

Abstract
Black women in the U.S. are disproportionately affected by early-onset, triple-negative breast cancer. DNA methylation has shown differences by race in healthy and tumor breast tissues. We examined associations between genome-wide DNA methylation levels in breast milk and breast cancer risk factors, including race, to explain how this reproductive stage influences a woman’s risk for – and potentially contributes to racial disparities in – breast cancer. Breast milk samples and demographic, behavioral, and reproductive data, were obtained from cancer-free, uniparous and lactating U.S. black (n=57) and white (n=82) women, ages 19 to 44. Genome-wide DNA methylation analysis was performed on extracted breast milk DNA using the Infinium HumanMethylation450K BeadChip. Statistically significant associations between breast cancer risk factors and DNA methylation beta values, adjusting for potential confounders, were determined using linear regression followed by Bonferroni Correction (P<1.4×10−7). Epigenetic analysis in breast milk revealed statistically significant associations with race and lactation duration. Of the 284 CpG sites associated with race, 242 were hypermethylated in black women. All 227 CpG sites associated with lactation duration were hypomethylated in women who lactated longer. IPA analysis of differentially methylated promoter region CpGs by race and lactation duration revealed enrichment for networks implicated in carcinogenesis. Associations between DNA methylation and lactation duration may offer insight on its role in lowering breast cancer risk. Epigenetic associations with race may mediate social, behavioral, or other factors related to breast cancer and may provide insight into potential mechanisms underlying racial disparities in breast cancer incidence.
Keywords: DNA methylation, race, breast feeding
Introduction
Black women are disproportionately affected by early-onset, highly aggressive breast cancer, more specifically triple negative breast cancer (TNBC) [1]. Black women also consistently have the highest breast cancer mortality rates compared to other races [1, 2]. It is not well-understood why black women have higher incidence rates of TNBC, however, differences in certain exposures, such as obesity or age at first live birth, may have differential effects on risk for different subtypes of breast cancer, and may contribute to the racial disparities identified in breast cancer [3]. Breastfeeding can also lower a woman’s risk of breast cancer, and black women tend to breastfeed at lower rates compared with white and Hispanic women [4, 5]. A better understanding of how race and lactation duration affect breast cancer risk among healthy women is greatly needed. A molecular understanding of how known breast cancer risk factors may influence the healthy breast microenvironment and potentially influence breast cancer development could provide important etiologic insights.
DNA methylation, the addition of a methyl group on a cytosine, can affect gene expression levels, and is thought to be linked to tumor development in the breast and other sites [6, 7]. DNA methylation is reversible, making it an ideal target for cancer prevention [8, 9]. DNA hypermethylation in promoter regions of tumor-suppressor genes may silence these genes in cancer, while DNA hypomethylation might increase oncogene expression [8, 10]. It is hypothesized that this modification occurs early in tumor development and could even affect tumor phenotypes and prognosis [10].
Previous work in healthy individuals has shown associations between tissue and blood DNA methylation and various clinical co-variates, such as race, age, BMI, alcohol consumption, and environmental exposures [11–14]. Researchers have also observed race-specific differences in DNA methylation at specific genes in healthy individuals that are also associated with various cancers, including breast, colorectal, pancreatic, and prostate cancers, all of which experience racial disparities in incidence or mortality [8, 11, 15, 16]. Because DNA methylation patterns are tissue- and site-specific, acquiring tissue specimens may require burdensome and invasive techniques, especially for healthy individuals.
Breast milk is one non-invasive specimen that represents the breast environment [17]. Breast milk contains epithelial cells, leucocytes, cytokines, proteins and hormones, each of which may be targeted and interrogated for understanding breast cancer development [17, 18]. Much of the work performed in breast milk has focused on better understanding its health benefits for newborn children [19, 20], with more recent findings correlating breast milk microbiome with maternal weight [21]. We and others have demonstrated the feasibility of measuring DNA methylation in breast milk [22–24] and identified associations between promoter DNA methylation levels and age [15].
The goal of the present study was to identify differences in genome-wide DNA methylation levels in breast milk of healthy lactating women by race and other breast cancer risk factors during a unique time in their reproductive life cycle (post-partum) that is known to affect breast cancer risk. Identifying epigenetic changes in breast milk may enable identification of biomarkers that are associated with breast cancer risk and that mediate risk factors and protective factors, including breastfeeding itself.
Materials and Methods
Study participants and collection of milk samples.
Lactating women (age 18 years and older) were recruited through national and local media to provide breast milk samples (approximately 100mL of pumped or hand-expressed breast milk), a health and lifestyle questionnaire, and written consent for research to the Breastmilk Laboratory at the University of Massachusetts at Amherst. Participants included 83 white and 61 black uniparous women who had not undergone a breast biopsy, were cancer-free at the time of donation, donated breast milk between 2006 and 2014, and had complete information from the questionnaire on pregnancy related variables as well as smoking and BMI.
Women who lived or visited within 100 miles of Amherst, Massachusetts had their breast milk samples and completed questionnaires collected by a researcher at home who immediately delivered the specimens to the laboratory at ambient temperature for processing. For women living outside of 100 miles of Amherst, Massachusetts, breast milk samples were shipped with an ice pack via a pre-paid UPS breast milk collection kit [17]. Fifty-two percent of milk samples were expressed in the morning (between 5am and 11:59am), and 20% were expressed at other times throughout the day. The remaining 28% of samples had no expression time data. All milk samples were shipped on the same day that they were expressed.
Covariate data were obtained from paper questionnaires completed at the time of donation and included questions about reproductive health (i.e. parity, breastfeeding history, and oral contraceptive use), general health (i.e. previous cancer diagnosis and subsequent cancer treatment, history of breast biopsy, prescription medication use, over-the-counter pain reliever use, over-the-counter vitamin or supplement use, and recent cold of flu symptoms), demographic information (i.e. smoking status, current age, race, ethnicity, occupation, income, current residence, current height and weight, general diet information, and general physical activity information), and family history of breast or ovarian cancer. Variables were selected a priori based on completeness as there were large amounts of missing data for some variables, including general diet information as well as occupation and income. Lactation duration was collected as the age of the current baby breastfed in days. This study was conducted in accordance with recognized ethnical guidelines (U.S. Common Rule) and approved by the institutional Review Boards of University of Massachusetts at Amherst and at the National Institutes of Health.
DNA Extraction.
DNA was extracted from milk samples using the phenol-chloroform method as described previously [15]. One mL from each milk sample was put into a 2.0 mL tube. Lysis buffer (100mM Tris, 1mM EDTA, 0.5% SDS, 200mM NaCl and 5 µL of 200 ng/mL Proteinase K) was added to each tube. All tubes were then placed in a 56°C water bath overnight. After an additional two-hour incubation at 56°C with 6µl extra 200 ng/mL proteinase K, each lysed sample was divided into two aliquots of 622 µl each. An equal volume of Phenol/Chloroform/Isoamyl Alcohol (25:24:1) was added to each tube. Samples were vortexed vigorously for 30 seconds and centrifuged at 15,000g for 10 minutes. The aqueous phase was transferred to two new tubes and an equal amount of chloroform/isoamyl alcohol (Sigma) was added. Samples were vortexed vigorously for 30 seconds and centrifuged at 15,000g for 10 minutes. The aqueous phase (1,200 µl) was transferred to three new tubes (400 µl in each tube) and 40 µl (0.1 volume) of 3M sodium acetate, 1ml (2.5 volumes) of ice-cold ethanol and 1µl of glycol blue were added to each tube. This was left overnight at −20°C. The precipitate for one tube of each sample was then spun at 15,000g for 10 minutes. The supernatant from the first tube of each sample was discarded and the precipitation mix of a second aliquot was added to this tube. Centrifugation was repeated. Supernatant of the 2nd tube was disposed then the 3rd aliquot was added, centrifuged once more, and the supernatant removed. Once the final supernatant was removed, the pellet was washed with 70% ethanol, dried and eluted in 22 µl of elution buffer (Qiagen).
Genomic DNA was quantified using a real-time TaqMan PCR assay targeting ALU repetitive elements. The forward primer sequence is: 5’-ATC ACG AGG TCA GGA GAT CGA G-3’; the reverse primer sequence is: 5’-CCG GCT AAT TTT TGT ATT TTT AGT AGA GA-3’, and the probe sequence is: 5’−6FAM-ATC CCG GCT AAC ACG GTG AAA CCC-BHQ-1-3’. Primer and probes were synthesized by Biosearch Technologies, Petaluma, CA, USA. Genomic DNA (1ul) of each sample in triplicate was evaluated using serial dilutions of white blood cell DNA (Promega, Madison, WI, USA) as a standard curve to determine DNA amounts. ALU PCRs were performed in a 30ul total reaction volume as described in Campan et al 2018 for MethyLight assays [25].
DNA Methylation Analysis.
Purified DNA from breast milk specimens was sent to the University of Southern California (USC) Molecular Genomics Core for Illumina HumanMethylation450 (HM450) BeadChip analysis. The total amount of DNA from each breast milk sample was bisulfite treated with the Zymo EZ DNA methylation kit (Zymo Research, Irvine, CA) and 1µl aliquots were used for MethyLight quality control (QC) analyses to determine the completeness of conversion and the amount of converted DNA available for the HM450 assay [26]. The remaining bisulfite converted DNA samples were further processed using the Illumina FFPE Restoration Solution (Illumina, San Diego, CA) as specified by the manufacturer. The Restoration Solution repairs degraded DNAs for use in genome-scale genotyping and DNA methylation assay platforms. The entire restored sample was then used as a substrate for the Illumina HM450 BeadArrays, as recommended by the manufacturer and described previously [27]. BeadArrays were scanned using Illumina iScan readers and the raw signal intensities were extracted from the *.IDAT files and normalized using the R package sesame [28, 29], a recently developed R package that masks problematic probes (i.e. probes for which DNA methylation is invalid because they overlap SNPs or repeats).
Statistical Analyses.
Subject characteristics were compared by race using a t-test for continuous variables and a chi-squared test for categorical variables.
Questionnaire and DNA methylation data were integrated into one file for statistical analysis in R (version 3.3.2). Of the original 144 women, 5 were removed because their overall analytical signal rates were below 85%. Of the 482,421 total probes on the HM450 Beadchip, 176,386 probes were excluded because they were (1) located at or within 10bp of known SNPs, (2) known to be cross-reactive[30], or (3) missing in 50% or more of the observations. After these exclusions 306,035 probes were included in the final analysis. Of these 306,035probes, 138,363 CpG probes in promoter regions (i.e. TSS200, TSS1500, 5’UTR, and 1st Exon), as defined by Sandoval et al., were used for promoter-specific analyses.
Generalized linear regression models were used to identify relationships between breast milk DNA methylation and race as well as other breast cancer risk factors. DNA methylation beta values were treated as a continuous outcome. Bonferroni corrections (P < 1.63×10−7 for the full list of probes and P < 3.61×10−7 for analysis restricted to probes in the promoter region) were used to adjust p-values unless otherwise noted. The adjusted model included race, lactation duration, age, BMI, smoking history and donation year. Principal component analysis of the 10,000 most variable methylated CpG sites did not reveal any batch effects, and surrogate variable analysis did not identify any additional variables to be included for adjustment in the final multivariable model. Black and white women differed by some covariates, including lactation duration, over-the-counter pain medication use, shipping status, and donation year, thus we performed analyses stratified by race to determine their effects.
Ingenuity Pathway Analysis.
Biological significance of the genes corresponding to the significantly differentially methylated probes from the promoter-based analysis was determined using the Ingenuity Pathway Analysis (IPA) software and knowledge base. Only significant probes with a mean beta value difference of 0.1 or greater were included in the IPA. This threshold was applied to filter out small differences reflecting minor shifts in the composition of originating cell type populations. For lactation duration, this difference was calculated between the first (<125 days) and last (>269 days) categories. P-values were calculated using the right-tailed Fisher’s Exact Test, which are then converted to p-scores (-log10(P-value)). For example, a p-value of 1×10−10 would be equivalent to a score of 10.
Results
Participant Characteristics.
The 139 healthy lactating women (82 white and 57 black) who donated milk for this study had a mean age of 30.2 years, ranging from 19 to 44 years (Table 1). Compared with white women, black women less frequently reported past week use of over-the-counter pain medications (p<0.01) and reported ever smoking less often (p=0.02). A higher percentage of black women were recruited >269 days after giving birth (p=0.03), were more likely to have had breast milk shipped to the laboratory (p<0.01) and to have donated after 2010 (p<0.01). Other factors assessed, including time of day of milk expression, did not differ significantly by race.
Table 1.
Demographics and characteristics of women included in the study
| All (n=139) | White (n=82) | Black (n=57) | p-value | |
|---|---|---|---|---|
| Characteristic | Mean(sd) | Mean(sd) | Mean(sd) | |
| Age at donation (years) | 30.2 (5.1) | 29.7 (5.2) | 30.9 (5.0) | 0.17 |
| Current BMI (kg/m2) | 26.5 (5.9) | 26.1 (5.9) | 27.0 (5.8) | 0.40 |
| N** (%) | N** (%) | N** (%) | ||
| Race | ||||
| White | 82 (59%) | |||
| Black | 57 (41%) | |||
| Age at Menarche (years) | ||||
| <13 | 82 (61%) | 47 (59%) | 35 (63%) | 0.20 |
| 13-14 | 41 (30%) | 23 (29%) | 18 (33%) | |
| >14 | 12 (9%) | 10 (12%) | 2 (4%) | |
| Age at first birth (years) | ||||
| <30 | 69 (49%) | 42 (51%) | 27 (47%) | 0.78 |
| 30+ | 70 (51%) | 40 (49%) | 30 (52%) | |
| Number of Pregnancies | ||||
| 1 | 91 (65%) | 58 (71%) | 33 (58%) | 0.24 |
| 2 | 34 (24%) | 18 (22%) | 16 (28%) | |
| 3+ | 14 (10%) | 6 (7%) | 8 (14%) | |
| Past week OTC pain medication use | ||||
| No | 105 (76%) | 55 (67%) | 50 (88%) | <0.01 |
| Yes | 34 (24%) | 27 (33%) | 7 (12%) | |
| Smoking | ||||
| Never | 98 (71%) | 51 (65%) | 47 (84%) | 0.02 |
| Ever | 37 (27%) | 28 (35%) | 9 (16%) | |
| Lactation duration (days) | ||||
| <125 | 48 (35%) | 32 (39%) | 16 (28%) | 0.03 |
| 125-269 | 45 (32%) | 30 (37%) | 15 (26%) | |
| >269 | 46 (33%) | 20 (24%) | 26 (46%) | |
| First degree family history of breast cancer**** | ||||
| No | 110 (85%) | 64 (83%) | 46 (88%) | 0.56 |
| Yes | 19 (15%) | 13 (17%) | 6 (12%) | |
| First degree family history of Ovarian cancer**** | ||||
| No | 125 (98%) | 76 (99%) | 49 (98%) | 0.76 |
| Yes | 2 (2%) | 1 (1%) | 1 (2%) | |
| Shipped Milk Sample | ||||
| No | 69 (49%) | 65 (79%) | 4 (7%) | <0.01 |
| Yes | 70 (51%) | 17 (21%) | 53 (93%) | |
| Year of Donation | ||||
| <2010 | 65 (47%) | 54 (66%) | 11 (19%) | <0.01 |
| 2010+ | 74 (53%) | 28 (34%) | 46 (81%) |
Number of women with whole milk sample tested
Ns are based on number of women. Numbers do not add to total due to missingness.
Only 5 current smokers
First degree: parent, sister, child
Note: p-value is t-test for continuous and chi-square for categorical variables.
DNA Methylation and Race.
DNA methylation levels significantly differed by race for 284 probes independent of lactation duration, age, smoking status, BMI, and donation year, and were scattered throughout the genome (Figure 1 and Supplemental Table 1). The top 10 CpG sites most significantly associated with race are listed in Table 2 along with gene annotation. A complete list of significant differentially methylated CpG sites by race is provided in Supplemental Table 2. Of the 284 significant CpG probes, 242 probes (85%) showed increased DNA methylation among black women (Supplemental Table 1). In addition, 80 (28%) probes were located in CpG islands, 84 (30%) were located in shores, 29 probes (10%) were located in shelves, and the remaining 91 (32%) probes were not located in or near CpG islands. Finally, 74 CpG sites were located in promoter region, of which 65 (86%) probes displayed increased DNA methylation among black women as compared to white women (Supplemental Table 3). A total of 116 (41%) CpG sites were located in the gene body regions, of which 106 (91%) probes displayed increased DNA methylation among black women compared to white women.
Figure 1.
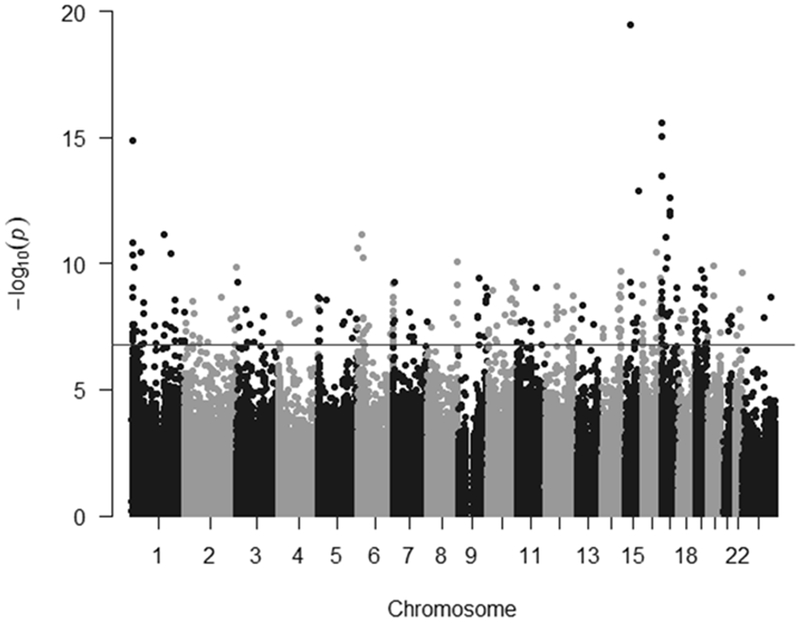
The significance −log10P-value of the associations with race by chromosome in a Manhattan plot. The genome-wide significance level of 1.63×10−7 is indicated by the horizonal line.
Table 2.
The 10 most significant CpG probes in relation to race in breast milk samples from black and white women
| IlmnID | Gene Name | Gene Description | Coefficienta | Mean beta value for black women | Mean beta value for white women | Corrected p-valueb | CHR | MAPINFO | UCSC Gene Group |
|---|---|---|---|---|---|---|---|---|---|
| cg21523688 | SORD | Sorbitol Dehydrogenase | −0.198 | 0.439 | 0.623 | 1.03E-14 | 15 | 45319037 | Body |
| cg17093615 | P2RX5 | Purinergic receptor P2X5 | 0.119 | 0.889 | 0.769 | 8.19E-11 | 17 | 3585069 | Body |
| cg23551198 | P2RX5 | Purinergic receptor P2X5 | 0.160 | 0.649 | 0.496 | 2.67E-10 | 17 | 3585166 | Body |
| cg00060374 | LOC441869 | 0.199 | 0.815 | 0.614 | 4.08E-10 | 1 | 1355235 | Body | |
| cg06468454 | P2RX5 | Purinergic receptor P2X5 | 0.172 | 0.515 | 0.340 | 1.03E-08 | 17 | 3591377 | Body |
| cg22812413 | 0.111 | 0.233 | 0.136 | 3.88E-08 | 15 | 81391742 | |||
| cg02228675 | DHX58 | DExH-Box Helicase 58 | −0.225 | 0.249 | 0.471 | 7.01E-08 | 17 | 40259724 | Body |
| cg00647820 | DHX58 | DExH-Box Helicase 58 | −0.275 | 0.245 | 0.513 | 2.50E-07 | 17 | 40259828 | Body |
| cg20291162 | DHX58 | DExH-Box Helicase 58 | −0.206 | 0.470 | 0.658 | 3.70E-07 | 17 | 40259547 | Body |
| cg23656322 | S100A2 | S100 Calcium Binding Protein A2 | 0.163 | 0.552 | 0.404 | 2.13E-06 | 1 | 153533922 | Body |
Multivariable GLM was performed using 306,035 probes from the Illumina HumanMethylation 450K Beadchip. The annotation “HumanMethylation450_15017582_v.1.2.csv” provided by Illumina was used to annotate the CpG loci.
Coefficients for the comparison of Black women to White women; positive coefficients indicate higher methylation in Black women compared to White women, while negative coefficients indicate higher methylation in White women compared to Black women.
The DNA methylation beta value was the outcome, race was the predictor variable, adjusted for lactation duration, age, BMI, smoking status, and donation year.
p-values after Bonferroni correction, 1.63×10−7
Analyses stratified by race revealed no statistically significant associations between DNA methylation and past week use of over-the-counter pain medication, smoking history, family history of breast cancer, age at first birth, number of pregnancies, menses age, or BMI, after the Bonferroni correction (1.63×10−7, Supplemental Table 5). There were also no significant associations between DNA methylation and shipping status for white women; there were not enough black women who did not ship their breast milk sample to evaluate the association between DNA methylation and shipping.
DNA Methylation and Lactation Duration.
We identified 227 CpG probes for which their DNA methylation levels were significantly and inversely associated with the lactation duration(Supplemental Table 1). These probes were independent of race, age, smoking status, BMI, and donation year, and were scattered throughout the genome (Figure 2). The top 10 CpG sites most significantly associated with lactation duration are listed in Table 3 along with gene annotation. A complete list of significant differentially methylated CpG sites by lactation duration is provided in Supplemental Table 4. Of these 227 significant CpG probes, 18 (8%) were located in CpG Islands, 59 (26%) were located in shores, 28 (12%) were located in shelves, while the remaining 122 (54%) probes were not located in or near CpG islands. Sixty-seven probes (30%) were in gene promoter regions, 111 probes were located in the gene body, and the remaining nine probes were in the 3’UTRs (Supplemental Table 3).
Figure 2.
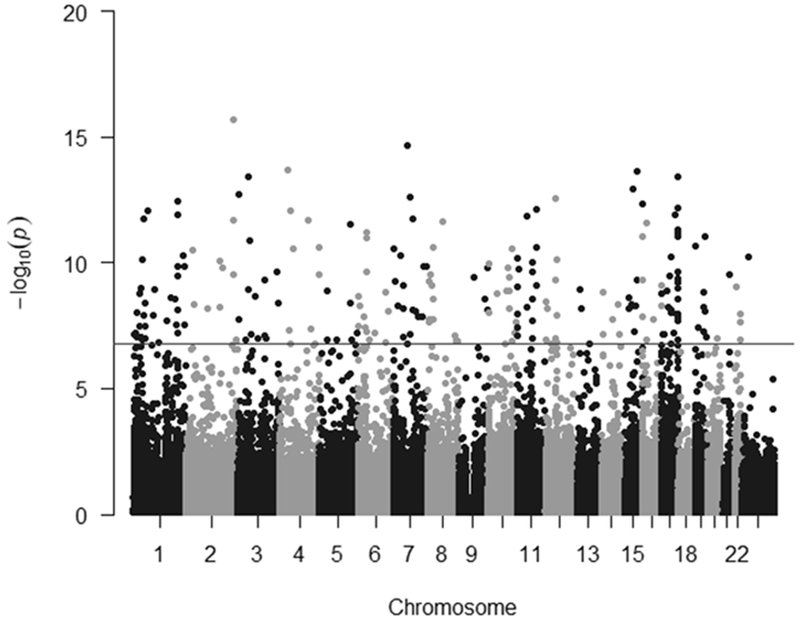
The significance −log10P-value of the associations with lactation duration by chromosome in a Manhattan plot. The genome-wide significance level of 1.63×10−7 is indicated by the horizonal line.
Table 3.
The 10 most significant CpG probes in relation to lactation duration in breast milk samples from black and white women
| IlmnID | Gene Name | Gene Description | Coefficienta | Mean beta value for baby age <125 days | Mean beta value for baby age 125-269 days | Mean beta value for baby age >269 days | Corrected p-valueb | CHR | MAPINFO | UCSC Gene Group |
|---|---|---|---|---|---|---|---|---|---|---|
| cg22891868 | MOGAT1 | Monoacylglycerol O-Acyltransferase 1 | 0.011 | 0.414 | 0.364 | 0.302 | 6.03E-11 | 2 | 223536069 | TSS1500 |
| cg00952162 | 0.053 | 0.889 | 0.689 | 0.595 | 6.55E-10 | 7 | 64711268 | |||
| cg09241455 | RELL1 | RELT like 1 precursor | 0.028 | 0.504 | 0.390 | 0.304 | 6.02E-09 | 4 | 37667583 | Body |
| cg13955984 | 0.064 | 0.873 | 0.766 | 0.672 | 7.06E-09 | 15 | 75022382 | |||
| cg07687398 | PRKCD | Protein kinase C delta | 0.080 | 0.785 | 0.617 | 0.531 | 1.10E-08 | 3 | 53198666 | 5’UTR |
| cg06463097 | FASN | Fatty acid synthase | 0.034 | 0.866 | 0.630 | 0.522 | 1.14E-08 | 17 | 80038921 | Body |
| cg16964728 | RORA | RAR Related Orphan Receptor A | 0.073 | 0.791 | 0.630 | 0.524 | 3.51E-08 | 15 | 61340524 | Body |
| cg06619959 | IL17RE | Interleukin 17 Receptor E | 0.054 | 0.576 | 0.505 | 0.461 | 6.13E-08 | 3 | 9956506 | Body |
| cg27457191 | PHTF2 | Putative homeodomain transcription factor 2 | 0.012 | 0.827 | 0.729 | 0.620 | 7.47E-08 | 7 | 77429766 | 5’UTR |
| cg20995304 | HDAC7 | Histone deacetylase 7 | 0.053 | 0.587 | 0.451 | 0.367 | 8.03E-08 | 12 | 48196167 | Body |
Multivariable GLM was performed using 306,035 probes from the Illumina HumanMethylation 450K Beadchip. The annotation “HumanMethylation450_15017582_v.1.2.csv” provided by Illumina was used to annotate the CpG loci.
Coefficients for comparison of older current age of baby breastfed (>269, or 125 to 269 days) to younger current age of baby breastfed (<125 days); positive coefficients indicate higher methylation values in older current age of baby breastfed compared to younger current age of baby breastfed, while negative coefficients indicate higher methylation values in younger current age of baby breastfed compared to older current age of baby breastfed.
The DNA methylation beta value was the outcome, current age of baby breastfed was the predictor variable, adjusted for race, age, BMI, smoking status, and donation year.
p-values after Bonferroni correction, 1.63×10−7
Analyses stratified by categorical lactation duration (<125 days, 125-269 days, and >270 days) revealed significant associations (after Bonferroni correction) between DNA methylation and race, and consistently showed increased DNA methylation in black women, which confirms the robustness of this finding in our adjusted model (Supplemental Table 1).
Promoter-based and Pathway Analysis.
DNA methylation levels varied by race at 94 probes targeting promoter regions, including 74 (79%) with higher levels among black women. For lactation duration, 75 probes showed decreased methylation (Supplemental Table 6). The full list of significant probes in promoter regions associated with race and lactation duration are presented in Supplemental Tables 7 and 8.
We performed Ingenuity Pathway Analysis (IPA) [31] on the gene lists from differentially methylated probes with a mean beta value difference of 0.1 or greater to identify their potential biological relevance. This analysis revealed 19 unique genes from the 19 differentially methylated probes that showed differential DNA methylation by race and met the difference threshold, which were enriched for the following networks: 1) Amino Acid Metabolism, Molecular Transport, Small Molecule Biochemistry (p-score = 3), 2) Cancer, Organismal Injury and Abnormalities, RNA Post-Transcriptional Modification (p-score = 3), and 3) Amino Acid Metabolism, Cancer, and Carbohydrate Metabolism (p-score = 3) (Supplemental Table 9a). Of these 19 genes, seven (SRMS, GSE1, ABCC4, DHRS4, STAB2, RPS16, and IFNGR2) had a Disease or Function Annotation Category that indicated “Cancer” in the Ingenuity Knowledge Base (IKB), however, none of them were for specific to breast cancer. Two additional proteins (from the full list of 94 promoter CpG sites), ALDH2 and EPAS1 were also identified as being associated with cancer according to the Cancer Gene Census (cancer.sanger.ac.uk)[32].
For lactation duration, IPA revealed 48 unique genes from the 56 differentially methylated probes by lactation duration that met the difference threshold and were enriched for the following networks: 1) Cellular Movement, Cellular Growth and Proliferation, Cell Signaling (p-score = 19), 2) Cell Death and Survival, Cellular Movement, Cardiac Enlargement (p-score = 17), and 3) Cellular Development, Cellular Growth and Proliferation, Hematological System Development and Function (p-score = 4) (Supplemental Table 9b). Of the 48 genes, eight (CYP19A1, G0S2, VEGFA, VDR, CDC42EP3, LIMA1, CD33, and PRKCD) had a Disease or Function Annotation Category that indicated “Cancer” in the IKB. Five (of the eight) proteins were implicated specifically in breast cancer: CYP19A1, G0S2, VEGFA, VDR, CDC42EP3. Two additional proteins (of the 48 genes), CANT1 and CREB3L1 were also identified as being associated with cancer according to the Cancer Gene Census (cancer.sanger.ac.uk)[32].
Discussion
In this first study to explore the relationship between genome-wide DNA methylation levels in breast milk and race as well as other breast cancer risk factors, we identified 284 CpG probes differentially methylated by race, and 227 CpG probes differentially methylated by lactation duration. Of the CpG probes differentially methylated by race, 85% of them indicated DNA hypermethylation associated with black race, while all probes differentially methylated as a function of lactation duration indicated reduced DNA methylation with increasing lactation duration, including when analyses were restricted to the promoter region. Furthermore, IPA analysis revealed networks believed to be important to the development of cancer.
Our results provide new evidence of the impact of race and lactation duration on breast milk DNA methylation which may inform breast cancer etiology. Previous studies have identified differentially methylated regions between breast tumors and healthy tissue [33], while other studies have identified differentially methylated regions by race and breast cancer subtype [34]. In particular, research has shown that there are more differentially methylated sites in ER-negative breast tumors in black women compared to breast tumors from white women [33–35], suggesting that DNA methylation could impact expression of genes involved in breast cancer subtype carcinogenesis and potentially explaining the racial disparities observed not only for breast cancer overall (i.e. highest incidence rates in white women [36] and highest mortality rates in black women [1]), but also in the proportion of breast cancer subtypes observed for these groups of women (i.e. highest proportion of triple-negative breast cancer in black women) [1]. Our findings suggest that DNA methylation states at this developmentally important time in a woman’s reproductive life cycle might contribute to our understanding of breast cancer etiology and/or racial disparities in breast cancer incidence.
In a previous study that explored racial differences (n=61 white, n=22 black) in genome-wide DNA methylation in breast tissues from women, undergoing breast reduction surgery, 485 CpG sites were differentially methylated between black and white women after adjusting for age and BMI, with 58% being hyper-methylated in black women [13]. These tissues were blunt dissected, prior to freezing, to remove adipose tissue. The mean age for black women was 34.4 years and 40.7 for white women. We compared our list of 284 CpG sites to their list of 485 and found 17 CpG sites in common, all of which showed DNA hypermethylation in the same direction, with all except one showing DNA hypermethylation in black women (Supplemental Table 10). Four CpG sites (out of the 17) were found in gene promoter regions. Some differences between the two studies that might account for such low concordance include, bio-specimen used (breast milk vs healthy breast tissue), the starting number of CpG sites tested (our 306,035 probes compared to their 247,456), our 139 (82 white and 57 black) samples compared to their 83 samples (61 white and 22 black), and the correction method for multiple tests (our Bonferroni, 1.63×10−7, compared to their Benjamin and Hochberg False Discovery Rate, 1.35×10−4). In addition, DNA methylation likely reflects a complex interplay between genetic and environmental exposures, which may be unaccounted for or different between our respective study populations. Despite these differences, we still observed some overlap between the IPA network results, which could indicate that the DNA methylation states in breast milk are robust, and in fact representative of the health of the breast tissue and not just the lactation state.
The IPA network results revealed that race and lactation length were associated with DNA methylation levels for genes in networks that may be relevant to carcinogenesis (i.e. cellular development, cellular growth and proliferation, etc.). These results suggest that race and lactation duration may affect DNA methylation states and, potentially, subsequently the expression of genes involved in cancer early in life. For race, there were seven genes (SRMS, GSE1, ABCC4, DHRS4, STAB2, RPS16, and IFNGR2) that the IKB [31] indicated were associated with cancer, but not specifically breast cancer. For lactation duration, there were eight genes (CYP19A1, G0S2, VEGFA, VDR, CDC42EP3, LIMA1, CD33, and PRKCD) that the IKB indicated were associated with cancer, with five (CYP19A1, G0S2, VEGFA, VDR, CDC42EP3) specifically related to breast cancer, and an additional two genes (CANT1 and CREB3L1) from the Cancer Gene Census. Whether differentially methylated probes and pathways affected by race have implications for early onset breast cancer is an area for future investigation.
Strengths of this study include the use of samples from cancer-free participants to understand epigenetic differences by exposure status and the use of novel and more rigorous normalization process to mask deleted and hyperpolymorphic regions. A special recruitment effort was employed to obtain samples from black women, which allowed us to oversample this demographic for this study and thus, provided additional power to detect differences between black and white women. However, some limitations to our study include the relatively small sample size and potential residual confounding as there are factors that may differ by race that we did not collect, such as diet, socio-economic information, and breastfeeding practices. Finally, we also recognize that DNA methylation levels in this study may reflect that of the lactation state rather than the long-term health of the breast, and additional studies comparing intra- and inter-woman variation in methylation profiles observed both during lactation as well as the post lactational period are needed. Despite these limitations, evaluating DNA methylation profiles among black and white women during a critical post-partum window may provide important etiologic information to better understand how lactation protects against breast carcinogenesis. Additionally, we were able to derive IPA networks consistent with a previous study performed in healthy breast tissue [13]. Confirmation and extension of these findings could provide insights into markers and mechanisms related to the effects of pregnancy and breastfeeding on breast cancer risk.
This study identified associations between genome-wide DNA methylation levels in breast milk and race and lactation duration, suggesting that these two exposures influence epigenetics of the healthy postpartum breast. It is well known that race and breastfeeding can influence breast cancer risk [3]; however, the mechanisms by which these two exposures play a role in breast carcinogenesis are not well understood. The epigenetic differences by race and lactation duration provide some clues as to how these exposures might affect breast cancer risk.
Supplementary Material
Acknowledgements
A special thank you to Dr. Mingyi Wang and Dr. Bin Zhu at the National Cancer Institute’s Cancer Genomics Research Laboratory for their lending their expertise in Bioinformatics. This research is supported by the Intramural Research Program and the Cancer Prevention Fellowship Program of the National Cancer Institute at the National Institutes of Health. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Funding
This study was funded, in part, by the Intramural Research Program of the National Cancer Institute and a National Institutes of Health Bench-to-Bedside Award from the Office of Research on Women’s Health.
List of Abbreviations
- IPA
Ingenuity Pathway Analysis
- CpG
Cytosine-phosphate-Guanosine
- BMI
Body mass index
- NCI
National Cancer Institute
Footnotes
Competing Interests
The following authors declare that they have no competing interests: BCDL, CB, RMP, HPY, HY, ML, MC, DJW, JM, JNS, EPB, DLA, MES, KFA, GLG.
PWL serves on the Scientific Advisory Board of AnchorDx and of Progenity, Inc.
DJW is a consultant for Zymo Research Corporation in Irvine, California.
References
- 1.DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A: Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA Cancer J Clin 2016, 66(1):31–42. [DOI] [PubMed] [Google Scholar]
- 2.DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A: Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J Clin 2017. [DOI] [PubMed] [Google Scholar]
- 3.Anderson KN, Schwab RB, Martinez ME: Reproductive risk factors and breast cancer subtypes: a review of the literature. Breast Cancer Res Treat 2014, 144(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones KM, Power ML, Queenan JT, Schulkin J: Racial and ethnic disparities in breastfeeding. Breastfeed Med 2015, 10(4):186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nichols HB, Schoemaker MJ, Cai J, Xu J, Wright LB, Brook MN, Jones ME, Adami HO, Baglietto L, Bertrand KA et al. : Breast Cancer Risk After Recent Childbirth: A Pooled Analysis of 15 Prospective Studies. Ann Intern Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan K, Flanagan JM: Is there a link between genome-wide hypomethylation in blood and cancer risk? Cancer Prev Res (Phila) 2012, 5(12):1345–1357. [DOI] [PubMed] [Google Scholar]
- 7.Das PM, Singal R: DNA methylation and cancer. J Clin Oncol 2004, 22(22):4632–4642. [DOI] [PubMed] [Google Scholar]
- 8.Mikeska T, Craig JM: DNA methylation biomarkers: cancer and beyond. Genes (Basel) 2014, 5(3):821–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsaprouni LG, Yang TP, Bell J, Dick KJ, Kanoni S, Nisbet J, Vinuela A, Grundberg E, Nelson CP, Meduri E et al. : Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 2014, 9(10):1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herman JG: Epigenetic changes in cancer and preneoplasia. Cold Spring Harb Symp Quant Biol 2005, 70:329–333. [DOI] [PubMed] [Google Scholar]
- 11.Xia YY, Ding YB, Liu XQ, Chen XM, Cheng SQ, Li LB, Ma MF, He JL, Wang YX: Racial/ethnic disparities in human DNA methylation. Biochim Biophys Acta 2014, 1846(1):258–262. [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Marioni RE, Hedman AK, Pfeiffer L, Tsai PC, Reynolds LM, Just AC, Duan Q, Boer CG, Tanaka T et al. : A DNA methylation biomarker of alcohol consumption. Mol Psychiatry 2018, 23(2):422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song MA, Brasky TM, Marian C, Weng DY, Taslim C, Dumitrescu RG, Llanos AA, Freudenheim JL, Shields PG: Racial differences in genome-wide methylation profiling and gene expression in breast tissues from healthy women. Epigenetics 2015, 10(12):1177–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christensen BC, Marsit CJ: Epigenomics in environmental health. Front Genet 2011, 2:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong CM, Anderton DL, Smith-Schneider S, Wing MA, Greven MC, Arcaro KF: Quantitative analysis of promoter methylation in exfoliated epithelial cells isolated from breast milk of healthy women. Epigenetics 2010, 5(7):645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumitrescu RG, Marian C, Krishnan SS, Spear SL, Kallakury BV, Perry DJ, Convit JR, Seillier-Moiseiwitsch F, Yang Y, Freudenheim JL et al. : Familial and racial determinants of tumour suppressor genes promoter hypermethylation in breast tissues from healthy women. J Cell Mol Med 2010, 14(6B):1468–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy J, Sherman ME, Browne EP, Caballero AI, Punska EC, Pfeiffer RM, Yang HP, Lee M, Yang H, Gierach GL et al. : Potential of breastmilk analysis to inform early events in breast carcinogenesis: rationale and considerations. Breast Cancer Res Treat 2016, 157(1):13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witkowska-Zimny M, Kaminska-El-Hassan E: Cells of human breast milk. Cell Mol Biol Lett 2017, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dave V, Street K, Francis S, Bradman A, Riley L, Eskenazi B, Holland N: Bacterial microbiome of breast milk and child saliva from low-income Mexican-American women and children. Pediatr Res 2016, 79(6):846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LaTuga MS, Stuebe A, Seed PC: A Review of the Source and Function of Microbiota in Breast Milk. Semin Reprod Med 2014, 32(1):68–73. [DOI] [PubMed] [Google Scholar]
- 21.Cabrera-Rubio R, Collado MC, Laitinen K, Salminen S, Isolauri E, Mira A: The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am J Clin Nutr 2012, 96(3):544–551. [DOI] [PubMed] [Google Scholar]
- 22.Shenker NS, Flower KJ, Wilhelm-Benartzi CS, Dai W, Bell E, Gore E, El Bahrawy M, Weaver G, Brown R, Flanagan JM: Transcriptional implications of intragenic DNA methylation in the oestrogen receptor alpha gene in breast cancer cells and tissues. BMC Cancer 2015, 15:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Browne EP, Punska EC, Lenington S, Otis CN, Anderton DL, Arcaro KF: Increased promoter methylation in exfoliated breast epithelial cells in women with a previous breast biopsy. Epigenetics 2011, 6(12):1425–1435. [DOI] [PubMed] [Google Scholar]
- 24.Browne EP, Dinc SE, Punska EC, Agus S, Vitrinel A, Erdag GC, Anderton DL, Arcaro KF, Yilmaz B: Promoter methylation in epithelial-enriched and epithelial-depleted cell populations isolated from breast milk. J Hum Lact 2014, 30(4):450–457. [DOI] [PubMed] [Google Scholar]
- 25.Campan M, Weisenberger DJ, Trinh B, Laird PW: MethyLight and Digital MethyLight. Methods Mol Biol 2018, 1708:497–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campan M, Weisenberger DJ, Trinh B, Laird PW: MethyLight. Methods Mol Biol 2009, 507:325–337. [DOI] [PubMed] [Google Scholar]
- 27.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL et al. : High density DNA methylation array with single CpG site resolution. Genomics 2011, 98(4):288–295. [DOI] [PubMed] [Google Scholar]
- 28.Zhou W, Laird PW, Shen H: Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res 2017, 45(4):e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou W, Triche TJ Jr., Laird PW, Shen H: SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res 2018, 46(20):e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R: Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8(2):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kramer A, Green J, Pollard J Jr., Tugendreich S: Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30(4):523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L et al. : COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Research 2017, 45(D1):D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ambrosone CB, Young AC, Sucheston LE, Wang D, Yan L, Liu S, Tang L, Hu Q, Freudenheim JL, Shields PG et al. : Genome-wide methylation patterns provide insight into differences in breast tumor biology between American women of African and European ancestry. Oncotarget 2014, 5(1):237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conway K, Edmiston SN, Tse CK, Bryant C, Kuan PF, Hair BY, Parrish EA, May R, Swift-Scanlan T: Racial variation in breast tumor promoter methylation in the Carolina Breast Cancer Study. Cancer Epidemiol Biomarkers Prev 2015, 24(6):921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mehrotra J, Ganpat MM, Kanaan Y, Fackler MJ, McVeigh M, Lahti-Domenici J, Polyak K, Argani P, Naab T, Garrett E et al. : Estrogen receptor/progesterone receptor-negative breast cancers of young African-American women have a higher frequency of methylation of multiple genes than those of Caucasian women. Clin Cancer Res 2004, 10(6):2052–2057. [DOI] [PubMed] [Google Scholar]
- 36.Davis Lynn BC, Rosenberg PS, Anderson WF, Gierach GL: Black-White Breast Cancer Incidence Trends: Effects of Ethnicity. J Natl Cancer Inst 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.