

Abstract
Background
The present study aims to summarize the clinical and genetic characteristics of ZTTK syndrome.
Methods
The clinical and genetic data of a Chinese girl with severe growth and development delay, intellectual disability, and facial features were analyzed. Original articles on ZTTK syndrome published up to November 20l8 were identified from PubMed, Human Gene Mutation Database, Online Mendelian Inheritance in Man, China National Knowledge Infrastructure, and WanFang databases using the keywords “ZTTK syndrome” and “SON”.
Results
The patient was born small for gestational age, and had poor academic performance, delayed language development, and motor retardation. The patient's height was 113 cm (less than −3 SD), and had moles on the back skin and possessed facial features. A novel heterozygous mutation c.394C>T (p.Q132X) of SON was found in this patient, but the parents were normal.
Conclusion
The patient's clinical phenotype was consistent with ZTTK syndrome. The novel heterozygous mutation c.394C>T (p.Q132X) of SON was its pathogenic mutation, which has not been reported at home and abroad.
Keywords: gene, mutation, SON, ZTTK syndrome
Short abstract
The patient's clinical phenotype was consistent with ZTTK syndrome. The novel heterozygous mutation c.394C>T (p.Q132X) of SON was its pathogenic mutation, which has not been reported at home and abroad.
1. INTRODUCTION
The ZTTK syndrome (Online Mendelian Inheritance in Man (OMIM 617144) is the abbreviation of the ZHU‐TOKITA‐TAKENOUCHI‐KIM syndrome. It is an autosomal dominant hereditary disease, and is a serious multisystem developmental disorder characterized by intellectual disability (ID). Its clinical features include facial features, hypotonia, malnutrition, and eye or visual abnormalities. Most patients have musculoskeletal abnormalities, and may be complicated with congenital heart and/or genitourinary system defects. Brain imaging usually reveals developmental abnormalities (e.g., cerebral cortical gyration changes, cortical and/or cerebellar atrophy, and thin corpus callosum).
This syndrome was first reported by Zhu et al. (2015). This is caused by a heterozygous mutation with a 4‐bp deletion in SON (OMIM accession number: 182465), which led to frameshift mutations and a premature termination codon. There are presently 30 cases of ZTTK syndrome reported worldwide, but there has been no report of ZTTK syndrome caused by SON mutation in Chinese patients. The clinical data and genetic analysis of a child with a new heterozygous mutation of SON, who was admitted to Jiangxi Children's Hospital in June 2018, were reported as follows. The relevant literatures were also reviewed.
2. SUBJECTS AND METHODS
2.1. Subjects
The proband was a 13‐year‐and‐2‐month‐old female patient when admitted to hospital. She was admitted to the hospital due to "growth retardation (GR) for several years." The patient's growth rate was <4 cm/year, the patient's intellectual development was obviously delayed, and the age at which the patient begin walking was late. The patient's mother had four times of gestation and four times of parturition. The brothers and sisters of the patient were healthy. The propositus was born at 40 weeks of gestation through spontaneous labor. The patient was a small‐for‐date infant, the birth weight was 1.5 kg. Furthermore, the patient has a history of hypoxia and asphyxia during delivery. Physical examination: The height of patient was 113 cm (standard deviation score [SDS]: −7.2), and the target height of the patient is 148.5 ± 8 cm ([father's height + mother's height – 13]/2 ± 8 cm). Physique was well‐proportioned, the forehead was protruding, and the anterior hairline was low. Furthermore, the patient presented with the following: transverse eyebrows, a slightly oblique cleft of the eye, adduction of the middle face, wide bridge of the nose and upturning wings of the nose, short nasolabial groove, high maxillary arch, low set ears, small mouth, and thin upper lip (Figure 1). The neck was not short, the lumbar spine was bent slightly toward the left, depression could be observed in the sacrococcygeal skin, which was similar to a sinus, and no secretion extravasation was found. The bilateral elbows exhibited ectropion. The father of the patient was 160 cm in height (standard deviation score [SDS]: −2SD), while the mother was 150 cm in height (standard deviation score [SDS]: −1SD). The child denied any history of inherited metabolic disease.
Figure 1.
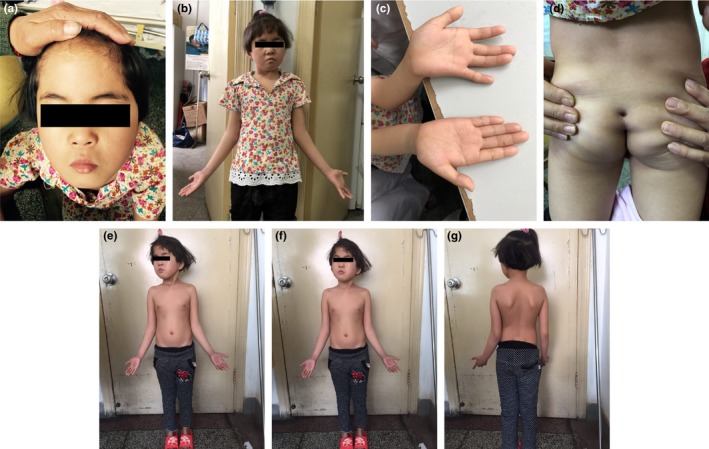
Physical characteristics of patient
Adjuvant examination results: The results of three major routine tests, a complete set of biochemical markers, and the thyroid function test were normal. The levels of both insulin‐like growth factor‐1 (IGF‐1) and BP3 decreased. The anterioposterior and lateral films of the whole spine revealed that the lumbar spine was bent slightly toward the left (Figure 2). Brain magnetic resonance imaging (MRI) revealed that white matter in the periventricular area and centrum semiovale slightly decreased, the corpus callosum was thin, and obstructive hydrocephalus (mild) was present (Figure 3). The bone age was 9.5 years old, which lagged behind the actual age. Color Doppler ultrasonography of the uterine and ovary revealed that the measured size values were small. However, the color Doppler ultrasound of the liver, spleen, pancreas, and adrenal glands revealed normal results. Furthermore, the color Doppler ultrasound of the heart was normal. The growth hormone provocation test revealed that a peak value of 7.37 ng/ml. The patient's chromosomes were normal. Furthermore, blood tandem mass spectrometry and urine gas chromatography were normal.
Figure 2.
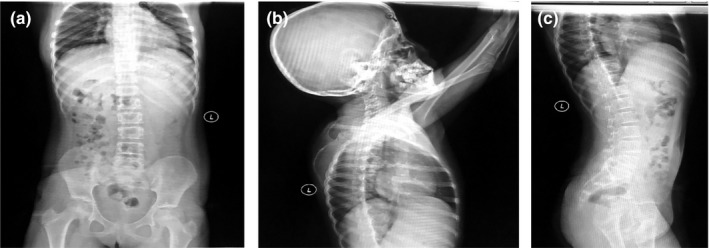
The result of the anterioposterior and lateral films of the whole spine
Figure 3.
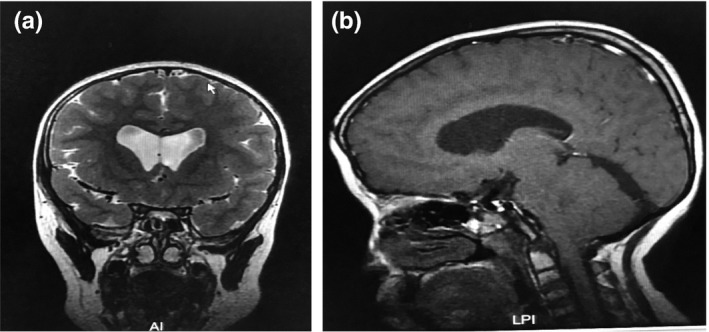
The result of Brain magnetic resonance imaging (MRI)
This study was conducted in accordance with the Declaration of Helsinki. This study was conducted with approval from the Ethics Committee of Jiangxi Provincial Children's Hospital. A written informed consent was obtained from legal guardians.
2.2. Methods
In the present study, the patient and her family were fully informed and provided signed informed consent. The present study was approved by the Ethics Committee of our hospital. Written consent was obtained to publish facial photographs from legal guardians (mother) of the participant.
2.2.1. Exon group capture and exome sequencing
EDTA anticoagulant tubes were used to collect 3 ml of peripheral blood from the child and her parents. Then, blood genomic DNA was routinely extracted, and sent to the Beijing MyGenostics Medical Laboratory for exon group capture and exome sequencing (brief process: DNA extraction → library establishment → targeted sequence capture → biological analysis → report issuance).
2.2.2. Protein function prediction
Protein function prediction software SIFT, PolyPhen_2, and REVEL were used to predict the protein function of SON mutation.
3. RESULTS
3.1. Exon group capture and exome sequencing results
There was a heterozygous mutation (c.394C>T, p.Q132X) in SON of the proband, and the mutation was a nonsense mutation. However, no mutation was detected in this locus of the parents’ DNA. Sanger sequencing was used to verify the mutation in the patient and the parents. The results suggested that this mutation was a new mutation. According to the guidelines of the American Society of Medical Genetics and Genomics (ASMG) in 2015, a nonsense mutation is a pathogenic mutation (Figure 4). No other pathogenic variants were present in the exome data.
Figure 4.
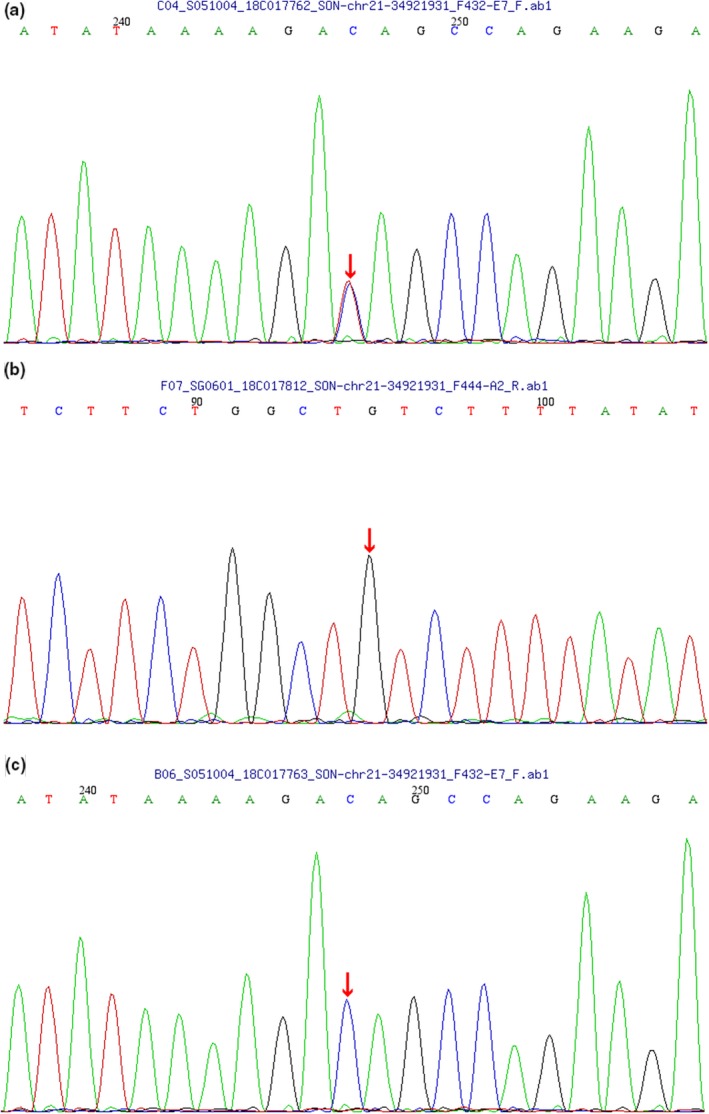
Results of the SON analysis in the child with ZTTK syndrome and the child's parents. (a) A heterozygous mutation (c.394C>T, p.Q132X) in SON was detected in the child; (b) No mutation in SON was detected in the father of the child; (c) No mutation in SON was detected in the mother of the child
3.2. Protein structure function prediction results
The results of the protein function prediction using software SIFT, PolyPhen_2, and REVEL were unknown, unknown and unknown, respectively.
4. LITERATURE RETRIEVAL RESULTS
With "ZTTK syndrome", "ZTTK syndrome", "SON gene" and "SON" as the retrieval words, data were retrieved from the PubMed, Online Mendelian Inheritance in Man (OMIM), China National Knowledge Infrastructure, and WanFang databases before 1 October 2018. A total of four literatures reported this syndrome, and all of which are non‐Chinese countries literatures (Kim et al., 2016; Takenouchi, Miura, Uehara, Mizuno, & Kosaki, 2016; Tokita et al., 2016; Zhu et al., 2015). However, no literature was found from China. The heterozygous mutation of SON (c.394C>T, p.Q132X) in the present case has not been reported in the abovementioned databases. The mutations of ZTTK syndrome reported in literatures are presented in Table 1. The clinical features of ZTTK syndrome are summarized in Table 2.
Table 1.
Mutations of the ZTTK syndrome reported in literatures to date
| Literatures | Nucleotide mutations | Protein mutations |
|---|---|---|
| Zhu et al. (2015), Takenouchi et al. (2016), Kim et al. (2016), Tokita et al. (2016) | c.5753_5756delTTAG | p. Val1918Glufs |
| Kim et al. (2016) | c.6002_6003insCC | p. Arg2002Glnfs |
| Kim et al. (2016) | c.4640delA | p. His1547Leufs |
| Kim et al. (2016) | c.5549_5550del | p. (Arg1850llefs) |
| Kim et al. (2016) | c.1881_1882del | p. (Val629Alafs) |
| Kim et al. (2016), Tokita et al. (2016) |
c.3852_3856del c.4999_5013del; |
p. (Met1284llefs) p. Asp1667_Asn1671del; |
| Kim et al. (2016) |
c.5031‐5032insAA c.4358_4359del |
Asp1678Lysfs) p. (Thr1453Serfs) |
| Kim et al. (2016) | c.6087del | p. (Ser2029Argfs) |
| Kim et al. (2016) | c.3597_3598dup | p. (Pro1200Argfs) |
| Kim et al. (2016) | c.4151_4174del24 | p. (Leu1384_Val1391del) |
| Kim et al. (2016) | c.2365del | p. (Ser789Alafs) |
| Kim et al. (2016) | c.3344C>T | p. (Arg1112) |
| Kim et al. (2016) | c.268del | p. (Ser90Valfs) |
| Kim et al. (2016) | c.4055del | p. (Pro1352Glnfs) |
| Kim et al. (2016) | c.4549dup | p. (Glu1517Glyfs) |
| Kim et al. (2016) | Whole gene deletiona | — |
| Kim et al. (2016) | ||
| Tokita et al. (2016) | c.286C>T | p. Gln96Ter |
| Tokita et al. (2016) | c.3073dupA | p. Met1025Asnfs |
| Tokita et al. (2016) | c.6233delC | p. Pro2078Hisfs |
| Mayo Clinic | c.3556C>Tb | p. Gln1186Ter |
| Present case | c.394C>T | p. Q132X |
This case carries a small deletion of copy number mutations, including SON and five other genes: [hg19] Chr21: g. (34877993_34894566‐3559909) del (ISCN arr 21q.22.11q22.11 (34894566–3527867) × 1 dn.
Mayo Clinic Heredity Laboratory, February 2018.
Table 2.
Clinical Features of the ZTTK syndrome reported in literatures to date
| Conditions | percentage | Literatures |
|---|---|---|
| Developmental delay/Intellectual disability | 100% | Zhu et al. (2015), Takenouchi et al. (2016), Kim et al. (2016), Tokita et al. (2016) |
| Brain malformation | 85% | Zhu et al. (2015), Kim et al. (2016), Tokita et al. (2016) |
| Ventricular enlargement | 50% | Kim et al. (2016) |
| Corpus callosum abnormality | 43% | Kim et al. (2016), Tokita et al. (2016) |
| Cortex malformation | 25% | Kim et al. (2016) |
| White matter abnormalities | 18% | Zhu et al. (2015), Kim et al. (2016) |
| Cerebellar abnormalities | 14% | Kim et al. (2016) |
| Other | 18% | Kim et al. (2016), Tokita et al. (2016) |
| Neurological features | 66% | Zhu et al. (2015), Takenouchi et al. (2016), Kim et al. (2016), Tokita et al. (2016) |
| Seizures | 50% | Zhu et al. (2015), Kim et al. (2016), Tokita et al. (2016) |
| Hypotonia | 75% | Takenouchi et al. (2016), Kim et al. (2016),, Tokita et al. (2016) |
| Musculoskeletal abnormalities | 79% | Kim et al. (2016), Tokita et al. (2016) |
| Hypermobility | 28% | Kim et al. (2016) |
| Scoliosis or kyphosis | 14% | Kim et al. (2016) |
| Hemivertebrae | 7% | Kim et al. (2016) |
| Contractures | 7% | Kim et al. (2016) |
| Other | 83% | Kim et al. (2016), Tokita et al. (2016) |
| Eye and/or vision abnormality | 71% | Kim et al. (2016), Tokita et al. (2016) |
| Strabismus | 54% | Kim et al. (2016), Tokita et al. (2016) |
| Suspicion of cortical visual impairment | 18% | Kim et al. (2016), Tokita et al. (2016) |
| Hypermetropia | 21% | Kim et al. (2016) |
| Heart defect | 30% | Zhu et al. (2015), Takenouchi et al. (2016), Kim et al. (2016), Tokita et al. (2016) |
| Gastrointestinal malformation | 39% | Zhu et al. (2015), Kim et al. (2016) |
| Urogenital malformation | 37% | Kim et al. (2016), Tokita et al. (2016) |
| Facial dysmorphism | 100% | Takenouchi et al. (2016), Kim et al. (2016), Tokita et al. (2016) |
| Short stature | 52% | Takenouchi et al. (2016), Kim et al. (2016), Tokita et al. (2016) |
| Craniosynostosis | 10% | Kim et al. (2016) |
5. DISCUSSION
The ZTTK syndrome is a syndrome with a series of clinical symptoms caused by heterozygous mutations of SON on autosomal 21q22.11. The first new mutation to truncate SON was detected from screening a large number of individuals with severe mental retardation (Gilissen et al., 2014). SON is located in the autosomal region 21q22.11, consists of 12 exons, and encodes the arginine/serine domain and two RNA binding regions (Ahn et al., 2011; Hickey, Kim, & Ahn, 2014; Sharma, Takata, Shibahara, Bubulya, & Bubulya, 2010). The prominent feature of the gene structure is the size of exon 3, which accounts for 82% of the whole coding region (Khan et al., 2014). SON is a nuclear spot localization protein, which has homology with pre‐RNA splicing cofactor (Wynn et al., 2000). It can splice DNA and RNA, has the function of RNA splicing and gene transcription, and regulates the cell cycle and maintains the function of embryonic stem cells (Ahn et al., 2013; Lu et al., 2013). The expression of genes involved in embryonic development, neuronal cell transition, metabolism, and mitochondrial function (TUBG1, FLNA, PNKP, WDR62, PSMD3, HDAC6, PCK2, PFKL, IDH2, ACY1, and ADA) significantly decrease when the dose of SON haploid is insufficient (Lu et al., 2013; Sharma et al., 2010). At present, more than 1,500 genetic changes are known to cause ID and GR (Lelieveld et al., 2016; Mefford, Batshaw, & Hoffman, 2012; Vissers, Gilissen, & Veltman, 2016). New single nucleotide and copy number mutations are important causes of severe ID and/or GR. Human neurodevelopment‐related genes are significantly down‐regulated when the SON haploid is insufficient. These results indicate that SON is the main regulator of essential genes in human neurodevelopment (Bilgüvar et al., 2010; Chen et al., 2014; Deciphering Developmental Disorders Study, 2015; Jamuar et al., 2014; Rauch et al., 2012).
In 2015, Zhu et al. (2015) reported a 5‐year‐old girl with developmental retardation, epilepsy, mild malformation, megalencephaly, white matter dysplasia, intestinal atresia, and ventricular septal defect, and a new frameshift mutation was detected in SON. In 2016, Takenouchi et al. (2016) reported a boy with postnatal developmental retardation postnatal, and the clinical characteristics of the boy were megalencephaly, hypotonia in infancy, severe ID, congenital heart disease, excessive joint extension, slender limbs, and smooth and soft skin. The boy's facial features were frontal bossing, curly hair, sparse eyebrows, towering folds, flat bridge of the nose, protruding ears, short nose, and full face. A frameshift mutation was detected at the same locus in SON in this boy, which is similar to the case reported by Zhu et al. (2015).
In 2016, Kim et al. (2016) reported 20 patients (including patients previously reported by Zhu et al.[, 2015]), and the clinical characteristics included mild to severe ID and complex neurodevelopmental disorders. A number of these patients had neonatal feeding difficulties and hypotonia, and facial features (facial asymmetry, adduction in the middle of the face, low set ears, eye cleft declivity, deep eyes, transverse frowns, broad nose bridge and/or flat nose bridge, and short philtrum). Furthermore, 15 patients had visual disorders, such as cortical visual impairment, hyperopia, optic atrophy and strabismus. In addition, brain imaging revealed that 89% of these patients had obvious abnormalities, including abnormal rotation patterns, ventriculomegaly, arachnoid cysts, callosal dysplasia, cerebellar dysplasia, and periventricular white matter deletion. Approximately half of these patients had epileptic seizures when they were 1–6 years old, approximately half of these patients presented short stature, and a number of patients had various musculoskeletal abnormalities, such as hemivertebra, scoliosis or kyphosis, contracture, hypermobility of the joints and small hands and feet. Some patients had congenital malformations, including urogenital system malformations (six patients), heart defects (five patients), intestinal malformation (three patients), premature closure of cranial suture (three patients), and high position or cleft palate (two patients). The identified new truncate mutations in these patients were located in SON. It was revealed by whole genome/exome sequencing that these mutations resulted from the lack of haploid dose, and four of these had the same frameshift mutation. Cells in the three patients with low haploid dose of SON were analyzed. The results revealed that the expression of mRNA and the splicing products of abnormal RNA could be reduced, and that these polymorphic gene products play an important role in neuronal cell migration, brain development, and metabolism.
In 2016, Tokita et al. (2016) carried out whole genome/exome sequencing for more than 6,000 patients (mainly children) with nervous system disease. As a result, a new heterozygous truncate mutation of SON was detected in seven patients. These patients had severe multisystem developmental disorders, feeding difficulty, dyspnea, and hypotonia shortly after birth. These patients also had facial features (frontal lobe protrusion, double temporal stenosis, sunken eyes, eyelids drooping, epicanthus, short philtrum, and thin lips) and developmental retardation, while some patients had developmental degeneration, autism, visual abnormality (strabismus, exotropia, esotropia, nystagmus, loss of vision and optic atrophy), musculoskeletal joint relaxation, scoliosis, hemivertebrae, abnormal ribs, and subarachnoid space. Among the six patients, the brain imaging of five patients revealed various abnormalities, including ventricular enlargement, thin corpus callosum, arachnoid cyst, and periventricular leukomalacia. The levels of IgA and/or IgG decreased in three patients. Abnormal coagulation occurred in two patients. Five patients had congenital abnormalities in other organ systems, such as the heart and urogenital system.
Kim et al. (2016) carried out SON knockout experiments in zebrafish. The results revealed that the haploid deficiency of SON led to a variety of developmental defects, including curving and shortening of the tail, eye malformation, microcephaly, and body curvature deformations of the body axis. Embryos with longer survival exhibited more severe phenotypes, including spinal malformation and cerebral edema.
The main clinical manifestations of the ZTTK syndrome presently reported are as follows: (1) short stature; (2) developmental delay and/or ID; (3) facial features: facial asymmetry, frontal uplift, midface depression, short nasolabial groove, low set ears, oblique cleft of eyes, sunken eyes, wide/low bridge of the nose, small mouth, thin upper lip, and high frontal arch and cleft palate; (4) combined various congenital malformations: (a) eyes: cortical visual impairment, hyperopia, optic atrophy, and strabismus; (b) tooth dysplasia; (c) congenital heart disease (for some of the patients); (d) gastrointestinal tract: feeding difficulty and gastrointestinal malformations (for some of the patients); (e) urogenital system (for some of the patients): single kidney, horseshoe kidney and kidney dysplasia; (f) bone dysplasia of the spine and limbs: small hands and feet, joint spasm, joint overextension, premature closure of cranial suture, scoliosis, kyphosis and hemivertebra; (5) nervous system defects: hypotonia, delayed development, IR, developmental regression, epilepsy (for some of the patients), abnormal cerebral cortical gyration, ventriculomegaly, thin corpus callosum, arachnoid cyst, cerebellar dysplasia, and white matter abnormality; (6) low level of immunoglobulin (for some of the patients).
In the present study, the gene locus in the child was 21q22.11, which was caused by the mutation of the DNA binding protein gene, SON. The stature and intellectual developments were obviously delayed, many naevi were found on the back skin, the anterior hairline was low, the eyebrows were transverse, the bridge of the nose was wide, and the oculi rimae were slightly oblique. Furthermore, the nasolabial groove was short, the maxillary arch was high, the neck was not short, and the lumbar spine was slightly bent to the left. In addition, depression could be observed in the sacrococcygeal skin, which is similar to a sinus, but no secretion was found. The bilateral elbow presented with ectropion. Anterioposterior and lateral films of the whole spine revealed that the lumbar spine was slightly bent to the left. Brain MRI revealed that the periventricular and centrum semiovale white matter slightly decreased, the corpus callosum was thin, and obstructive hydrocephalus (mild) was present. The exon group capture and exome sequencing of the child revealed a heterozygous mutation detected in SON, c.394C>T (cytosine was substituted by thymine in the 394th nucleotide in the coding region), causing an amino acid change, p.Q132X, which was a nonsense mutation. According to the ASMG guidelines, the mutation was preliminarily identified as a pathogenic mutation. The mutation meets the criterion to be identified as a pathogenic mutation, including PVS, PS2, and PM2. PVS: The mutation was a zero effect mutation (nonsense mutation), which may lead to loss of gene function. PS2: The family validation analysis revealed that no mutation was detected in the locus in the parents of the patient, and the mutation was a spontaneous mutation. PM2: The frequency in the normal population database was negative, and the mutation was a low frequency mutation. There is no related report on this locus in the GMD database. The family validation analysis revealed that no mutation was detected in this locus in the father of the child, and no mutation was also detected in this locus in the mother of the child. On the basis of clinical manifestations, it was considered as a pathogenic mutation.
In summary, the present study reports a child with ZTTK syndrome carrying the c.394C>T mutation of SON (NM_001291411.1). A ZTTK syndrome case caused by a SON mutation was reported for the first time in the Chinese population, which enriches clinical and genetic data.
CONFLICT OF INTEREST
The author reports no conflicts of interest in this work.
ACKNOWLEDGMENTS
This work was supported in part by the National Natural Science Foundation of China (No. 81460501).
Yang Y, Xu L, Yu Z, Huang H, Yang L. Clinical and genetic analysis of ZTTK syndrome caused by SON heterozygous mutation c.394C>T. Mol Genet Genomic Med. 2019;7:e953 10.1002/mgg3.953
REFERENCES
- Ahn, E. Y. , DeKelver, R. C. , Lo, M. C. , Nguyen, T. A. , Matsuura, S. , Boyapati, A. , … Zhang, D. E. (2011). SON controls cell‐cycle progression by coordinated regulation of RNA splicing. Molecular Cell, 42(2), 185–198. 10.1016/j.molcel.2011.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, E. E. , Higashi, T. , Yan, M. , Matsuura, S. , Hickey, C. J. , Lo, M. C. , … Zhang, D. E. (2013). SON protein regulates GATA‐2 through transcriptional control of the microRNA 23a∼27a∼24‐2 cluster. Journal of Biological Chemistry, 288(8), 5381–5388. 10.1074/jbc.M112.447227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilgüvar, K. , Oztürk, A. K. , Louvi, A. , Kwan, K. Y. , Choi, M. , Tatli, B. , … Günel, M. (2010). Whole‐exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature, 467(7312), 207–210. 10.1038/nature09327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. F. , Zhang, Y. , Wilde, J. , Hansen, K. C. , Lai, F. , & Niswander, L. (2014). Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nature Communications, 5, 3885 10.1038/ncomms4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study . (2015). Large‐scale discovery of novel genetic causes of developmental disorders. Nature, 519(7542), 223–228. 10.1038/nature14135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen, C. , Hehir‐Kwa, J. Y. , Thung, D. T. , van de Vorst, M. , van Bon, B. W. , Willemsen, M. H. , … Veltman, J. A. (2014). Genome sequencing identifies major causes of severe intellectual disability. Nature, 511(7509), 344–347. 10.1038/nature13394 [DOI] [PubMed] [Google Scholar]
- Hickey, C. J. , Kim, J. H. , & Ahn, E. Y. (2014). New discoveries of old SON: A link between RNA splicing and cancer. Journal of Cellular Biochemistry, 115(2), 224–231. 10.1002/jcb.24672 [DOI] [PubMed] [Google Scholar]
- Jamuar, S. S. , Lam, A.‐T. , Kircher, M. , D’Gama, A. M. , Wang, J. , Barry, B. J. , … Walsh, C. A. (2014). Somatic mutations in cerebral cortical malformations. New England Journal of Medicine, 371(21), 733–743. 10.1056/NEJMoa1314432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, I. M. , Fisher, R. A. , Johnson, K. J. , Bailey, M. E. , Siciliano, M. J. , Kessling, A. M. , … Buluwela, L. (2014). The SON gene encodes a conserved DNA binding protein mapping to human chromosome 21. Annals of Human Genetics, 58(1), 25–34. [DOI] [PubMed] [Google Scholar]
- Kim, J. H. , Shinde, D. N. , Reijnders, M. R. F. , Hauser, N. S. , Belmonte, R. L. , & Wilson, G. R. , … Ahn, E. Y. E. (2016). De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual‐disability syndrome. American Journal of Human Genetics, 99(3), 711–719. 10.1016/j.ajhg.2016.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelieveld, S. H. , Reijnders, M. R. , Pfundt, R. , Yntema, H. G. , Kamsteeg, E. J. , de Vries, P. , … Gilissen, C. (2016). Meta‐analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nature Neuroscience, 19(9), 1194–1196. 10.1038/nn.4352 [DOI] [PubMed] [Google Scholar]
- Lu, X. , Göke, J. , Sachs, F. , Jacques, P. E. , Liang, H. , Feng, B. , … Ng, H. H. (2013). SON connects the splicing‐regulatory network with pluripotency in human embryonic stem cells. Nature Cell Biology, 15(10), 1141–1152. 10.1038/ncb2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford, H. C. , Batshaw, M. L. , & Hoffman, E. P. (2012). Genomics, intellectual disability, and autism. New England Journal of Medicine, 366(8), 733–743. 10.1056/NEJMra1114194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch, A. , Wieczorek, D. , Graf, E. , Wieland, T. , Endele, S. , Schwarzmayr, T. , … Strom, T. M. (2012). Range of genetic mutations associated with severe non‐syndromic sporadic intellectual disability: An exome sequencing study. Lancet, 380(9854), 1674–1682. 10.1016/S0140-6736(12)61480-9 [DOI] [PubMed] [Google Scholar]
- Sharma, A. , Takata, H. , Shibahara, K. , Bubulya, A. , & Bubulya, P. A. (2010). Son is essential for nuclear speckle organization and cell cycle progression. Molecular Biology of the Cell, 21(4), 650–663. 10.1091/mbc.e09-02-0126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenouchi, T. , Miura, K. , Uehara, T. , Mizuno, S. , & Kosaki, K. (2016). Establishing SON in 21q22.11 as a cause a new syndromic form of intellectual disability: Possible contribution to Braddock‐Carey syndrome phenotype. American Journal of Medical Genetics. Part A, 170(10), 2587–2590. 10.1002/ajmg.a.37761 [DOI] [PubMed] [Google Scholar]
- Tokita, M. J. , Braxton, A. A. , Shao, Y. , Lewis, A. M. , Vincent, M. , Küry, S. , … Walkiewicz, M. A. (2016). De novo truncating variants in SON cause intellectual disability, congenital malformations, and failure to thrive. American Journal of Human Genetics, 99(3), 720–727. 10.1016/j.ajhg.2016.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers, L. E. , Gilissen, C. , & Veltman, J. A. (2016). Genetic studies in intellectual disability and related disorders. Nature Reviews Genetics, 7(1), 9–18. 10.1038/nrg3999 [DOI] [PubMed] [Google Scholar]
- Wynn, S. L. , Fisher, R. A. , Pagel, C. , Price, M. , Liu, Q. Y. , Khan, I. M. , … Buluwela, L. (2000). Organization and conservation of the GART/SON/DONSON locus in mouse and human genomes. Genomics, 68(1), 57–62. 10.1006/geno.2000.6254 [DOI] [PubMed] [Google Scholar]
- Zhu, X. , Petrovski, S. , Xie, P. , Ruzzo, E. K. , Lu, Y. F. , McSweeney, K. M. , … Goldstein, D. B. (2015). Whole‐exome sequencing in undiagnosed genetic diseases: Interpreting 119 trios. Genetics in Medicine, 17(10), 774–781. 10.1038/gim.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]