

Abstract
Purpose:
Neuroendocrine prostate cancer (NEPC), an aggressive variant of CRPC, often emerges after AR-targeted therapies such as enzalutamide (ENZ) or de novo, via trans-differentiation process of neuroendocrine differentiation. The mechanistic basis of NED is poorly understood, contributing to lack of effective predictive biomarkers and late disease recognition. The purpose of present study was to examine the role of novel pro-neural POU-domain transcription factors (TFs) in NEPC and examine their potential as non-invasive predictive biomarkers.
Experimental Design:
PCa patient-derived xenograft models, clinical samples and cellular NED models were employed to determine the expression of TFs BRN1 and BRN4. BRN4 levels were modulated in PCa cell lines followed by functional assays. Further, EVs were isolated from patient samples and cell culture models, characterized by Nanoparticle Tracking analyses, Western blotting and real time PCR.
Results:
We identify for the first time that: (i) BRN4 is amplified and overexpressed in NEPC clinical samples and that BRN4 overexpression drives NED via its interplay with BRN2, a TF that was previously implicated in NEPC; (ii) BRN4 and BRN2 mRNA are actively released in PCa EVs upon NED induction; (iii) ENZ treatment augments release of BRN4 and BRN2 in PCa EVs, promoting NED induction.
Conclusions:
Our study identifies a novel TF that drives NEPC and suggests that as adaptive mechanism to ENZ treatment, PCa cells express and secrete BRN4 and BRN2 in EVs that drive oncogenic reprogramming of PCa cells to NEPC. Importantly, EV-associated BRN4 and BRN2 are potential novel non-invasive biomarkers to predict NED in CRPC.
Keywords: BRN4, neuroendocrine differentiation, castration-resistant prostate cancer, BRN2, extracellular vesicles
INTRODUCTION
Prostate cancer (PCa), a leading cause of male cancer-related mortality in the US (1), is dependent on Androgen receptor (AR) signaling. Therefore, ablation of AR signaling by androgen deprivation is the goal of first-line therapy (2) that initially results in cancer regression. However, 2-3 years after androgen deprivation in 25-40% of cases, the disease develops into castration-resistant prostate cancer CRPC (3) that has limited therapeutic options (3). CRPC patients are treated with AR pathway inhibitors (API) such as Enzalutamide (MDV3100/ENZ) and abiraterone (ABI) as a second-line of therapy that improves survival initially (2,4). However, CRPC patients develop drug resistance over a certain period owing to heterogenous molecular mechanisms such as AR bypass signaling or complete AR independence (5,6). A subset of API resistant tumors undergo a reversible trans-differentiation process known as neuroendocrine differentiation (NED), that is associated with altered expression of lineage markers such as decreased expression of AR and increased expression of neuroendocrine (NE) lineage markers including enolase 2 (ENO2), chromogranin A (CHGA) and synaptophysin (SYP) (7,8). Due to lack of AR signaling, these PCa variants, referred to as neuroendocrine prostate cancer (NEPC), are impervious to anti-androgen therapy and constitute an aggressive variant of advanced CRPC with shorter survival times and limited therapeutic options (7). Though NEPC is thought to arise late in the disease course subsequent to treatment with ENZ and ABI, this variant can also arise de novo in metastatic CRPC after primary docetaxel therapy or early on after API treatment (7–9). Further, it is not clear if therapy-induced NED is the same disease as de novo small cell PCa that emerges from rare neuroendocrine cell populations in the prostate (10). NEPC variants are associated with the presence of visceral metastasis to liver, lung and central nervous system, in addition to lytic bone metastases and low serum PSA levels relative to disease burden (7). The molecular mechanistic basis of NED is poorly understood that contributes to lack of effective predictive biomarkers and late recognition of the disease. The genetic and epigenetic alterations underlying NED has been investigated (11–15) recently that shows that these states are derived via clonal evolution from adenocarcinomas (11). The key genetic events driving this transition include loss of the tumor suppressors retinoblastoma (RB1), tumor protein 53 (TP53), phosphatase and tensin homolog (PTEN), (11–15), frequent TMPRSS2-ERG gene rearrangements (14), EZH2 overexpression and amplifications of NMYC and Aurora Kinase A (AURKA) (11–13,15). AURKA is a cell cycle kinase that stabilizes N-Myc oncoprotein and prevents N-Myc degradation (12,13,16). Disruption of the molecular interaction between AURKA and N-Myc via a therapeutic agent Alisertib is being examined as a therapeutic modality for NEPC (17) with promising results. Further, the upregulation of Delta-like protein 3 (DLL3) has been reported in NEPC cases as compared to CRPC-adenocarcinomas. Infact, DLL3 expression can be exploited for therapeutic targeting of NEPC by employing a humanized DLL3 antibody (18–20). Though these studies have characterized the key alterations driving NED, we are still far from understanding the genetic alterations driving this transition.
POU (Pit-Oct-Unc) -domain/Oct proteins are a set of reprogramming transcription factors (TFs) that are critical regulators of gene expression programs determining cellular identities and play important roles in neurogenesis (21–23). Out of six classes, Class III POU genes (POU3F1/OCT6, POU3F2/BRN2, POU3F3/BRN1, POU3F4/BRN4) are considered to be crucial for neurogenesis (24). POU3F2/BRN2 was recently reported as a AR-repressed, master neural TF that drives PCa NED by controlling SOX2 expression (25). BRN3a has also been reported to be upregulated with NEPC (26). However, the roles of other Class III pro-neural TFs have not been unequivocally studied in PCa. Here we report for the first time, that BRN4 is a novel driver of NED states in CRPC. BRN4 (Brain4) is located on X-chromosome and is involved in the patterning of the neural tube, paraventricular and supraoptic nuclei of the hypothalamus in the developing embryo (27). BRN4 mutations have been linked to X-linked non-syndromic deafness (28). However, its involvement in PCa has never been studied.
Recently, extracellular vesicles (EVs)/exosomes have emerged as key regulators of cancer progression and metastasis. Exosomes (exos) are small membranous EVs, typically between 30-150 nm in size, (29) that are gaining significant interest as alternate disease biomarkers. EVs can be detected non-invasively in biological fluids such as serum (30) and can be used as a liquid biopsy for PCa (31–33). EVs mediate intercellular communication by transferring their cargo such as mRNA and proteins to recipient cells to modulate target cell functions (34,35). We hypothesized that in addition to cell intrinsic genetic determinants of NED, tumor exosomes/EVs (referred to as EVs subsequently) are important determinants that facilitate this trans-differentiation by mediating intercellular communication between cancer cells via horizontal transfer of functional neuronal factors. In this study, we validated our hypothesis and demonstrate for the first time that EV-mediated signaling is important for NED induction via the release of BRN4 and BRN2 mRNA in PCa EVs.
MATERIALS AND METHODS
Cell lines and cell culture
Nonmalignant prostate epithelial cell line RWPE-1 and PCa cell lines (LNCaP, Du145, PC3, C42B, NCI-H660 (36)) were obtained from the American Type Culture Collection (ATCC) and cultured under recommended conditions. All cell lines were maintained in an incubator with a humidified atmosphere of 95% air and 5% CO2 at 37°C. The experiments with cell lines were performed within 6 months of their procurement/resuscitation. Prostate cell lines were authenticated by DNA short-tandem repeat analysis. Cell lines were checked at periodic intervals for mycoplasma contamination by DAPI staining.
Clinical Samples
The study was conducted in accordance with ethical guidelines of US Common Rule and was approved by the UCSF Committee on Human Research. Written informed consent was obtained from patients. Serum samples (0.5ml-1ml) from PCa patients and deidentified clinical information were obtained from Prostate Cancer Biorepository Network (PCBN) and stored at −80°C till processed. Cohort included metastatic human CRPC clinical samples with adenocarcinoma features (CRPC-adeno) vs those with neuroendocrine features (CRPC-NE) (Table S1). CRPC-adeno included patients with no evidence of NED while CRPC-NE included AR-patients with therapy-induced NED with features of small cell/large cell NE carcinoma. Follow up information included prior therapies for all the clinical samples.
Isolation of EVs
Serum derived EVs were isolated from 250-500μL of serum using the Total exosome isolation reagent (Life Technologies, Cat. No. 4478360) as per manufacturer’s instructions and as described in (37). For isolation of EVs from cell culture media, cells were grown in recommended media with exosome-depleted FBS for 48 hours. Conditioned media were collected and EVs were isolated with exosome isolation reagent (Life Technologies, Cat. No. 4478359) as per manufacturer’s instructions.
Statistics
All quantified data represents an average of triplicate samples or as indicated. All experiments with cell lines included at least three biological replicates. Data are represented as mean ± S.E.M or as indicated. Statistical significance between groups was assessed by Student’s t-test. Mann-Whitney U test was used to assess the difference between mRNA expression in independent test/control samples. Correlations between mRNA expression and clinicopathological parameters were assessed using Chi squared test. Receiver Operating Characteristic (ROC) curves were generated based on dCt values of test mRNA. Statistical analyses were performed using MedCalc version 10.3.2. Results were considered statistically significant at P ≤ 0.05.
RESULTS
POU-domain TFs BRN1 and BRN4 are highly expressed in NEPC cell line models and enzalutamide-resistant cells
We hypothesized that multiple POU-domain TFs act in concert to promote PCa NED. To test our hypothesis, we examined the copy number alterations (CNAs) of these factors in CRPC patients with adenocarcinoma (CRPC-Adeno) and those with NE features (CRPC-NE) by querying SUC2C/PCF Dream Team (38) and Beltran et al (11) cohorts using cBioPortal (39,40) (Fig. 1A). We found that Class III TFs are frequently altered in these cases, with BRN4 and BRN1 alterations (mostly amplifications) found in 11% and 4% of cases, respectively as compared to 5% in BRN2. Importantly, BRN4, BRN1 and BRN2 amplifications were present in ~20%, 9% and 16% of CRPC-NE cases in these two cohorts suggestive of a potential role of these genes in NEPC (Fig. 1A, lower panels). While BRN4 amplifications were also significantly increased in CRPCs as compared to prostate adenocarcinomas (PRAD), BRN2 and BRN1 amplifications were more prevalent in NEPC as compared to CRPC and PRAD. Importantly, BRN2 alterations were found to significantly co-occur with BRN1 and BRN4 alterations suggesting that they may act in a concerted manner to drive NED (Fig. 1A). Correlation analyses of CNAs with corresponding mRNA expression of these factors (Fig. 1B) showed that BRN4 mRNA is significantly positively correlated with BRN4 CNAs while BRN2 mRNA showed an inverse correlation and BRN1 mRNA showed no significant correlation. Further, we identified that BRN1 and BRN4 are upregulated upon ENZ treatment of PCa cell lines and are associated with PCa NED (Fig. 1C–1E). We examined the expression of BRN1, BRN4 alongwith BRN2 in normal immortalized prostate epithelial cell line (RWPE-1), benign non-transformed prostate epithelial cell line (BPH1) and PCa cell lines (PC3, Du145, LNCaP and NCI-H660) (Fig. 1C). Our analyses showed that similar to BRN2, BRN1 and BRN4 mRNA levels are specifically significantly upregulated in NE cell line, NCI-H660 (36). We also examined the levels of these factors in an inducible NE cell line model (Fig. S1 and 1D). LNCaP cells were grown under androgen depleted conditions by culturing in RPMI medium with 10% C/D FBS for 4 days. These conditions have been shown to induce NED in LNCaP cells (41) following which cells were treated with DMSO/20µM ENZ for 48 hrs. As a negative control, LNCaP cells grown under regular conditions (RPMI+10% FBS) were included and positive control included NCiH660 cell line. Both androgen depletion by C/D FBS (middle panel) and ENZ treatment (right panel) led to induction of NE morphology (Fig. S1A) as assessed by phase contrast microscopy concomitant with induction of neuronal markers BRN2, SYP, ENO2 as assayed by RT-PCR (Fig. S1B) and BRN2, SYP, CHGA protein as assessed by Western blotting (Fig. S1C). Interestingly, we found that BRN1 and BRN4 mRNAs were upregulated along with ENO2 and BRN2 in LNCaP cells induced to undergo NED via growth under C/D FBS/ENZ treatment vs control cells (Fig. 1D). While BRN2 expression was induced 7.7- and 7-fold upon androgen withdrawal and ENZ treatment respectively, BRN1 induction was ~23- and 17-fold and that of BRN4 was ~36- and 45-fold, respectively under same conditions (Fig. 1D). This suggests that androgen withdrawal/enzalutamide treatment induces the expression of these TFs. To gain better insight into the potential regulation of these factors by enzalutamide, we examined these TFs in ENZ-resistant LNCaP-AR cell line. As a control, LNCaP cells stably expressing AR (LNCaP-AR) and parental LNCaP (Fig. 1E) cells were used. We found that BRN1, BRN2 and BRN4 mRNAs were upregulated in LNCaP-AR-ENZR cells suggesting the potential association of these factors with enzalutamide resistance in CRPC (Fig. 1E, left panel). BRN4 protein was found to be upregulated in LNCaP-AR-ENZR cells as compared to LNCaP-AR/LNCaP cells alongwith induction of BRN2, CHGA and ENO2 (Fig. 1E, right panel).
Fig. 1. POU-domain TFs BRN1 and BRN4 are highly expressed in neuroendocrine PCa cell line models and enzalutamide resistant cells.
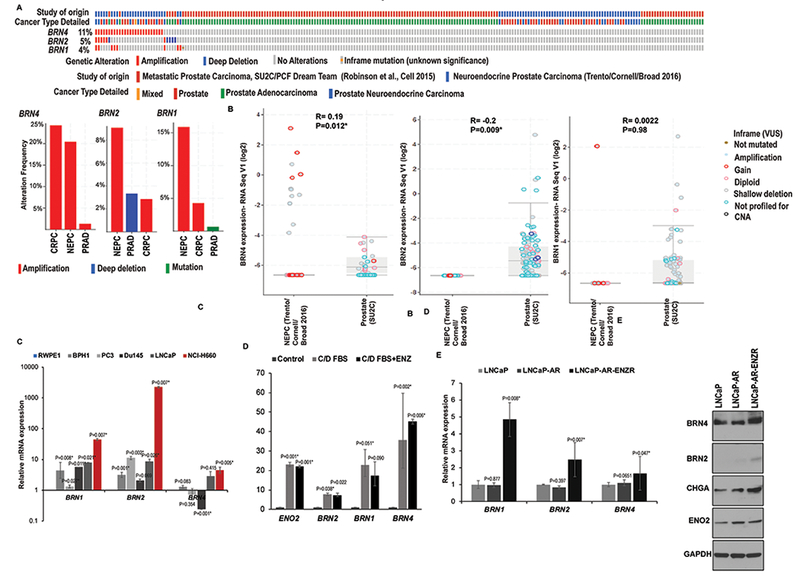
A. Genomic alterations in Class III POU-domain genes in prostate neuroendocrine carcinomas (CRPC-NE), mixed small cell carcinoma-adenocarcinomas and adenocarcinomas in SUC2C/PCF Dream Team (38) and Beltran et al (11) cohorts as analyzed by cBioPortal. Lower panel shows the relative frequencies of these alterations in PRAD, CRPC and NEPC cases within these two cohorts.
B. Correlation analyses of BRN1, BRN2 and BRN4 mRNA and CNAs in the two cohorts.
C. Relative mRNA levels of BRN1, BRN2 and BRN4 in normal immortalized prostate epithelial cell line (RWPE-1), benign non-transformed prostate epithelial cell line (BPH1) and PCa cell lines (PC3, Du145, LNCaP and NCI-H660) as assessed by real-time PCR. Data were normalized to GAPDH control and represented as mean ± SEM.
D. Relative mRNA levels of BRN11, BRN2 and BRN4 in LNCaP cells cultured in regular media (control), androgen-depleted media (C/D FBS) and 20 µM ENZ in C/D FBS media as assessed by real-time PCR. Data were normalized to GAPDH control and represented as mean ± SEM.
E. Left panel: Relative mRNA levels of BRN1, BRN2 and BRN4 in LNCaP, LNCaP-AR and ENZ-resistant LNCaP-AR cells as assessed by real-time PCR. Data were normalized to GAPDH control and represented as mean ± SEM. Right panel: Western blot analyses of BRN4, BRN2 and indicated neuronal markers in LNCaP, LNCaP-AR and ENZ-resistant LNCaP-AR cells. GAPDH was used as a loading control.
BRN4 expression is selectively upregulated in CRPC-NE patient-derived xenograft (PDX) models, clinical samples and cellular NED models
In view of our data showing induction of BRN1 and BRN4 mRNA in NE cellular models, we examined the clinical relevance of these alterations using PDX models (Fig. 2A). We assessed their levels in PDX models with CRPC-Adenocarcinoma characteristics (LuCaP 70, 78, 81 and 92) vs those with CRPC-NE alterations (LuCaP 49, 145.1 and 145.2) (42) by real-time PCR analyses. While the expression of BRN1 was not significantly different between CRPC-Adeno vs CRPC-NE PDXs (Fig. 2A), BRN4 expression was significantly upregulated in CRPC-NE xenografts LuCaP 49, 145.1 and 145.2 (Fig. 2B). These data suggest that BRN4 upregulation is a clinically relevant alteration associated with transition of adenocarcinomas to NE states. In view of these data, we focused on BRN4. To validate the association of BRN4 with NEPC, we examined BRN4 mRNA alterations in Beltran et al (11) cohort using cBioPortal (39,40). We found that BRN4 mRNA is significantly upregulated in NEPC cases (Fig. 2C). Further, we examined BRN4 expression in inducible cellular NED models (Fig. 2D–F). MYCN has been implicated as a critical gene that drives NEPC (12,16,43). We generated stable clones from the LNCaP/AR and C42B cell lines overexpressing MYCN construct/control vector (Fig. 2D). Upon NMYC overexpression (Fig. 2D), we observed an increase in BRN4 protein levels concomitant with induction of neuronal markers BRN2, CHGA and ENO2 (Fig. 2D, lower panels). Similarly, knockdown of TP53 and RB1 has been shown to induce NE phenotype in LNCaP/AR cells (44). shRNA-mediated TP53 and RB dual knockdown in LNCaP/AR cells led to BRN4 mRNA induction (Fig. 2E). BRN4 induction with BRN2 and other NE markers by androgen withdrawal was confirmed in additional PCa cell lines, C42B and 22Rv1 (Fig. 2F), consolidating the association of BRN4 expression with induction of PCa NED.
Fig. 2. BRN4 expression is selectively upregulated in CRPC-NE patient-derived xenograft (PDX) models, clinical samples and cell line models.
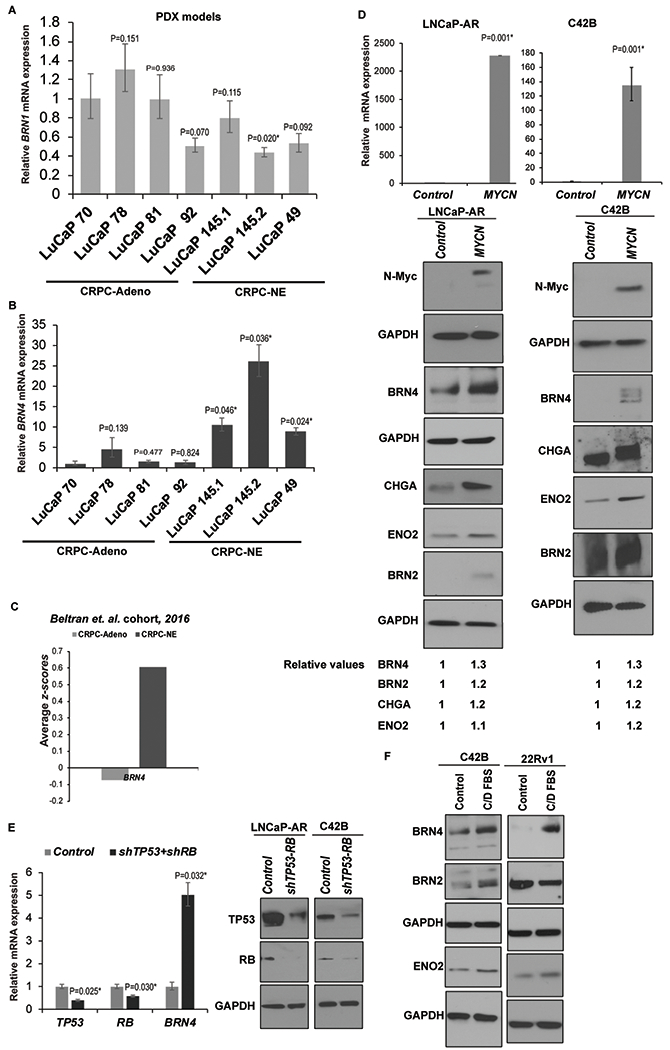
A. Relative BRN1 mRNA and;
B. BRN4 mRNA expression in PDX models with CRPC-Adenocarcinoma characteristics (LuCaP 70, 78, 81 and 92) vs those with CRPC-NE alterations (LuCaP 49, 145.1 and 145.2) as assessed by real-time PCR analyses. Data were normalized to GAPDH control and represented as mean ± SEM. P-values were calculated relative to LuCaP 70 PDX.
C. Average z-scores for BRN4 mRNA expression in CRPC-Adeno vs CRPC-NE tissues in Beltran et al (11) cohort.
D. LNCaP-AR and C42B cells were stably transfected with control/NMYC overexpression constructs followed by real time PCR analyses of NMYC mRNA (upper panel) and Western blot analyses of N-Myc, BRN4 and neuronal markers BRN2, CHGA and ENO2 (lower panels). Band intensities were quantified by Image J, relative values were calculated for indicated proteins after normalizing to corresponding GAPDH values and are represented below blots.
E. Left panel: LNCaP-AR cells were transfected with control shRNA/shRNA targeting TP53 and RB1 followed by real time PCR analyses of TP53, RB and BRN4 mRNA. Right panel: Western blot analyses confirming TP53 and RB knockdown following shRNA transfections. GAPDH was used as a loading control.
F. C42B and 22Rv1 cell lines were grown under androgen depleted conditions (RPMI medium with 10% C/D FBS) for 5 days followed by Western blot analyses of BRN4, BRN2 and indicated neuronal markers. GAPDH was used as a loading control.
BRN4 interplays with BRN2 and regulates SOX2 expression
To understand the mechanistic role of BRN4 in NEPC, we overexpressed control/BRN4 construct in LNCaP-AR and C42B cell lines (Fig. 3A) followed by expression analyses of NE markers. Since BRN2 was recently implicated as a principal driver of NEPC (25), we included BRN2 overexpression as a positive control. Interestingly, we found that BRN4 overexpression led to SOX2 overexpression in both cell lines compared to control and BRN2 overexpression (Fig. 3B) concomitant with induction of ENO2, a NE marker. Since SOX2 is a critical TF that has been implicated in NEPC (45) and BRN2 has been reported to an upstream regulator of SOX2 in driving NEPC (25), we sought to determine if there is potential interaction between BRN4 and BRN2. We performed co-immunoprecipitation (co-IP) with BRN4 in LNCaP-AR and C42B cell lines followed by Western blotting for BRN2 (Fig. 3C). In a converse approach, we pulled down BRN2 with BRN2 Ab and probed for BRN4 (Fig. 3C). Our data shows that these two TFs interact directly suggesting that they may act in concert in driving NEPC. To further understand the interplay between these factors, we examined BRN4 levels upon BRN2 overexpression in LNCaP-AR and C42B cell lines (Fig. 3D). Interestingly, we found that BRN2 overexpression led to BRN4 upregulation in both cell lines. In a converse approach, we knocked down BRN2 expression in NE cell line NCI-H660 and C42B cells and observed low BRN4 expression concomitant with BRN2 knockdown (Fig. 3E). These data suggest that BRN4 interplays with BRN2. We further examined the correlation of BRN4 with BRN2 expression in PCTA (Prostate Cancer Transcriptome Analyses) dataset (46), a large cohort of mCRPC patients (n=260) and observed a significant positive correlation between BRN4 and BRN2 in mCRPC (P<0.001) (Fig. 3F). These data validate BRN4 as a crucial TF that interacts with master neural TF BRN2 in PCa (25). We also examined the correlation of BRN4 with AR in the PCTA cohort and observed a negative correlation (Fig. 3G) suggesting that BRN4 is expressed upon AR downregulation. We further examined the effects of BRN4 overexpression on LNCaP-AR cells grown in regular media/androgen-depleted media + ENZ (Fig. 3H). We observed that BRN4 overexpression confers a growth advantage to cells grown in regular media/androgen depleted media suggesting that BRN4 is oncogenic and confers resistance to ENZ. Further, BRN4 overexpression led to augmentation of in vitro migration and invasive abilities of LnCaP-AR and C42B cell lines as compared to corresponding controls (Fig. 3I) suggesting that BRN4 controls PCa aggressiveness.
Fig. 3. BRN4 regulates SOX2 expression and controls expression of NE genes.
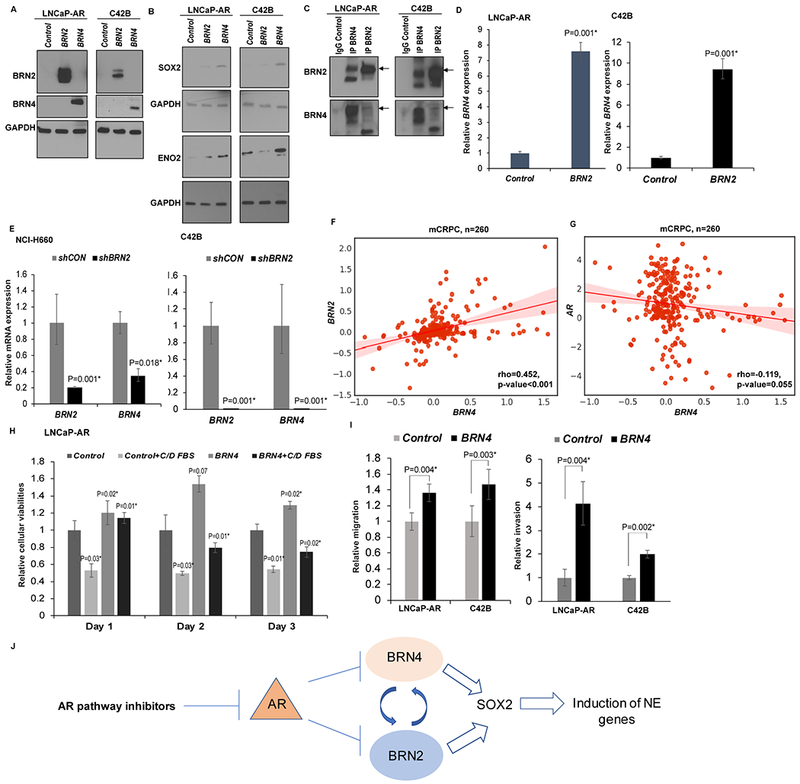
A-B. LNCaP-AR and C42B cells were stably transfected with control/BRN2/BRN4 overexpression constructs followed by Western blot analyses of A. BRN2 and BRN4 protein levels; B. SOX2 and ENO2 protein levels. GAPDH was used as a loading control.
C. LNCaP-AR and C42B cells were used to perform co-IP with control IgG, BRN4 antibody and BRN2 antibody followed by Western blot analyses for BRN2 and BRN4.
D. LNCaP-AR and C42B cells were stably transfected with control/BRN2 overexpression constructs followed by real time PCR analyses of BRN4 mRNA. GAPDH was used as an endogenous control.
E. NCI-H660 and C42B cells were stably transfected with control/BRN2 shRNA constructs followed by real time PCR analyses of BRN2 and BRN4 mRNA. GAPDH was used as an endogenous control.
F. Correlation between BRN2 and BRN4 expression; and G. AR and BRN4 expression in PCTA (Prostate Cancer Transcriptome Analyses) dataset of mCRPC patients (n=260).
H. Cellular viabilities of LNCaP-AR cells transfected with control/BRN4 grown in regular media/androgen-depleted media + ENZ.
I. Transwell in vitro migration and invasion assays upon control/BRN4 expression in LNCaP-AR and C42B cell lines.
J. Schematic representation showing the proposed role of BRN4 in NEPC.
Alterations in EV secretion pathways upon induction of NED states in PCa
Recent results from our laboratory suggest that EVs play a key role in mediating NED states in advanced PCa. We propose that ENZ resistance is mediated via EVs whereby these vesicles act as vehicles for exchange of neuronal TFs between heterogenous populations of tumor cells, promoting NED and engendering a transmitted API resistance. Towards this, we assayed EVs released from clinical samples (CRPC-Adeno, n=42 vs CRPC-NE, n=6, Table S1). Isolated exosomal preparations were comprehensively characterized by electron microscopy, NTA (Fig. 4A) and immunoblot analyses for presence of multiple exosomal markers (CD9, CD63, TSG101) and absence of contaminating proteins such as GRP94 (Fig. 4B). NTA analyses showed that the average particle size (Fig. 4A, lower left panel) and numbers (Fig. 4A, lower right panel) were not significantly different between CRPC-Adeno and CRPC-NE, though CRPC-NE samples trended towards higher particle number and size. While we confirmed EV markers by Western blotting (Fig. 4B), we found that CD9 expression is variable, decreasing predominantly in CRPC-NE cases. To gain further insights, we examined CD9 alterations in Beltran et al (11) cohort using cBioPortal (39,40). We found that CD9 is amplified in 4% cases of CRPC-NE vs ~13% in other PCa cases while CD63 amplification frequency in NEPC is not significantly different (Fig. 4C, left panel). In concordance with amplifications, average CD9 mRNA expression was found to be ~2.5-fold lower in CRPC-NE vs CRPC-Adeno while CD63 is not altered significantly (Fig. 4C, right panel). These data suggest that PCa NED is associated with alterations in EV secretion pathways.
Fig. 4. Alterations in EV secretion pathways upon induction of neuroendocrine differentiation states in prostate cancer and release of BRN2 and BRN4 mRNA in PCa EVs upon ENZ treatment.
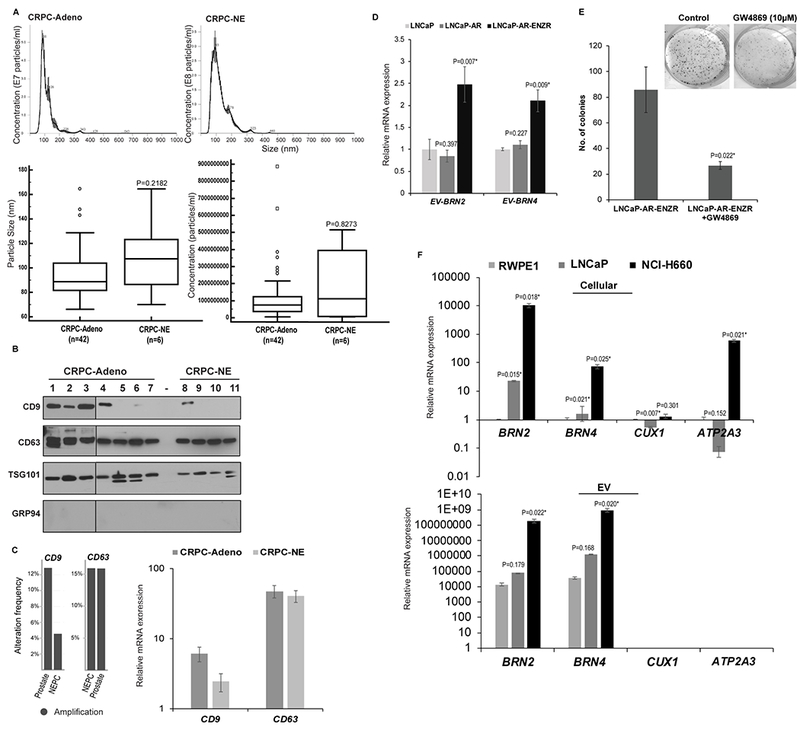
A. EVs were isolated from sera of PCa patients with CRPC-Adeno, n=42 and CRPC-NE, n=6. Nanoparticle Tracking Analyses (NTA) of representative CRPC-Adeno (upper left panel) and CRPC-NE (upper right panel) cases showing size and concentration of isolated particles. Lower left: Average particle size and; Lower right: Particle concentration in CRPC-Adeno vs CRPC-NE cases as determined by NTA analyses.
B. Western blot analyses for EV markers CD9, CD63, TSG101 and negative marker GRP94 to confirm the integrity of EVs isolated from sera of CRPC-Adeno (n=7) and CRPC-NE (n=4) cases.
C. Genomic alteration frequencies (left panel) and relative mRNA expression (right panel) for CD9 and CD63 in CRPC-Adeno and CRPC-NE cases in Beltran et al (11) cohort. mRNA data is represented as mean ± SEM.
D. EVs were extracted from conditioned media of LNCaP, LNCaP-AR and ENZ-R cell line followed by RNA isolation and real-time PCR based expression profiling for EV-associated BRN2 and BRN4 mRNA. Data were normalized to GAPDH control and represented as mean ± SEM.
E. LNCaP-AR ENZR cell line was treated with exosome inhibitor GW4869 (20µM) for 48 hours followed by clonogenicity assay. Representative images from control/GW4869 treated cells are shown above.
F. Expression of indicated genes in cellular (upper) and EV (lower) fractions from RWPE-1, LNCaP and NCI-H660 cells. Data were normalized to GAPDH control and represented as mean ± SEM.
BRN2 and BRN4 mRNA are released in PCa EVs upon ENZ treatment
Importantly, we identified that BRN4 and BRN2 mRNA are specifically released into PCa EVs. We extracted EVs from conditioned media of LNCaP, LNCaP-AR and LNCaP-AR-ENZR cell line followed by expression profiling (Fig. 4D) and found that BRN2 and BRN4 mRNA (denoted as EV-BRN2 and EV-BRN4, respectively) were significantly increased in PCa EVs isolated from LNCaP-AR-ENZR cell line. These data point to an association of ENZ resistance to an increased secretion of these TFs in EVs. We hypothesize that secretion of these factors in PCa EVs in CRPC underlie enzalutamide resistance and may be an adaptive mechanism for PCa cells to survive under the selective pressure of APIs. Further, treatment of LNCaP-AR-ENZR cell line with exosome inhibitor GW4869 (Fig. 4E) could partially restore the sensitivity of this cell line to ENZ as monitored by clonogenicity assay. This supports a key role of exosome-mediated intercommunication in ENZ resistance. We also examined the expression of BRN4 and BRN2 in cells (Fig. 4F, upper panels) and EVs (Fig. 4F, lower panels) from RWPE-1 cells vs PCa cell lines and found that their levels are specifically upregulated in EVs from NE cell line NCI-H660 while the increases in LNCaP EVs were statistically insignificant. To confirm the specific release of BRN4 and BRN2 in EVs in NEPC, we examined the expression of additional genes- CUX1 and ATP2A3- in cells and corresponding EVs from PCa cell lines (Fig. 4F). CUX1 encodes Cut-like homeobox 1 TF while ATP2A3 encodes a Calcium-Translocating P-Type ATPase that has been shown to be regulated in prostate cancer (47). While CUX1 and ATP2A3 were found to be expressed in LNCaP and NCI-H660 (Fig. 4F, upper panel), their expression in EVs were undetected in all analyzed cell lines (Fig. 4F, lower panel). These observations validated our findings on EV-BRN2 and -BRN4.
EV-associated BRN4 and BRN2 are upregulated in sera from NEPC patients and can predict NED induction non-invasively
In view of our data showing presence of BRN4 and BRN2 mRNA in EVs and increase in their levels upon ENZ treatment and elevated levels in NE cell line, we asked if these factors could be used as non-invasive markers to predict PCa NED (Fig. 5). EVs were extracted from the sera of CRPC-Adeno and CRPC-NE cases (Cohort 1, Table S1). Following extensive characterization of EVs by NTA analyses and Western blotting for positive and negative EV markers (4A-B), vesicle-associated RNAs were extracted and profiled by real-time PCR. EV-BRN4 was significantly upregulated (~7-fold) in CRPC-NE compared to CRPC-Adeno cases (Fig. 5A). To assess the potential of EV-BRN4 to be a diagnostic biomarker for assessing NED, we performed Receiver Operating Characteristic (ROC) curve analyses based on dCt values in CRPC-Adeno and CRPC-NE (Fig. 5B) cases. Our analyses showed that EV-BRN4 expression is an excellent marker to diagnose NED in CRPC cases with an AUC of 1 (P<0.0001) (95% CI: 0.832-1.000), 100% specificity and 100% sensitivity. Further, in view of our data with cellular models showing release of BRN2 mRNA in EVs, we also evaluated its levels in sera of CRPC-Adeno and CRPC-NE patients (Fig. 5C). Similar to BRN4, EV-BRN2 was found to be significantly higher (~4-fold) in CRPC-NE as compared to CRPC-Adeno. ROC curve analyses for EV-BRN2 (Fig. 5D) showed that it can diagnose NED with an AUC of 0.944 (P<0.0001) (95% CI: 0.782-0.998), 94.4% specificity and 100% sensitivity. These data demonstrate the promising potential of EV-associated BRN4 and BRN2 to predict NED in CRPC patients non-invasively.
Fig. 5. EV-associated BRN4 and BRN2 are upregulated in sera from neuroendocrine PCa patients and can predict NED induction in PCa patients non-invasively.
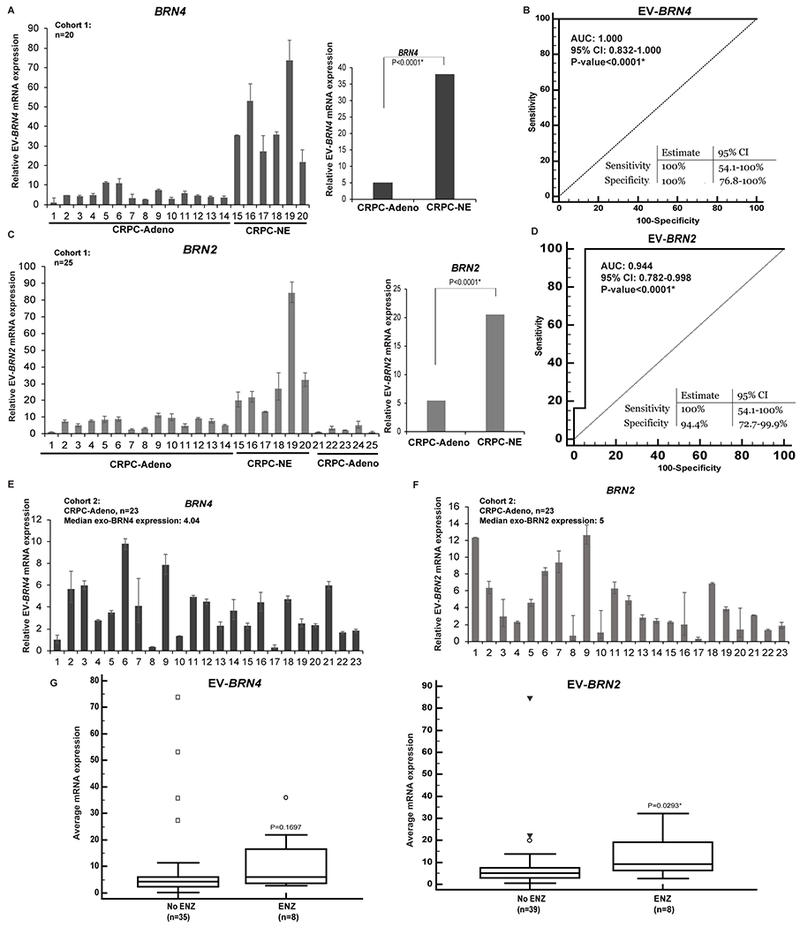
A. Relative EV-BRN4 levels in CRPC-Adeno (n=14) and CRPC-NE samples (n=4) as assessed by real-time PCR. Data were normalized to GAPDH control and represented as mean ± SEM (left). Average EV-BRN4 expression in CRPC-Adeno vs CRPC-NE cases (right).
B. Receiver Operating Characteristic (ROC) curve analyses for EV-BRN4 as a parameter to discriminate between non-NE and NE cases based on dCt values in CRPC-Adeno vs CRPC-NE cases.
C. Relative EV-BRN2 levels in CRPC-Adeno (n=19) and CRPC-NE samples (n=4) as assessed by real-time PCR (left). Average EV-BRN2 expression in CRPC-Adeno vs CRPC-NE cases (right). Data were normalized to GAPDH control and represented as mean ± SEM.
D. ROC curve analyses for EV-BRN2 based on dCt values in CRPC-Adeno (n=19) and CRPC-NE (n=4).
E. Relative EV-BRN4 levels and,
F. EV-BRN2 levels in sera of cohort 2 of CRPC-Adeno patients (n=23) as assessed by real-time PCR. Data were normalized to GAPDH control and represented as mean ± SEM.
G. Median EV-BRN4 (left panel) and EV-BRN2 expression (right panel) in CRPC-Adeno cases treated with/without ENZ. P-values are based on Mann-Whitney U test.
Enzalutamide treatment increases EV-associated BRN4 and BRN2 levels in CRPC patients
In view of our preceding results, we analyzed the expression of EV-BRN4 and EV-BRN2 in additional CRPC patients (Cohort 2, n=23) (Table S1 and Fig. 5E–F). While this cohort did not include patients with proven NED, it included CRPC-Adeno patients with/without ENZ treatment. The levels of BRN4 (Fig. 5E) and BRN2 (Fig. 5F) was found to range from low to high. We further examined if EV-associated BRN4 and BRN2 levels in CRPC-Adeno patients (cohort 1+2) are correlated with clinicopathological parameters (Fig. S2). Based on median expression of EV-BRN4 (4.04) and EV-BRN2 (5) in CRPC-Adeno patients, these patients were stratified into two groups- (≤median and >median expression). While no correlations were observed with Gleason score of primary tumor, age at diagnosis or final serum PSA, EV-BRN4 and EV-BRN2 were higher in CRPC-Adeno patients treated with ENZ (67% and 83%, respectively) vs those that were non-ENZ treated (48% and 43%, respectively) (Fig. S2) though it failed to reach statistical significance. Further, the median expression levels of EV-BRN4 and EV-BRN2 were found to be higher in ENZ-treated cases vs non-ENZ CRPC (adeno+NE) cases (Fig. 5G), with the levels of EV-BRN2 levels significantly higher (~2 fold higher, P=0.029*) in ENZ treated cases (Fig. 5G, right panel). These data suggest that these factors are increasingly released in EVs upon ENZ treatment. We propose that their release in EVs promote ENZ-induced NED in CRPC patients.
BRN4 and BRN2 protein are selectively released in PCa EVs and their release increases upon NED induction
In addition to BRN4 and BRN2 mRNA, we found that EVs contain BRN4 and BRN2 protein (Fig. 6A). EVs were isolated from control/ENZ treated LNCaP and NCI-H660 cells and subjected to Western blot analyses. We found that BRN4 (Fig. 6A, left panels) and BRN2 (25) (Fig. 6A, right panels) are selectively released in PCa EVs with their release increasing upon ENZ treatment and enrichment in EVs derived from the NE cell line NCI-H660. To validate our EV preparations, we performed Western blot analyses for EV markers CD9 and CD63 (Fig. 6A). While we confirmed the purity of our preparations as they stained positive for these markers, we found that CD9-containing vesicles decrease significantly upon NED induction as compared to CD63-positive vesicles, further supporting our data suggesting that alterations in EV secretion pathways occur with NED induction (Fig. 4B–C). To validate the release of BRN2 and BRN4 into PCa EVs, we inhibited EV release in our cellular LNCaP NED model (Fig. S1) by treatment with inhibitor GW4869 followed by BRN2/4 expression analyses in cellular and EV fractions (Fig. 6B). EV secretion inhibition increased cellular BRN2 levels and decreased EV-associated BRN2 upon NED induction by ENZ treatment and/or androgen depletion. Similarly, increased BRN4 was observed in EVs upon androgen depletion and/or ENZ treatment and these increases were attenuated by GW4869 treatment. Interestingly, probing for BRN4 in EVs yield a higher band in addition to expected size which may reflect glycosylated protein. Further, GW4869 treatment led to increased cellular BRN4 in control and C/D FBS treated cells while expected increase was not observed in combined treatment with ENZ suggesting a different regulatory control of BRN4 and BRN2 secretion and expression. We also assayed the release of BRN4 and BRN2 in EVs derived from RWPE-1/BPH1 cells and PCa cell lines (Fig. 6C) and found that these proteins are specifically expressed in EVs derived from PCa cell lines LNCaP, PC3, Du145 while EVs from RWPE-1 and BPH1 cells had undetectable levels. Treatment of PCa cell lines PC3 and LNCaP with EV inhibitor GW4869 led to decreased EV-associated BRN2 and BRN4 mRNA concomitant with increased cellular levels (Fig. 6D).
Fig. 6. BRN4 and BRN2 are selectively released in PCa EVs upon NED induction that mediate neuroendocrine differentiation states in prostate cancer.
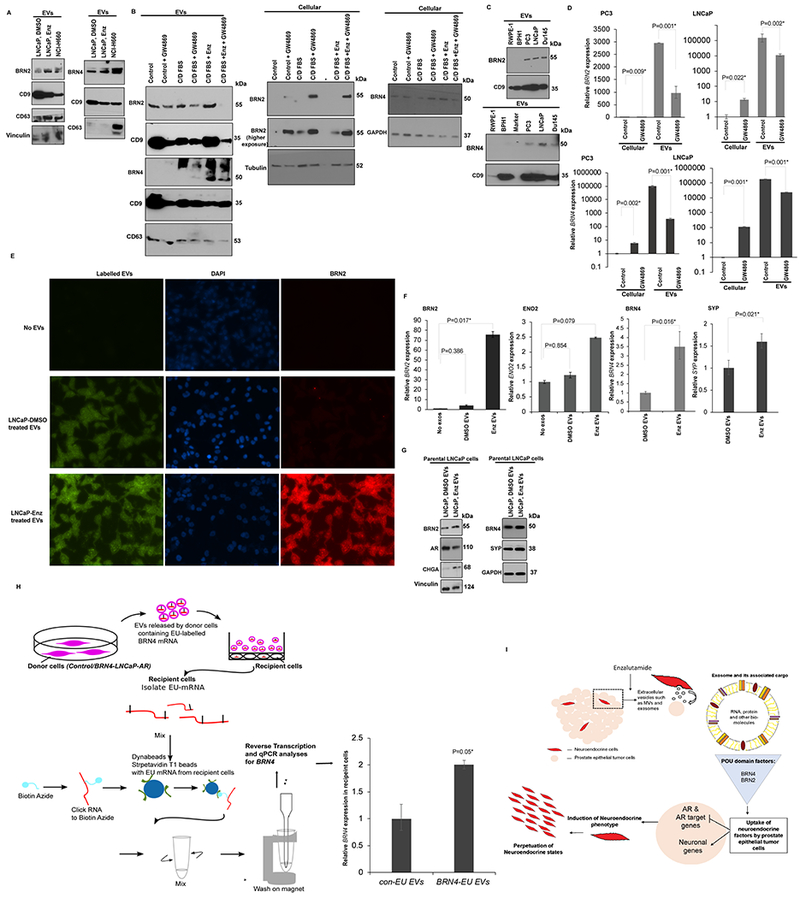
A. EVs were isolated from control (DMSO)/1μM ENZ treated LNCaP cells and NCI-H660 cells and subjected to Western blot analyses for indicated proteins.
B. LNCaP cells were cultured under regular conditions (control) or under androgen depleted conditions (C/D FBS) or treated with 20μM ENZ in C/D FBS media. These treatments were followed by treatment with exosome inhibitor GW4869 for 48 hrs. Cellular and EV fractions were extracted after various treatments followed by Western blot analyses for BRN2 and BRN4 in the two fractions. CD9, CD63 were used as controls for EV while tubulin/GAPDH were used as controls for cellular fractions.
C. BRN2 and BRN4 protein levels in EVs derived from normal immortalized (RWPE-1)/benign prostate epithelial (BPH1) cells and PCa cell lines (PC3, LNCaP, Du145). CD9 was used as an exosomal control.
D. Relative BRN2 mRNA (upper panels) and BRN4 mRNA (lower panels) expression in cellular and EV fractions of PC3 and LNCaP cell lines with/without exosome inhibitor GW4869 treatment as assessed by real time PCR. Data were normalized to Vinculin control and represented as mean ± SEM.
E-G. E-G’Uptake experiment’ with labelled EVs in parental LNCaP cells. EVs were isolated from control (DMSO)/1μM ENZ treated LNCaP cells, labelled with SYTO RNA Select green fluorescent stain followed by incubation of labelled EVs (40µg/ml) with parental LNCaP cells. As a negative control, parental LNCaP cells were incubated with media with no EVs.
E. Fluorescence microscopy analyses to confirm uptake of labelled EVs (green, left panels), DAPI staining (blue, middle panels) and BRN2 IF staining (red, right panels) after EV treatment.
F. Relative cellular BRN2, ENO2, BRN4 and SYP expression in EV treated/control LNCaP cells as assessed by real-time PCR. Data was normalized to GAPDH control and represented as mean ± SEM.
G. Western blot analyses for indicated proteins after ‘uptake assay’. Vinculin/GAPDH were used as loading controls.
H. Control/BRN4 expressing LNCaP-AR cells (donor cells) were grown in the presence of 5EU for 24 hours to label nascent RNA transcripts. EVs released by donor cells after labelling were isolated, characterized and applied to parental LNCaP-AR cells (recipient, non-EU labelled) for 48 hours. Total RNA was extracted from recipient cells followed by purification of EU-labelled mRNA from recipient cells as shown schematically using Click-iT Nascent RNA Capture Kit (#C10365, ThermoFisher) following the manufacturer’s protocol. Purified labeled RNA was used for real time PCR based analyses of labelled BRN4 in recipient cells. Data was normalized to GAPDH control and represented as mean ± SEM.
I. Schematic representation depicting proposed role of EV-associated BRN4 and BRN2 in inducing reprogramming in PCa cells to NE states. We propose that as an adaptive mechanism to androgen deprivation conditions/ENZ treatment, PCa cells express and secrete BRN2 and BRN4 in EVs/exosomes that in turn, drives oncogenic reprogramming of PCa cells. We propose that these reprogramming TFs are selectively sorted into PCa EVs/exosomes upon NED induction that mediates intercellular communication between PCa cells leading to perpetuation of NE states. EV-associated BRN2 and BRN4 are taken up by neighboring ‘non-NE’ PCa epithelial cells leading to suppression of AR and AR target genes and induction of neuronal genes.
EV-associated BRN4 and BRN2 mediates NED states in prostate cancer
In view of our results showing the release of BRN factors in EVs upon ENZ treatment, we hypothesized that as an adaptive mechanism to androgen deprivation conditions/ENZ treatment, PCa cells express and secrete these factors in EVs that act in a paracrine manner on neighboring cancer cells to drive oncogenic reprogramming to NE-like states (Fig. 6I). To test our hypothesis, we performed ‘uptake experiments’ (Fig. 6E–H). EVs were isolated from control/ENZ-treated LNCaP cells, labelled with SYTO RNASelect green fluorescent stain (Thermofisher Scientific) (48) followed by incubation with parental LNCaP cells. As a negative control, parental LNCaP cells were incubated with EV-free media. After 48 hours, parental LNCaP cells were harvested and analyzed. Cellular uptake of labelled EVs was confirmed by fluorescence microscopy (Fig. 6E, left panels). We performed BRN2 IF staining on parental LNCaP cells (Fig. 6E, right panels) which showed augmented cellular BRN2 staining upon treatment with ENZ EVs, suggesting that BRN2 protein is increasingly released in PCa EVs upon ENZ treatment and horizontally transferred to neighboring cancer cells. Interestingly, BRN2 protein showed as tiny speckles inside the cells, co-localized with EVs (green label) validating the transfer of BRN2 protein in EVs. Real time PCR analyses of parental LNCaP cells after ‘uptake experiment’ showed an induction of BRN2, BRN4 alongwith NE genes ENO2 and SYP by real-time PCR (Fig. 6F). Western blot analyses after ‘uptake experiment’ (Fig. 6G) showed that treatment of parental LNCaP cells with ENZ EVs led to a significant increase in the expression of BRN2, BRN4, CHGA and SYP with a concomitant decrease in AR expression. We examined the effects of ENZ EVs on AR target genes (NKX3.1 and KLK3) and NE/stem cell marker CD44 in LNCaP-AR cells (Fig. S3A) and found a significant repression of NKX3.1 and KLK3 while CD44 expression was upregulated. To further consolidate the transfer of BRN4 in EVs, we labelled nascent RNA in donor cells (control/BRN4 overexpressing LNCaP-AR cells) with 5-Ethynyl Uridine (5EU) followed by tracking of EU-labelled EV RNA release and uptake in recipient parental LNCaP-AR cells (non-EU labeled), to determine if labelled BRN4 mRNA transferred from donor cells could be detected in recipient cells (Fig. 6H, left panel). Interestingly, we found that parental cells treated with EU-labelled EVs from BRN4 expressing cells showed higher expression as compared to corresponding control EV treated cells (Fig. 6H, right panel). This data validates our hypothesis of transfer of BRN4 via EVs.
Since BRN2 was reported as a key neuronal factor driving PCa NED (25), we further tested the role of EV-associated BRN2 in PCa NED (Fig. S3B). We performed shRNA-mediated stable BRN2 knockdown in NCI-H660 cells (Fig. S3B, left panels). EVs were harvested from conditioned media of control shRNA (shCON) vs shBRN2 transfected cells (Fig. S3B, right panels) and used in an ‘uptake experiment’ with parental LNCaP cells incubated with shCON or shBRN2 EVs (Fig. S3C). Our data shows that treatment of parental LNCaP cells with shBRN2 EVs led to decreased expression of BRN2 and CD44 concomitant with AR upregulation. Collectively, these data support a role of EV-associated BRN2/4 in inducing NE states (Fig. 6I).
DISCUSSION
While the widespread use of enzalutamide and other second generation APIs has led to transformative impact in the management of metastatic CRPC patients (2,4), API resistance (5,6) is near-universal leading to significantly increased incidences of therapy-induced NED (9) with aggressive clinical course. Therapy-induced NED emerges in mCRPC patients either late upon treatment with ENZ/ABI, but is also believed to occur early in disease course upon treatment with these APIs or even de novo post docetaxel therapy (9,10). The genetic and epigenetic changes underlying this trans-differentiation process has been investigated and have been reported to involve key events including loss of the tumor suppressors retinoblastoma (RB1), tumor protein 53 (TP53) and NMYC overexpression amongst others (11–15). Here we show that BRN4, encoding a pro-neural TF, is a crucial factor that is upregulated in NEPC. While both BRN1 and BRN4 were induced upon ENZ treatment of PCa cell lines alongwith BRN2, BRN4 alterations were found to be associated more specifically with NEPC. In this regard, we identified that (i) In addition to BRN2, BRN4 is upregulated upon ENZ treatment/androgen withdrawal of PCa cell lines and in ENZ-resistant PCa cell line; (ii) BRN4 is selectively upregulated in CPC-NE PDX models as compared to CRPC-Adeno PDXs (42); (iii) BRN4 is upregulated in inducible cellular NED models including MYCN overexpression and dual TP53 and RB1 knockdown (11,12,16,44) (iv) analyses in Beltran et al (11) cohort showed that BRN4 mRNA is upregulated in NEPC. In view of these lines of evidence, we propose that BRN4 upregulation is a clinically relevant alteration associated with transition of CRPC-adenocarcinomas to CRPC-NE and that BRN4 may play a critical role in reprogramming PCa cells to NE states. In agreement with this hypothesis, BRN4 overexpression led to upregulation of neuronal markers including SOX2, a critical TF that drives NEPC (44) and ENO2, a canonical NE marker. BRN2 was previously reported to be an AR-repressed and an upstream regulator of SOX2 in driving NEPC (25). In view of our results showing an interplay between BRN2 and BRN4, we speculate that BRN4 and BRN2 act synergistically to control SOX2 expression in regulating NEPC (Fig. 3J). Our co-IP data showing that BRN4 directly interacts with BRN2 support our hypothesis. Overexpression/knockdown of BRN2 led to corresponding changes in BRN4 levels. We speculate that in response to APIs such as ENZ treatment, BRN4 and BRN2 are upregulated that work together to initiate a pro-neural program that drives PCa NED (Fig. 3J). Future mechanistic studies mapping the binding sites and regulation of BRN4 are warranted and is the subject of ongoing investigations in our laboratory. Interestingly, our preliminary analyses suggest that BRN4 promoter possess multiple SOX2 and AR binding sites suggesting a potential regulatory interplay between these TFs in driving NEPC.
It has been suggested and supported that NEPC transformation is a potentially reversible epigenetic phenomenon (49). We hypothesized that EVs mediate intercellular signaling in NEPC and plays a role in oncogenic reprogramming of CRPC-Adeno to CRPC-NE states via the transfer of functional TFs. Importantly, treatment of ENZ resistant cell line with EV inhibitor could restore the sensitivity of these cells to ENZ, supporting a key role of EVs in imparting ENZ resistance and suggesting EV inhibition as a potential strategy to reverse ENZ resistance. Our data lend support to our hypothesis that EVs are crucial to NED induction and that key oncogenic factors including BRN2 and BRN4 are released in PCa EVs. We found that ENZ causes alterations in vesicular sorting pathways such as increased release of BRN2 and BRN4 mRNA in EVs. Our data suggests that EV-associated BRN4 and BRN2 are horizontally transferred to neighboring cancer cells to propagate NED states. We propose that as an adaptive mechanism to APIs, PCa cells express BRN2 and BRN4, that in turn, drives oncogenic reprogramming of PCa cells. Further, these reprogramming TFs are selectively sorted into PCa EVs to mediate intercellular communication between PCa cells, leading to induction of neuronal genes, thereby promoting perpetuation of NED states (Fig. 6I). Since amplification of N-Myc and overexpression of EZH2 have been identified as key oncogenic factors in NEPC (11–13), we also assayed these factors in EVs (Fig. S4). We found that EZH2 and N-Myc are released in EVs by PCa cells undergoing NED (Fig. S4), lending support to our hypothesis that NED induction is associated with release of oncogenic TFs that perpetuate these states in advanced PCa.
Importantly, we identified that: there is increased expression of EV-BRN4 and EV-BRN2 in CRPC-NE cases as compared to CRPC-Adeno and that EV-BRN4 and EV-BRN2 have promising potential as non-invasive diagnostic/predictive biomarkers for NEPC/NED. Rigorous validation of the proposed EV-associated BRN4 and BRN2 as novel, non-invasive markers for detection and prediction of NEPC in larger cohorts are warranted. If validated, these markers can provide a significant advancement over existing methods of assessing NED based on histopathological criteria that are often flawed owing to the heterogeneity of NED (7,8). Further, we found that CD9 containing vesicles decrease significantly upon ENZ treatment and that NEPC is associated with lower CD9 amplification, lower mRNA expression and low CD9-positive vesicles suggesting that the amount of CD9 positive vesicles may act as an indicator of NED induction in CRPC. Previous studies have associated CD9-positive vesicles with advanced metastatic PCa (50). A limitation of our study was limited number of CRPC-NE samples. Our present findings need to be validated in larger cohorts.
In conclusion, our study has important clinical implications and transformative potential as it identifies BRN4 as an important player in NEPC/therapy-induced NED. We propose that selective modulation of BRN4 can be exploited to prevent NED induction. Importantly, we identified novel, non-invasive, EV biomarkers for detection and NED prediction that can potentially improve clinical management of CRPC.
Supplementary Material
TRANSLATIONAL RELEVANCE.
The emergence of neuroendocrine differentiation (NED) in castration resistant prostate cancer (CRPC) poses significant clinical challenge as the survival rates are extremely poor. The molecular basis of this trans-differentiation process is poorly understood, contributing to a lack of robust molecular biomarkers for its diagnosis and prediction. This study identifies for the first time, BRN4 as a novel transcription factor that drives neuroendocrine differentiation in prostate cancer and interplays with a reported master neural transcription factor, BRN2. Importantly, this study demonstrates that BRN4 and BRN2 mRNA are actively released in PCa extracellular vesicles (EVs) upon NED induction and that EV-associated BRN4 and BRN2 are potential novel non-invasive biomarkers to predict NED in CRPC patients.
ACKNOWLEDGEMENTS
We thank Dr. Roger Erickson for his support and assistance with preparation of the manuscript. We acknowledge Michael Liston for his help with graphical representation in Fig. 6.
Financial support: This work is supported by the US Army Medical Research Acquisition Activity (USAMRAA) Prostate Cancer Research Program Award No W81XWH-18-1-0303 and National Cancer Institute of the National Institutes of Health (NIH) under Award Number R01CA177984 and UO1CA184966. Additionally, supported by Award no. K6BX004473 (Department of Veterans Affairs) and W81XWH-18-2-0015, W81XWH-18-2-0016, W81XWH-18-2-0017, W81XWH-18-2-0018, and W81XWH-18-2-0019 Prostate Cancer Biorepository Network (PCBN). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH and DoD.
Footnotes
Conflicts of interest: The authors declare no potential conflicts of interest
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69(1):7–34 doi 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res 2009;15(15):4792–8 doi 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev 2010;24(18):1967–2000 doi 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367(13):1187–97 doi 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 5.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 2015;15(12):701–11 doi 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Culig Z Molecular Mechanisms of Enzalutamide Resistance in Prostate Cancer. Curr Mol Biol Rep 2017;3(4):230–5 doi 10.1007/s40610-017-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aggarwal R, Zhang T, Small EJ, Armstrong AJ. Neuroendocrine prostate cancer: subtypes, biology, and clinical outcomes. J Natl Compr Canc Netw 2014;12(5):719–26. [DOI] [PubMed] [Google Scholar]
- 8.Aggarwal RR, Small EJ. Small-cell/neuroendocrine prostate cancer: a growing threat? Oncology (Williston Park) 2014;28(10):838–40. [PubMed] [Google Scholar]
- 9.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 2018;36(24):2492–503 doi 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aparicio AM, Shen L, Tapia EL, Lu JF, Chen HC, Zhang J, et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin Cancer Res 2016;22(6):1520–30 doi 10.1158/1078-0432.CCR-15-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22(3):298–305 doi 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 2011;1(6):487–95 doi 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016;30(4):563–77 doi 10.1016/j.ccell.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, Chaux A, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol 2011;24(6):820–8 doi 10.1038/modpathol.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maina PK, Shao P, Liu Q, Fazli L, Tyler S, Nasir M, et al. c-MYC drives histone demethylase PHF8 during neuroendocrine differentiation and in castration-resistant prostate cancer. Oncotarget 2016;7(46):75585–602 doi 10.18632/oncotarget.12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016;29(4):536–47 doi 10.1016/j.ccell.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beltran H, Oromendia C, Danila DC, Montgomery B, Hoimes C, Szmulewitz RZ, et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin Cancer Res 2019;25(1):43–51 doi 10.1158/1078-0432.CCR-18-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Protein Delta-like 3 Is a Target in Neuroendocrine Prostate Cancer. Cancer Discov 2019;9(5):577 doi 10.1158/2159-8290.CD-RW2019-047. [DOI] [Google Scholar]
- 19.Puca L, Gavyert K, Sailer V, Conteduca V, Dardenne E, Sigouros M, et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci Transl Med 2019;11(484) doi 10.1126/scitranslmed.aav0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thoma C Targeting DLL3 in neuroendocrine prostate cancer. Nat Rev Urol 2019;16(6):330 doi 10.1038/s41585-019-0190-6. [DOI] [PubMed] [Google Scholar]
- 21.Chang YK, Srivastava Y, Hu C, Joyce A, Yang X, Zuo Z, et al. Quantitative profiling of selective Sox/POU pairing on hundreds of sequences in parallel by Coop-seq. Nucleic Acids Res 2017;45(2):832–45 doi 10.1093/nar/gkw1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishii J, Sato H, Yazawa T, Shishido-Hara Y, Hiramatsu C, Nakatani Y, et al. Class III/IV POU transcription factors expressed in small cell lung cancer cells are involved in proneural/neuroendocrine differentiation. Pathol Int 2014;64(9):415–22 doi 10.1111/pin.12198. [DOI] [PubMed] [Google Scholar]
- 23.Jerabek S, Merino F, Scholer HR, Cojocaru V. OCT4: dynamic DNA binding pioneers stem cell pluripotency. Biochim Biophys Acta 2014;1839(3):138–54 doi 10.1016/j.bbagrm.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Andersen B, Rosenfeld MG. POU domain factors in the neuroendocrine system: lessons from developmental biology provide insights into human disease. Endocr Rev 2001;22(1):2–35 doi 10.1210/edrv.22.1.0421. [DOI] [PubMed] [Google Scholar]
- 25.Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov 2017;7(1):54–71 doi 10.1158/2159-8290.CD-15-1263. [DOI] [PubMed] [Google Scholar]
- 26.Diss JK, Faulkes DJ, Walker MM, Patel A, Foster CS, Budhram-Mahadeo V, et al. Brn-3a neuronal transcription factor functional expression in human prostate cancer. Prostate Cancer Prostatic Dis 2006;9(1):83–91 doi 10.1038/sj.pcan.4500837. [DOI] [PubMed] [Google Scholar]
- 27.Mathis JM, Simmons DM, He X, Swanson LW, Rosenfeld MG. Brain 4: a novel mammalian POU domain transcription factor exhibiting restricted brain-specific expression. EMBO J 1992;11(7):2551–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sobol SE, Teng X, Crenshaw EB, 3rd. Abnormal mesenchymal differentiation in the superior semicircular canal of brn4/pou3f4 knockout mice. Arch Otolaryngol Head Neck Surg 2005;131(1):41–5 doi 10.1001/archotol.131.1.41. [DOI] [PubMed] [Google Scholar]
- 29.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol 2002;2(8):569–79 doi 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 30.Giusti I, Dolo V. Extracellular vesicles in prostate cancer: new future clinical strategies? BioMed research international 2014;2014:561571 doi 10.1155/2014/561571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hessvik NP, Sandvig K, Llorente A. Exosomal miRNAs as Biomarkers for Prostate Cancer. Front Genet 2013;4:36 doi 10.3389/fgene.2013.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valentino A, Reclusa P, Sirera R, Giallombardo M, Camps C, Pauwels P, et al. Exosomal microRNAs in liquid biopsies: future biomarkers for prostate cancer. Clin Transl Oncol 2017. doi 10.1007/s12094-016-1599-5. [DOI] [PubMed] [Google Scholar]
- 33.Duijvesz D, Luider T, Bangma CH, Jenster G. Exosomes as biomarker treasure chests for prostate cancer. Eur Urol 2011;59(5):823–31 doi 10.1016/j.eururo.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 34.Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008;10(12):1470–6 doi 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007;9(6):654–9 doi 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 36.Lai SL, Brauch H, Knutsen T, Johnson BE, Nau MM, Mitsudomi T, et al. Molecular genetic characterization of neuroendocrine lung cancer cell lines. Anticancer Res 1995;15(2):225–32. [PubMed] [Google Scholar]
- 37.Bhagirath D, Yang TL, Bucay N, Sekhon K, Majid S, Shahryari V, et al. microRNA-1246 Is an Exosomal Biomarker for Aggressive Prostate Cancer. Cancer Res 2018;78(7):1833–44 doi 10.1158/0008-5472.CAN-17-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015;162(2):454 doi 10.1016/j.cell.2015.06.053. [DOI] [PubMed] [Google Scholar]
- 39.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2(5):401–4 doi 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6(269):pl1 doi 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan TC, Veeramani S, Lin FF, Kondrikou D, Zelivianski S, Igawa T, et al. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocr Relat Cancer 2006;13(1):151–67 doi 10.1677/erc.1.01043. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen HM, Vessella RL, Morrissey C, Brown LG, Coleman IM, Higano CS, et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease an--d Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017;77(6):654–71 doi 10.1002/pros.23313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res 2014;20(11):2846–50 doi 10.1158/1078-0432.CCR-13-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355(6320):84–8 doi 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu X, Cates JM, Morrissey C, You C, Grabowska MM, Zhang J, et al. SOX2 expression in the developing, adult, as well as, diseased prostate. Prostate Cancer Prostatic Dis 2014;17(4):301–9 doi 10.1038/pcan.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rotinen M, You S, Yang J, Coetzee SG, Reis-Sobreiro M, Huang WC, et al. ONECUT2 is a targetable master regulator of lethal prostate cancer that suppresses the androgen axis. Nat Med 2018;24(12):1887–98 doi 10.1038/s41591-018-0241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Li F, Liu L, Jiang H, Hu H, Du X, et al. Salinomycin triggers endoplasmic reticulum stress through ATP2A3 upregulation in PC-3 cells. BMC Cancer 2019;19(1):381 doi 10.1186/s12885-019-5590-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicola AM, Frases S, Casadevall A. Lipophilic dye staining of Cryptococcus neoformans extracellular vesicles and capsule. Eukaryot Cell 2009;8(9):1373–80 doi 10.1128/EC.00044-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wadosky KM, Ellis L, Goodrich DW. Evasion of targeted cancer therapy through stem-cell-like reprogramming. Mol Cell Oncol 2017;4(2):e1291397 doi 10.1080/23723556.2017.1291397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soekmadji C, Corcoran NM, Oleinikova I, Jovanovic L, Australian Prostate Cancer Collaboration B, Ramm GA, et al. Extracellular vesicles for personalized therapy decision support in advanced metastatic cancers and its potential impact for prostate cancer. Prostate 2017;77(14):1416–23 doi 10.1002/pros.23403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.