

Abstract
Background
Pediatric myelodysplastic syndromes (MDS) display clonal genomic instability that can lead to acquisition of other hematological disorders, usually by loss of heterozygosity. Immunodeficiency caused by uniparental disomy (UPD) has not previously been reported.
Methods
We investigated a 13‐year‐old boy who suffered from recurrent infections and pancytopenia for 1 year. Both the comet assay and chromosome breakage analysis were normal, but the bone marrow showed evidence of dysplasia characteristic of MDS. With his normal sister as donor, he underwent failed hematopoietic stem cell transplantation (HSCT) with reduced intensity conditioning (RIC) followed by successful HSCT with myeloablative conditioning (MAC). We used single nucleotide polymorphism (SNP) array, targeted gene panel, and whole exome sequencing to investigate the etiology of his disease.
Results
The molecular analyses revealed multiple regions of homozygosity, one region encompassing a homozygous missense variant of recombination activating gene 1 (RAG1) which was previously associated with severe immunodeficiency in infancy. This RAG1 mutation was heterozygous in the proband’s fingernail DNA, but was changed to homozygous in the proband’s marrow by somatic acquisition of UPD event. No other pathogenic driver mutation for MDS‐related genes was identified.
Conclusion
The hematological phenotype, somatic genomic instability, and response to HSCT MAC but not HSCT RIC deduced to a diagnosis of MDS type refractory cytopenia of children in this patient. His immunodeficiency was secondary to MDS due to somatic acquisition of homozygosity for known pathogenic RAG1 mutation.
Keywords: acquired UPD, immunodeficiency, myelodysplastic syndrome, RAG1
1. INTRODUCTION
Myelodysplastic syndromes (MDS:OMIM # 614,286) are a heterogeneous group of hematological conditions characterized by abnormal differentiation and maturation of myeloid cells, bone marrow failure, and genetic instability with enhanced risk to transform to acute myeloid leukemia (Greenberg et al., 2017). In children, MDS is rare and more related to inherited genetic bone marrow failure than adult MDS. Previous surveys have showed the incidence of inherited bone marrow failure is 1.8% in children with MDS, and 13.6% in children and young adult with MDS (Alabbas et al., 2016; Keel et al., 2016). It is sometimes difficult to diagnose the pediatric MDS in question, especially in the absence of typical clinical characteristics or definitive cytogenetic and/or molecular changes. Here, we used multiple genomic techniques including single nucleotide polymorphism (SNP) array, targeted gene panel sequencing, and whole exome sequencing to investigate the unusual coincidence of MDS and late‐onset immunodeficiency in a 13‐year‐old product of a consanguineous mating. We integrated these molecular genetic analyses with detailed clinical phenotype and the patient’s response to treatments to conclude that MDS‐associated genomic instability produced an acquired uniparental disomy (UPD) region that somatically reduced heterozygous pathogenic variant of recombination activating gene 1 (RAG1: OMIM# 179,615; NM_000448.2) to homozygosity. This case report demonstrates that in addition to hematological disorders, MDS progression via somatic UPD can lead to a late‐onset immunological disorder.
2. MATERIAL AND METHODS
2.1. Ethical compliance
This study was performed in accordance with the Declaration of Helsinki and approved by the ethics committee of Capital Institute of Pediatrics. Written informed consent was obtained from the patient’s parents for the publication of clinical information and accompanying images.
2.2. SNP array analysis, targeted gene panel sequencing, and whole exome sequencing
Genomic DNAs were extracted from the blood or bone marrow using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) or from the fingernail using a standard Phenol/Chloroform method. SNP chip (Affymetrix, CytoScan 750) was used to detect genomic copy number variants, UPD or loss of heterozygosity (LOH).
Two targeted gene sequencing panels (290 genes for congenital hematological disease and 202 genes for immunological diseases, covering all exons and known intronic alleles with high coverage) were performed for DNA extracted from his bone marrow (Table S1) (Itan & Casanova, 2015; Keel et al., 2016; Muramatsu et al., 2017). Whole exome sequencing was performed only for DNA extracted from his fingernail after hematopoietic stem cell transplantation (HSCT) when his remained DNA from target sequencing is not enough and his parents refused to donate dermal fibroblasts. DNA were captured and amplified for either target genes or whole exome using a commercial enrichment kit (SureSelect, V6 kit, Agilent Technologies Inc., Cedar Creek, TX), and sequenced by 100 bp paired‐end reads on the Illumina (Hiseq X10) platform (Illumina, San Diego, California). Raw image files were processed by the Illumina Pipeline for base calling using default parameters.
Raw data were mapped to the reference human genome version hg19 (200,902 release, http://genome.ucsc.edu/), and variants were visualized using NextGENe software version 2.1.1.1 (SoftGenetics, State College, PA) and analyzed using Ingenuity Variant Analysis software (http://www.ingenuity.com). Single nucleotide variants (SNVs) with minor allele frequency (MAF) >1% in dbSNP (http://www.ncbi.nlm.nih.gov/snp), the 1,000 Genomes dataset (http://browser.1000genomes.org/index.html), or the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) were not recognized as rare SNVs. The Exome Aggregation Consortium database (ExAC, http://exac.broadinstitute.org) was used to confirm the novelty of rare SNVs.
2.3. Validation of the RAG1 mutation using Sanger and amplicon‐based deep sequencing
For Sanger sequencing, two unique Polymerase Chain Reaction (PCR) products (514 and 690bp) containing the RAG1 variant (Table S2) were amplified using standard touch‐down PCR. For amplicon‐based deep sequencing, two short unique amplicons (182 and 214bp) containing the RAG1 variant were generated and sequenced on an Ion Torrent Personal Genome Machine (Life Technologies, Carlsbad, CA) using our described method (Jiang et al., 2017). Amplicon‐based deep sequencing was performed for both proband’s bone marrow and fingernail, or for family member’s blood.
3. RESULTS
3.1. Clinical report
The proband was a 13‐year‐old boy who had suffered from severe and recurrent infections in multiple systems for one year, including pneumonia, stomatitis, perianal infection, bloodstream infection, septic shock, posthitis, and gingivitis. He was the product of the fourth pregnancy of first‐cousin parents whose previous pregnancies had produced two healthy girls and a hydatidiform mole. The proband received regular immunizations from birth without side effects or complications. No severe fever or infections occurred prior to age 12 years. At 11 years of age, he had a transcatheter closure operation for an atrial septal defect. No environmental risk factors, such as human immunodeficiency virus or drugs were recorded in his medical history but prior investigations from other hospitals showed positivity for Epstein‐Barr virus, cytomegalovirus, and Pseudomonas aeruginosa.
Physical examination confirmed short stature (weight was 24 kg and height was 123 cm, both <3rd percentile for boys of his age), pallor and mild hepatosplenomegaly. According to the patient’s medical record and parental recall, he had a normal neurodevelopmental trajectory and learned normally in school. An special finger morphology was noted (Figure S1). No other skeletal or organ malformations/dysmorphisms were found. Routine blood tests revealed pancytopenia, with neutropenia and lymphocytopenia being particularly severe (neutrophils: 0.07–0.13 × 109/L, lymphocytes: 0.1–1.0 × 109/L, hemoglobin: 80–105 g/L, and platelet count: 49–150 × 109/L). Rheumatoid factor, Coomb’s test, anti‐cardiolipin antibody, antinuclear antibody, and double‐stranded DNA were all negative. The absolute numbers of T cells, B cells, and NK cells, and the subsets of T cells (CD4+, CD8+) were extremely low (Table 1). Immunological function testing was normal except for Ig A (IgA:0.2g/L, IgM:1.12g/L, IgG:9.35g/L, IgG1:14g/L, IgG2:2.8g/L, IgG3:0.153g/L, IgG4:0.115g/L). Bone marrow morphology showed dysplastic abnormalities without excessive blast cells but with hypoplasia of the granulocyte series (16% granulocyte lineage) (Figure 1). Flow cytometry detected abnormal bone marrow granulocyte differentiation (CD13+/CD16+, CD34+/CD117+) and increased myeloid precursor cells with abnormal expression of CD34+/CD117+/CD13+/CD33+/CD38+ (Figure 2). The percentage of erythroid cells and granulocytes in peripheral blood expressing CD55+and CD59+ was normal. The karyotyping analysis of bone marrow was normal. A comet assay and chromosome breakage analysis were performed on peripheral blood lymphocytes during his 1st hospitalization and recorded as normal. In summary, his clinical manifestations were highly suggestive of immunodeficiency of late‐onset since his severe lymphocytopenia occurred in the absence of any recurrent fever or severe infections prior to age 12. Meanwhile, both morphology and flow cytometry analysis of the bone marrow showed evidence of dysplasia, which is an important characteristic of MDS.
Table 1.
Lymphocyte subsets in the patient’s peripheral blood samples
| Type of lymphocytes | Absolute value (×109/L) | Normal ranges (×109/L) |
|---|---|---|
| T cell/ CD3+ | 0.26 | 1.57–3.72 |
| CD4+ T cell/CD4+ | 0.1 | 0.391–1.421 |
| CD8+T cell/CD8+ | 0.08 | 0.366–1.091 |
| B cell/CD19+ | 0.0066 | 0.141–0.534 |
| NK cell/CD16+/CD56+ | 0.05 | 0.121–1.064 |
Figure 1.
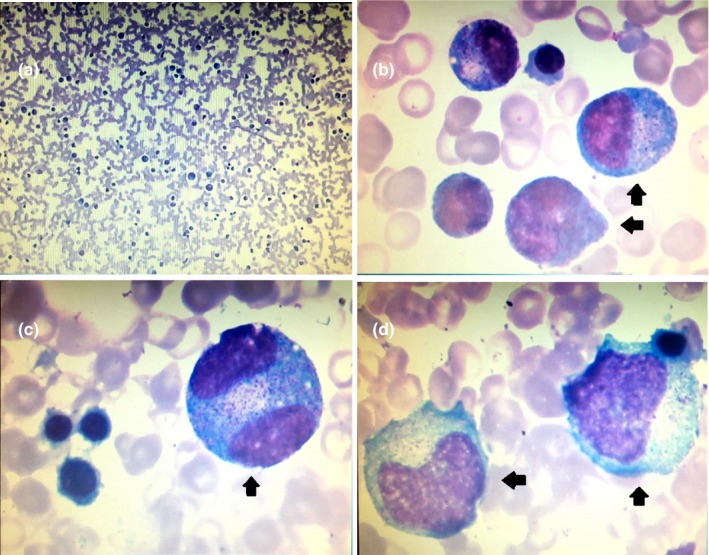
Bone marrow aspiration smear of the patient (Wright‐Giemsa stain). Panel a shows granulocyte series hypoplasia with slightly increasing proportion of myeloblasts and neutrophilic promyelocyte and scarity of bands and segmented neutrophils. Panels b, c and d show abnormal promyelocyte with immature nuclear and aging cytoplasm (arrowhead). The promyelocyte in Panel c is binuclear (arrowhead). Bone marrow aspiration smear showed the percentages of different lineage: 2% myeloblasts, 5% promyelocytes, 6.5% myelocytes, 1.5% metamyelocytes, 0.5% bands, 1% segmented neutrophils, 47.5% lymphocytes, 0.5% normoblasts, 6% rubricytes, 22.5% metarubricytes, and 2.5% monocytes
Figure 2.
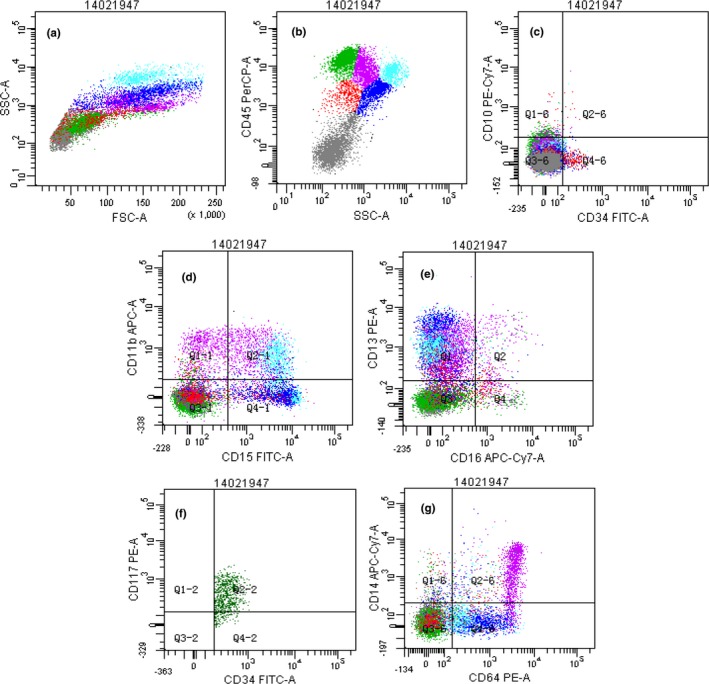
Immunophenotyping of patient’s bone marrow sample by flow cytometry. Panel a shows a global overview of bone marrow cellular compartments projected on SSC graphs, including lymphocyte (green, 25.0%), monocyte (purple, 15.1%), granulocyte (blue, 14.4%), eosinophil (light blue, 8.0%), blast cell or pre‐B cell (red, 4.7%) and erythroblast (gray, 32.8%). The myeloid cells had reduced CD45 and SSC expression (Panel b), and also lacked the expression of CD10, CD11b and CD16 (Panels c, d, e). The myeloid cells expressing CD13+/CD16+ showed an abnormal distribution pattern, indicating irregular myeloid differentiation (Panel e). The myeloblasts with expression of CD34 and CD117 (CD34+/CD117+) accounted for approximate 1% of all the nucleated cells observed, indicating a slight increase of the ratio (Panel f). The monocytes accounted for approximate 15.1% of all the nucleated cells indicative of an increase of the ratio and did not express significant abnormality (Panel g). The fluorochrome‐conjugated monoclonal antibodies (PerCP, FITC, PE‐Cy7, PE, APC‐Cy7 and APC) were used to stain the following antigens: HLA‐DR, CD2, CD3, CD4, CD5, CD7, CD8, CD10, CD11b, CD13, CD14, CD15, CD16, CD19, CD20, CD22, CD33, CD34, CD38, CD56, CD64, CD71, CD117, Kappa, Lambda, MPO, TdT, cCD3, cIgM and CD45
The patient underwent salvage HSCT due to uncontrolled infections. No matched unrelated donor was found, so his healthy eldest sister, who had a normal karyotype, was chosen to be his human leukocyte antigen (HLA)‐haploidentical donor. We performed a physical examination, bone marrow aspiration, SNP array analysis and genetic counselling for his sister. Her evaluation indicated a normal, healthy donor. In view of his severe immunodeficiency, we chose a reduced intensity conditioning (RIC) regimen including cyclophosphamide (CTX) 140 mg/kg, cytarabine 4 g/m2, fludarabine 150 mg/m2, and rabbit anti‐human thymocyte globulin (ATG) 10 mg/kg. Prophylaxis for Graft versus Host Disease (GvHD) included mycophenolate mofetil, tacrolimus and short course methotrexate. The number of infused mononuclear cells and CD34+ cells were 13.23 × 108/kg and 2.86 × 106/kg, respectively. Unfortunately, the first transplantation ended with graft failure. Then, the patient underwent a second HSCT from the same donor. This time a myeloablative conditioning regimen (MAC) including cytarabine 4 g/m2, busulfan 12 mg/kg, CTX 3.6 g/m2, and semustine 250 mg/m2 was pursued. He had delayed neutrophil and platelet engraftment (Day + 29 and Day + 30, respectively), and mixed chimerism with 50% donor cells on Day + 30. After four infusions of donor Granulocyte Colony Stimulating Factor (G‐CSF) stimulated lymphocytes, the patient has achieved sustained complete donor chimerism for nearly 3 years and returned to normal (hemoglobin:13.8 g/dl; platelet count: 119,000/µl; white blood cell count: 7200/µl; neutrophil count: 4400/µl; lymphocyte count: 2100/µl).
3.2. Genetic analysis
SNP array analysis permits a genome‐wide assessment of large regions of homozygosity (ROH), which can result from three potential causes: (a) identity by descent (IBD) in a consanguineous family; (b) constitutional UPD due to a meiotic mistake in the germline or to an early mitotic error after fertilization; and (c) somatically acquired UPD (aUPD) in deleterious cancer cells (Makishima & Maciejewski, 2011). Extensive genome‐wide ROH implies consanguinity or aUPD in malignancy while ROH limited to a single chromosome or region implies a meiotic/mitotic mistake in a germ/zygote cell, so information such as pedigree analysis, disease history and onset age can be useful in distinguishing different sources of ROH. For example, ROH due to consanguinity or meiotic mistakes in germ cells often produce disease at birth or early in life while aUPD is most often seen at an older age or along with cancer progression. The SNP array analysis from the proband’s bone marrow before HSCT detected extensive ROH (369Mb/2850Mb = 12.9%) throughout the autosomes (1p, 1q, 2p, 3q, 4p, 4q, 6p, 7q, 8q, 9p, 9q, 11p, 11q, 12q, and 13q) Table 2) (Gondek et al., 2008). No pathogenic copy number variant was identified. Since the proband was the product of a consanguineous mating, we presumed that the extensive ROH represented IBD and therefore initially sought an MDS‐associated mutation in these regions by targeted gene panel sequencing.
Table 2.
ROH regions and copy number variant (CNV) in the patient identified from SNP chip
| Chromosome abnormalities | Size (kb) | Significance |
|---|---|---|
| ROH (1p35.2‐pter) | 30,576 | These regions are associated with hematologic malignancies |
| ROH (6p22.3‐p21.2) | 20,445 | |
| ROH (11p15.3‐p11.12) | 39,506 | |
| ROH (11q11‐q24.2) | 70,522 | |
| ROH (12q14.1‐q23.1) | 38,426 | |
| ROH (3q25.2‐q27.1) | 30,577 | These regions are possibly associated with hematologic malignancies |
| ROH (7q11.21‐q31.32) | 58,588 | |
| ROH (13q33.1‐qter) | 13,391 | |
| Gain (14q32.33) | 634 | CNV polymorphism |
| ROH (1q32.1‐q32.2) | 6,655 | Significance of the variations are unknown |
| ROH (2p23.3‐p22.3) | 5,796 | |
| ROH (2p25.3‐p25.1) | 6,277 | |
| ROH (4p16.3‐p16.1) | 5,166 | |
| ROH (4q13.3‐q21.23) | 13,119 | |
| ROH (8q24.22‐q24.23) | 8,055 | |
| ROH (9p21.3‐p13.1) | 16,999 | |
| ROH (9q21.11‐q21.13) | 5,802 |
Mean sequence coverage across the targeted gene panel was 300×. No potentially pathogenic variant in any known inherited hematological disease gene was seen, except a homozygous missense mutation (rs199474676, chr11:36,596,949, C>T, c.2095C>T, p. Arg699Trp, R699W, UCSC version hg19) in exon 2 of RAG1 (Recombination activating gene 1, NM_000448.2). No pathogenic mutation for RPS genes of Diamond‐Blackfan anemia (RPS10, PRS17, RPS19, RPS24, RPS26, RPS7, RPL5, RPL11, RPL35a) was detected. The R699W mutation of RAG1 is a very low‐MAF variant (T = 0.00002). This variant is absent in the ExAC Asian population (exac.broadinstitute.org/), and is predicted to be deleterious by two widely‐used analysis software programs (SIFT = 0.00, PolyPhen = 0.99). It has been reported previously as causing with severe combined immunodeficiency (SCID) or Omenn syndrome (OS) in Western and Chinese patients (Avila et al., 2010; Lee et al., 2014; Reiff et al., 2013; Zhang et al., 2011). According to the standards and guidelines for the interpretation of sequence variants from the American College of Medical Genetics (Richards et al., 2015), this variant was classified as pathogenic (PS1 + PM2+PM3 + PP3+PP4). The proband’s parents and donor sister were all heterozygous carriers of this variant (Figure 3). Mean coverage across whole exome sequencing was generally 100× with 97% achieving > 20× coverage. Some candidate genetic variants were chosen for Sanger sequencing and inheritance analysis in family members. Like the results of the targeted gene panel, no pathogenic variant except the heterozygous RAG1 mutation was seen in the whole exome sequencing that could explain the proband’s pancytopenia, bone marrow failure, or short stature. None of any pathogenic mutation in series of RPS genes was identified from this exome sequencing.
Figure 3.
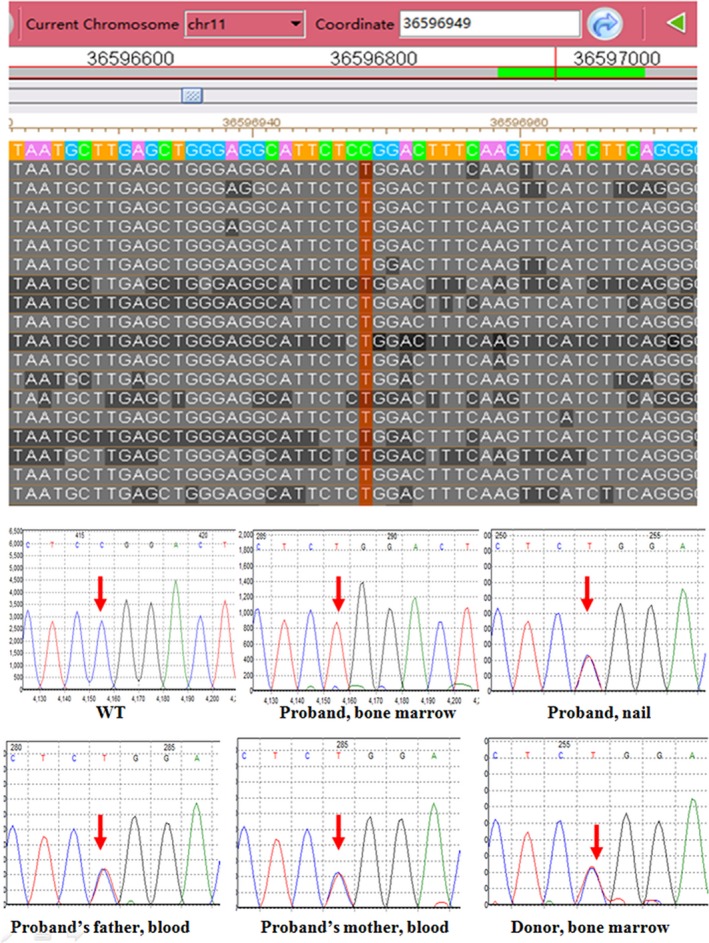
Sequencing of RAG1 from patient's bone marrow and nail sample, his family member's samples (blood or bone marrow). The upper panel presents the targeted gene panel sequencing data for the patient’s bone marrow in the Integrative Genomics Viewer. The sequencing coverage of this allele was at 700×. The text column in red shows the homozygous mutation (c.2095C>T, p.R699W) in exon 2 of RAG1. The bottom panels show the Sanger sequencing data of this RAG1 mutation in the indicated samples from the patient and family members
Sanger sequencing of DNA from the proband's bone marrow and blood confirmed homozygosity for the RAG1 mutation. Surprisingly, his nail sample showed it to be heterozygous, suggesting that the ROH in the bone marrow encompassing RAG1 might not be due to consanguinity as initially assumed. To confirm the nail DNA result, we again designed two independent sets of PCR primers and used amplicon‐based deep sequencing to call the precise allelic ratio at the mutant site. The deep sequencing revealed the clear heterozygosity for the RAG1 mutation in the proband’s fingernail DNA (Figure 4). The real‐time PCR further confirmed two copies of mutant RAG1 in this sample (Figure S2). Heterozygosity in the nail DNA versus homozygosity in the bone marrow DNA suggested that the ROH harboring mutant RAG1 in the bone marrow was somatically acquired via a UPD event.
Figure 4.
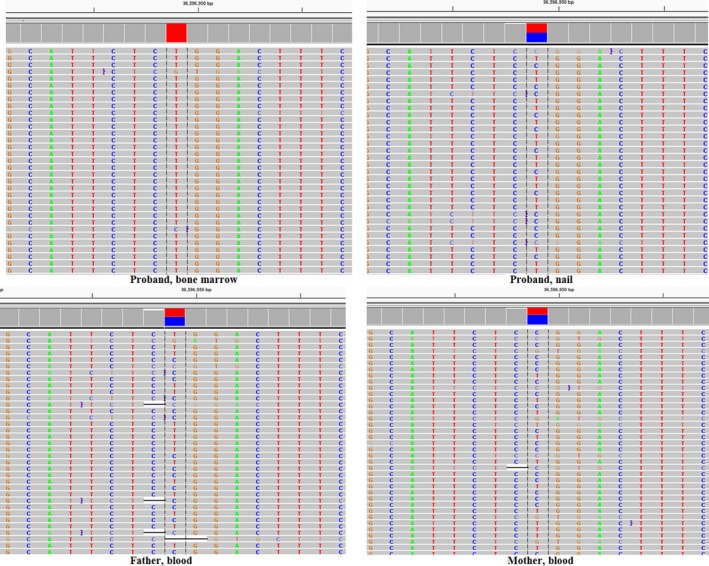
Validation of RAG1 mutation using amplicon‐based deep sequencing. Four panels present the next‐generation sequencing data of RAG1 mutation (c.2095C>T, p.R699W) from the family members. The general coverage of the amplicon‐based deep sequencing was more than 8000×. The mutation frequency of the proband's marrow, the proband's nail, father’s blood and mother's blood was 99% (coverage = 13280×), 51% (coverage = 12958×), 49% (coverage = 12363×), and 46% (coverage = 8717×), respectively. The mutations from the proband's nail, father and mother all were categorized as germline heterozygous rather than somatic/mosaics mutation according to the normal distribution curve of germline heterozygous mutations frequency (Jiang et al., 2017)
4. DISCUSSION
Mutations in RAG1 can cause immunodeficiency with an autosomal recessive inheritance pattern (OMIM 603,554), appearing early in life, resulting in increased susceptibility to severe infections and leading to early death unless treated by HSCT. Infants with these disorders typically experience pneumonia and chronic diarrhea beginning as neonates. Most patients are diagnosed before 6 months of age due to severe or persistent infection and generally survive only until age 1 or 2 (Kalman et al., 2004). Vaccination with live attenuated vaccines, such as oral poliovirus vaccine, smallpox, tuberculosis, measles, mumps, rubella, varicella, and typhoid can cause fatal infections and should be avoided (Kalman et al., 2004). Late‐onset (atypical) severe immunodeficiency caused by hypomorphic RAG1 mutation has been described previously (Patiroglu et al., 2014; Schuetz et al., 2008; Sharapova et al., 2013). Compared with classic SCID, late‐onset immunodeficiency usually has a milder clinical course and delayed presentation and retention of a partially functioning B‐lymphocyte lineage with continued capacity for antibody production has been reported (Patiroglu et al., 2014). In addition, these cases lack severe life‐threatening infections while granuloma formation and autoimmunity are common symptoms (Rossi et al., 2004).
The patient in this study presented with late‐onset immunodeficiency for 1 year and suspected MDS. We performed SNP array analysis and targeted gene panel sequencing for his bone marrow/blood to identify the fundamental etiology of MDS and immunodeficiency. A homozygous RAG1 mutation detected in his bone marrow and blood reasonably explained the immunodeficiency, but two important issues remained. First, after retrieval of the detailed clinical information for all carriers of the R699W mutation in the literature (Avila et al., 2010; Lee et al., 2011, 2014; Reiff et al., 2013; Zhang et al., 2011), we found that all suffered definitive or severe but early‐onset immunodeficiency or autoimmune symptoms in infancy. In contrast, our patient was a 13‐year‐old boy without either severe infections or autoimmune history in infancy. He did not overreact to any vaccination. The extraordinary late onset of his immunodeficiency made us question why the apparent homozygosity for the RAG1 mutation had not caused a penetrant immunodeficiency phenotype in infancy. Second, we performed an HLA‐haploidentical transplantation. In general, a successful engraftment of HSCT depends on HLA matching, transfused CD34+ cell count, the level of donor‐specific HLA antibodies and the condition regimen. The number of infused CD34+ cells from donor bone marrow was over the lower threshold required for engraftment, and donor‐specific HLA antibodies were absent, yet the RIC HSCT failed for this patient. Instead, MAC HSCT successfully treated his dysplasia. Allogeneic HSCT with MAC regimen is nearly the only treatment option with realistic curative potential for MDS, especially for children and adolescents, so his successful cure by MAC HSCT suggested a diagnosis of MDS in this patient.
All the above phenomena including late onset of immunodeficiency, persistent hematological abnormality, bone marrow morphology, MDS‐associated ROH, initial failed HSCT, and subsequent success MAC HSCT prompted us to consider a causal relationship between MDS and late‐onset immunodeficiency in this patient. We suspected that the homozygous RAG1 mutation of his bone marrow sample might have been due to acquired UPD resulting from the genomic instability seen in MDS rather than from constitutional IBD. Consequently, we chose his fingernail as the matched sample, and performed Sanger and deep sequencing for the mutated RAG1 allele. The heterozygous mutation status in his nail confirmed that his immunodeficiency was acquired due to a somatic event, which explained why he had no history of recurrent infection before 12 years of age. Although we failed to identify the driver mutation explaining the patient’s pancytopenia, his symptom evolution, bone marrow phenotypes, and responses to treatment allowed us to make a diagnosis according to the updated international criteria of MDS type refractory cytopenia of children (RCC), based upon the following lines of evidence (Greenberg et al., 2017): constant cytopenia, dysplastic abnormalities 16% in the granulocyte lineage and somatic regional chromosomal change.
Hematological disorders occurring due to MDS progression, such as acquired thalassemia (Brunner & Steensma, 2016), acquired Bernard–Soulier‐like platelet dysfunction have been previously reported (Frigeni & Galli, 2014). Our case demonstrates that an acquired immunological disorder, in addition to the known hematological disorders, can also be seen in a subject with MDS. In this instance, a somatic UPD event reducing a known SCID‐causing RAG1 mutation to homozygosity provides a reasonable explanation for the acquired immunodeficiency. Notably, most of the acquired hematological disorders reported previously arose from acquired LOH rather than from aUPD in patients with MDS (Frigeni & Galli, 2014). Recently, somatically acquired UPD has been recognized in cancer as an important and common mechanism by which adventitious mutations are amplified, leading to a growth advantage of these mutant cells (Makishima & Maciejewski, 2011). This mechanism is particularly common in both hematologic and solid tumors. The most well‐known example of aUPD involved in hematologic malignancies is UPD9p, which led to the identification of the JAK2 V617F mutation in myeloproliferative disorders (Kralovics et al., 2005). Other homozygous mutations such as in WT1, FLT3, CEBPA, and RUNX1 by aUPD have also been reported (Fitzgibbon et al., 2005). Our report suggests that aUPD can have pathogenic effects similar to LOH in hematological disorders. In addition, since blood is the most readily available and commonly used sample for genetic testing, especially for hematological diseases where it can directly reflect causative changes, our report also emphasizes that clinicians should consider and confirm a mutation’s origin as a germline or somatic when a seemingly hereditary hematological disease presents with a late‐onset.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHOR CONTRIBUTIONS
Xiaoli Chen and Xiaodong Shi designed the study. Junhui Li and Juanjuan Li performed all clinical diagnosis and treatments and collected detailed clinical information. Hailan Yao helped in data analysis of flow cytometry. Jianguo Li helped in immunological data analysis and diagnosis. Juanjuan Li performed routine experiments (DNA extraction, PCR, and Sanger sequencing). Fang Liu performed the target panel and amplicon‐based deep sequencing. Xiaoli Chen analyzed all genetic data. Xiaodong Shi provided advice and instructions on HSCT treatment and James F. Gusella provided advice on genetic interpretation. Juanjuan Li and Xiaoli Chen drafted the manuscript. Junhui Li, Xiaoli Chen and James F. Gusella were involved in manuscript revision.
Supporting information
ACKNOWLEDGMENTS
We sincerely thank the patient and his family members who contributed to the study. We also thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Li J, Li J, Li J, et al. A rare case of acquired immunodeficiency associated with myelodysplastic syndrome. Mol Genet Genomic Med. 2019;7:e923 10.1002/mgg3.923
Juanjuan Li and Junhui Li contributed equally to this work and should be considered co‐first authors
Funding information
This work is supported by grants from CAMS Innovation Fund for Medical Sciences (2016‐I2M‐1‐008), the Beijing Natural Science Foundation (7162029 to Xiaoli Chen), the Chinese National Nature Science Fund (31671310 to Xiaoli Chen), the U.S. National Institute of General Medical Sciences (GM061354) to James F. Gusella and the advanced Personnel Training Program of Beijing Municipal Health Bureau to Xiaoli Chen.
Contributor Information
Xiaodong Shi, Email: xsusan28@sina.com.
Xiaoli Chen, Email: cxlwx@sina.com.
REFERENCES
- Alabbas, F. , Weitzman, S. , Grant, R. , Bouffet, E. , Malkin, D. , Abla, O. , & Dror, Y. (2016). Underlying undiagnosed inherited marrow failure syndromes among children with cancer. Pediatric Blood & Cancer, 64(2), 302–305. 10.1002/pbc.26120 [DOI] [PubMed] [Google Scholar]
- Avila, E. M. , Uzel, G. , Hsu, A. , Milner, J. D. , Turner, M. L. , Pittaluga, S. , … Holland, S. M. (2010). Highly variable clinical phenotypes of hypomorphic RAG1 mutations. Pediatrics, 126(5), e1248–e1252. 10.1542/peds.2009-3171 [DOI] [PubMed] [Google Scholar]
- Brunner, A. M. , & Steensma, D. P. (2016). Myelodysplastic syndrome associated with acquired beta thalassemia: “BTMDS”. American Journal of Hematology, 91(8), E325–327. 10.1002/ajh.24400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgibbon, J. , Smith, L.‐L. , Raghavan, M. , Smith, M. L. , Debernardi, S. , Skoulakis, S. , … Young, B. D. (2005). Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Research, 65(20), 9152–9154. 10.1158/0008-5472.CAN-05-2017 [DOI] [PubMed] [Google Scholar]
- Frigeni, M. , & Galli, M. (2014). Childhood myelodysplastic syndrome associated with an acquired Bernard‐Soulier‐like platelet dysfunction. Blood, 124(16), 2609 10.1182/blood-2014-08-587832 [DOI] [PubMed] [Google Scholar]
- Gondek, L. P. , Tiu, R. , O'Keefe, C. L. , Sekeres, M. A. , Theil, K. S. , & Maciejewski, J. P. (2008). Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS‐derived AML. Blood, 111(3), 1534‐ 10.1182/blood-2007-05-092304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg, P. L. , Stone, R. M. , Al‐Kali, A. , Barta, S. K. , Bejar, R. , & Bennett, J. M. (2017). Myelodysplastic syndromes, version 2.2017, NCCN clinical practice guidelines in oncology. Journal of the National Comprehensive Cancer Network, 15(1), 60–87. [DOI] [PubMed] [Google Scholar]
- Itan, Y. , & Casanova, J. L. (2015). Novel primary immunodeficiency candidate genes predicted by the human gene connectome. Frontiers in Immunology, 6, 142 10.3389/fimmu.2015.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Q. , Liu, F. , Miao, C. , Li, Q. , Zhang, Z. , & Xiao, P. (2017). RET somatic mutations are underrecognized in Hirschsprung disease. Genetics in Medicine, 20(7), 770‐777 10.1038/gim.2017.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalman, L. , Lindegren, M. L. , Kobrynski, L. , Vogt, R. , Hannon, H. , Howard, J. T. , & Buckley, R. (2004). Mutations in genes required for T‐cell development: IL7R, CD45, IL2RG, JAK3, RAG1, RAG2, ARTEMIS, and ADA and severe combined immunodeficiency: HuGE review. Genetics in Medicine, 6(1), 16–26. 10.1097/01.GIM.0000105752.80592.A3 [DOI] [PubMed] [Google Scholar]
- Keel, S. B. , Scott, A. , Sanchez‐Bonilla, M. , Ho, P. A. , Gulsuner, S. , Pritchard, C. C. , … Shimamura, A. (2016). Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica, 101(11), 1343–1350. 10.3324/haematol.2016.149476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovics, R. , Passamonti, F. , Buser, A. S. , Teo, S. S. , Tiedt, R. , & Passweg, J. R. (2005). A gain‐of‐function mutation of JAK2 in myeloproliferative disorders. New England Journal of Medicine, 352(17), 1779–1790. 10.1056/NEJMoa051113 [DOI] [PubMed] [Google Scholar]
- Lee, P. P. W. , Chan, K.‐W. , Chen, T.‐X. , Jiang, L.‐P. , Wang, X.‐C. , Zeng, H.‐S. , … Lau, Y.‐L. (2011). Molecular diagnosis of severe combined immunodeficiency–identification of IL2RG, JAK3, IL7R, DCLRE1C, RAG1, and RAG2 mutations in a cohort of Chinese and Southeast Asian children. Journal of Clinical Immunology, 31(2), 281–296. 10.1007/s10875-010-9489-z [DOI] [PubMed] [Google Scholar]
- Lee, Y. N. , Frugoni, F. , Dobbs, K. , Walter, J. E. , Giliani, S. , & Gennery, A. R. (2014). A systematic analysis of recombination activity and genotype‐phenotype correlation in human recombination‐activating gene 1 deficiency. The Journal of Allergy and Clinical Immunology, 133(4), 1099–1108. 10.1016/j.jaci.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima, H. , & Maciejewski, J. P. (2011). Pathogenesis and consequences of uniparental disomy in cancer. Clinical Cancer Research, 17(12), 3913–3923. 10.1158/1078-0432.CCR-10-2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu, H. , Okuno, Y. , Yoshida, K. , Shiraishi, Y. , Doisaki, S. , & Narita, A. (2017). Clinical utility of next‐generation sequencing for inherited bone marrow failure syndromes. Genetics in Medicine, 19(7), 796–802. 10.1038/gim.2016.197 [DOI] [PubMed] [Google Scholar]
- Patiroglu, T. , Akar, H. H. , Gilmour, K. , Ozdemir, M. A. , Bibi, S. , Henriquez, F. , … Unal, E. (2014). Atypical severe combined immunodeficiency caused by a novel homozygous mutation in Rag1 gene in a girl who presented with pyoderma gangrenosum: A case report and literature review. Journal of Clinical Immunology, 34(7), 792–795. 10.1007/s10875-014-0077-5 [DOI] [PubMed] [Google Scholar]
- Reiff, A. , Bassuk, A. G. , Church, J. A. , Campbell, E. , Bing, X. , & Ferguson, P. J. (2013). Exome sequencing reveals RAG1 mutations in a child with autoimmunity and sterile chronic multifocal osteomyelitis evolving into disseminated granulomatous disease. Journal of Clinical Immunology, 33(8), 1289–1292 10.1007/s10875-013-9953-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, G. , Zecca, M. , Giorgiani, G. , Bonetti, F. , De Stefano, P. , & Locatelli, F. (2004). Non‐myeloablative stem cell transplantation for severe combined immunodeficiency–Omenn syndrome. British Journal of Haematology, 125(3), 406–407. 10.1111/j.1365-2141.2004.04906.x [DOI] [PubMed] [Google Scholar]
- Schuetz, C. , Huck, K. , Gudowius, S. , Megahed, M. , Feyen, O. , & Hubner, B. (2008). An immunodeficiency disease with RAG mutations and granulomas. New England Journal of Medicine, 358(19), 2030–2038. 10.1056/NEJMoa073966 [DOI] [PubMed] [Google Scholar]
- Sharapova, S. O. , Migas, A. , Guryanova, I. , Aleshkevich, S. , Kletski, S. , & Durandy, A. (2013). Late‐onset combined immune deficiency associated to skin granuloma due to heterozygous compound mutations in RAG1 gene in a 14 years old male. Human Immunology, 74(1), 18–22. 10.1016/j.humimm.2012.10.010 [DOI] [PubMed] [Google Scholar]
- Zhang, Z.‐Y. , Zhao, X.‐D. , Jiang, L.‐P. , Liu, E.‐M. , Cui, Y.‐X. , Wang, M. O. , … Yang, X.‐Q. (2011). Clinical characteristics and molecular analysis of three Chinese children with Omenn syndrome. Pediatric Allergy and Immunology, 22(5), 482–487. 10.1111/j.1399-3038.2010.01126.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials