H, ArH), 7.60 (d, J = 6.5 Hz, 1H, ArH), 8.01 (m, 2H, ArH), 7.35 (m, 6H, ArH), 7.06 (d, J = 8.3 Hz, 1H, ArH), 3.88 (s, 3H, OCH3), 3.81 (s, 3H, OCH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 149.33, 148.49, 147.24, 143.12, 131.42, 131.27, 131.17, 129.60, 128.80, 128.13, 124.93, 123.48, 120.29, 119.44, 112.48, 111.11, 95.31, 90.75, 83.53, 55.82, 55.73 ppm. HRMS m/z [M+H]+ calcd for C23H18N2O2 355.14409; found 355.1435.
5-(3,4-Dimethoxyphenyl)-3-(pyridin-3-ylethynyl)-1H-pyrrolo[2,3-b]pyridine (21b)
The title compound was synthesized according to the general procedure using 5-bromo-3-(pyridin-3-ylethynyl)-1H-pyrrolo[2,3-b]pyridine 20b (140 mg, 0.470 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 9:1) as mobile phase, yielding the title compound as a white solid (75 mg, 44%). Purity of 98% 1H NMR (300 MHz, DMSO-d6) δ 12.30 (s, 1H, NH), 8.81 (s, 1H, ArH), 8.63 (d, J = 2.0 Hz, 1H, ArH), 8.55 (d, J = 4.9 Hz, 1H, ArH), 8.30 (d, J = 1.9 Hz, 1H, ArH), 8.01 (m, 2H, ArH), 7.46 (m, 1H, ArH), 7.31 (m, 2H, ArH), 7.07 (d, J = 8.3 Hz, 1H), 3.89 (s, 3H, OCH3), 3.81 (s, 3H, OCH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 151.46, 149.33, 148.52, 148.34, 147.24, 143.24, 138.19, 131.78, 131.33, 129.73, 125.03, 123.68, 120.59, 120.28, 119.46, 112.47, 111.13, 94.77, 87.69, 86.87, 55.83, 55.74 ppm. HRMS m/z [M+H]+ calcd for C22H17N3O2 356.13934; found 356.1393.
5-(3,4-Dimethoxyphenyl)-3-(3-methoxyphenylethynyl)-1H-pyrrolo[2,3-b]pyridine (21c)
The title compound was synthesized according to the general procedure using 3-(3-methoxyphenylethynyl)-1H-pyrrolo[2,3-b]pyridine 20c (110 mg, 0.353 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 9:1). Further purification by preparative TLC (the mobile phase being a mixture of nitromethane:toluene in a ratio of 6:4) yielded the title compound as a white solid (25 mg, 20%). Purity of 97%. 1H NMR (300 MHz, DMSO-d6) δ 12.21 (s, 1H, NH), 8.61 (d, J = 1.9 Hz, 1H, ArH), 8.25 (d, J = 1.9 Hz, 1H, ArH), 7.96 (d, J = 2.6 Hz, 1H, ArH), 7.32 (m, 3H, ArH), 7.16 (m, 2H, ArH), 7.07 (d, J = 8.3 Hz, 1H, ArH), 6.96 (m, 1H, ArH), 3.89 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 3.80 (s, 3H, OCH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 159.33, 149.33, 148.50, 147.24, 143.12, 131.41, 131.34, 129.91, 129.60, 124.97, 124.56, 123.61, 120.28, 119.44, 115.95, 114.58, 112.49, 111.12, 95.24, 90.74, 83.43, 55.82, 55.75, 55.34 ppm. HRMS m/z [M+H]+ calcd for C24H20N2O3 385.15465; found 385.1541.
5-(3,4-Dimethoxyphenyl)-3-(p-tolylethynyl)-1H-pyrrolo[2,3-b]pyridine (21d)
The title compound was synthesized according to the general procedure using 3-(p-tolylethynyl)-1H-pyrrolo[2,3-b]pyridine 20d (110 mg, 0.353 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 9:1) as mobile phase, yielding the title compound as a white solid (25 mg, 20%). Purity of 95%. 1H NMR (300 MHz, DMSO-d6) δ 12.18 (bs, 1H, NH), 8.60 (d, J = 2.0 Hz, 1H, ArH), 8.22 (d, J = 1.8 Hz, 1H, ArH), 7.93 (d, J = 2.6 Hz, 1H, ArH), 7.49 (d, J = 8.0 Hz, 2H, ArH), 7.27 (m, 4H, ArH), 7.03 (m, 2H, ArH), 3.88 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 2.34 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ 149.33, 148.48, 147.23, 143.07, 137.78, 131.45, 131.11, 131.04, 129.54, 129.42, 124.91, 120.46, 120.27, 119.43, 112.49, 111.11, 95.49, 90.81, 82.77, 55.83, 55.74, 21.14. HRMS m/z [M+H]+ calcd for C24H20N2O2 369.15974; found 369.1593.
5-(3,4-Dimethoxyphenyl)-3-(4-fluorophenylethynyl)-1H-pyrrolo[2,3-b]pyridine (21e)
The title compound was synthesized according to the general procedure using 3-(4-fluorophenylethynyl)-1H-pyrrolo[2,3-b]pyridine 20e (80 mg, 0.269 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 9:1), yielding the desired compound as a brown solid (34 mg, 36%). Purity of 98%. 1H NMR (300 MHz, DMSO-d6) δ 12.21 (bs, 1H, NH), 8.61 (s, 1H, ArH), 8.25 (s, 1H, ArH), 7.95 (d, J = 2.4 Hz, 1H, ArH), 7.65 (m, 2H, ArH), 7.28 (m, 4H, ArH), 7.06 (d, J = 8.5 Hz, 1H, ArH), 3.88 (s, 3H, OCH3), 3.81 (s, 3H, OCH3) ppm. 13C NMR (150 MHz, DMSO-d6) δ 161.68 (d, JCF = 247 Hz) 149.27, 148.44, 147.17, 143.07, 133.36 (d, JCF = 8.3 Hz), 131.35, 131.20, 129.54, 124.91, 120.23, 119.90, 119.89, 119.38, 115.95 (d, JCF = 22 Hz), 112.42, 111.05, 95.11, 89.60, 83.20, 55.76, 55.68 ppm. HRMS m/z [M+H]+ calcd for C23H17FN2O2 373.13467; found 373.1333.
3-((5-(3,4-Dimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)ethynyl)benzonitrile (21f)
The title compound was synthesized according to the general procedure using 3-(3-cyanophenylethynyl)-1H-pyrrolo[2,3-b]pyridine 20f (100 mg, 0.310 mmol). The crude residue was purified by silica gel flash column chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 9:1) as mobile phase, yielding the title compound as a brown solid (44 mg, 37%). Purity of 98%. 1H NMR (300 MHz, DMSO-d6) δ 12.31 (bs, 1H, NH), 8.62 (d, J = 2.0 Hz, 1H, ArH), 8.32 (d, J = 1.9 Hz, 1H, ArH), 8.11 (s, 1H, ArH), 8.00 (d, J = 2.5 Hz, 1H, ArH), 7.91 (d, J = 7.9 Hz, 1H, ArH), 7.82 (d, J = 7.9 Hz, 1H, ArH), 7.62 (t, J = 7.9 Hz, 2H, ArH), 7.31 (m, 2H, ArH), 7.07 (d, J = 8.3 Hz, 1H, ArH), 3.89 (s, 3H, OCH3), 3.81 (s, 3H, OCH3). 13C NMR (75 MHz, DMSO-d6) δ 149.33, 148.54, 147.25, 143.28, 135.52, 134.37, 131.91, 131.43, 131.35, 130.10, 129.81, 125.11, 124.93, 120.34, 119.51, 118.33, 112.48, 112.17, 111.17, 94.64, 89.01, 86.05, 55.84, 55.75. HRMS m/z [M+H]+ calcd for C24H17N3O2 380.13934; found 380.1390.
5-Bromo-1-methyl-1H-pyrrolo[2,3-b]pyridine (22)
To a solution of 5-bromo-1H-pyrrolo[2,3-b]pyridine 4 (200 mg, 1.02 mmol) in THF (5 ml) at 0°C was added 60% NaH on mineral oil (49 mg, 1.22 mmol). The reaction was stirred at 0°C for 1.5 hour. Methyl iodide (70 μl, 1.12 mmol) was added and the mixture was allowed to warm at ambient temperature and then stirred at room temperature for 3 hours. After completion, the mixture was quenched with water and extracted three times with ethyl acetate. The combined organic layers were washed with brine, dried over MgSO4, filtered and evaporated. The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 8:2) as mobile phase, yielding the title compound as an oil that subsequently crystallized to an off-white solid (160 mg, 75%). 1H NMR (300 MHz, DMSO-d6) δ 8.31 (d, J = 2.07 Hz, 1H, HetH), 8.20 (d, J = 2.07 Hz, 1H, HetH), 7.59 (d, J = 3.39 Hz, 1H, HetH), 6.46 (d, J = 3.42 Hz, 1H, HetH), 3.81 (s, 3H, CH3) ppm. HRMS m/z [M+H]+ calcd for C8H7N2Br 210.98658; found 210.9866.
5-Bromo-1-methyl-3-nitro-1H-pyrrolo[2,3-b]pyridine (23)
5-Bromo-1H-pyrrolo[2,3-b]pyridine 22 (130 mg, 0.615 mmol) was added portionwise to a stirring solution of fuming nitric acid (1 ml) at 0°C over 10 minutes. The reaction was allowed to stir for 30 minutes at 0°C. The mixture was poured out in ice water and the formed precipitate was collected via vacuum filtration. The filter cake was washed generously with water and heptane yielding the title compound as a pink solid (155 mg, 98%). 1H NMR (300 MHz, DMSO-d6) δ 8.99 (s, 1H, HetH), 8.61 (s, 1H, HetH), 8.57 (m, 1H, HetH), 3.92 (s, 3H, CH3) ppm.
5-(3,4-Dimethoxyphenyl)-1-methyl-3-nitro-1H-pyrrolo[2,3-b]pyridine (24)
To a solution of 5-bromo-1-methyl-3-nitro-1H-pyrrolo[2,3-b]pyridine 23 (100 mg, 0.391 mmol) in dioxane (4 ml) was added 3,4-dimethoxyphenylboronic acid (69 mg, 0.469 mmol) and 1 ml of a K2CO3 solution (161 mg, 1.17 mmol). The system was purged three times with argon and heated to 105°C. After stirring for 10 minutes, Pd(PPh3)4 (10 mol%) was added and the reaction was purged once more with argon. The reaction mixture was stirred at 105°C overnight. After completion, the reaction mixture was cooled to room temperature and filtered through Celite. Extraction was performed with water and ethyl acetate. The combined organic layers were washed with brine, dried over MgSO4 and evaporated. Purification by silica gel flash chromatography using a mixture of dichloromethane and ethyl acetate (in a ratio of 9:1) as mobile phase yielded the title compound as a yellow solid (90 mg, 83%). 1H NMR (300 MHz, DMSO-d6) δ 8.96 (s, 1H, HetH), 8.79 (d, J = 2.07 Hz, 1H, HetH), 8.56 (d, J = 2.01 Hz, 1H, HetH), 7.30 (m, 2H, PhH), 7.10 (d, J = 8.22 Hz, 1H, PhH), 3.96 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.82 (s, 3H) ppm.
N-(5-(3,4-Dimethoxyphenyl)-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)nicotinamide (26)
The title compound was synthesized according to the general procedure described for the synthesis of compounds 8a-h, starting from 5-(3,4-dimethoxyphenyl)-1-methyl-3-nitro-1H-pyrrolo[2,3-b]pyridine 24 (90 mg, 0.287 mmol). Purification by silica gel flash column chromatography using a mixture of dichloromethane and methanol (in a ratio gradually ranging from 98:2 to 95:5) yielded the title compound as a red solid (22 mg, 20%). Purity of 98%. 1H NMR (300 MHz, DMSO-d6) δ 10.60 (bs, 1H, NH), 9.19 (bs, 1H, NH), 8.77 (d, J = 2.6 Hz, 1H, ArH), 8.60 (s, 2H, ArH), 8.36 (d, J = 8.0 Hz, 1H, ArH), 8.07 (s, 1H, ArH), 7.59 (m, 1H, ArH), 7.26 (m, 2H, ArH), 7.08 (d, J = 8.3 Hz, 1H, ArH), 3.87 (s, 6H, OCH3/CH3), 3.80 (s, 3H, OCH3/CH3). 13C NMR (75 MHz, DMSO-d6) δ 163.33, 152.03, 149.33, 148.87, 148.39, 144.19, 142.14, 135.65, 131.81, 130.43, 127.77, 125.22, 123.63, 121.31, 119.21, 113.40, 113.14, 112.54, 110.93, 55.83, 55.75, 30.91 ppm. HRMS m/z [M+H]+ calcd for C22H20N4O3 389.16080; found 389.1604.
2-Chloro-5-(3,4-dimethoxyphenyl)nicotinonitrile (28)
To a solution of 3,4-dimethoxyphenylboronic acid (460 mg, 2.53 mmol) and 5-bromo-2-chloronicotinonitrile 27 (500 mg, 2.3 mmol) in isopropanol (12 ml) was added a solution of K2CO3 (953 mg, 6.9 mmol) in water (4 ml). The reaction mixture was flushed three times with argon and heated to 90°C. Then, Pd(PPh3)4 (10 mol%) was added and the system was flushed once more with argon. After stirring at 90°C for 2.5 hours, the reaction was concentrated under reduced pressure. The crude residue was partitioned between ethyl acetate and water and extracted three times. The combined organic phases were washed with brine and dried over MgSO4. The solvent was removed under reduced pressure and the crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethylacetate (in a ratio of 7:3) as mobile phase. This was followed by a second purification by silica gel flash column chromatography using a mixture of dichloromethane, heptane and ethylacetate (in a ratio of 70:25:5) as mobile phase, yielding the title compound as a white solid (320 mg, 50%). 1H NMR (300 MHz, DMSO-d6) δ 9.06 (d, J = 2.5 Hz, 1H, HetH), 8.85 (d, J = 2.5 Hz, 1H, HetH), 7.41 (m, 2H, PhH), 7.10 (m, 1H, PhH), 3.87 (s, 3H, OCH3), 3.82 (s, 3H, OCH3).
5-(3,4-Dimethoxyphenyl)-1H-pyrazolo[3,4-b]pyridin-3-amine (29)
To a solution of 2-chloro-5-(3,4-dimethoxyphenyl)nicotinonitrile 28 (100 mg, 0.364 mmol) in pyridine (3 ml) was added a hydrazine monohydrate solution (65% in water, 103 μl, 0.728 mmol). The reaction was refluxed overnight and after completion the solvent was evaporated. The crude residue was purified by silica gel flash column chromatography using a mixture of dichloromethane and methanol (in a ratio of 95:5) yielding the title compound as a yellow solid (90 mg, 91%). 1H NMR (300 MHz, DMSO-d6) δ 11.97 (bs, 1H, NH), 8.66 (d, J = 2.1 Hz, 1H, HetH), 8.36 (d, J = 2.0 Hz, 1H, HetH), 7.21 (m, 2H, PhH), 7.07 (d, J = 8.4 Hz, 1H, PhH), 3.86 (s, 3H, OCH3), 3.80 (s, 3H, OCH3).
N-(5-(3,4-Dimethoxyphenyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)nicotinamide (30)
To a solution of 5-(3,4-dimethoxyphenyl)-1H-pyrazolo[3,4-b]pyridin-3-amine 29 (90 mg, 0.332 mmol) in pyridine (3 ml) at 0°C was added nicotinoyl chloride hydrochloride (71 mg, 0.400 mmol). The reaction was stirred at 0°C for 1 hour and then stirred at room temperature overnight. After reaction completion, water was added and the mixture was extracted three times with ethyl acetate. The combined organic phases were washed with brine, dried over MgSO4 and evaporated to dryness. The crude residue was purified by silica gel flash chromatography using a mixture of dichloromethane and methanol (in a ratio gradually ranging from 98:2 to 96:4) yielding the title compound as a beige solid (76 mg, 61%). Purity of 99%. 1H NMR (300 MHz, DMSO-d6) δ 13.47 (s, 1H), 11.33 (s, 1H), 9.24 (s, 1H), 8.85 (d, J = 2.1 Hz, 1H), 8.79 (d, J = 3.4 Hz, 1H), 8.50 (d, J = 2.0 Hz, 1H), 8.42 (d, J = 8.1 Hz, 1H), 7.59 (dd, J = 7.7, 4.8 Hz, 1H), 7.28 (s, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 3.86 (s, 3H), 3.80 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 164.30, 152.57, 151.36, 149.35, 149.22, 148.89, 148.67, 139.54, 135.92, 130.86, 129.45, 129.25, 129.13, 123.66, 119.50, 112.54, 111.08, 108.70, 55.84, 55.74. HRMS m/z [M+H]+ calcd for C20H17N5O3 376.14040; found 376.1400.
2-Amino-3,5-dibromopyrazine (32)
To a solution of aminopyrazine 31 (1 g, 10.52 mmol) in DMSO (10 ml) was added N-bromosuccinimide (3.94 g, 22.08 mmol) portionwise over 45 minutes. The resulting mixture was stirred for 3 hours at room temperature. The reaction was poured in ice water and extracted five times with ethyl acetate. The combined organic phases were washed with brine, dried over MgSO4 and evaporated to dryness. The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 7:3) as mobile phase, yielding the desired compound as a fluffy white solid (1.94 g, 73%). 1H NMR (300 MHz, DMSO-d6) δ 8.13 (s, 1H, ArH), 6.99 (bs, 2H, NH2) ppm. HRMS m/z [M+H]+ calcd for C4H3N3Br2 251.87675; found 251.8765.
5-Bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine (33)
To a solution of 2-amino-3,5-dibromopyrazine 32 (1 g, 3.95 mmol) in degassed THF (15 ml) was added copper iodide (7.53 mg, 0.040 mmol), Pd(PPh3)2Cl2 (45mg, 0.040 mmol) and triethylamine (1.65 ml, 11.86 mmol). The system was flushed with nitrogen and trimethylsilylacetylene (534 μl, 1.91 mmol) was added dropwise over 5 minutes. The reaction was stirred overnight at room temperature. After reaction completion, the solvent was evaporated and water was added. The resulting suspension was extracted three times with ethyl acetate. The combined organic phases were washed with water and brine, dried over MgSO4 and evaporated to dryness. The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethylacetate (in a ratio of 80:20) as mobile phase, yielding the title compound as a bright yellow solid (769 mg, 72%). 1H NMR (300 MHz, DMSO-d6) δ 8.12 (s, 1H, ArH), 6.82 (s, 2H, NH2), 0.27 (s, 9H, Si(CH3)3) ppm.
2-Bromo-5H-pyrrolo[2,3-b]pyrazine (34)
To a solution of 5-bromo-3-((trimethylsilyl)ethynyl)pyridin-2-amine 33 (1 g, 3.7 mmol) in dry NMP (10 ml) was added portionwise KOtBu (498 mg, 4.44 mmol). The reaction mixture was flushed with nitrogen and stirred for 3 hours at 80°C. After reaction completion, the mixture was extracted three times with water and ethyl acetate. The combined organic layers were washed twice with water and once with brine, dried over MgSO4 and evaporated in vacuo. The crude residue was then purified by silica gel flash column chromatography using a mixture of heptane/ethyl acetate (in a ratio of 7:3) as mobile phase, yielding the title compound as a yellow solid (502 mg, 69%). 1H NMR (300 MHz, DMSO-d6) δ 12.42 (br s, 1H, NH), 8.35 (s, 1H, ArH), 7.97 (d, J = 3.48 Hz, 1H, ArH), 6.63 (d, J = 3.48 Hz, 1H, ArH) ppm.
5-Bromo-3-iodo-1H-pyrrolo[2,3-b]pyrazine (35)
To a solution of 5-bromo-1H-pyrrolo[2,3-b]pyrazine 34 (500 mg, 2.52 mmol) in a minimal volume of acetone was added portionwise N-iodosuccinimide (625 g, 2.78 mmol). The reaction mixture was stirred for 2 hours at room temperature. The formed precipitate was collected via filtration, yielding the title product as an off-white solid (408 mg, 50%). %). 1H NMR (300 MHz, DMSO-d6) δ 12.82 (br s, 1H, NH), 8.40 (d, J = 1.77 Hz, 1H, ArH), 8.19 (s, 1H, ArH) ppm.
2-Bromo-7-(pyridin-3-ylethynyl)-5H-pyrrolo[2,3-b]pyrazine (36)
To a degassed solution of 5-bromo-3-iodo-1H-pyrrolo[2,3-b]pyrazine 35 (200 mg, 0.617 mmol) in THF (10 ml) were added triethylamine (238 μl, 1.85 mmol), Pd(PPh3)2Cl2 (4.33 mg, 1mol%) and CuI (2.35 mg, 2 mol%). The resulting mixture was stirred under inert atmosphere for 10 minutes at room temperature. Then, a solution of 3-ethynylpyridine (61 mg, 0.586 mmol) in THF (1 ml) was added to the reaction mixture. The reaction was stirred for 4 hours at room temperature. After completion, the reaction was filtered through Celite, extracted with water and ethyl acetate, washed with brine, dried over MgSO4 and evaporated in vacuo. The crude residue was purified by silica gel flash column chromatography using a mixture of heptane and ethyl acetate (in a ratio of 6:4) as mobile phase, yielding the title compound as a white solid (130 mg, 71%). 1H NMR (300 MHz, DMSO-d6) δ 12.94 (bs, 1H, NH), 8.76 (s, 1H), 8.59 (d, J = 3.2 Hz, 1H, ArH), 8.51 (s, 1H, ArH), 8.45 (s, 1H, ArH), 7.99 (dt, J = 7.9, 1.8 Hz, 1H, ArH), 7.47 (dd, J = 7.8, 4.8 Hz, 1H, ArH). HRMS m/z [M+H]+ calcd for C13H7N4Br 298.99273; found 298.9930.
2-(3,4-Dimethoxyphenyl)-7-(pyridin-3-ylethynyl)-5H-pyrrolo[2,3-b]pyrazine (37)
To a solution of 2-bromo-7-(pyridin-3-ylethynyl)-5H-pyrrolo[2,3-b]pyrazine 36 (110 mg, 0.368 mmol) in dioxane (4 ml) was added 3,4-dimethoxyphenylboronic acid (80 mg, 0.441 mmol) and 1 ml of a K2CO3 solution (152 mg, 1.1 mmol). The system was purged three times with argon and heated to 105°C. After stirring for 10 minutes, Pd(PPh3)4 (10 mol%) was added and the reaction was purged once more with argon. The reaction mixture was stirred at 105°C for 3 hours. After completion, the reaction mixture was cooled to room temperature and filtered through Celite. The filtrate was extracted with water and ethyl acetate. The combined organic layers were washed with brine, dried over MgSO4 and evaporated in vacuo. Purification by silica gel flash column chromatography using a mixture of dichloromethane and ethylacetate (in a ratio of 1:1) as mobile phase, yielded the title compound as a white solid (37 mg, 28%). Purity of 98%. 1H NMR (300 MHz, DMSO-d6) δ 12.63 (bs, 1H, NH), 8.96 (s, 1H, ArH), 8.78 (s, 1H, ArH), 8.58 (d, J = 3.3 Hz, 1H, ArH), 8.35 (s, 1H, ArH), 8.00 (d, J = 7.8 Hz, 1H, ArH), 7.76 (m, 2H, ArH), 7.48 (m, 1H, ArH), 7.11 (d, J = 9.0 Hz, 1H, ArH), 3.90 (s, 3H, OCH3), 3.83 (s, 3H, OCH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ 151.44, 149.97, 149.32, 148.57, 146.70, 139.95, 138.27, 137.69, 136.06, 135.59, 130.21, 123.77, 120.46, 119.60, 112.33, 110.46, 95.69, 88.10, 85.92, 55.88, 55.81 ppm. HRMS m/z [M+H]+ calcd for C21H16N4O2 357.13459; found 357.1346.
Binding-displacement assay for NAK family selectivity
Inhibitor binding to NAK family kinase domain proteins was determined using a binding-displacement assay which tests the ability of the inhibitors to displace a fluorescent tracer compound from the ATP binding site of the kinase domain. Inhibitors were dissolved in DMSO and dispensed as 16-point, 2x serial dilutions in duplicate into black multiwell plates (Greiner) using an Echo dispenser (Labcyte Inc). Each well contained 1 nM biotinylated AAK1, BMP2K, GAK or STK16 kinase domain protein ligated to streptavidin-Tb-cryptate (Cisbio), either 12.5 nM (for AAK1 or BMP2K) or 25 nM (for GAK or STK16) Kinase Tracer 236 (ThermoFisher Scientific), 10 mM Hepes pH 7.5, 150 mM NaCl, 2 mM DTT, 0.01% BSA, 0.01% Tween-20. Final assay volume for each data point was 5 μL, and final DMSO concentration was 1%. The plate was incubated at room temperature for 1.5 hours and then read using a TR-FRET protocol on a PheraStarFS plate reader (BMG Labtech). The data was normalized to 0% and 100% inhibition control values and fitted to a four parameter dose-response binding curve in GraphPad Software. For the purpose of estimating the selectivity between each kinase domain, the determined IC50 values were converted to Ki values using the Cheng-Prusoff equation and the concentration and KD values for the tracer (previously determined).
AAK1 expression, purification and crystallization
AAK1 (UniProtKB: Q2M2I8) residues T27-A365 was cloned, expressed and purified as described39, except that the protein was expressed in a cell line together with lambda phosphatase to produce the unphosphorylated protein. The purified protein was concentrated to 12 mg/mL and compound 1 dissolved in 100% DMSO was added to a final concentration of 1.5 mM (3% final DMSO concentration). The protein-ligand solution was incubated on ice for 30 minutes, then centrifuged at 14,000 rpm for 10 minutes, 4 °C, immediately prior to setting up sitting-drop vapour diffusion crystallization plates. The best-diffracting crystals of the AAK1 - compound 1 complex were obtained using a reservoir solution containing 26% PEG 3350, 0.1 M Bis-Tris pH 5.5 by spiking drops with 20 nL of seed-stock solution immediately prior to incubation at 18°C. Seed stock was prepared from poorly-formed crystals of AAK1-compound 1 grown during previous rounds of crystal optimization, which were diluted in 50–100 μL reservoir solution and vortexed for 2 min in an Eppendorf containing a seed bead. A 1:1 dilution series of seeds was prepared in order to find the optimal seed concentration. Prior to mounting, crystals were cryo-protected in situ by addition of reservoir solution containing an additional 25% ethylene glycol. Crystals were then flash frozen in liquid nitrogen.
Data collection, structure solution and refinement
Data was collected at Diamond beamline I02 using monochromatic radiation at wavelength 0.9795 Å. Diffraction data were processed using XDS46 as part of the xia2 pipeline47 and scaled using AIMLESS48, molecular replacement was carried out in Phaser49 with PDB ID 4WSQ as a search model. Data processing and refinement statistics are given in Table 1 from the Supporting Information. REFMAC550, PHENIX51 and Coot52 were used for model building and refinement. Coordinates were submitted to the PDB under accession code 5L4Q.
Virus construct
DENV2 (New Guinea C strain)53,54Renilla reporter plasmid used for in vitro assays was a gift from Pei-Yong Shi (The University of Texas Medical Branch). DENV 16681 plasmid (pD2IC-30P-NBX) used for ex vivo experiments was a gift from Claire Huang (CDC)55.
Cells
Huh7 (Apath LLC) cells were grown in DMEM (Mediatech) supplemented with 10% FBS (Omega Scientific), nonessential amino acids, 1% L-glutamine, and 1% penicillin-streptomycin (ThermoFisher Scientific) and maintained in a humidified incubator with 5% CO2 at 37 °C. MDDCs were prepared as described with slight modifications.56 Buffy coats were obtained from the Stanford Blood Center. CD14+ cells were purified by EasySep™ Human Monocyte Enrichment Kit without CD16 Depletion (Stemcell Technologies). Cells were seeded in 6-well plates (2 ×106 cells per well), stimulated with 500 U/ml granulocyte-macrophage colony-stimulating and 1,000 U/ml interleukin-4 (Pepro tech), and incubated at 37 °C for 6 days prior to DENV infection (MOI 1).
Virus Production
DENV2 RNA was transcribed in vitro using mMessage/mMachine (Ambion) kits. DENV was produced by electroporating RNA into BHK-21 cells, harvesting supernatants on day 10 and titering via standard plaque assays on BHK-21 cells. In parallel, on day 2 post-electroporation, DENV-containing supernatant was used to inoculate C6/36 cells to amplify the virus. EBOV (Kikwit isolate) was grown in Vero E6 cells, supernatants were collected and clarified and stored at −80°C until further use. Virus titers were determined via standard plaque assay on Vero E6 cells.
Infection assays
Huh7 cells were infected with DENV in replicates (n = 5) at a multiplicity of infection (MOI) of 0.05. Overall infection was measured at 48 hours using a Renilla luciferase substrate. MDDCs were infected with DENV2 (16881) at an MOI of 1. Standard plaque assays were conducted following a 72-hour incubation. Huh7 cells were infected with EBOV at an MOI of 1 or 0.1 under biosafety level 4 conditions. Forty-eight hours after infection, supernatants were collected and stored at −80 °C until further use. Cells were formalin-fixed for twenty-four hours prior to removal from biosafety level 4. Infected cells were detected using an EBOV glycoprotein specific monoclonal antibody (KZ52), and quantitated by automated fluorescence microscopy using an Operetta High Content Imaging System and the Harmony software package (Perkin Elmer). For select experiments, supernatants were assayed by standard plaque assay, as described previously. Briefly, supernatants were thawed, serially diluted in growth media, added to VeroE6 cells and incubated for 1 hour at 37°C in a humidified 5% CO2 incubator, prior to being overlaid with agarose. Infected cells were incubated for 7 days, then stained with neutral red vital dye (Gibco). Plaques were counted, and titers were calculated.
Viability assays
Viability was assessed using AlamarBlue® reagent (Invitrogen) or Cell-Titer-Glo® reagent (Promega) assay according to manufacturer’s protocol. Fluorescence was detected at 560 nm on InfiniteM1000 plate reader and luminescence on InfiniteM1000 plate reader (Tecan) or a Spectramax 340PC.
Effect of compounds 1, 8g and 21b on AP-2 phosphorylation
Huh7 cells were kept in serum free medium for 1 hour and then treated with the compounds or DMSO in complete medium for 4 hours at 37 °C. To allow capturing of phosphorylated AP2M1, 100 nM of the PP2A inhibitor calyculin A (Cell Signalling) was added 30 min prior to lysis in M-PER lysis buffer (ThermoFisher Scientific) with 1X Halt Protease & phosphatase inhibitor cocktail (ThermoFisher Scientific). Samples were then subjected to SDS-PAGE and blotting with antibodies targeting phospho-AP2M1 (Cell Signaling), total AP2M1 (Santa Cruz Biotechnology), and actin (Sigma-Aldrich). Band intensity was measured with NIH ImageJ.
AAK1 LanthaScreen™ Eu binding assay
The compounds were subjected to a LanthaScreen™ binding assay in which 10 titrations of dissolved test compound in DMSO are transferred to a 384-well plate. Sequential addition of the kinase buffer (50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl and 1mM EGTA), the 2X kinase antibody (Eu Anti GST) mixture and the 4X Tracer 222 solution was performed. After shaking for 30 seconds and a one hour incubation period at room temperature, the plate was read on a fluorescence plate reader. When the bound tracer in the active site was displaced by the test compound, fluorescence was not observed. The collected data were then compared to a 0% displacement control with pure DMSO and a 100% displacement control with sunitinib, a known inhibitor of AAK1, and plotted against the logarithmic concentration parameter. The IC50 was subsequently extracted.
AAK1 Kd assay
KD values for AAK1 were determined as previously described.40 Briefly, the DNA-tagged AAK1, an immobilized ligand on streptavidin-coated magnetic beads, and the test compound were combined. When binding occurred between AAK1 and a test compound, no binding can occurred between AAK1 and the immobilized ligand. Upon washing, the compound-bound, DNA-tagged AAK1 was washed away. The beads carrying the ligands were then resuspended in elution buffer and the remaining kinase concentration was measured by qPCR on the eluate. KD values were determined using dose-response curves.
Kinase Selectivity assay
Compound 21b was screened against a diverse panel of 468 kinases (DiscoveRX, KinomeScan) using an in vitro ATP-site competition binding assay at a concentration of 10 μM. The results are reported as the percentage of kinase/phage remaining bound to the ligands/beads, relative to a control. High affinity compounds have % of control values close to zero, while weaker binders have higher % control values.
Statistical analysis
All data were analyzed with GraphPad Prism software. Fifty percent effective concentration (EC50, EC90 and CC50) values were measured by fitting data to a three-parameter logistic curve. P values were calculated by two-way ANOVA with Bonferroni’s multiple comparisons tests or by 2-tailed unpaired t test.
Molecular modelling
Docking was initiated from the AAK1 PDB structure 5L4Q, from which first all ligands and water molecules were removed. The remaining structure was prepared by AutodockTools57 as a receptor: polar hydrogens were added, Gasteiger charges were defined, the AD4 atom type was assigned and finally everything saved in a pdbqt file. The original inhibitor lkb (identical to compound 1) from the PDB file 5L4Q was extracted, processed by AutodockTools and saved in the pdbqt format. Compounds 8g and 21b were drawn in Chemdraw and a 3D structure was generated by Chem3D.58 The amide conformation in compound 8g was initially chosen having an anti orientation. Autodocktools was used again to prepare the corresponding pdbqt files. Autodock Vina was used for the docking experiments.42 A cubic box was defined 60×60×60 units (0.375 Å/unit) and the box was centred at the ALA72:CB atom in the first kinase domain (chain A). The docking process used variable dihedral angles in the ligand while the receptor was defined as rigid. Amide bonds in the ligands were not allowed to rotate.
A control docking was executed using the original inhibitor lkb present in the 5l4q PDB file. Vina reproduced the original X-ray position very well. For the three docked systems with the best Vina docking score a molecular dynamics simulation using the Amber 18 software.43 Enzyme parameters and charges were taken from the default amber ff14sb force field. Parameters and atomic charges were calculated by antechamber (gaff2). The barrier of the dihedral torsion parameters for the amide bond was increased from 2.6 (from antechamber) to 10.0 (in line with a peptide bond amide in the standard amber force field).
Three molecular systems (AAK1/compound 1, AAK1/compound 8g and AAK1/compound 21b) were solvated with TIP3P and neutralized in charge. Standard NPT simulations of 40 ns were started (300K, periodic conditions with PME, cutoff 10.0 Å, shake for H bonds constraints, 2 fs time step). Overall root mean square deviation curves (RMSD) of the MD trajectories are shown in supplemental Figure S1 (see Supporting Information). The last 4 ns of the MD trajectories were used in the MM/PBSA MM/GBSA calculations.
Supplementary Material
Scheme 6.

Synthesis of pyrazolo[3,4-b]pyridine 30
Reagents and conditions. a) 3,4-dimethoxyphenylboronic acid, Pd(PPh3)4, K2CO3, H2O, dioxane, 105°C ; b) 35% hydrazine hydrate, EtOH, 80°C ; c) nicotinoyl chloride, pyridine, rt.
Acknowledgments
This work was supported by award number W81XWH-16-1-0691 from the Department of Defense (DoD), Congressionally Directed Medical Research Programs (CDMRP) to S.E, P.H; Grant 12393481 from the Defense Threat Reduction Agency (DTRA), Fundamental Research to Counter Weapons of Mass Destruction to S.E, P.H; and seed grant from the Stanford SPARK program. S.V. is the recipient of a doctoral fellowship from the Research Foundation - Flanders (1S00116N). S.P. was supported by the Child Health Research Institute, Lucile Packard Foundation for Children’s Health and the Stanford CSTA (grant number UL1 TR000093). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z].
Abbreviations
- AAK1
adaptor-associated kinase 1
- AP
adaptor protein
- BOP
benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- CC50
half-maximal cytotoxic concentration; CCV, clathrin-coated vesicle
- DENV
Dengue virus; EBOV, Ebola virus
- EC50
half-maximal effective concentration
- EC90
90% effective concentration
- EGFR
epidermal growth factor receptor
- GAK
Cyclin G-associated kinase
- HCV
hepatitis C virus
- KD
dissociation constant
- MM/GBSA
molecular mechanics with a generalized Born surface area continuum solvation model
- MM/PBSA
molecular mechanics energies combined with the Poisson–Boltzmann surface area continuum solvation model
- NAK
numb-associated kinase
- PDB
protein data bank
- RMSD
root mean square deviation
- TGN
trans-Golgi network
- WHO
World Health Organization.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Copies of 1H and 13C NMR spectra of intermediates and final compounds
Details of the kinase selectivity profiling of compound 21b
Data collection and refinement statistics for cocrystallography of compound 1 with AAK1.
Molecular formula strings (CSV)
Accession Codes
The coordinates have been deposited in the PDB with accession code 5L4Q.
Authors will release the atomic coordinates and experimental data upon article publication.
References
- (1).Anne NE Epidemiology of Dengue : Past, Present and Future Prospects. Clin. Epidemiol. 2013, 5, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).WHO | Epidemiology http://www.who.int/denguecontrol/epidemiology/en/ (accessed Nov 15, 2018).
- (3).Weekly Epidemiological Record. World Heal. Organ. 2016, 91 (30), 349–364. [Google Scholar]
- (4).Sanyal S; Sinha S; Halder KK Pathogenesis of Dengue Haemorrhagic Fever. J Indian Med Assoc 2013, 89 (6), 152–153. [PubMed] [Google Scholar]
- (5).Sullivan N; Yang Z; Nabel GJ Ebola Virus Pathogenesis : Implications for Vaccines and Therapies MINIREVIEW. J. Virol. 2003, 77 (18), 9733–9737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).WHO. Ebola virus disease http://www.who.int/news-room/fact-sheets/detail/ebola-virus-disease (accessed Aug 7, 2018).
- (7).Byrd CM; Dai D; Grosenbach DW; Berhanu A; Jones KF; Cardwell KB; Schneider C; Wineinger KA; Page JM; Harver C; Stavale E; Tyavanagimatt S; Stone MA; Bartenschlager R; Scaturro P; Hruby DE; Jordan R A Novel Inhibitor of Dengue Virus Replication That Targets the Capsid Protein. Antimicrob. Agents Chemother. 2013, 57 (1), 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bixler SL; Duplantier AJ; Bavari S Discovering Drugs for the Treatment of Ebola Virus. Curr. Treat. options Infect. Dis. 2017, 9 (3), 299–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bekerman E; Neveu G; Shulla A; Brannan J; Pu S-Y; Wang S; Xiao F; Barouch-Bentov R; Bakken RR; Mateo R; Govero J; Nagamine CM; Diamond MS; Jonghe S. De; Herdewijn P; Dye JM; Randall G; Einav S Anticancer Kinase Inhibitors Impair Intracellular Viral Trafficking and Exert Broad-Spectrum Antiviral Effects. J. Clin. Invest. 2017, 127 (4), 1338–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bekerman E; Einav S Combating Emerging Viral Threats. Science. 2015, 348 (6232), 282–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Grove J; Marsh M The Cell Biology of Receptor-Mediated Virus Entry. J. Cell Biol. 2011, 195 (7), 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Neveu G; Ziv-Av A; Barouch-Bentov R; Berkerman E; Mulholland J; Einav S AP-2-Associated Protein Kinase 1 and Cyclin G-Associated Kinase Regulate Hepatitis C Virus Entry and Are Potential Drug Targets. J. Virol. 2015, 89 (8), 4387–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Neveu G; Barouch-Bentov R; Ziv-Av A; Gerber D; Jacob Y; Einav S Identification and Targeting of an Interaction between a Tyrosine Motif within Hepatitis C Virus Core Protein and AP2M1 Essential for Viral Assembly. PLoS Pathog. 2012, 8 (8), e1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Xiao F; Wang S; Barouch-Bentov R; Neveu G; Pu S; Beer M; Schor S; Kumar S; Nicolaescu V; Lindenbach BD; Randall G; Einav S Interactions between the Hepatitis C Virus Nonstructural 2 Protein and Host Adaptor Proteins 1 and 4 Orchestrate Virus Release. MBio 2018, 9 (2), 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pu S-Y; Xiao F; Schor S; Bekerman E; Zanini F; Barouch-Bentov R; Nagamine CM; Einav S Feasibility and Biological Rationale of Repurposing Sunitinib and Erlotinib for Dengue Treatment. Antiviral Res. 2018, 155, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kostich W; Hamman BD; Li Y-W; Naidu S; Dandapani K; Feng J; Easton A; Bourin C; Baker K; Allen J; Savelieva K; Louis JV; Dokania M; Elavazhagan S; Vattikundala P; Sharma V; Das ML; Shankar G; Kumar A; Holenarsipur VK; Gulianello M; Molski T; Brown JM; Lewis M; Huang Y; Lu Y; Pieschl R; OMalley K; Lippy J; Nouraldeen A; Lanthorn TH; Ye G; Wilson A; Balakrishnan A; Denton R; Grace JE; Lentz KA; Santone KS; Bi Y; Main A; Swaffield J; Carson K; Mandlekar S; Vikramadithyan RK; Nara SJ; Dzierba C; Bronson J; Macor JE; Zaczek R; Westphal R; Kiss L; Bristow L; Conway CM; Zambrowicz B; Albright CF Inhibition of AAK1 Kinase as a Novel Therapeutic Approach to Treat Neuropathic Pain. J. Pharmacol. Exp. Ther. 2016, 358 (3), 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bronson J; Chen L; Ditta J; Dzierba CD; Jalagam PR; Luo G; Macor J; Maishal TK; Nara SJ; Rajamani R; Sistla RK; Thangavel S Biaryl Kinase Inhibitors. WO 2017/059085, 2017. [Google Scholar]
- (18).Kuai L; Ong S-E; Madison JM; Wang X; Duvall JR; Lewis TA; Luce CJ; Conner SD; Pearlman DA; Wood JL; Schreiber SL; Carr SA; Scolnick EM; Haggarty SJ AAK1 Identified as an Inhibitor of Neuregulin-1/ErbB4-Dependent Neurotrophic Factor Signaling Using Integrative Chemical Genomics and Proteomics. Chem. Biol. 2011, 18 (7), 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bamborough P; Drewry D; Harper G; Smith GK; Schneider K Assessment of Chemical Coverage of Kinome Space and Its Implications for Kinase Drug Discovery. J. Med. Chem. 2008, 51 (24), 7898–7914. [DOI] [PubMed] [Google Scholar]
- (20).Barl NM; Sansiaume-Dagousset E; Karaghiosoff K; Knochel P Full Functionalization of the 7-Azaindole Scaffold by Selective Metalation and Sulfoxide/Magnesium Exchange. Angew. Chemie Int. Ed. 2013, 52 (38), 10093–10096. [DOI] [PubMed] [Google Scholar]
- (21).Brumsted CJ; Moorlag H; Radinov RN; Ren Y; Waldmeier P Method for Preparation of N-{3-[5-(4-Chlorophenyl)-1H-Pyrrolo[2,3-b]Pyridine-3-Carbonyl]-2,4-Difluorophenyl} Propane-1-Sulfonamide. WO 2012/010538 A2, 2012. [Google Scholar]
- (22).Han C; Green K; Pfeifer E; Gosselin F Highly Regioselective and Practical Synthesis of 5 ‑ Bromo-4-Chloro-3- Nitro-7-Azaindole. Org. Process Res. Dev. 2017, 21 (4), 664–668. [Google Scholar]
- (23).Stokes S; Graham CJ; Ray SC; Stefaniak EJ 1H-Pyrrolo[2,3-b]Pyridine Derivatives and Their Use as Kinase Inhibitors. WO 2013/114113 A1, 2013. [Google Scholar]
- (24).Gao L; Kovackova S; Ramadori AT; Jonghe S De; Herdewijn, P. Discovery of Dual Death-Associated Protein Related Apoptosis Inducing Protein Kinase 1 and 2 Inhibitors by a Scaffold Hopping Approach. J. Med. Chem. 2014, 57 (18), 7624–7643. [DOI] [PubMed] [Google Scholar]
- (25).Le Huerou Y; Blake J; Gunwardana I; Mohr P; Wallace E; Wang B; Chicarelli M; Lyon M Pyrrolopyridines as Kinase Inhibitors. WO 2009/140320, 2009. [Google Scholar]
- (26).Stavenger R; Witherington J; Rawlings D; Holt D; Chan G CHK1 Kinase Inhibitors. WO 03/028724, 2003. [Google Scholar]
- (27).Bahekar RH; Jain MR; Jadav PA; Prajapati VM; Patel DN; Gupta AA; Sharma A; Tom R; Bandyopadhya D; Modi H; Patel PR Synthesis and Antidiabetic Activity of 2,5-Disubstituted-3-Imidazol-2-Yl-Pyrrolo[2,3-b]Pyridines and Thieno[2,3-b]Pyridines. Bioorg. Med. Chem. 2007, 15 (21), 6782–6795. [DOI] [PubMed] [Google Scholar]
- (28).Chavan NL; Nayak SK; Kusurkar RS A Rapid Method toward the Synthesis of New Substituted Tetrahydro α-Carbolines and α-Carbolines. Tetrahedron 2010, 66 (10), 1827–1831. [Google Scholar]
- (29).Chinta BS; Baire B Reactivity of Indole-3-Alkoxides in the Absence of Acids: Rapid Synthesis of Homo-Bisindolylmethanes. Tetrahedron 2016, 72 (49), 8106–8116. [Google Scholar]
- (30).McCoull W; Hennessy EJ; Blades K; Box MR; Chuaqui C; Dowling JE; Davies CD; Ferguson AD; Goldberg FW; Howe NJ; Kemmitt PD; Lamont GM; Madden K; McWhirter C; Varnes JG; Ward RA; Williams JD; Yang B Identification and Optimisation of 7-Azaindole PAK1 Inhibitors with Improved Potency and Kinase Selectivity. Medchemcomm 2014, 5 (10), 1533–1539. [Google Scholar]
- (31).Gourdain S; Dairou J; Denhez C; Bui LC; Rodrigues-Lima F; Janel N; Delabar JM; Cariou K; Dodd RH Development of DANDYs, New 3,5-Diaryl-7-Azaindoles Demonstrating Potent DYRK1A Kinase Inhibitory Activity. J. Med. Chem. 2013, 56 (23), 9569–9585. [DOI] [PubMed] [Google Scholar]
- (32).Gelbard H; Dewhurst S; Goodfellow V; Wiemann T; Ravula S; Loweth C Bicyclic Heteroaryl Kinase Inhibitors and Methods of Use. WO 2011/149950, 2011. [Google Scholar]
- (33).Knauber T; Tucker J Palladium Catalyzed Monoselective α-Arylation of Sulfones and Sulfonamides with 2,2,6,6-Tetramethylpiperidine·ZnCl·LiCl Base and Aryl Bromides. J. Org. Chem. 2016, 81 (13), 5636–5648. [DOI] [PubMed] [Google Scholar]
- (34).Zhao B; Li Y; Xu P; Dai Y; Luo C; Sun Y; Ai J; Geng M; Duan W Discovery of Substituted 1 H -Pyrazolo[3,4- b ]Pyridine Derivatives as Potent and Selective FGFR Kinase Inhibitors. ACS Med. Chem. Lett. 2016, 7 (6), 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Shi J; Xu G; Zhu W; Ye H; Yang S; Luo Y; Han J; Yang J; Li R; Wei Y; Chen L Design and Synthesis of 1,4,5,6-Tetrahydropyrrolo[3,4-c]Pyrazoles and Pyrazolo[3,4-b]Pyridines for Aurora-A Kinase Inhibitors. Bioorganic Med. Chem. Lett. 2010, 20 (14), 4273–4278. [DOI] [PubMed] [Google Scholar]
- (36).Van Mileghem S; Egle B; Gilles P; Veryser C; Van Meervelt L; De Borggraeve WM Carbonylation as a Novel Method for the Assembly of Pyrazine Based Oligoamide Alpha-Helix Mimetics. Org. Biomol. Chem. 2017, 15 (2), 373–378. [DOI] [PubMed] [Google Scholar]
- (37).Maccormick S; Storck P-H; Mertimore M; Charrier J-D; Knegtel R; Young S; Pinder J; Durrant S Compounds Useful as Inhibitors of ATR Kinase. WO 2012/178123, 2012. [Google Scholar]
- (38).Gelbard H; Dewhurst S; Goodfellow V; Wiemann T; Bennet D MLK Inhibitors and Methods of Use. WO 2010/068483, 2010. [Google Scholar]
- (39).Sorrell FJ; Szklarz M; Azeez KRA; Elkins JM; Sorrell FJ; Szklarz M; Azeez KRA; Elkins JM; Knapp S Family-Wide Structural Analysis of Human Numb- Associated Protein Kinases Article. Struct. Des. 2016, 24 (3), 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Fabian MA; Biggs WH; Treiber DK; Atteridge CE; Azimioara MD; Benedetti MG; Carter TA; Ciceri P; Edeen PT; Floyd M; Ford JM; Galvin M; Gerlach JL; Grotzfeld RM; Herrgard S; Insko DE; Insko MA; Lai Andiliy G; Lélias; Mehta SA; Milanov ZV; Velasco AM; Wodicka LM; Patel HK; Zarrinkar PP; Lockhart DJ A Small Molecule–kinase Interaction Map for Clinical Kinase Inhibitors. Nat. Biotechnol. 2005, 23 (3), 329–336. [DOI] [PubMed] [Google Scholar]
- (41).Karaman MW; Herrgard S; Treiber DK; Gallant P; Atteridge CE; Campbell BT; Chan KW; Ciceri P; Davis MI; Edeen PT; Faraoni R; Floyd M; Hunt JP; Lockhart DJ; Milanov ZV; Morrison MJ; Pallares G; Patel HK; Pritchard S; Wodicka LM; Zarrinkar PP A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2008, 26 (1), 127–132. [DOI] [PubMed] [Google Scholar]
- (42).Trott O; Olson AJ AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Case DA; Ben-Shalom I.; Brozell SR; Cerutti DS; Cheatham T.; Cruzeiro VWD; Darden TA; Duke RA; Ghoreishi D; Gilson MK; Gohlke H; Goetz AW; Greene D; Harris R; Homeyer N; Huang Y; Izadi S; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein DJ; Merz KM; Miao Y; Monard G; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Smith J; SalomonFerrer R; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xiao L; York DM; Kollman PA AMBER 2018; San Francisco, 2018. [Google Scholar]
- (44).Genheden S; Ryde U The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10 (5), 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera?A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25 (13), 1605–1612. [DOI] [PubMed] [Google Scholar]
- (46).Kabsch W XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Winter G Xia2 : An Expert System for Macromolecular Crystallography Data Reduction. J. Appl. Crystallogr. 2010, 43, 186–190. [Google Scholar]
- (48).Winn MD; Charles C; Cowtan KD; Dodson EJ; Leslie AGW; Mccoy A; Stuart J; Garib N; Powell HR; Randy J Overview of the CCP 4 Suite and Current Developments. 2011, D67, 235–242. [Google Scholar]
- (49).Mccoy AJ; Grosse-kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software Research Papers. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Murshudov GN; Vagin AA; Dodson EJ Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53 (3), 240–255. [DOI] [PubMed] [Google Scholar]
- (51).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX : A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Emsley P; Lohkamp B; Scott WG; Cowtan K; IUCr. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Perera R; Khaliq M; Kuhn RJ Closing the Door on Flaviviruses: Entry as a Target for Antiviral Drug Design. Antiviral Res. 2008, 80 (1), 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Xie X; Gayen S; Kang C; Yuan Z; Shi P-Y Membrane Topology and Function of Dengue Virus NS2A Protein. J. Virol. 2013, 87 (8), 4609–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Huang CY-H; Butrapet S; Moss KJ; Childers T; Erb SM; Calvert AE; Silengo SJ; Kinney RM; Blair CD; Roehrig JT The Dengue Virus Type 2 Envelope Protein Fusion Peptide Is Essential for Membrane Fusion. Virology 2010, 396 (2), 305–315. [DOI] [PubMed] [Google Scholar]
- (56).Rodriguez-Madoz JR; Bernal-Rubio D; Kaminski D; Boyd K; Fernandez-Sesma A Dengue Virus Inhibits the Production of Type I Interferon in Primary Human Dendritic Cells. J. Virol. 2010, 84 (9), 4845–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Evans DA History of the Harvard ChemDraw Project. Angew. Chemie Int. Ed. 2014, 53 (42), 11140–11145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.