

Abstract
Background
β‐thalassemia is one of the most common monogenic diseases in the world. Southeast China is a highly infected area affected by four β‐thalassemia mutation types (HBB:c.‐78A>G, HBB:c.52A>T, HBB:c.126_129delCTTT, and HBB:c.316‐197C>T). Relative haplotype dosage (RHDO), a haplotype‐based approach, has shown promise as an application for noninvasive prenatal diagnosis (NIPD); however, additional family members (such as the proband) are required for haplotype construction. The abovementioned circumstances make RHDO‐based NIPD cost prohibitive; additionally, the genetic information of the proband is not always available. Thus, it is necessary to find a practical method to solve these problems.
Methods
Targeted sequencing was applied to sequence parental genomic DNA and cell‐free fetal DNA (cffDNA). Parental haplotypes were constructed with the SHAPEIT software based on the 1000 Genomes Project (1000G) Phase 3 v5 Southern Han Chinese (CHS) haplotype dataset. Single‐nucleotide polymorphisms (SNPs) in the target region were called and classified, and the fetal mutation inheritance status was deduced using the RHDO method.
Results
Construction of the parental haplotypes and detection of the inherited parental mutations were successfully achieved in five families, despite a suspected recombination event. The status of the affected fetuses is consistent with the results of traditional reverse dot blot (RDB) diagnosis.
Conclusion
This research introduced SHAPEIT into the classical RHDO workflow and proved that it is applicable to construct parental haplotypes without information from other family members.
Keywords: cell‐free fetal DNA, haplotype, noninvasive prenatal diagnosis, relative haplotype dosage, β‐thalassemia
1. INTRODUCTION
β‐thalassemia, which is caused by mutations in the HBB gene located on chromosome 11, is one of the most common monogenic diseases in the world (Higgs, Engel, & Stamatoyannopoulos, 2012). These variants result in reduced or absent hemoglobin tetramer β‐globin chain synthesis (Rund & Rachmilewitz, 2005). Over 200 different β‐thalassemia mutations have been identified to date, with the majority being point mutations, deletions, or insertions in the HBB gene or its immediate flanking region (Giardine et al., 2014). In China, a large number of carriers live in the southeastern region, and the following four mutations in the HBB gene comprise nearly 87% of all β‐thalassemia mutations in this area: ‐28A>G (HBB: c.‐78A>G), CD17A>T (HBB: c.52A>T), CD41‐42(‐TTCT) (HBB: c.126_129delCTTT), and IVS‐II‐654C>T (HBB:c.316‐197C>T) (Lai, Huang, Su, & He, 2017; Li et al., 2014; Lin et al., 2014; Yin et al., 2014).
If both parents are carriers, they have a 25% risk of giving birth to an affected child, who will need frequent blood transfusions and iron chelation; the ultimate cure requires stem‐cell transplantation and gene therapy (Higgs et al., 2012). The prevention of this disease in newborns is a significant issue for the parents and the government because the treatment is painful for the patients and places a heavy burden on society. Therefore, the most effective strategy is to reduce the incidence of this genetic disease, which can be accomplished by utilizing appropriate prenatal diagnosis in clinical practice. However, traditional prenatal diagnosis, which includes invasive procedures such as amniocentesis and chorionic villus sampling (CVS), increases the risk of miscarriage (Akolekar, Beta, Picciarelli, Ogilvie, & D'Antonio, 2015; Han et al., 2014). Noninvasive prenatal diagnosis (NIPD) offers an excellent option.
The discovery of cell‐free fetal DNA (cffDNA) in maternal plasma by Lo et al. shows excellent potential for NIPD (Lo et al., 1997). To date, the performance of NIPD in the screening of common fetal chromosomal aneuploidies has been reported with high levels of accuracy, resulting in a decline in the number of invasive diagnostic procedures (Chiu et al., 2008; Gil, Accurti, Santacruz, Plana, & Nicolaides, 2017; Wong & Lo, 2015). In addition to detecting fetal aneuploidies, researchers have focused on monogenic disease diagnosis via this discovery (Chitty & Lo, 2015; Daley, Hill, & Chitty, 2014; Parks et al., 2017; Vermeulen et al., 2017).
The most common strategy for the noninvasive determination of autosomal dominant diseases is the identification of a paternal mutation in maternal plasma, which can prove the presence of an affected fetus. For example, NIPD procedures for Huntington disease (Gonzalez‐Gonzalez et al., 2008), achondroplasia (Chitty et al., 2011), and myotonic dystrophy (Amicucci, Gennarelli, Novelli, & Dallapiccola, 2000) have shown great success in at‐risk couples. For autosomal recessive inheritance, if the parents carry different mutations, the absence of the paternal allele in maternal plasma would exclude the presence of an affected fetus, and thus, a subset of pregnant women could avoid invasive diagnostic procedures. Various diseases for which this NIPD strategy has been adopted include cystic fibrosis (Bustamante‐Aragones et al., 2008), congenital adrenal hyperplasia (CAH) (Chiu et al., 2002), and thalassemia (Xiong et al., 2015; Zafari et al., 2016).
Although NIPD for monogenic recessive disease shows a bright future in clinical application, it still faces several technical challenges. The contamination of cffDNA by maternal plasma is one of the major obstructions to overcome incorrectly identifying the fetal disease‐related genotype, particularly when both parents carry the same variant. In the last decade, using massively parallel sequencing to conduct genome‐wide genotyping for the disease‐causing gene or its surrounding single‐nucleotide polymorphisms (SNPs) has provided new possibilities for inferring fetal SNP alleles.
For NIPD of β‐thalassemia, Lo et al. first demonstrated the feasibility of a haplotype‐based method, termed relative haplotype dosage (RHDO) analysis, for deducing the fetal inheritance of the maternally transmitted haplotype (Lo et al., 2010). Further successful cases have shown that researchers can establish NIPD with high detection rates in other monogenic diseases, such as Duchenne muscular dystrophy (DMD) (Xu et al., 2015), spinal muscular atrophy (SMA) (Parks et al., 2017), and CAH (New et al., 2014), with this strategy. However, the process of haplotype construction requires either the use of other family members as probands or complex bioinformatics analysis, which causes difficulties in translation to clinical practice.
Technical complexity and the high cost are two constraints in NIPD for single gene disorders. In this study, we aimed to develop a fetal haplotype phase strategy without relying on proband information and confirm the practicability of the RHDO method for detecting fetal mutations inherited from at‐risk parents who carry the same types of mutant alleles, which can decrease testing cost and enhance the application range.
2. MATERIALS AND METHODS
2.1. Ethical approval and sample collection
This study was approved by the Ethics Committee of the Third Affiliated Hospital of Guangzhou Medical University (code:2017‐052). Five couples at risk of β‐thalassemia were recruited (shown in Table 1) after appropriate counseling and provided written informed consent. Fifteen milliliters of peripheral blood was collected from each pregnant woman before she underwent prenatal diagnosis, and 5 ml of peripheral blood was collected from each of their husbands. The maternal blood samples were collected at gestational ages from 18+3 weeks to 28+0 weeks (median, 20+4 weeks) in EDTA tubes. Plasma samples were separated from whole blood by centrifugation at 1,600× g for 10 min and then at 16,000× g for 10 min, both at 4°C. Finally, the plasma samples were stored in 2 ml aliquots at −80°C until cffDNA extraction.
Table 1.
Parental genotypes and sequencing data
| Family | Maternal genotype | Paternal genotype | Gestational age | Maternal sequencing reads | Maternal sequencing depth (×) | Paternal sequencing reads | Paternal sequencing depth (×) |
|---|---|---|---|---|---|---|---|
| 1 | IVS‐II‐654/N | CD41‐42/N | 22 + 2 | 3501017 | 634 | 3145580 | 571 |
| 2 | CD17/N | IVS‐II‐654/N | 28 + 0 | 3608357 | 653 | 3608421 | 654 |
| 3 | CD41‐42/N | ‐28/N | 18 + 3 | 3186049 | 583 | 3464331 | 626 |
| 4 | CD41‐42/N | CD41‐42/N | 20 + 4 | 3027789 | 548 | 3580031 | 651 |
| 5 | CD41‐42/N | CD41‐42/N | 20 + 3 | 3890924 | 707 | 3243197 | 589 |
Abbreviations: ‐28, HBB: c.‐78A>G; CD17, HBB: c.52A>T; CD41‐42, HBB: c.126_129delCTTT; IVS‐II‐654, HBB:c.316‐197C>T; N, wild type.
The fetal DNA extracted from the amniocentesis samples was genotyped by the PCR‐based reverse dot blot (RDB) method, which was used to validate the cffDNA results.
2.2. DNA extraction
The cffDNA was extracted from 2 ml of thawed plasma using the Qiagen Circulating Nucleic Acid Kit (Qiagen) according to the manufacturer's instructions. Parental genomic DNA was extracted from buffy coat samples using the Qiagen DNA Mini Kit (Qiagen) following the manufacturer's protocol.
2.3. Probe design
We designed probes covering approximately 800 kb of the target region, which contained the HBB gene and its upstream and downstream regions at human genome position chr11: 4846695‐5648301 (hg19, NCBI37). Other probes targeted to chromosome 16 for other studies were also present in the custom‐designed NimbleGen EZ array (Roche NimbleGen) used in this study. All available probes were used to calculate the concentration of the fetal fraction in the maternal plasma. The probes were obtained from Roche Sequencing (Roche NimbleGen) with biotinylation.
2.4. Targeted capture sequencing
The genomic DNA from the parents was sonicated randomly into fragments of ~200 bp. The size distribution of the sonicated DNA was confirmed using a DNA 1000 Assay on a Bioanalyzer Model 2100 (Agilent), and the concentration was determined using a Qubit double‐stranded DNA High Sensitivity Assay Kit and Qubit fluorometer (Life Technologies). Two micrograms of fragmented DNA and cffDNA were used as inputs for library preparation. All steps were carried out according to the NimbleGen SeqCap EZ HyperCap Workflow User's Guide (Version 1.0), including precapture library synthesis, hybridization to custom‐designed NimbleGen sequence capture probes, washing, recovery, and amplification of captured DNA. Next, the postcapture cffDNA libraries were gel selected to 180–200 bp (Li et al., 2005). Finally, the DNA libraries were quantified by a Qubit fluorometer and Bioanalyzer Model 2100.
The postcapture libraries were sequenced on an Illumina HiSeqTM 2000 (Illumina) according to the manufacturer's recommendations using a paired‐end 150 bp protocol.
2.5. Alignment and calling of SNPs and indels
The raw data were first aligned to the human reference genome (hg19, NCBI37) using the BWA (version 0.7) software with default parameters. SAMtools (version 1.2) was used to convert, sort, and index the alignment “.bam” files. To remove the effects of duplicate reads, we used the Picard (version 1.93) software to mark duplicates and reduce PCR errors. After these steps, SNPs and indels calling was performed using GATK. The presence of genomic DNA mutations was confirmed by visual inspection of the mapped reads in Integrative Genomics Viewer (IGV) (Thorvaldsdottir, Robinson, & Mesirov, 2013).
2.6. Estimation of the fetal fraction concentration
For the captured SNPs on chromosomes 11 and 16, the concentration of the fetal fraction was estimated. SNPs that were homozygous for different alleles in each parent were used to calculate the cffDNA concentration in maternal plasma (f). The formula was , where p indicates the read count of the fetal‐specific allele and q indicates the depth of the other allele, which was shared by the maternal and fetal genomes.
2.7. Construct haplotype information and informative SNP selection
To construct the parental haplotypes without requiring the genotypes of other family members, we selected informative SNPs in the target region. Next, the parental haplotypes were constructed using the SHAPEIT software (O'Connell et al., 2014) from the informative SNPs and reference data of the 1000 Genome Project CHS haplotype dataset.
2.8. Informative SNP category and fetus haplotype deduction
For RHDO analysis, the informative SNPs selected previously were classified into two categories as reported previously (Lo et al., 2010). To deduce the maternally inherited allele, SNPs which are heterozygous in the mother and homozygous in the father were selected first. Then, these selected SNPs were further grouped into two categories. An SNP located in the maternal haplotype I (Hap I) and same as the paternal allele was denoted as a type A SNP. In contrast, an SNP in the maternal haplotype II (Hap II) and identical to the paternal allele was denoted as type B SNPs. If the fetus inherited Hap I, type A SNPs would be examined as an overrepresentation. Otherwise, if the fetus inherited Hap II, alleles on Hap I and Hap II would be equally represented. Similarly, if the fetus inherited Hap II, type B SNPs would be overrepresented. While if Hap I was transmitted to the fetus, alleles on Hap I and Hap II would be equally represented. The haplotypes inherited by the fetus can be deduced by calculating the allelic imbalance using the sequential probability ratio test (SPRT) (El, Zhou, & Whittemore, 2006; Lo et al., 2010). After identifying the haplotype (Hap I or Hap II) inheritance of the fetus, we checked whether the haplotype was mutation located or not, which would then determine the genetic situation of the fetus.
2.9. Data analysis workflow
The analytical pipeline was written in Python version 2.7 in the Linux environment and aimed to efficiently diagnose the fetal inherited disease status using parental DNA and cffDNA. The parental haplotypes were phased using SHAPEIT (Delaneau, Coulonges, & Zagury, 2008), and the RHDO method was subsequently applied (Lo et al., 2010) to predict the maternal haplotype most likely inherited by the fetus, as has been reported previously (Lo et al., 2010).
The workflow analysis consists of the following main steps (Figure 1, with a detailed description in the Supplementary Note): (1) detection of parental SNP loci; (2) classification of cffDNA loci; (3) estimation of fetal fraction concentration; (4) determination of parental haplotypes with SHAPEIT; (5) RHDO prediction of the most likely inherited maternal haplotype; (6) prediction of the most likely inherited paternal mutation; (7) deduction of inherited mutations; and (8) validation.
Figure 1.
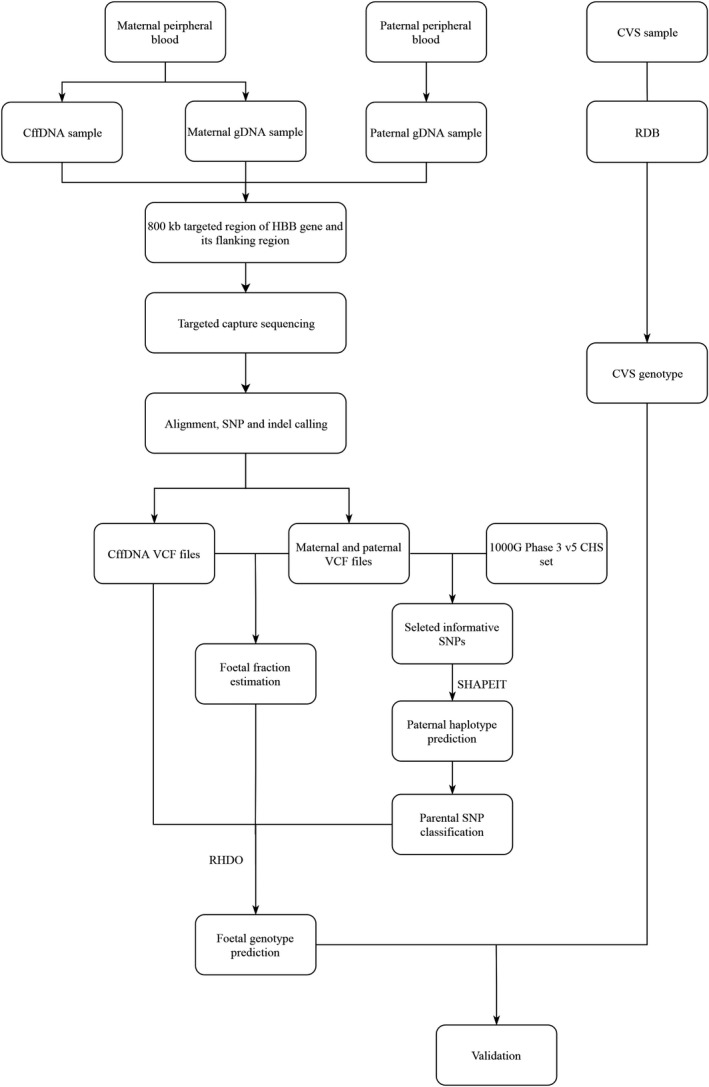
General workflow for β‐thalassemia diagnosis using the traditional method (RBD) and the noninvasive prenatal method. gDNA, genomic DNA; cffDNA, cell‐free fetal DNA; CVS, chorionic villus sampling; RDB, reverse dot blot; RHDO, relative haplotype dosage
3. RESULTS
3.1. General workflow and sample characteristics
The overview workflow is shown in Figure 1. A target region capture strategy was used for DNA sequencing, both for parental genomic DNA and maternal cffDNA. Parental genomic DNA sequencing information was used to construct the parental haplotypes, and maternal DNA sequencing results were applied to predict fetal genotypes. CVS samples were tested by using a reverse dot plot to compare results obtained from the sequence‐based method.
Five couples at risk for β‐thalassemia who attended a clinic for prenatal diagnosis were recruited. The five couples are all carriers of β‐thalassemia mutations, but their mutations were different, except for the two couples from families 4 and 5 (Table 1). Both couples from families 4 and 5 carried the CD41‐42 (HBB:c.126_129delCTTT) mutation. Four common Southeast Chinese point mutations in the β‐thalassemia gene were identified in five couples, which were confirmed by the sequencing data. Figures 2 and 3 show the two mutations carried by the couple in family 1.
Figure 2.

The family 1 maternal mutation in the genomic DNA. Integrative Genomics Viewer (IGV) shows the point mutation (IVS‐II‐654, HBB:c.316‐197C>T) at chromosome 11:52947153, which was G in the reference. The SNP at chromosome 11:5247141 was linked with the mutation in the same haplotype. The green letters in the gray alignments represent the bases that did not match the reference base. M, mutation
Figure 3.

The family 1 paternal mutation in the genomic DNA. Integrative Genomics Viewer (IGV) shows the deletion (CD41‐42, HBB:c.126_129delCTTT) at chromosome 11:52947993 to 52947996, which was AAAG in the reference. The short horizontal black lines in the gray alignments represent 4‐nucleotide deletions. DEL, deletion
3.2. Sequencing data characteristics
Parental genomic DNA and cffDNA were sequenced separately. The sequencing data from the parental genomic samples in the target region produced a mean of 3,425,569.6 reads per sample with a mean depth of 621.6‐fold (Table 1). With a mean sequencing depth of 835.58‐fold, each cffDNA sample produced a mean of 4,603,777 reads (Table 2). The fetal DNA concentration was measured and is listed in Table 2. The fetal fraction ranged from 17% to 34%, with an average of 26.2% (Table 2).
Table 2.
Fetal genotypes and cffDNA sequencing data
| Family | Fetal genotype | SNPs used for RHDO | Fetal fraction (%) | CffDNA sequencing reads | CffDNA sequencing depth (×) | Predicted result | SNPs used for parental haplotype | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Maternal | Paternal | |||||||||
| Type A | Type B | Type A | Type B | |||||||
| 1 | CD41‐42/IVS‐II‐654 | 72 | 8 (6) | — | — | 19 | 3982453 | 727 | Accordance | 134 |
| 2 | CD17/IVS‐II‐654 | 101 | 8 | — | — | 34 | 5881100 | 1067 | Accordance | 198 |
| 3 | ‐28/N | 24 | 12 | — | — | 31 | 4312775 | 776 | Accordance | 127 |
| 4 | CD41‐42/CD41‐42 | 0 | 56 | 21 | 45 (42) | 17 | 4428868 | 807 | Accordance | 146 |
| 5 | CD41‐42/N | 16 | 6 | 24 | 13 | 30 | 4413689 | 802 | Accordance | 127 |
In family 1, six type B SNPs supported the inherence of one haplotype, while two type B SNPs support the inherence of the other haplotype. The same phenomenon was found in family 5, in which three type B SNPs showed a negative result among the 45 type B SNPs used for the paternally inherited haplotype deduction.
Abbreviations: ‐28, HBB: c.‐78A>G; CD17, HBB: c.52A>T; CD41‐42, HBB: c.126_129delCTTT; IVS‐II‐654, HBB:c.316‐197C>T; N, wild type.
3.3. Construction of parental haplotypes
Before deducing the fetal haplotype, the parental haplotypes were first phased. Without the proband genotype, parental haplotypes were constructed using a computational phase method that took advantage of the 1000G database CHS haplotype dataset using the SHAPEIT software. In total, 340 SNPs were selected as a candidate SNP (informative SNPs) set for constructing the parental haplotypes (Supplementary File S1), which contained four SNP categories (detailed in the Methods). Informative SNPs were then used to construct parental haplotypes of each family separately. The parental haplotypes of the five families are presented in Supplementary File S2–S6. The number of informative SNPs used to construct the haplotypes of each couple ranged from 127 to 198, with a median value of 146 (Table 2, Supplementary File S2–S6), among which 28.3% to 83.6% of the applied SNPs were used for fetus haplotype deduction (Table 2).
3.4. Deduction of the fetus haplotype in family 1 to family 3
The parents from families 1 to 3 carry different HBB mutations (Table 1); thus, paternally inherited mutations can be detected by testing the paternal mutation loci in maternal plasma DNA through sequencing reads. We correctly identified the paternal‐inherited mutations in family 1, family 2, and family 3, with a frequency of 4.99%, 9.64%, and 11.85% in the cffDNA sequencing data, respectively, which indicated that all three fetuses inherited their father's mutant locus (Table 3).
Table 3.
The percentage of paternal mutations in maternal plasma DNA and the same loci in maternal genomic DNA
| Family | In maternal genomic DNA (%) | In maternal plasma DNA (%) | Reference readsa | Mutant readsb |
|---|---|---|---|---|
| 1 | 0.11 | 4.99 | 724 | 38 |
| 2 | 0.19 | 9.64 | 703 | 75 |
| 3 | 0 | 11.85 | 632 | 85 |
Reads supporting the reference allele in the maternal plasma DNA sequencing data.
Reads supporting the mutation in the maternal plasma DNA sequencing data.
Maternally inherited mutations were detected through haplotype deduction based on the RHDO strategy (Lo et al., 2010). In family 1, 72 type A SNPs and 6 type B SNPs supported that the fetus inherited one of the two maternal haplotypes, and it was then identified as a mutation‐located haplotype (Table 2). The fetal haplotypes of family 2 and family 3 were deduced by the same RHDO method as family 1. The results showed that the family 2 fetus inherited a maternal mutation‐linked haplotype and the family 3 fetus inherited a maternal wild‐type allele (Table 2).
A problem was encountered when constructing the maternal haplotype of family 1. The mother of family 1 was a carrier of the IVS‐II‐654 (HBB:c.316‐197C>T) mutation (Figure 2). However, there were no reference loci available in the 1000G CHS haplotype dataset, which made it difficult to determine to which haplotype the SNPs belonged. Fortunately, an SNP locus (base A) at chr11: 5247141 was identified upstream of the mutant allele. After data analysis, we inferred that both the chr11: 5247141 loci and the mutation loci were likely to be present in the same haplotype (Figure 2). A total of 119 reads supported the linkage of the base A with the mutant allele in the same haplotype, which included 59 identified reads showing the mutation linked with this SNP and 60 reads consistent with the reference sequence. Therefore, we assumed that the mutant‐linked haplotype contained this SNP and that the SNP could represent the mutation allele.
In families 1, 2, and 3, the fetal genotype was successfully deduced. The fetal genotype obtained from the RHDO‐based method was consistent with CVS–RDB‐based experimental results (Table 2, Figure 4).
Figure 4.
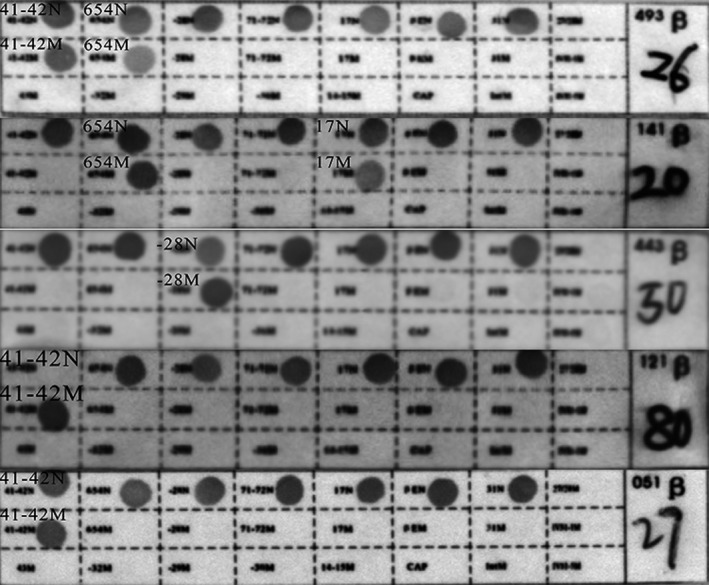
Results for CVS samples via the RBD method. The RBD results for “26,” “20,” “50,” “80,” and “27” represent the fetal genotypes of families 1–5, which were CD41‐42/IVS‐II‐654, CD17/IVS‐II‐654, ‐28/N, CD41‐42/CD41‐42, and CD41‐42/N, respectively. ‐28, HBB: c.‐78A>G; CD17, HBB: c.52A>T; CD41‐42, HBB: c.126_129delCTTT; IVS‐II‐654, HBB:c.316‐197C>T; N, wild type
3.5. Deduction of the fetus haplotype in family 4 and family 5
The parents in family 4 and family 5 carried the same kind of mutation (Table 1). For couples who carried the same mutation, the paternally inherited haplotype was identified using the RHDO strategy. This strategy was the same as the process used to determine the maternal haplotype, except that the SNP definition was different. To deduce paternally inherent haplotype, paternal heterozygous and maternally homozygous SNPs were selected. In family 4, 56 type B SNPs supported the inheritance of the maternal mutant haplotype and 21 type A and 42 type B SNPs supported the inherence of paternal mutant haplotype (Table 2). The family 4 fetus was affected and inherited mutant alleles from the parents. The results showed that the family 5 fetus was a carrier who inherited one mutant allele and one normal allele (Table 2). These deducted results were identical to that of the CVS–RDB‐based results (Table 2, Figure 4).
Finally, in these cases, the test sensitivity, specificity, NPV, PPV, and overall accuracy were calculated (Table 4). All of these cases were identified and identical with the CVS–RDB‐based results for all five cases.
Table 4.
Sensitivity, specificity, NPV, PPV, and accuracy of the five cases
| Percentage (95% CI) | |
|---|---|
| Sensitivity | 100% (0.2924018, 1.0000000) |
| Specificity | 100% (0.1581139, 1.0000000) |
| PPV | 100% (0.2924018, 1.0000000) |
| NPV | 100% (0.1581139, 1.0000000) |
| Accuracy | 100% (0.4781762, 1.0000000) |
Abbreviations: PPV, positive predictive value; NPV, negative predictive value.
4. DISCUSSION
Since the discovery that fetal DNA circulates in the plasma of pregnant women, the application of NIPD has rapidly spread around the world. Detection of the most common chromosome aneuploidies has been shown with high sensitivity, specificity, and positive predictive values, globally revolutionizing prenatal care (Hui & Bianchi, 2017). However, when applied to single gene disorders (SGDs), NIPD tests have been limited by both technical problems and high costs (Parks et al., 2017).
In previous studies, NIPD for SGD has focused on detecting fetus‐specific mutations in maternal plasma, such as paternal mutant alleles, which aimed to eliminate the need for additional diagnostic testing of fetuses who do not inherit the paternal mutation when the couple is carriers of different thalassemia variants (Lam et al., 2012; Xiong et al., 2015). Thus, some at‐risk pregnancies could still not avoid undergoing invasive procedures. Another limitation is related to the haplotype strategy. Generally, an affected proband is necessary to construct parental haplotypes before deducing the fetal haplotype from cffDNA information (New et al., 2014; Parks et al., 2017; Xu et al., 2015), which means the test can be applied only to those families that had a previously affected fetus. This method cannot cover all at‐risk pregnancies and has a high cost.
In the present study, we have explored an efficient protocol based on target region capture sequencing to identify the fetal genotype of β‐thalassemia noninvasively without proband information. Moreover, our method is practical for detecting maternally inherited mutations. We focused on the four most common β‐thalassemia mutations in Southeast China and performed targeted sequencing on two families where the parents were carriers of the same mutation and on three families where the parental alleles differed. Previous studies have shown the successful identification of fetal mutations in cases where the parents shared genetic variants (Barrett, McDonnell, Chan, & Chitty, 2012; Saba et al., 2017). However, in most cases, identifying the maternal mutation inherited by the fetus in the plasma was relatively tricky because of the overwhelming amount of maternal DNA compared to the amount of cffDNA. The RHDO method, which we took advantage of in this research, has paved a way to detect the maternal origin β‐thalassemia allele (Lam et al., 2012; Lo et al., 2010), though parents from the two recruited families carried different kinds of mutations in previous studies.
In this research, we verified the applicability of RHDO using two recruited families in which the parents carried the same mutation (Table 1). The haplotype‐based approach provides adequate information for constructing parental haplotypes and detecting the fetal genotype regarding the disease through qualitative and quantitative analysis of the SNPs in the genomic region of interest.
To construct the parental haplotypes without analyzing other family members to deduce the fetal genotype, we developed a computational haplotype phase strategy. We first selected an approximately 800 kb region containing the HBB gene cluster via targeted sequencing of the DNA for each trio of samples. Then, we selected informative SNPs in the β‐globin gene cluster based on the 1000G CHS haplotype dataset and SHAPEIT2. Informative SNP selection was the most crucial step in the bioinformatic analysis, which was needed to further process the cffDNA data. The informative SNPs selected in this research included four categories that defined in the previous study (Lo et al., 2010), of which three kinds of informative SNPs were used here. Parental homozygous alleles were used to evaluate the fetal DNA fraction. An allele that was paternal homozygous and maternal heterozygous was used to deduce the maternally inherited haplotype, whereas a maternal homozygous and paternal heterozygous allele was applied to deduce the paternally inherited haplotype. Lam et al. used parental heterozygous alleles because the maternal homozygous and paternal heterozygous allele count was insufficient (Lam et al., 2012). Although our study did not have this problem, it is an important point when applying this method to a large cohort study. Additionally, parental heterozygous alleles would be practical in consanguineous families (Lam et al., 2012; Parks et al., 2017).
Furthermore, finding more informative SNPs can be helpful to exclude false positives. In a recent study, Parks et al. developed an effective NIPD strategy for spinal muscular atrophy (SMA) disease in which they selected informative SNPs before target sequencing. They reported that the more informative SNPs, the less fetal fraction DNA was required (Parks et al., 2017). Otherwise, the haplotype‐based approach that we used to construct the parental haplotypes and detect the fetal haplotype is based on the SNP and fetal concentration. Figure 5 shows that there is a significant positive correlation between the fetal concentration and accumulated sequencing depth, which means a lower accumulated sequencing depth depends on the SNP reads for each haplotype if the sample has a higher fetal concentration. In other words, the required minimum number of SNPs is based on the fetal concentration (Lo et al., 2010). In our research, the fetal fraction, upon which the number of SNPs depends, is relatively high for NIPD, which may result from the long pregnancy weeks for the recruited samples. The earlier that clinical screening can be performed, the better. Thus, different kinds of samples, such as low gestational weeks, should be studied before clinical practice.
Figure 5.
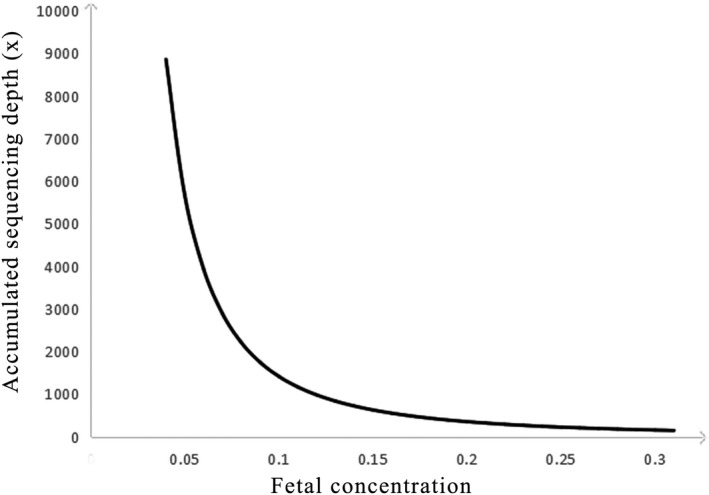
The correlation between the fetal concentration and the accumulated sequencing depth
To avoid relying on proband information, we used a computational haplotype phase method that took advantage of the 1000G CHS haplotype dataset. However, the IVS‐II‐654 (HBB: c.316‐197C>T) mutation is not found in the 1000G CHS haplotype dataset; consequently, we could not distinguish the wild‐type‐linked and mutant‐linked haplotypes using this technique. Fortunately, 119 reads in the sequencing data BAM files indicated that an SNP allele (base A) was located 12 bp upstream in linkage with the mutant allele, which means that we could identify the mutant haplotype using this SNP allele. However, in cases where no available SNP is linked with the mutant allele of interest, such as ‐28A>G (HBB: c.‐78A>G), the fetal inheritance of the maternal mutant‐linked haplotype cannot be determined. This case represents a limitation of this approach because the 1000G CHS haplotype dataset does not contain these mutations as reference alleles. Therefore, additional sequencing data from the Chinese population are still needed to supplement the data in the 1000 Genomes Project before our method can achieve complete and ideal predictive and practical utility. Moreover, the experimental method is also usable for haplotype phasing without relying on proband information. Lam et al. used digital PCR to successfully construct parental haplotypes, but this method was costly, and the experimental design was complicated, which were additional limitations (Lam et al., 2012). Computational and experimental methods are two main haplotype phasing strategies (Browning & Browning, 2011). Despite the pros and cons of these two methods, abundant population‐based haplotype repositories are required.
The parents in family 5 are both CD41‐42 (HBB: c.126_129delCTTT) carriers, and the RHDO results showed that the fetus inherited one mutant allele from the paternal mutation‐located haplotype. The shortcomings of this research are that we have no other evidence to confirm this result. Thus, it would be better to recruit families with proband information in further studies.
Furthermore, inconsistent results were found in family 1 and family 4. In family 1, six type B SNPs supported the inheritance of one haplotype, while only two type B SNPs supported the inherence of the other haplotype. The same phenomenon was observed in family 5, in which three type B SNPs showed negative results among the 45 type B SNPs used to deduce the paternally inherited haplotype. We speculated that a recombination event might have occurred. When meiotic recombination has rearranged the target region inside this relatively small segment, an inconclusive result would be obtained rather than a false deduction. Another possible explanation is the presence of an unexpected SNP imbalance at some sites sequenced in the cffDNA samples could cause the false identification of the inherited maternal haplotype. If the NIPD result is inconclusive in such a situation, the couple must undergo traditional diagnosis by CVS or amniocentesis. Parks et al. also mentioned that recombination could influence the test accuracy and efficiency, primarily because if it occurs in highly repeated sequences. Selecting haplotype block loci at downstream or upstream of the target region is a suitable solution (Parks et al., 2017), which was confirmed in this research.
In conclusion, we have developed a strategy for NIPD of β‐thalassemia in at‐risk couples without proband information that shows high sensitivity, specificity, NPV, PPV, and accuracy. The haplotype‐based approach provided a new idea for the field of NIPD for SGD, and it improves the detection of both parental haplotypes as inherited by the fetus. However, this method requires more improvements to increase its practicality, and additional β‐thalassemia variants and many more samples must be tested in the future. Overall, our results have proven that NIPD can be used in couples who carry the four most common β‐thalassemia mutations in Southeast China, including those who share the same mutation.
CONFLICT OF INTEREST
The authors have declared that they have no conflicts of interest related to this work.
Supporting information
ACKNOWLEDGMENTS
This work was supported in part by the Guangdong Province Science and Technology Project under grant number 2016B030229008, Project of Science and Technology of Guangzhou City under grant number 201400000004‐4, 201400000003‐4, the Science and Information Technology of Guangzhou Key Project under grant number 201508020258, the Foundation of Guangzhou Science and Information Technology of Guangzhou Key Project under grant number 201803040009, and the National Natural Science Foundation of China under grant number 81270745. The biobank of the Third Affiliated Hospital of Guangzhou Medical University provided the CVS samples.
Li H, Du B, Jiang F, et al. Noninvasive prenatal diagnosis of β‐thalassemia by relative haplotype dosage without analyzing proband. Mol Genet Genomic Med. 2019;7:e963 10.1002/mgg3.963
Contributor Information
Weiqiang Liu, Email: liuweiqiang@gzhmu.edu.cn.
Xiaofang Sun, Email: xiaofangsun@gzhmu.edu.cn.
REFERENCES
- Akolekar, R. , Beta, J. , Picciarelli, G. , Ogilvie, C. , & D'Antonio, F. (2015). Procedure‐related risk of miscarriage following amniocentesis and chorionic villus sampling: A systematic review and meta‐analysis. Ultrasound in Obstetrics and Gynecology, 45(1), 16–26. 10.1002/uog.14636 [DOI] [PubMed] [Google Scholar]
- Amicucci, P. , Gennarelli, M. , Novelli, G. , & Dallapiccola, B. (2000). Prenatal diagnosis of myotonic dystrophy using fetal DNA obtained from maternal plasma. Clinical Chemistry, 46(2), 301–302. [PubMed] [Google Scholar]
- Barrett, A. N. , McDonnell, T. C. , Chan, K. C. , & Chitty, L. S. (2012). Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clinical Chemistry, 58(6), 1026–1032. 10.1373/clinchem.2011.178939 [DOI] [PubMed] [Google Scholar]
- Browning, S. R. , & Browning, B. L. (2011). Haplotype phasing: Existing methods and new developments. Nature Reviews Genetics, 12(10), 703–714. 10.1038/nrg3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante‐Aragones, A. , Gallego‐Merlo, J. , Trujillo‐Tiebas, M. J. , de Alba, M. R. , Gonzalez‐Gonzalez, C. , Glover, G. , … Ramos, C. (2008). New strategy for the prenatal detection/exclusion of paternal cystic fibrosis mutations in maternal plasma. Journal of Cystic Fibrosis, 7(6), 505–510. 10.1016/j.jcf.2008.05.006 [DOI] [PubMed] [Google Scholar]
- Chitty, L. S. , Griffin, D. R. , Meaney, C. , Barrett, A. , Khalil, A. , Pajkrt, E. , & Cole, T. J. (2011). New aids for the non‐invasive prenatal diagnosis of achondroplasia: Dysmorphic features, charts of fetal size and molecular confirmation using cell‐free fetal DNA in maternal plasma. Ultrasound in Obstetrics and Gynecology, 37(3), 283–289. 10.1002/uog.8893 [DOI] [PubMed] [Google Scholar]
- Chitty, L. S. , & Lo, Y. M. (2015). Noninvasive prenatal screening for genetic diseases using massively parallel sequencing of maternal plasma DNA. Cold Spring Harbor Perspectives in Medicine, 5(9), a23085 10.1101/cshperspect.a023085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, R. W. K. , Chan, K. C. A. , Gao, Y. , Lau, V. Y. M. , Zheng, W. , Leung, T. Y. , … Lo, Y. M. D. (2008). Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proceedings of the National Academy of Sciences of the United States of America, 105(51), 20458–20463. 10.1073/pnas.0810641105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, R. W. , Lau, T. K. , Cheung, P. T. , Gong, Z. Q. , Leung, T. N. , & Lo, Y. M. (2002). Noninvasive prenatal exclusion of congenital adrenal hyperplasia by maternal plasma analysis: A feasibility study. Clinical Chemistry, 48(5), 778–780. [PubMed] [Google Scholar]
- Daley, R. , Hill, M. , & Chitty, L. S. (2014). Non‐invasive prenatal diagnosis: Progress and potential. Archives of Disease in Childhood. Fetal and Neonatal Edition, 99(5), F426–F430. 10.1136/archdischild-2013-304828 [DOI] [PubMed] [Google Scholar]
- Delaneau, O. , Coulonges, C. , & Zagury, J. F. (2008). Shape‐IT: New rapid and accurate algorithm for haplotype inference. BMC Bioinformatics, 9, 540 10.1186/1471-2105-9-540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El, K. N. , Zhou, W. , & Whittemore, A. S. (2006). Getting more from digital SNP data. Statistics in Medicine, 25(18), 3124–3133. 10.1002/sim.2379 [DOI] [PubMed] [Google Scholar]
- Giardine, B. , Borg, J. , Viennas, E. , Pavlidis, C. , Moradkhani, K. , Joly, P. , … Patrinos, G. P. (2014). Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Research, 42(D1), D1063–D1069. 10.1093/nar/gkt911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil, M. M. , Accurti, V. , Santacruz, B. , Plana, M. N. , & Nicolaides, K. H. (2017). Analysis of cell‐free DNA in maternal blood in screening for aneuploidies: Updated meta‐analysis. Ultrasound in Obstetrics and Gynecology, 50(3), 302–314. 10.1002/uog.17484 [DOI] [PubMed] [Google Scholar]
- González‐González, M. C. , Garcia‐Hoyos, M. , Trujillo‐Tiebas, M. J. , Bustamante Aragonés, A. , Rodriguez de Alba, M. , Alvarez, D. D. , … Ramos, C. (2008). Improvement in strategies for the non‐invasive prenatal diagnosis of Huntington disease. Journal of Assisted Reproduction and Genetics, 25(9–10), 477–481. 10.1007/s10815-008-9256-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J. , Pan, M. , Zhen, L. , Yang, X. , Ou, Y.‐M. , Liao, C. , & Li, D.‐Z. (2014). Chorionic villus sampling for early prenatal diagnosis: Experience at a mainland Chinese hospital. Journal of Obstetrics and Gynaecology, 34(8), 669–672. 10.3109/01443615.2014.920793 [DOI] [PubMed] [Google Scholar]
- Higgs, D. R. , Engel, J. D. , & Stamatoyannopoulos, G. (2012). Thalassaemia. Lancet, 379(9813), 373–383. 10.1016/S0140-6736(11)60283-3 [DOI] [PubMed] [Google Scholar]
- Hui, L. , & Bianchi, D. W. (2017). Noninvasive prenatal DNA testing: The vanguard of genomic medicine. Annual Review of Medicine, 68, 459–472. 10.1146/annurev-med-072115-033220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, K. , Huang, G. , Su, L. , & He, Y. (2017). The prevalence of thalassemia in mainland China: Evidence from epidemiological surveys. Scientific Reports, 7(1), 920 10.1038/s41598-017-00967-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, K. W. , Jiang, P. , Liao, G. J. , Chan, K. C. , Leung, T. Y. , Chiu, R. W. , & Lo, Y. M. (2012). Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma: Application to beta‐thalassemia. Clinical Chemistry, 58(10), 1467–1475. [DOI] [PubMed] [Google Scholar]
- Li, B. , Zhang, X.‐Z. , Yin, A.‐H. , Zhao, Q.‐G. , Wu, L. I. , Ma, Y.‐Z. , … Yu, S.‐Y. (2014). High prevalence of thalassemia in migrant populations in Guangdong Province, China. BMC Public Health, 14, 905 10.1186/1471-2458-14-905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Di Naro, E. , Vitucci, A. , Zimmermann, B. , Holzgreve, W. , & Hahn, S. (2005). Detection of paternally inherited fetal point mutations for beta‐thalassemia using size‐fractionated cell‐free DNA in maternal plasma. JAMA, 293(7), 843–849. [DOI] [PubMed] [Google Scholar]
- Lin, M. , Zhong, T.‐Y. , Chen, Y.‐G. , Wang, J.‐Z. , Wu, J.‐R. , Lin, F. , … Yang, L.‐Y. (2014). Molecular epidemiological characterization and health burden of thalassemia in Jiangxi Province, P. R. China. PLoS ONE, 9(7), e101505 10.1371/journal.pone.0101505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, Y. M. D. , Chan, K. C. A. , Sun, H. , Chen, E. Z. , Jiang, P. , Lun, F. M. F. , … Chiu, R. W. K. (2010). Maternal plasma DNA sequencing reveals the genome‐wide genetic and mutational profile of the fetus. Science Translational Medicine, 2(61), 61r–91r. 10.1126/scitranslmed.3001720 [DOI] [PubMed] [Google Scholar]
- Lo, Y. M. D. , Corbetta, N. , Chamberlain, P. F. , Rai, V. , Sargent, I. L. , Redman, C. W. , & Wainscoat, J. S. (1997). Presence of fetal DNA in maternal plasma and serum. The Lancet, 350(9076), 485–487. 10.1016/s0140-6736(97)02174-0 [DOI] [PubMed] [Google Scholar]
- New, M. I. , Tong, Y. K. , Yuen, T. , Jiang, P. , Pina, C. , Chan, K. C. A. , … Lo, Y. M. (2014). Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell‐free fetal DNA in maternal plasma. Journal of Clinical Endocrinology and Metabolism, 99(6), E1022–E1030. 10.1210/jc.2014-1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell, J. , Gurdasani, D. , Delaneau, O. , Pirastu, N. , Ulivi, S. , Cocca, M. , … Marchini, J. (2014). A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genetics, 10(4), e1004234 10.1371/journal.pgen.1004234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, M. , Court, S. , Bowns, B. , Cleary, S. , Clokie, S. , Hewitt, J. , … Allen, S. (2017). Non‐invasive prenatal diagnosis of spinal muscular atrophy by relative haplotype dosage. European Journal of Human Genetics, 25(4), 416–422. 10.1038/ejhg.2016.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rund, D. , & Rachmilewitz, E. (2005). Beta‐thalassemia. New England Journal of Medicine, 353(11), 1135–1146. 10.1056/nejmra050436 [DOI] [PubMed] [Google Scholar]
- Saba, L. , Masala, M. , Capponi, V. , Marceddu, G. , Massidda, M. , & Rosatelli, M. C. (2017). Non‐invasive prenatal diagnosis of beta‐thalassemia by semiconductor sequencing: A feasibility study in the sardinian population. European Journal of Human Genetics, 25(5), 600–607. 10.1038/ejhg.2017.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir, H. , Robinson, J. T. , & Mesirov, J. P. (2013). Integrative genomics viewer (IGV): High‐performance genomics data visualization and exploration. Briefings in Bioinformatics, 14(2), 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen, C. , Geeven, G. , de Wit, E. , Verstegen, M. J. A. M. , Jansen, R. P. M. , van Kranenburg, M. , … de Laat, W. (2017). Sensitive monogenic noninvasive prenatal diagnosis by targeted haplotyping. American Journal of Human Genetics, 101(3), 326–339. 10.1016/j.ajhg.2017.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, A. I. , & Lo, Y. M. (2015). Noninvasive fetal genomic, methylomic, and transcriptomic analyses using maternal plasma and clinical implications. Trends in Molecular Medicine, 21(2), 98–108. 10.1016/j.molmed.2014.12.006 [DOI] [PubMed] [Google Scholar]
- Xiong, L. , Barrett, A. N. , Hua, R. , Tan, T. Z. , Ho, S. S. , Chan, J. K. , … Choolani, M. (2015). Non‐invasive prenatal diagnostic testing for beta‐thalassaemia using cell‐free fetal DNA and next generation sequencing. Prenatal Diagnosis, 35(3), 258–265. 10.1002/pd.4536 [DOI] [PubMed] [Google Scholar]
- Xu, Y. , Li, X. , Ge, H.‐J. , Xiao, B. , Zhang, Y.‐Y. , Ying, X.‐M. , … Ji, X. (2015). Haplotype‐based approach for noninvasive prenatal tests of Duchenne muscular dystrophy using cell‐free fetal DNA in maternal plasma. Genetics in Medicine, 17(11), 889–896. 10.1038/gim.2014.207 [DOI] [PubMed] [Google Scholar]
- Yin, A. , Li, B. , Luo, M. , Xu, L. , Wu, L. , Zhang, L. , et al. (2014). The prevalence and molecular spectrum of alpha‐ and beta‐globin gene mutations in 14,332 families of Guangdong Province, China. PLoS ONE, 9(2), e89855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafari, M. , Kosaryan, M. , Gill, P. , Alipour, A. , Shiran, M. , Jalalli, H. , … Fatahi, F. (2016). Non‐invasive prenatal diagnosis of beta‐thalassemia by detection of the cell‐free fetal DNA in maternal circulation: A systematic review and meta‐analysis. Annals of Hematology, 95(8), 1341–1350. 10.1038/gim.2014.207 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials