

Abstract
Background
Inducing apoptosis in cancer cells is an important step for the successful treatment of cancer patients. Bcl‐2 is an antiapoptotic protein which determines apoptosis by interacting with proapoptotic members of the Bcl‐2 family. Exome sequencing has identified Bcl‐2 and Bax missense mutations in more than 40 cancer types. However, a little information is available about the functional impact of each Bcl‐2 and Bax mutation on the pathogenesis of cancer.
Methods
The mutational data from cancer tissues and cell lines were retrieved from the cBioPortal web resource. The 13 mutated Bcl‐2 and wild‐type Bax complexes with experimentally verified binding were identified from previous studies wherein, binding for all complexes was reportedly disrupted except one. Several protein–protein docking methods such as ClusPro, HDOCK, PatchDock, FireDock, InterEVDock2 and several mutation prediction methods such as PolyPhen‐2, SIFT, and OncoKB have been used to predict the effect of mutation to disrupt the binding between Bcl‐2 and Bax. The result obtained was compared with the known experimental data.
Results
The protein–protein docking method, ClusPro, employed in the present study confirmed that the binding affinity of 11 out of 13 complexes decreases. Similarly, binding affinity computed for all the 10 wild‐type Bcl‐2 and mutated Bax complexes agreed with experimentally verified results.
Conclusion
Several methods like PolyPhen‐2, SIFT, and OncoKB have been developed to predict cancer‐associated or deleterious mutations, but no method is available to predict apoptosis‐inducing mutations. Thus, in this study, we have examined the mutations in Bcl‐2 and Bax proteins that disrupt their binding, which is crucial for inducing apoptosis to eradicate cancer. This study suggests that protein–protein docking methods can play a significant role in the identification of hotspot mutations in Bcl‐2 or Bax that can disrupt their binding with wild‐type partner to induce apoptosis in cancer cells.
Keywords: apoptosis, Bax, Bcl‐2, cancer, docking, mutation
1. INTRODUCTION
Apoptosis is a tightly regulated cell death process that operates along two major pathways, the extrinsic and intrinsic (Fruehauf & Meyskens, 2007; Hancock, Desikan, & Neill, 2001). The intrinsic pathway is crucially regulated by B‐cell lymphoma‐2 (Bcl‐2), an antiapoptotic protein, which contains four Bcl‐2 homology (BH) domains (BH1–BH4) (Raghav, Verma, & Gangenahalli, 2012a). However, Bcl‐2 is commonly associated with several malignancies, but evidences of its all binding sites are not fully understood because of the presence of a flexible loop domain (Zacarías‐Lara, Correa‐Basurto, & Bello, 2016).
The proto‐oncogene Bcl‐2 inhibits apoptosis and encourages tumor progression (Rupnarain, Dlamini, Naicker, & Bhoola, 2004) while, Bax regulates apoptosis by contributing to tumor regression (Backus et al., 2002). The later phenomenon is accomplished by interaction between BH3‐cleft of Bcl‐2 and the BH3 domain of Bax which inhibits apoptosis (Raghav, Verma, & Gangenahalli, 2012b). This inhibition occurred by preventing cytochrome c release from mitochondria, or its binding to Apaf‐1 (Gupta, 2003; Korsmeyer, Yin, Oltvai, Veis‐Novack, & Linette, 1995; Luanpitpong et al., 2013). Evidently, Bcl‐2 interacts with a higher affinity with Bax among the proapoptotic family members (Backus et al., 2002; Raghav et al., 2012b).
Several diagnosed cases and deaths have been reported due to the malignancy of B‐cell lymphocytes (Singh & Briggs, 2016). Among these cancers, follicular lymphoma (FL) and large B‐cell lymphoma (DLBCL) are the two most common types of non‐Hodgkin lymphoma (NHL) detected frequently (Morin et al., 2011; Schuetz et al., 2012). It has been reported that Bcl‐2 is the primary target of FL and has a central role in the inhibition of apoptosis (Perini, Ribeiro, Pinto Neto, Campos, & Hamerschlak, 2018; Vaux, Cory, & Adams, 1988). Even though the occurrence of variations in Bcl‐2 is a well‐known event in DLBCL, the relevance of these mutations in the pathogenesis of lymphoma is a matter of debate (Correia et al., 2015; McDonnell & Korsmeyer, 1991; Reed & Tanaka, 1993). Prediction of the functional impact of Bcl‐2 mutation is difficult, owing to the pleiotropic nature of the protein and the heterogeneity in its mutational profile (Singh & Briggs, 2016). Therefore, characterization of nonsynonymous mutations in Bcl‐2 requires further study to address the role of Bcl‐2 mutations in the etiology of the disease, pathways of its pathogenesis, and effect of these mutations on drug response.
The mutations in the Bcl‐2 and Bax protein are often detected in several types of cancers, indicating both their significance and occurrence. Cancer genome sequencing has identified several mutations having an essential role in cellular mapping pathways leading to tumorigenesis (Lawrence et al., 2014; Leiserson et al., 2015). Also, it has played a crucial role in elucidating molecular mechanisms that drive oncogenic transformation (Alexandrov et al., 2013) and drug resistance (Buljan, Blattmann, Aebersold, & Boutros, 2018).
Moreover, several anti‐Bcl‐2 inhibitors induce apoptosis in tumor cells (Lu et al., 2016), though no molecule has been reported which targets the Bcl‐2 mutations. The elucidation of Bcl‐2 mutation with wild‐type Bax, and wild‐type Bcl‐2 with mutated Bax binding and their interacting domain impact is important to regulate apoptosis. Therefore, primarily it is required to study the effect of mutations which disrupt the binding of the mutated Bcl‐2 with wild‐type Bax and vice‐versa. There are numerous methods available to predict the functional impact of mutations but so far, no method has been developed to predict the impact of the mutation on binding of Bcl‐2 and Bax. Hence, this study emphasizes the likely consequences of Bcl‐2 or Bax mutations on binding with their wild‐type antagonist. To understand this we collected the Bcl‐2 and Bax mutation data of patient samples and cell lines from cBioPortal and then performed sequence and structural analysis (Figure 1). These results revealed an in‐depth focus to evaluate the functional effect of missense mutations of Bcl‐2 and Bax associated with different types of cancers. Subsequently, sequence and structure‐based docking were performed using several docking web‐servers. Among all docking tools used, ClusPro predicted the binding impact which was found close to the experimental data curated from the literature. Finally, in this study, complexes (mutated Bcl‐2 and wild‐type Bax; wild‐type Bcl‐2 and mutated Bax) possessing higher binding affinity have been identified, which can be further targeted to release Bax as a free protein leading to increased apoptosis in cancers.
Figure 1.
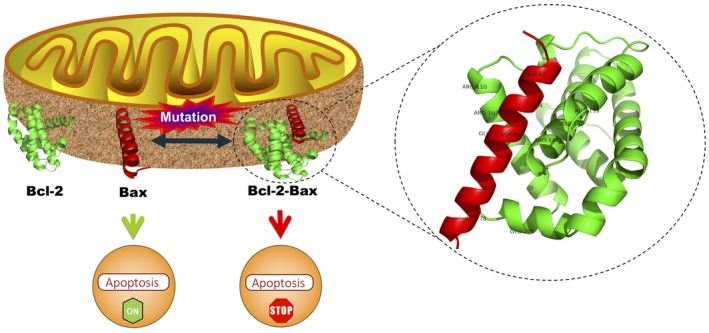
Represents the overall impact of wild‐type and mutated, Bcl‐2 and Bax, respectively, and vice‐versa interaction on apoptosis. The ribbon shaped three‐dimensional Bcl‐2 (green) and Bax (red) structures are present on the outer mitochondrial membrane. Depth cueing shows interacting residues of Bcl‐2 (R139, D140, R146, R107, R110, and E200) with Bax (E61, R64, D68, E69, and R79). This Bcl‐2‐Bax heterodimer inhibits apoptosis while free Bax promotes apoptosis
2. MATERIALS AND METHODS
2.1. Data curation
The sample having Bcl‐2 and Bax missense mutations in the different types of cancers were retrieved from cBioPortal up to December 2018 (Gao et al., 2013). Total 234 missense mutations of which 119 found in patient samples and 5 in cell lines, were collected from cBioportal. The BH3 cleft of Bcl‐2 is an important site for binding of Bax (Raghav et al., 2012b) therefore, all the 70 mutations found in BH3 cleft of Bcl‐2 of 54 sample ids were examined. Further, total 38 mutants of Bax, occured in 45 sample ids were gathered from cBioPortal. Finally, 16 mutations of Bax BH3 domain from 18 sample ids proceeded for mutational analysis. Besides, the information on experimantally known impact of 13 Bcl‐2 mutations on its binding with wild‐type Bax was gathered by literature review (Barretina et al., 2010, 2012; Bell et al., 2011; Cancer Genome Atlas Research Network, 2012; Koboldt et al., 2012; McLendon et al., 2008; Network, 2012; Taylor et al., 2010). Similarly, the impact of experimental known 10 Bax mutation with wild‐type Bcl‐2 was collected from the literature (Czabotar et al., 2011; Ku, Liang, Jung, & Oh, 2011; Meijerink et al., 1998; Meijerink, Smetsers, Slöetjes, Linders, & Mensink, 1995).
2.2. Preprocessing of data
The missense mutations of BH3 cleft of Bcl‐2, and BH3 domain of Bax, retrieved from cBioPortal were further preprocessed. The frequency of sample id, frequency of each mutation, and generation of single and multiple mutational sequences of Bcl‐2 and Bax were obtained using in‐house R scripts.
2.3. Methods used for identifying mutations responsible for changing structure and function
The implications of mutations were analyzed by considering the structural and functional effects on Bcl‐2 and Bax protein. The former primarily affects attributes, such as the stability and fold of the protein product, and the latter affects functional sites (Ferrer‐Costa, Orozco, Cruz, & de., 2002; Fischer, Greenman, & Mustonen, 2011; Miller & Kumar, 2001; Sitbon & Pietrokovski, 2007; Stitziel et al., 2003; Sunyaev, Ramensky, & Bork, 2000; Todd, Orengo, & Thornton, 2002; Valdar, 2002). The impact of each Bcl‐2 mutation on apoptosis in cancer was identified using OncoKB (Chakravarty et al., 2017), Cancer Hotspot (Chang et al., 2018), 3D Hotspot (Gao et al., 2017), MutationAssessor (Merid, Goranskaya, & Alexeyenko, 2014), Sorting Intolerant From Tolerant (SIFT) (Ng & Henikoff, 2003; Vaser, Adusumalli, Leng, Sikic, & Ng, 2016), and PolyPhen‐2 (Adzhubei, Jordan, & Sunyaev, 2013). Specifically, SIFT, MutationAssessor, and PolyPhen‐2 accessed the functional impact of mutation. These tools assigned a score to each Bcl‐2 and Bax mutation based on the physical‐chemical properties or evolutionary conservation of the amino acid sequences affected by the modification.
2.4. Three‐dimensional (3D) structure modeling
The 3D structure of wild‐type and mutated, Bcl‐2, and Bax protein was generated using SWISS‐MODEL (Schwede, Kopp, Guex, & Peitsch, 2003). To generate Bcl‐2 3D structures of wild‐type and mutants, 1GJH (NMR) template was chosen by SWISS‐MODEL tool except for mutant 37, 39 and 53 where, these structures were generated on 5VAU (X‐ray) template. Further, 2K7W (NMR) template was used to generate 3D structures of Bax mutants except for mutant 7 and 8 which were based on 1F16 (NMR) while 3D structure of wild‐type and mutant 14 were based on 2LR1 (NMR). The 3D model having highest sequence identity was considered further for docking studies.
2.5. Docking
The binding of mutated Bcl‐2 with wild‐type Bax and vice‐versa was determined for patient sample data, retrieved from cBioPortal. The sequence and structure‐based docking approaches were used to evaluate the binding affinity between the mutated Bcl‐2 and wild‐type Bax and vice‐versa. Sequence‐based docking was performed using the sequence as input whereas, structure‐based docking refers to a 3D structure as the input. The PPA‐Pred2 (Yugandhar & Gromiha, 2014), ISLAND (Abbasi, Hassan, & Yaseen, 2017), HDOCK (Yan, Zhang, Zhou, Li, & Huang, 2017), InterEVDock2 (Quignot et al., 2018), SOAP_PP (Quignot et al., 2018), FRODOCK2 (Ramírez‐Aportela, López‐Blanco, & Chacón, 2016) docking programs were used to perform the sequence‐based docking. Besides, ZDOCK (Pierce et al., 2014), ClusPro (Kozakov et al., 2017), HDOCK (Yan et al., 2017), PatchDock (Mashiach et al., 2010), FireDock (Mashiach, Schneidman‐Duhovny, Andrusier, Nussinov, & Wolfson, 2008), InterEvDock2, SOAP PP, and FRODOCK2 tools were used to perform structure‐based docking. The various tools used to perform docking were PPA‐Pred2 and ISLAND to predict the ΔG (binding free energy) and Kd (dissociation constant) respectively; HDOCK to calculate negative docking score and ligand rmsd (Å); ZDOCK to calculate positive docking score; ClusPro to calculate center‐weighted score; PatchDock to calculate positive score; FireDock to obtain global energy and obtained solution number of complex used in PatchDock; InterEvDock2 to compute SOAP_PP (negative score), and InterEvScore and FRODOCK2, (positive scores).
3. RESULTS AND DISCUSSION
3.1. Data analysis
In this work, the analysis of missense mutations within different cancer types was mainly considered. The mutation analysis used in this study helped identify passenger mutations and cancer driver mutations that drive carcinogenesis. Among all Bcl‐2 mutations from cBioPortal, single point mutations were identified in 56 sample ids and multiple mutations in 63 sample ids (Table S1). The sample ids, P‐0003578‐T01‐IM5, P‐0007086‐T02‐IM5, P‐0008460‐T02‐IM5, and P‐0011441‐T01‐IM5 exhibited the highest frequency of occurrence in cBioPortal with six multiple mutations.
Similarly, DLBCL‐Ls3309 sample id with a frequency of 5 was identified for DLBCL (Table S1). All these sample ids are associated with the cancer type, DLBCL which suggests that this cancer type is generally caused due to multiple mutations. Among cancer types, DLBCL was identified as the most frequently occuring cancer type in 36 samples. This analysis signifies that Bcl‐2 mutations mostly occur in DLBCL. In contrast, anaplastic oligodendroglioma, bladder urothelial carcinoma, cervical squamous cell carcinoma, hepatocellular carcinoma, head and neck squamous cell carcinoma, histiocytic dendritic cell sarcoma, intestinal‐type stomach adenocarcinoma, mixed ovarian carcinoma, and small cell lung cancer were identified as least frequently occuring cancer type with a frequency of one. These cancer types imply that Bcl‐2 mutations rarely occur.
Moreover, 51 out of 150 Bcl‐2 mutations occurred frequently, suggesting that these mutations would be cancer hotspots (Table S2). These cancer hotspots demonstrated that these mutations are possible cancer drivers compared to a mutation which occurs at low frequency, signifying nonhotspots. These low‐frequency mutations can be used to prioritize the cancer drivers. The amino acid, A131 attained highest frequency with an occurrence of 12 wherein, mutations A131D, A131V, and A131T had a frequency of 7, 3, and 2 respectively. This analysis demonstrated that A131 is a common site of mutation which would likely be targeted to design a marker or drug against it.
Furthermore, 41 single, 10 double, and three triple mutations were identified in BH3 cleft of Bcl‐2 (Table S3). Multiple Bcl‐2 mutations were noticed in DLBCL, FL, and germinal center B‐cell (GCB) type cancers. The mutations, A131D, A131T and A131V were associated with other multiple mutations observed in six out of 13 sample ids, and three single mutations identified in six out of 47 sample ids. This result shows that these mutations are frequently occurring mutations in BH3 cleft of Bcl‐2 as single and multiple mutations in DLBCL, B‐Cell Lymphoma, Glioblastoma Multiforme, FL, Cutaneous Squamous Cell Carcinoma, and GCB type cancers.
Additionally, 17 out of 59 mutations have multiple frequencies in sample ids, wherein A131D and R98H attained a higher frequency of occurrence in the BH3 cleft of Bcl‐2 (Table S4). These mutations would be the target sites to eradicate the progression of cancer through target‐based drug discovery. In addition, all five cell lines having Bcl‐2 mutations originated from hematopoietic and lymphoid tissues. This showed that Bcl‐2 mutants, S117R, R129C, V156F, R106L, and L95V would likely be the targets of hematopoietic originated cancers.
Furthermore, the highest mutation frequency (8) was observed in Bax, in each colorectal and uterine carcinoma, whereas two variants at each E44 and G67 residues were obtained in two different sample ids respectively (Table S5). These mutations would be a target to prevent the progression of their respective cancer types. Nonetheless, double variation (G67R and L63I) in Bax was found in TCGA‐A5‐A0G2‐01 sample id.
The mutation R89Q occurred with the highest frequency of five sample ids from cBioPortal (Table S6). Interestingly, it has been perceived that no mutations occurred in DLBCL as obtained in Bcl‐2.
Instead of all residues of Bax, the BH3 domain (59–73 residues) and some nearby residues (41–58 residues, and 74–93 residues) were considered for further mutational analysis (Table S7). The foremost mutation site appeared for residue E44 in colorectal adenocarcinoma, G67 in uterine serous carcinoma/uterine papillary serous carcinoma and cutaneous melanoma, A82 in esophageal adenocarcinoma and hepatocellular carcinoma, P88 in cutaneous melanoma, and R89 in breast invasive ductal carcinoma, uterine endometrioid carcinoma, uterine endometrioid carcinoma, and cutaneous squamous cell carcinoma. Specifically, the Bax BH3 domain mutant, G67R was obtained from the hematopoietic and lymphoid tissue cell line.
3.2. Mutation impact identifies the cancer hotspots
The impact analysis helped explore the effects of missense mutations in the Bcl‐2 protein associated with cancer. Different types of disease‐causing mutations have been studied including germline diseases, somatic cancer mutations in oncogenes and tumor‐suppressors, along with known hotspots in Bcl‐2. New approaches were explored for determining the impacts of disease‐associated mutations on Bcl‐2 structure and function. The present work is mainly focused on the investigating cancer mutations which could be further applied to analyze other types of disease causing mutations. Mutation hotspots identified in this study would serve as valuable resources in the selection of functional driver mutations and associated genes. However, properties associated with a residue's potential to drive tumorigenesis on modification have not yet been systematically examined before.
Here, using a novel analysis approach, we identify and characterize the effect of 81 hotspot mutations in BH3 cleft of Bcl‐2 which occur with a significant frequency and are likely to have a functionally relevant impact (Table S8). The proximity of disease‐associated mutations has been analyzed to predict the structural and functional impact using OncoKB, 3DHotspot, MutationAssessor, SIFT, and PolyPhen‐2. These tools identified driver mutations and passenger mutations, the former comes under positive selection pressure in tumor evolution, thus promoting oncogenesis and drug resistance (Baudot, Real, Izarzugaza, & Valencia, 2009; Gong & Blundell, 2010; Schmitt, Loeb, & Salk, 2016; Talavera, Taylor, & Thornton, 2010), whereas later are under neutral selection pressure which confer no survival advantage to the tumor (Nussinov & Tsai, 2015). The identification of functional impact carried out by MutationAssessor, SIFT, and PolyPhen‐2, revealed that the damaging effects of mutations has implications on Bcl‐2 function.
The mutant A131D in DLBCL, B‐Cell lymphoma and glioblastoma multiforme; A131T in FL; A131V in cutaneous squamous cell carcinoma, GCB type, and DLBCL; N172Q in DLBCL; and N172S in DLBCL and colon adenocarcinoma were predicted oncogenic using OncoKB. These mutations demonstrated their oncogenic effect mainly in DLBCL. CancerHotspot also corroborated this result by predicting the oncogenic mutation as cancer hotspots.
Further, MutationAssessor assessed that 21 mutations had medium score, 23 mutations had low functional impact and 14 muation were neutral. MutationAssessor has been shown to be an effective method for predicting SNP deleteriousness by predicting a medium impact score for 21 variations. The mutant A131D was predicted medium impact whereas, A131V, N172S, and A131T predicted neutral. This functional impact score of modification was calculated by averaging a conservation score in a protein family, and specificity score in the subfamily. Besides, R129C, V156F, R106L, and L95V mutations of cell lines possessed low impact.
SIFT predicted the tolerated impact of 28 mutations with a score ranging from 0.05 to 0.97, and the deleterious impact for 31 variations with a different score from 0 to 0.04. The “tolerated” impact demonstrated that the substitution is predicted to be neutral affect, whereas “deleterious” means that the substitution affected Bcl‐2 function. SIFT scores the pathogenicity of mutation wherein scores below 0.05 are predicted to be deleterious based on a change to a completely different amino acid at positions with a conserved alignment of homologous sequences, obtained from SwissProt/TrEMBL (Nussinov & Tsai, 2015). The earlier discussed structural impact of N172Q, A131D, A131T, A131V, and N172S mutations were predicted to have tolerated impact by SIFT. The functional effect of cell line mutant, R129C and V156F was found deleterious whereas, S117R, R106 L, and L95V have tolerated impact.
PolyPhen‐2 analyzed the effect of mutations on various structural features (solvent accessibility, B‐factor, CpG context, position of modification within a Pfam domain, change in residue volume and the difference in Position specific independent count scores) between wild‐type and mutated residue (Nussinov & Tsai, 2015), which is used in probabilistic Naive Bayes classifier to build the SNP predictor PolyPhen‐2 (Nussinov & Tsai, 2015). The 18 mutations were identified benign with a score between 0.003 and 0.442, 12 modifications as possibly damaging acquired a between 0.477 and 0.90, and 29 probably damaging mutations with a score ranging 0.929–1. Based on classifier prediction these 29 Probable damaging mutations are the most confident predictions to have the highest effect to change the Bcl‐2 function. The R129C, and V156F mutation in cell line was detected as probably damaging, though R106L had a low confidence score and was predicted as possibly damaging, while the L95V mutant was confirmed as benign.
Furthermore, no cancer hotspots and oncogenic Bax mutations in patient samples and cell lines were recognized by 3D Hotspot and OncoKB, respectively (Table S9). This result revealed that the Bax BH3 domain mutations are not cancer hotspots. However, seven variations have a low impact, and six were found neutral, while three medium impacts were characterized in patient samples using MutationAssessor. The neutral impact possessed a score between 0.345 and 0.76; low, 0.895 and 1.845; and medium, 2.24 and 2.915. The mutations of BH3 region, E44K, Q52H, A54V, P88S, P88L, R89Q, and R89L were observed with low impact, while E41K, A42S, E44Q, A82T, A82S, and F93L were identified with neutral impact. On the contrary, the mutants, L63I, G67R, and G67V have medium effect which determines that these mutations could be the driver mutations. Likewise, the cell line mutant, V83M has a low impact, while medium impact was predicted for G67R and E75K mutants.
Additionally, SIFT predicted a deleterious impact with a score 0–0.05 for six mutants and tolerated impact for 10 mutants with score 0.1–0.85. The mutants, E41K, Q52H, L63I, G67R, G67V, and P88L have a deleterious effect. On the contrary, mutants A42S, E44K, E44Q, A54V, A82T, A82S, P88S, R89Q, R89L, and F93L have tolerated impact.
The PolyPhen‐2 predicted the functional impact, benign for 12 Bax mutations, two possibly damaging, and two probably damaging which signifies that the mutants G67V and P88L affect the function of Bax. Although, L63I, and P88S mutants have low confidence score which suggests that these mutations perhaps associated with disease. However, E41K, A42S, E44K, E44Q, Q52H, A54V, G67R, A82T, A82S, R89Q, R89L, and F93L mutations were found benign, indicating that these mutations do not affect Bax function.
The cell line mutants, V83M have low impact, while medium impact was identified using MutationAssessor for mutants G67R, and E75K (Table S9). The tolerated impact of mutant V83M, and deleterious impact of mutant G67R, and E75K was identified using SIFT. PolyPhen‐2 identified the mutant E75K which possibly damaged the function of Bax, though no effect on function was observed for G67R. Whilst, the mutant, V83M disturbed the function of Bax. Eventually, the function of Bax was altered by mutant E75K which was mutually confirmed using MutationAssessor, SIFT, and PolyPhen‐2.
Besides, no significant structural impact was observed for Bcl‐2 mutations predicted by OncoKB and CancerHotspot (Table 1). MutationAssessor predicted medium impact for Bcl‐2 mutant, G145A, G145E, and Q190L while, low for D191A and neutral for E200A. The deleterious impact was identified using SIFT for W144A, G145A, G145E, R146A, W188A, and F104C Bcl‐2 mutants, though Q190L, D191A, N192A, E200A, and F104L were found tolerated. PolyPhen‐2 identified the probable damaging effect of Bcl‐2 mutants, W144A, G145A, G145E, R146A, W188A, Q190L, D191A, N192A, and F104C. This result revealed that the maximum functional overlap was observed between MutationAssessor, SIFT, and PolyPhen‐2 methods for G145A and G145E mutants which are shown agree with the known experimental impact. In contrast, higher inconsistencies were observed between these methods for Q190L, D191A, and N192A mutants.
Table 1.
Structural and functional impact of experimental known Bcl‐2 mutations
| Complexes | Bcl−2 mutations | Experimental Impact of Bcl−2 mutants with Bax wild‐type | Pubmed ID | OncoKB | Cancer Hotspot | Mutation Assessor (impact and score) | SIFT (impact and score) | PolyPhen−2 (impact and score) |
|---|---|---|---|---|---|---|---|---|
| 1‐Bax | Wild‐type | Binds with Bax | 22155216 | — | — | — | — | — |
| 2‐Bax | FRDG138‐141AAAA | Complete Loss of Bax‐binding | 8183370 | Unknown | No | Undefined | Undefined | Not Predicted |
| 3‐Bax | W144A | Complete Loss of Bax‐binding | 8183370 | Unknown | No | Undefined | Deleterious; 0 | Probably Damaging; 1 |
| 4‐Bax | G145A | Complete Loss of Bax‐binding | 8183370; 17418785; 25703009; 9202007 | Unknown | No | Medium; 2.79 | Deleterious; 0 | Probably Damaging; 1 |
| 5‐Bax | G145E | Complete Loss of Bax‐binding | 8183370; 17418785; 20382739 | Unknown | No | Medium; 3.34 | Deleterious; 0 | Probably Damaging; 1 |
| 6‐Bax | R146A | Complete Loss of Bax‐binding | 8183370 | Unknown | No | Undefined | Deleterious; 0 | Probably Damaging; 1 |
| 7‐Bax | W188A | Complete Loss of Bax‐binding | 8183370 | Unknown | No | Undefined | Deleterious; 0 | Probably Damaging; 1 |
| 8‐Bax | Q190L | Partial loss of Bax‐binding | 8183370 | Unknown | No | Medium; 2.195 | Tolerated; 0.18 | Probably Damaging; 0.989 |
| 9‐Bax | D191A | No effect on Bax‐binding | 8183370 | Unknown | No | Low; 1.15 | Tolerated; 0.11 | Probably Damaging; 0.960 |
| 10‐Bax | N192A | Partial loss of Bax‐binding | 8183370 | Unknown | No | Undefined | Tolerated; 0.07 | Probably Damaging; 0.990 |
| 11‐Bax | E200A | Partial loss of Bax‐binding | 8183370 | Unknown | No | Neutral; 0.345 | Tolerated; 0.34 | Benign; 0.056 |
| 12‐Bax | F104C | Complete Loss of ABT−199 binding | 24786774 | Unknown | No | Undefined | Deleterious; 0.00 | Probably Damaging; 1 |
| 13‐Bax | F104L | Complete Loss of ABT−199 binding | 24786774 | Unknown | No | Undefined | Tolerated; 0.08 | Possibly Damaging; 0.866 |
| 14‐Bax | MISSING 194–197 | Complete Loss of Bax‐binding | 8183370 | Unknown | No | Undefined | Undefined | Not Predicted |
Similarly, no difference for the structural impact of Bax mutation was identified by OncoKB and 3D Hotspot (Table 2). The Bax mutants, G67R, E61A, D68A, and E69A, attained medium impact as predicted by MutationAssessor. SIFT noticed deleterious effect for Bax mutants, M74D, M74E, G67R, and E69A but E61A, R65A, D68A, R78A, E61A/ R65A/R78A, and I66E/D68R mutants displayed tolerated impact. The Bax mutant, M74D, and M74E were found benign, though E61A, E69A, and R78A were identified as possibly damaging by PolyPhen‐2. On the contrary, the mutants G67R, R65A, D68A, and I66E/D68R have a damaging effect which was predicated on the highest score. Jointly, these results strongly proved that mutant G67R had a functional damaging effect predicted simultaneously by MutationAssessor, SIFT and PolyPhen‐2. Nevertheless, paradox prediction existed between these methods for mutants, M74D, M74E, E61A, R65A, D68A, E69A, R78A, and I66E/D68R.
Table 2.
Structural and functional impact of experimental known Bax mutations
| Complexes | Bax mutations | Experimental Impact of Bax mutants with Bcl‐2 wild‐type | Pubmed ID | OncoKB | 3D Hotspot | Mutation Assessor (impact and score) | SIFT (impact and score) | PolyPhen‐2 (impact and score) |
|---|---|---|---|---|---|---|---|---|
| 1‐Bcl‐2 | Wild‐type | Bind with Bcl‐2 | 22155216 | — | — | — | — | — |
| 2‐Bcl‐2 | M74D | Strongly reduced interaction with Bcl‐2 | 21199865 | NA | No | Undefined | Deleterious; 0.01 | Benign; 0.091 |
| 3‐Bcl‐2 | M74E | Strongly reduced interaction with Bcl‐2 | 21199865 | NA | No | Undefined | Deleterious; 0.02 | Benign; 0.004 |
| 4‐Bcl‐2 | G67R | Loss of heterodimerization with Bcl‐2 | 7475270; 9531611 | NA | No | Medium; 2.915 | Deleterious; 0.01 | Probably Damaging; 0.997 |
| 5‐Bcl‐2 | E61A | Reduced the affinities with Bcl‐2 | 21060336 | NA | No | Medium; 2.075 | Tolerated; 0.73 | Possibly Damaging; 0.530 |
| 6‐Bcl‐2 | R65A | Reduced the affinities with Bcl‐2 | 21060336 | NA | No | Undefined | Tolerated; 0.27 | Probably Damaging; 0.972 |
| 7‐Bcl‐2 | D68A | Greatly reduced the affinities with Bcl‐2 | 21060336 | NA | No | Medium; 2.84 | Tolerated; 0.08 | Probably Damaging; 0.998 |
| 8‐Bcl‐2 | E69A | Reduced the affinities with Bcl‐2 | 21060336 | NA | No | Medium; 2.305 | Deleterious; 0.00 | Possibly Damaging; 0.839 |
| 9‐Bcl‐2 | R78A | Reduced the affinities with Bcl‐2 | 21060336 | NA | No | Undefined | Tolerated; 0.28 | Possibly Damaging; 0.671 |
| 10‐Bcl‐2 | E61A/ R65A/R78A | Greatly reduced the affinities with Bcl‐2 | 21060336 | NA | No | Medium; 2.075/Undefined/Undefined | Tolerated; 0.73/ Tolerated; 0.27/ | Possibly Damaging; 0.530/ Probably Damaging; 0.972/ Possibly Damaging; 0.671 |
| 11‐Bcl‐2 | I66E/D68R | Completely lost the binding | 25703009 | NA | No | Undefined/ Undefined | Tolerated; 0.11/ Deleterious; 0.01 | Probably Damaging; 1/ Probably Damaging; 1 |
3.3. The mutation affects the binding affinity of the complex
Variations can affect specific functional sites of Bcl‐2 and Bax, resulting in loss of function can lead to disease (Nussinov & Tsai, 2015). Also, impact analysis identified that mutations within earlier discussed critical residues possessed significantly higher pathogenicity scores reported by PolyPhen‐2 as compared to noncritical residues.
We considered mutations in Bcl‐2 which would interfere with the interactions with Bax and vice‐versa. Accordingly, docking was performed to examine the impact of the interaction between them. Jointly, docking results suggest that disruption and dysregulation of Bcl‐2 and Bax interactions can be involved in switching functions of cancer proteins and activating downstream changes.
We first gathered the information from literature for experimental impact on binding of Bcl‐2 mutants with wild‐type Bax (Table 3). This result suggested that Bcl‐2 mutants FRDG138‐141AAAA, W144A, G145A, G145E, R146A, W188A, F104C, F104L, and MISSING194‐197 completely disrupt the binding with wild‐type Bax. This result revealed that these mutants lead to structural changes in Bcl‐2 which in turn disrupt the binding with Bax that promotes apoptosis. Nevertheless, Bcl‐2 mutants, Q190L, N192A, and E200A partially lost the binding with wild‐type Bax which demonstrates that these mutants have increased binding affinity with Bax. However, D191A had the opposite effect with Bax, suggesting that this mutation has a neutral impact on binding with Bax. Afterward, these known impacts of coupling were further validated using sequence and structure‐based docking.
Table 3.
Impact of experimental known Bcl‐2 mutations with wild‐type Bax
| Complexes | Bcl‐2 mutations | Experimental Impact of Bcl‐2 mutants with Bax wild‐type | Pubmed ID | Sequence‐Based Docking | Structure‐Based Docking | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PPA‐Pred2 ΔG (kcal/mol); Kd (M) | ISLAND ΔΔG (kcal/mol); Kd (M) | HDOCK score and ligand rmsd (Å) | InterEvDock Score | SOAP_PP Score | FRODOCK2 Score | ZDOCK score | ClusPro score Score | HDOCK score and ligand rmsd (Å) | PatchDock score | FireDock (sol no; global energy) | InterEvDock score | SOAP_PP score | FRODOCK2 score | ||||
| 1‐Bax | Wild‐type | Binds with Bax | 22155216 | −10.63; 1.60e‐08 | −11.08; 7.4604444936e‐09 | −259.86; 0.70 | 696.4 | −12906.73 | 1701.52 | 1,670.276 | −969.9 | −256.32; 0.65 | 17,778 | 5; −3.91 | 542.35 | −14018.72 | 1958.41 |
| 2‐Bax | FRDG138‐141AAAA | Complete Loss of Bax‐binding | 8183370 | −10.80; 1.20e‐08 | −11.112; 7.06580309294e‐09 | −264.73; 1.57 | 510.54 | −12813.01 | 1601.06 | 1767.023 | −1096 | −224.21; 49.19 | 17,874 | 8; −38.13 | 586.46 | −13775.67 | 1911.1 |
| 3‐Bax | W144A | Complete Loss of Bax‐binding | 8183370 | −10.05; 4.24e‐08 | −11.058; 7.74229308501e‐09 | −257.73; 0.88 | 510.54 | −12813.01 | 1601.06 | 1808.146 | −836 | −252.14; 55.91 | 19,970 | 1; −2.23 | 580.92 | −13894.81 | 1991.23 |
| 4‐Bax | G145A | Complete Loss of Bax‐binding | 8183370; 17418785; 25703009; 9202007 | −8.96; 2.66e‐07 | −11.08; 7.4604444936e‐09 | −355.61; 36.65 | 548.35 | −12489.31 | 1616.96 | 1767.024 | −909.9 | −259.09; 1.91 | 18,422 | 8; −7.10 | 601.84 | −14169.27 | 1992.47 |
| 5‐Bax | G145E | Complete Loss of Bax‐binding | 8183370; 17418785; 20382739 | −10.98; 8.81e‐09 | −11.074; 7.53261663003e‐09 | −281.92; 40.41 | 475.4 | −12654.42 | 1559.74 | 1735.535 | −825.2 | −245.00; 0.65 | 19,564 | 3; −22.81 | 739.89 | −14144.15 | 2049.74 |
| 6‐Bax | R146A | Complete Loss of Bax‐binding | 8183370 | −10.73; 1.35e‐08 | −11.094; 7.28756127346e‐09 | −272.73; 3.76 | 558.55 | −12898.91 | 1737.22 | 1652.922 | −993.2 | −232.13; 1.43 | 17,126 | 6; −1.46 | 531.36 | −14327.42 | 1956.29 |
| 7‐Bax | W188A | Complete Loss of Bax‐binding | 8183370 | −9.54; 1.00e‐07 | −11.059; 7.73769580621e‐09 | −346.02; 0.23 | 511.99 | −12900.89 | 1566.34 | 1702.357 | −882.9 | −263.68; 0.68 | 17,904 | 4; −7.14 | 524.01 | −13998.5 | 1862.52 |
| 8‐Bax | Q190L | Partial loss of Bax‐binding | 8183370 | −10.07; 4.13e‐08 | −11.048; 7.88061661122e‐09 | −276.34; 0.63 | 573.68 | −12959.51 | 1652.88 | 1705.53 | −915.5 | −248.37; 0.89 | 17,768 | 9; −20.98 | 527.54 | −14070 | 1939.9 |
| 9‐Bax | D191A | No effect on Bax‐binding | 8183370 | −9.54; 1.00e‐07 | −11.071; 7.57463963234e‐09 | −261.66; 0.82 | 550.7 | −12894.51 | 1698.28 | 1669.818 | −968.3 | −240.08; 2.50 | 16,914 | 3; −10.21 | 527.54 | −14070 | 1939.9 |
| 10‐Bax | N192A | Partial loss of Bax‐binding | 8183370 | −11.35; 4.78e‐09 | −11.038; 8.00847792497e‐09 | −268.03; 0.55 | 545.87 | −12986.21 | 1599.07 | 1,630.672 | −854 | −272.98; 3.10 | 18,300 | 1; −3.14 | 544 | −13902.16 | 1949.79 |
| 11‐Bax | E200A | Partial loss of Bax‐binding | 8183370 | −10.19; 3.37e‐08 | −11.097; 7.2561981079e‐09 | −269.79; 0.96 | 574.19 | −12905.27 | 1523.53 | 1857.958 | −844.8 | −280.46; 0.69 | 18,472 | 9; −10.58 | 608.72 | −14036.8 | 2011.58 |
| 12‐Bax | F104C | Complete Loss of ABT‐199 binding | 24786774 | −9.61; 8.95e‐08 | −11.221; 5.87634318994e‐09 | −277.79; 59.34 | 537.47 | −12650.5 | 1646.94 | 1627.871 | −929.3 | −251.54; 37.48 | 16,946 | 10; −19.93 | 555.54 | −13739.84 | 1977.78 |
| 13‐Bax | F104L | Complete Loss of ABT‐199 binding | 24786774 | −11.05; 7.86e‐09 | −11.08; 7.4604444936e‐09 | −250.22; 70.08 | 535.97 | −12872.47 | 1,570.45 | 1537.393 | −843.0 | −234.76; 48.47 | 17,936 | 5; −28.12 | 561.97 | −13744.5 | 1,770.14 |
| 14‐Bax | MISSING 194‐197 | Complete Loss of Bax‐binding | 8183370 | −9.20; 1.78e‐07 | −11.143; 6.71211168528e‐09 | −253.52; 60.27 | 407.88 | −12192.09 | 1,458.03 | 1651.886 | −896.3 | −253.17; 49.06 | 17,396 | 1; 3.54 | 624.87 | −13848.6 | 1926.85 |
In sequence‐based docking, PPA predicted higher affinity (ΔG) for five Bcl‐2 mutants, FRDG138‐141AAAA, G145E, R146A, N192A, and F104L with wild‐type Bax while, found lower in eight mutants (W144A, G145A, W188A, Q190L, D191A, E200, F104C, and MISSING194‐197) as compared to control (Table 3). Similarly, ISLAND predicted higher affinity for five out of 13 mutants (FRDG138‐141AAAA, R146A, E200A, F104C, MISSING194‐197); G145A and F104L have neutral effects but W144A, G145E, W188A, Q190L, D191A, and N192A have disrupted effect on binding with Bax. Nonetheless, HDOCK identified W144A, F104L, and MISSING194‐197 have decreased binding affinity. However, all mutants have reduced binding affinity with Bax as compared to control as predicted by InterEVDock. Besides, mutants Q190L and N192A identified by SOAP_PP; and R146A identified using FRODOCK2 possessed a higher affinity with Bax. These docking results demonstrated that these Bcl‐2 mutants which have high affinity with wild‐type Bax would not leave Bax free, resulting in diminutive apoptosis. Instead, lower or no binding affinity of Bcl‐2 mutants with Bax, makes Bax free which leads to increased apoptosis. Furthermore, the cumulative effect of all the sequence‐based docking results exhibited that Bcl‐2 mutation, W144A reduced the binding affinity with Bax which also supported the experimental known results.
Collectively, the Bcl‐2 mutants and Bax wild‐type interactions in 13 Bcl‐2 mutants were identified disrupted using sequence‐based docking, PPA‐Pred2 for eight complexes; ISLAND for six complexes; HDOCK for three complexes; InterEvDock for all 13 complexes; SOAP_PP for 11 complexes; and FRODOCK2 for 12 complexes (Figure 2).
Figure 2.
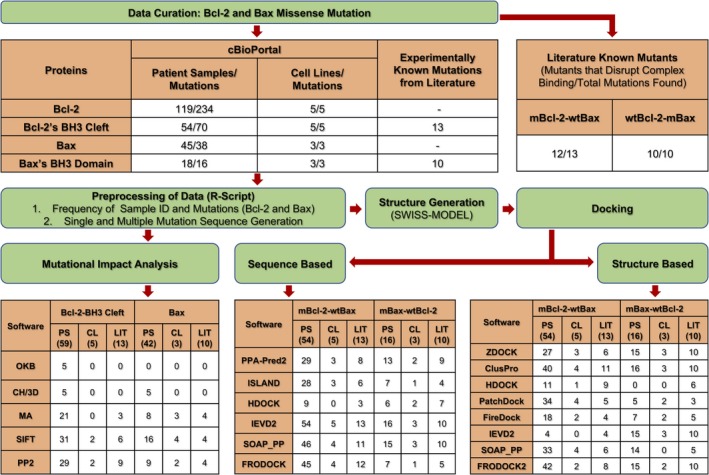
Work flow of the study represents total Bcl‐2 mutations, 234 from 119 patient samples, five from five cell lines, though, Bcl‐2’s BH3 cleft mutations, 70 from 54 patient samples, and five from five cell lines were curated from cBioPortal. Likewise, overall 38 Bax mutations from 45 patient samples, and three from three cell lines whereas, 16 from 18 patient samples were curated. Moreover, experimental known binding impact of 13 mutants of Bcl‐2’s BH3 cleft, and 10 mutants of Bax's BH3 domain were retrieved from literature. Among these 13 Bcl‐2 mutants, the binding were reported disrupted for 12 mutants with wild‐type Bax, but in case of 10 Bax mutants the binding of all 10 were found disrupted with wild‐type Bcl‐2. Further, preprocessing of the data was performed using in‐house R scripts wherein, frequency of sample ID, Bcl‐2 and Bax mutations, and their single and multiple mutated sequences were generated. The Bcl‐2 and Bax mutation impact, oncogenic, cancer hotspots, medium, deleterious, and probably damaging was predicted by OncoKB (OKB), Cancer Hotspot/ 3D Hotspot (CH/3D), Mutation Assessor (MA), SIFT, and PolyPhen‐2 (PP2) for respective total numbers of patient samples (PS), cell lines (CL), literature (LIT). The 3D structures of mutated and wild‐type sequences of Bcl‐2 and Bax were generated using SWISS‐MODEL web‐server. Subsequently, sequence and structure‐based dockings were performed, and further obtained the number of disrupted mutated Bcl‐2 and wild‐type Bax (mBcl‐2‐wtBax) and vice‐versa (wtBcl‐2‐mBax) complexes among total number of samples
Structure‐based docking by ZDOCK score predicted the six mutants, R146A, D191A, N192A, F104C, F104L and MISSING194‐197 which have reduced binding affinity with Bax compared to wild‐type complex (Table 3). Likewise, decreased binding affinity of wild‐type Bax was identified with Bcl‐2 mutants, W144A, G145A, G145E, W188A, Q190L, and D191A by ClusPro; FRDG138‐141AAAA, W144A, G145E, R146A, Q190L, D191A, F104C, F104L and MISSING194‐197 in HDOCK; R146A, Q190L, D191A, F104C, and MISSING194‐197 in PatchDock; W144A, R146A, N192A, and MISSING194‐197 in FireDock; R146A, W188A, Q190L, D191A by InterEVDock score; FRDG138‐141AAAA, W144A, W188A, F104C, F104L and MISSING194‐197 by SOAP_PP score; and FRDG138‐141AAAA, R146A, W188A, Q190L, D191A, N192A, F104L, and MISSING194‐197 in FRODOCK2 score. Collectively, all structure‐based docking of Bcl‐2 mutants displayed inconsistencies for increased and decreased binding affinity with Bax.
Among 13 Bcl‐2 mutants, only 6, 11, 9, 5, 4, 4, 6, and 8 retrieved from literature disrupt interaction with wild‐type Bax, identified by ZDOCK, ClusPro, HDOCK, PatchDock, FireDock, InterEvDock, SOAP_PP, and FRODOCK2, respectively (Figure 2).
The sequence‐based docking of Bcl‐2 mutants retrieved from cBioPortal with wild‐type Bax was identified by PPA‐Pred2 in which 30 patient samples were found to be decreased, and 22 samples increased the binding affinity (Table S10). The mutant, E160D and T178N was found to have a neutral effect. Similarly, ISLAND predicted 28 samples with decreased binding affinity, and increased affinity for Bax was obtained for 12 samples whereas, 14 samples do not affect Bax binding. HDOCK score predicted nine samples with reduced binding affinity and 45 samples have higher affinity with Bax. Interestingly, InterEvDock identified all samples having Bcl‐2 mutations with a lower binding affinity for Bax. Nonetheless, among 54 Bcl‐2 mutants, SOAP_PP predicted 46 samples, and FRODOCK2 identified 45 samples which decrease binding affinity with wild‐type Bax. Also, the cell line having Bcl‐2 mutations R129C, V156F, and R106L (PPA‐Pred2); S117R, and V156F, and R106L (ISLAND); none (HDOCK); R129C, L95V, S117R, V156F, and R106L (InterEvDock); S117R, and R129C (SOAP_PP); S117R, R129C, V156F, L95V (FRODOCK2) predicted with lower binding affinity for Bax. However, complex, 46‐Bax and 50‐Bax of the patient sample having Bcl‐2 mutations A113G/V133A/A131D, and T132M/V134L/A131D respectively, exhibit lower binding affinity was confirmed by all sequence‐based six docking methods.
Collectively, the Bcl‐2 mutants and Bax wild‐type interactions in 54 patient samples and five cell lines were identified disrupted by InterEvDock for all 54 and five complexes; SOAP_PP for 47 and four complexes; FRODOCK2 for 46 and four complexes; PPA‐Pred2 for 30 and three complexes; ISLAND for 28 and three complexes; and HDOCK for nine and zero complexes (Figure 2).
Structure‐based docking for patient samples categorized 26 complexes associated with reduced binding affinity, and 28 complexes resulted in higher binding affinity using ZDOCK score (Table S11). Nonetheless, ClusPro predicted 40 complexes having a lower affinity, one with neutral impact, and 13 were associated with higher affinity. HDOCK identified 11 complexes having a lower affinity, while 43 possessed higher affinity compared to wild‐type complex. PatchDock predicted 34 complexes which have lower, and 20 have higher binding affinity compared to control. Although, FireDock classified 18 complexes which have lower and 36 complexes into higher binding affinity.
Further, InterEVDock2 identified four complexes with decreased, one neutral, and 49 have increased binding affinity compared to wild‐type. SOAP_PP projected 33 complexes which have reduced, and 21 complexes to have enhanced binding affinity. Nevertheless, the majority of the complexes, 41 have decreased, and 12 complexes identified with increased binding affinity.
Likewise, cell lines having Bcl‐2 mutations, R129C, V156F, and L95V identified low while, S117R and R106L possessed higher affinity for Bax by ZDOCK (Table S11). Nevertheless, S117R, R129C, V156F, and R106L have decreased binding affinity except for L95V that showed increased binding impact identified using ClusPro. HDOCK identified four mutants with increased binding affinity whereas, R129C have decreased affinity. In contrast, PatchDock and SOAP_PP predicted lower binding affinity to all mutants except S117R and R106L respectively. FireDock score assigned S117R, V156F, and L95V mutants with increased binding affinity while R129C and R106L have decreased affinity. Although, all docked complexes were shown to have higher binding affinity identified by InterEvDock and FRODOCK2.
The cumulative effect of structure‐based docking in patient samples demonstrated that increased binding affinity of Bcl‐2 mutants, S105P, Q118H, and G145E was corroborated maximally by seven docking methods (Table S11). Moreover, S117R exhibited higher binding affinity in both patient sample and cell lines which was confirmed by six docking methods. In contrast, the Bcl‐2 mutants R129C, N163S, and N172Q in patient samples have a lower binding affinity, predicted by seven docking tools. Jointly, Bcl‐2 mutants that disrupt interaction with wild‐type Bax among 54 total patient samples and five cell lines were identified by ZDOCK, 28 and three; ClusPro, 41 and four; HDOCK, 12 and one; PatchDock, 35 and four; FireDock, 19 and two; InterEvDock, four and zero; SOAP_PP, 34 and four; and FRODOCK2, 43 and two, respectively (Figure 2). This result showed the close resemblance between the scoring of ClusPro, PatchDock, SOAP_PP, and FRODOCK2.
Moreover, no common sample id was identified where both mutations fall in Bcl‐2 or Bax. The experimental effect of Bax mutants on wild‐type Bcl‐2 was collected from literature (Table 4). Subsequently, Sequence‐based dockings were performed wherein, PPA‐Pred2 predicted the lowest binding affinity for all complexes excluding R78A mutant. Based on ΔΔG, ISLAND identified E61A, R65A, D68A, E69A, R78A, and E61A/R65A/R78A mutants having a higher binding affinity. The mutant M74D, M74E, and R78A possessed higher binding affinity while, G67R, E61A, R65A, D68A, E69A, E61A/R65A/R78A, and I66E/D68R mutants have disrupted the binding with wild‐type complex, predicted by HDOCK. Remarkably, no high‐affinity complex was noted in InterEvDock and SOAP_PP docking. The higher binding affinity was identified by FRODOCK2 for M74D, G67R, E61A, R65A, and D68A mutants, though M74E, E69A, R78A, E61A/R65A/R78A, and I66E/D68R mutants showed lower binding affinity.
Table 4.
Impact of known experimental mutation of Bax with wild‐type Bcl‐2
| Complexes | Bax mutations | Experimental Impact of Bax mutants with Bcl‐2 wild‐type | Pubmed ID | Sequence‐based docking | Structure‐based docking | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PPA‐Pred2 ΔG | ISLAND(ΔΔG; Kd) | HDOCK score and ligand rmsd (Å) | InterEvDock score | SOAP_PP score | FRODOCK2 score | ZDOCK score | ClusProscore | HDOCK score | PatchDock score | FireDock | InterEvDock score | SOAP_PP score | FRODOCK2 score | ||||
| 1‐Bcl‐2 | Wild‐type | Bind with Bcl‐2 | 22155216 | −10.63; 1.60e‐08 | −11.08; 7.4604444936e‐09 | −228.19; 56.74 | 696.4 | −12906.73 | 1701.52 | 1,670.275 | −969.9 | −226.16; 43.58 | 17,778 | 5; −3.91 | 542.35 | −14018.72 | 1958.41 |
| 2‐Bcl‐2 | M74D | Strongly reduced interaction with Bcl‐2. | 21199865 | −9.15; 1.96e‐07 | −11.027; 8.16707446983e‐09 | −228.99; 59.18 | 672.77 | −12711.00 | 1702.02 | 1,376.78 | −836.9 | −220.78; 30.61 | 18,188 | 1; −2.51 | 493.65 | −14141.73 | 1892.05 |
| 3‐Bcl‐2 | M74E | Strongly reduced interaction with Bcl‐2. | 21199865 | −8.88; 3.08e‐07 | −11.027; 8.16707446983e‐09 | −229.58; 39.73 | 575.82 | −12652.06 | 1697.73 | 1,376.937 | −840.3 | −221.52; 30.64 | 17,258 | 8; −7.49 | 493.29 | −14046.23 | 1900.81 |
| 4‐Bcl‐2 | G67R | Loss of heterodimerization with Bcl‐2 | 7475270; 9531611 | −9.60; 9.04e‐08 | −10.993; 8.64245933479e‐09 | −220.05; 61.09 | 409.64 | −12411.27 | 1744.87 | 1593.659 | −787.5 | −242.72; 42.28 | 17,352 | 2; −28.87 | 452.62 | −14178.76 | 1681.72 |
| 5‐Bcl‐2 | E61A | Reduced the affinities with Bcl‐2 | 21060336 | −10.05; 4.24e‐08 | −11.185; 6.25205707374e‐09 | −226.30; 55.71 | 585.11 | −12649.85 | 1734.13 | 1,376.703 | −852.4 | −224.40; 44.75 | 17,066 | 4; −10.56 | 489.33 | −13894.91 | 1818.77 |
| 6‐Bcl‐2 | R65A | Reduced the affinities with Bcl‐2 | 21060336 | −8.96; 2.67e‐07 | −11.139; 6.75043626242e‐09 | −226.67; 29.40 | 589.99 | −12564.84 | 1736.22 | 1,460.780 | −895.3 | −227.49; 33.15 | 19,412 | 8; −2.79 | 479.66 | −13993.99 | 1812.4 |
| 7‐Bcl‐2 | D68A | Greatly reduced the affinities with Bcl‐2 | 21060336 | −6.99; 7.45e‐06 | −11.12; 6.97977542734e‐09 | −224.38; 40.15 | 507.53 | −12754.91 | 1,850.12 | 1,375.982 | −849.1 | −232.13; 44.42 | 19,330 | 10; −29.97 | 498.68 | −13907.78 | 1,880.18 |
| 8‐Bcl‐2 | E69A | Reduced the affinities with Bcl‐2 | 21060336 | −8.50; 5.81e‐07 | −11.092; 7.31216923735e‐09 | −221.72; 42.42 | 513.82 | −12679.25 | 1681.43 | 1,370.099 | −855 | −251.21; 44.67 | 18,508 | 10; −9.31 | 479.66 | −13858.86 | 1952.09 |
| 9‐Bcl‐2 | R78A | Reduced the affinities with Bcl‐2 | 21060336 | −10.73; 1.35e‐08 | −11.188; 6.22064695229e‐09 | −241.20; 28.87 | 581.95 | −12891.89 | 1608.04 | 1,376.562 | −841.2 | −225.76; 46.04 | 17,840 | 10; 0.88 | 520.86 | −14024.04 | 1853.4 |
| 10‐Bcl‐2 | E61A/R65A/R78A | Greatly reduced the affinities with Bcl‐2 | 21060336 | −9.80; 6.48e‐08 | −11.355; 4.6932701273e‐09 | −224.17; 40.30 | 638.81 | −12616.22 | 1681.58 | 1,461.352 | −816.8 | −224.04; 46.12 | 18,510 | 8; 0.29 | 530.58 | −14014.14 | 1832.93 |
| 11‐Bcl‐2 | 166E/D68R | Completely lost the binding | 25703009 | −8.45; 6.36e‐07 | −11.075; 7.52808568847e‐09 | −224.95; 55.08 | 551.8 | −12430.25 | 1677.31 | 1,373.619 | −778 | −221.59; 30.63 | 18,176 | 10; 1.87 | 469.45 | −14031.22 | 1916.36 |
Correspondingly, in structure‐based docking by ZDOCK, ClusPro, InterEvDock and FRODOCK2 wherein, all complexes belong to lower binding affinity. The mutant G67R, R65A, D68A, and E69A in HDOCK displayed higher binding affinity (Table 4). PatchDock showed a higher binding affinity for M74D, R65A, D68A, E69A, R78A, E61A/R65A/R78A, and I66E/D68R mutants but M74E, G67R, and E61A mutants displayed lower affinity. Similarly, FireDock identified mutant M74E, G67R, E61A, D68A, and E69A exhibited higher binding affinity while M74D, R65A, R78A, E61A/R65A/R78A, and I66E/D68R showed lower binding affinity. The higher binding score of SOAP_PP was predicted for M74D, M74E, G67R, R78A, and I66E/D68R, though E61A, R65A, D68A, E69A, and E61A/R65A/R78A mutants disturbed the binding. Thus experimental evidence was substantiated with sequence‐based docking tools (InterEvDock and SOAP_PP) and structure‐based docking tools (ZDOCK, ClusPro, and FRODOCK2).
Jointly, Bax mutants that disrupt interaction with wild‐type Bcl‐2 among 10 total experimental known mutations from the literature were identified by sequence‐based docking, PPA‐Pred2, nine; ISLAND, four; HDOCK, seven; InterEvDock, 10; SOAP_PP, 10; and FRODOCK2, five. Similarly, in structure‐based docking, the Bax mutants which disrupted the binding with wild‐type Bcl‐2 were identified, ZDOCK, 10; ClusPro, 10; HDOCK, six; PatchDock, three; FireDock, five; InterEvDock, 10; SOAP_PP, five; and FRODOCK2, 10, respectively (Figure 2). This result revealed that the correct prediction was displayed by sequence‐based InterEvDock and SOAP_PP docking. Likewise, ZDOCK, ClusPro, InterEvDock, and FRODOCK2 structure‐based docking agreed with the experimental validated binding impact of Bax mutants with wild‐type Bcl‐2. Thus, this result revealed that these methods have higher sensitivity as compared to other methods.
Additionally, higher binding affinity has been identified for wild‐type Bcl‐2 with Bax mutants, R89Q and R89L among total 16 mutations in patient sample by sequence‐based docking using PPA‐Pred2 (Table S12). The affinity of R89L, A42S, E44Q, and R89Q has been identified as higher according to ISLAND △△G score. However, higher binding affinity was recognized by HDOCK maximally by 10 mutants (F93L, A54V, R89Q, A82T, G67V, R89L, Q52H, E44K, A82S, and P88S). Conversely, InterEvDock displayed a lower binding affinity to all mutants, though highest affinity was exhibited by E41K mutant using SOAP_PP. Also, nine Bax mutants have higher affinity but E41K, E44K, Q52H, G67V, A82S, R89Q, and R89L mutants disturbed the binding affinity as compared to wild‐type predicted by FRODOCK2.
Likewise, sequence‐based docking of Bax mutants, occured in the cell lines, was performed with wild‐type Bcl‐2 wherein, PPA‐Pred2 predicted a decreased binding affinity of mutant G67R, and V83M (Table S12). Nevertheless, a lower binding affinity was anticipated by ISLAND for Bax mutant, G67R; and HDOCK for G67R, and V83M mutants. InterEvDock, and SOAP_PP identified that all Bax mutants disrupted their binding with wild‐type Bcl‐2. However, FRODOCK2 identified the lower affinity of V83M Bax mutants with wild‐type Bcl‐2.
Communally, 16 and three BH3 domains of Bax mutants that disrupt interaction with wild‐type Bcl‐2 in patient samples and cell lines, respectively, were identified by sequence‐based docking, PPA‐Pred2, 13 and two; ISLAND, seven and one; HDOCK, six and two; InterEvDock, 16 and three; SOAP_PP, 15 and three; and FRODOCK2, seven and one, respectively (Figure 2).
The structure‐based docking of Bax mutants with wild‐type Bcl‐2 in patient samples was analyzed (Table S13). The docking score of mutants, R89Q showed highest binding affinity by ZDOCK, InterEvDock, SOAP_PP, and FRODOCK2. Nevertheless, ClusPro indicated all mutants have lesser binding affinity compared to control. In contrast, HDOCK exposed that all mutants have a higher binding affinity as compared to wild‐type complex. Besides, 11 Bax mutants (F93L, Q52H, E41K, E44Q, A54V, A42S, P88L, E44K, A82T, A82S, and G67V) in PatchDock, and nine (P88S, G67R, E44Q, E44K, L63I‐G67R, Q52H, G67V, R89L, and R89Q) among 16 mutants in FireDock displayed higher binding affinity.
In cell lines, all mutants have lower binding affinity identified by ZDOCK, ClusPro and InterEvDock score (Table S13). Contrariwise, HDOCK and SOAP_PP identified a higher binding affinity for all Bax mutants which implies that no mutant has disruptive effect. Nonetheless, all mutants have reduced affinity predicted by PatchDock except V83M. The Bax mutants, E75K, and V83M disrupted the binding with wild‐type Bcl‐2 identified by FireDock, while G67R increased the affinity. Although, G67R, and E75K have a lower binding affinity identified by FRODOCK2 whereas, V83M increased the affinity. Suggestively, these data convinced that HDOCK, ClusPro, and InterEvDock2 have similar docking impact for structure‐based docking for Bax mutant, while reverse effects were noted for ZDOCK and SOAP_PP.
Unequivocally, the Bax mutants 16 in patient samples and three in cell lines which disrupted the binding with wild‐type Bcl‐2 were identified by structure‐based docking, ZDOCK, 15 and three; ClusPro, 16 and three; HDOCK, 0 and 0; PatchDock, five and two; FireDock, seven and two; InterEvDock, 15 and three; SOAP_PP, 14 and 0; and FRODOCK2, 15 and two, respectively (Figure 2).
4. CONCLUSION
The mutation impact prediction tools used in this study have identified the impact of missense mutations, which would be used to develop a method to predict cancer‐causing mutations. Therefore, identification of driver and passenger mutations would probably be robust candidate biomarkers for personalized cancer therapies. Using several tools, we identified Bcl‐2 and Bax mutation hotspots as well as new ones and further showed the potential utility of mutation hotspot and evaluation of cancer drivers. Whereas, the most common Bcl‐2 mutation hotspots need to be explored for the development of potentially curative combination therapies and stratified cancer care. Thus, mutations which enhance the binding affinity of the Bcl‐2 mutant with wild‐type Bax or vice‐versa would be the targets to prevent cancer progression. Conclusively, this study revealed that docking is a crucial step to identify the driver mutation based on binding affinity which would likely be used to eradicate cancers.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
The authors are thankful to the funding agencies, Department of Science and Technology‐Science and Engineering Research Boards (DST‐SERB), the Council of Scientific and Industrial Research (CSIR), University Grant Commission (UGC), and DST Govt. of India for fellowships. PKR would like to acknowledge DST‐SERB, India for providing a financial grant (PDF/2016/003387) for this work.
Raghav PK, Kumar R, Kumar V, Raghava GPS. Docking-based approach for identification of mutations that disrupt binding between Bcl-2 and Bax proteins: Inducing apoptosis in cancer cells. Mol Genet Genomic Med. 2019;7:e910 10.1002/mgg3.910
Contributor Information
Pawan Kumar Raghav, Email: pwnrghv@gmail.com.
Gajendra P. S. Raghava, Email: raghava@iiitd.ac.in.
REFERENCES
- Abbasi, W. A. , Hassan, F. U. , Yaseen, A. , & Minhas, F. U. A. A. (2017). ISLAND: In‐Silico prediction of proteins binding affinity using sequence descriptors . Retrieved from https://arxiv.org/abs/1711.10540 [DOI] [PMC free article] [PubMed]
- Adzhubei, I. , Jordan, D. M. , & Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen‐2. Current Protocols in Human Genetics, 76:7.20.1–7.20.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov, L. B. , Nik‐Zainal, S. , Wedge, D. C. , Aparicio, S. A. J. R. , Behjati, S. , Biankin, A. V. , … Stratton, M. R. (2013). Signatures of mutational processes in human cancer. Nature, 500, 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus, H. H. J. , Van, G. C. J. , Vos, W. , Dukers, D. F. , Bloemena, E. , Wouters, D. , … Peters, G. J. (2002). Differential expression of cell cycle and apoptosis related proteins in colorectal mucosa, primary colon tumours, and liver metastases. Journal of Clinical Pathology, 55, 206–211. 10.1136/jcp.55.3.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina, J. , Caponigro, G. , Stransky, N. , Venkatesan, K. , Margolin, A. A. , Kim, S. , … Garraway, L. A. (2012). The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–607. 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina, J. , Taylor, B. S. , Banerji, S. , Ramos, A. H. , Lagos‐Quintana, M. , DeCarolis, P. L. , … Singer, S. (2010). Subtype‐specific genomic alterations define new targets for soft‐tissue sarcoma therapy. Nature Genetics, 42, 715–721. 10.1038/ng.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudot, A. , Real, F. X. , Izarzugaza, J. M. , & Valencia, A. (2009). From cancer genomes to cancer models: Bridging the gaps. EMBO Reports, 10, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, D. , Berchuck, A. , Birrer, M. , Chien, J. , Cramer, D. W. , Dao, F. , … Gross, J. (2011). Integrated genomic analyses of ovarian carcinoma. Nature, 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buljan, M. , Blattmann, P. , Aebersold, R. , & Boutros, M. (2018). Systematic characterization of pan‐cancer mutation clusters. Molecular Systems Biology, 14, e7974 10.15252/msb.20177974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network . (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature, 489, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty, D. , Gao, J. , Phillips, S. , Kundra, R. , Zhang, H. , Wang, J. , … Schultz, N. (2017). OncoKB: A precision oncology knowledge base. JCO Precision Oncology, 1, 1–16. 10.1200/PO.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, M. T. , Bhattarai, T. S. , Schram, A. M. , Bielski, C. M. , Donoghue, M. T. A. , Jonsson, P. , … Taylor, B. S. (2018). Accelerating discovery of functional mutant alleles in cancer. Cancer Discovery, 8, 174–183. 10.1158/2159-8290.CD-17-0321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia, C. , Schneider, P. A. , Dai, H. , Dogan, A. , Maurer, M. J. , Church, A. K. , … Kaufmann, S. H. (2015). BCL2 mutations are associated with increased risk of transformation and shortened survival in follicular lymphoma. Blood, 125, 658–667. 10.1182/blood-2014-04-571786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar, P. E. , Lee, E. F. , Thompson, G. V. , Wardak, A. Z. , Fairlie, W. D. , & Colman, P. M. (2011). Mutation to Bax beyond the BH3 domain disrupts interactions with pro‐survival proteins and promotes apoptosis. Journal of Biological Chemistry, 286, 7123–7131. 10.1074/jbc.M110.161281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer‐Costa, C. , Orozco, M. , & de la Cruz, X. (2002). Characterization of disease‐associated single amino acid polymorphisms in terms of sequence and structure properties 1 1Edited by. Journal of Molecular Biology, 315, 771–786. 10.1006/jmbi.2001.5255 [DOI] [PubMed] [Google Scholar]
- Fischer, A. , Greenman, C. , & Mustonen, V. (2011). Germline fitness‐based scoring of cancer mutations. Genetics, 188, 383–393. 10.1534/genetics.111.127480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruehauf, J. P. , & Meyskens, F. L. (2007). Reactive oxygen species: A breath of life or death? Clinical Cancer Research, 13, 789–794. 10.1158/1078-0432.CCR-06-2082 [DOI] [PubMed] [Google Scholar]
- Gao, J. , Aksoy, B. A. , Dogrusoz, U. , Dresdner, G. , Gross, B. , Sumer, S. O. , … Schultz, N. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signalling, 6, pl1 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, J. , Chang, M. T. , Johnsen, H. C. , Gao, S. P. , Sylvester, B. E. , Sumer, S. O. , … Sander, C. (2017). 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Medicine, 9, 4 10.1186/s13073-016-0393-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, S. , & Blundell, T. L. (2010). Structural and functional restraints on the occurrence of single amino acid variations in human proteins. PLoS One, 5, e9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, S. (2003). Molecular signaling in death receptor and mitochondrial pathways of apoptosis (Review). International Journal of Oncology, 22, 15–20. 10.3892/ijo.22.1.15 [DOI] [PubMed] [Google Scholar]
- Hancock, J. T. , Desikan, R. , & Neill, S. J. (2001). Role of reactive oxygen species in cell signalling pathways. Biochemical Society Transactions, 29, 345–350. 10.1042/bst0290345 [DOI] [PubMed] [Google Scholar]
- Koboldt, D. C. , Fulton, R. S. , McLellan, M. D. , Schmidt, H. , Kalicki‐Veizer, J. , McMichael, J. F. , … Ally, A. (2012). Comprehensive molecular portraits of human breast tumours. Nature, 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer, S. J. , Yin, X. M. , Oltvai, Z. N. , Veis‐Novack, D. J. , & Linette, G. P. (1995). Reactive oxygen species and the regulation of cell death by the Bcl‐2 gene family. Biochimica et Biophysica Acta, 1271, 63–66. 10.1016/0925-4439(95)00011-R [DOI] [PubMed] [Google Scholar]
- Kozakov, D. , Hall, D. R. , Xia, B. , Porter, K. A. , Padhorny, D. , Yueh, C. , … Vajda, S. (2017). The ClusPro web server for protein–protein docking. Nature Protocols, 12, 255–278. 10.1038/nprot.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku, B. , Liang, C. , Jung, J. U. , & Oh, B.‐H. (2011). Evidence that inhibition of BAX activation by BCL‐2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Research, 21, 627–641. 10.1038/cr.2010.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, M. S. , Stojanov, P. , Mermel, C. H. , Robinson, J. T. , Garraway, L. A. , Golub, T. R. , … Getz, G. (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature, 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson, M. D. M. , Vandin, F. , Wu, H.-T. , Dobson, J. R. , Eldridge, J. V. , Thomas, J. L. , … Raphael, B. J. (2015). Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nature Genetics, 47, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Y. , Wu, S. , Yue, Y. , He, S. , Li, J. , Tang, J. , … Zhou, H. B. (2016). Gossypol with hydrophobic linear esters exhibits enhanced antitumor activity as an inhibitor of antiapoptotic proteins. ACS Medicinal Chemistry Letters, 7, 1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luanpitpong, S. , Chanvorachote, P. , Stehlik, C. , Tse, W. , Callery, P. S. , Wang, L. , & Rojanasakul, Y. (2013). Regulation of apoptosis by Bcl‐2 cysteine oxidation in human lung epithelial cells. Molecular Biology of the Cell, 24, 858–869. 10.1091/mbc.e12-10-0747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiach, E. , Schneidman‐Duhovny, D. , Andrusier, N. , Nussinov, R. , & Wolfson, H. J. (2008). FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Research, 36, W229–W232. 10.1093/nar/gkn186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiach, E. , Schneidman‐Duhovny, D. , Peri, A. , Shavit, Y. , Nussinov, R. , & Wolfson, H. J. (2010). An integrated suite of fast docking algorithms. Proteins: Structure, Function, and Bioinformatics, 78, 3197–3204. 10.1002/prot.22790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell, T. J. , & Korsmeyer, S. J. (1991). Progression from lymphoid hyperplasia to high‐grade malignant lymphoma in mice transgenic for the t(14;18). Nature, 349, 254–256. 10.1038/349254a0 [DOI] [PubMed] [Google Scholar]
- McLendon, R. , Friedman, A. , Bigner, D. , Van, M. E. G. , Brat, D. J. , Mastrogianakis, G. , … Bogler, O. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijerink, J. P. , Mensink, E. J. , Wang, K. , Sedlak, T. W. , Slöetjes, A. W. , de Witte, T. , … Korsmeyer, S. J. (1998). Hematopoietic malignancies demonstrate loss‐of‐function mutations of BAX. Blood, 91, 2991–2997. [PubMed] [Google Scholar]
- Meijerink, J. P. , Smetsers, T. F. , Slöetjes, A. W. , Linders, E. H. , & Mensink, E. J. (1995). Bax mutations in cell lines derived from hematological malignancies. Leukemia, 9, 1828–1832. [PubMed] [Google Scholar]
- Merid, S. , Goranskaya, D. , & Alexeyenko, A. (2014). Distinguishing between driver and passenger mutations in individual cancer genomes by network enrichment analysis. BMC Bioinformatics, 15, 308 10.1186/1471-2105-15-308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M. P. , & Kumar, S. (2001). Understanding human disease mutations through the use of interspecific genetic variation. Human Molecular Genetics, 10, 2319–2328. 10.1093/hmg/10.21.2319 [DOI] [PubMed] [Google Scholar]
- Morin, R. D. , Mendez‐Lago, M. , Mungall, A. J. , Goya, R. , Mungall, K. L. , Corbett, R. D. , … Marra, M. A. (2011). Frequent mutation of histone‐modifying genes in non‐Hodgkin lymphoma. Nature, 476, 298–303. 10.1038/nature10351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network, T. C. G. A. (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature, 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, P. C. , & Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Research, 31, 3812–3814. 10.1093/nar/gkg509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov, R. , & Tsai, C.‐J. (2015). ‘Latent drivers’ expand the cancer mutational landscape. Current Opinion in Structural Biology, 32, 25–32. 10.1016/j.sbi.2015.01.004 [DOI] [PubMed] [Google Scholar]
- Perini, G. F. , Ribeiro, G. N. , Pinto Neto, J. V. , Campos, L. T. , & Hamerschlak, N. (2018). BCL‐2 as therapeutic target for hematological malignancies. Journal of Hematology & Oncology, 11, 65 10.1186/s13045-018-0608-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce, B. G. , Wiehe, K. , Hwang, H. , Kim, B.‐H. , Vreven, T. , & Weng, Z. (2014). ZDOCK server: Interactive docking prediction of protein‐protein complexes and symmetric multimers. Bioinformatics, 30, 1771–1773. 10.1093/bioinformatics/btu097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignot, C. , Rey, J. , Yu, J. , Tufféry, P. , Guerois, R. , & Andreani, J. (2018). InterEvDock2: An expanded server for protein docking using evolutionary and biological information from homology models and multimeric inputs. Nucleic Acids Research, 46, W408–W416. 10.1093/nar/gky377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghav, P. K. , Verma, Y. K. , & Gangenahalli, G. U. (2012a). Peptide screening to knockdown Bcl‐2’s anti‐apoptotic activity: Implications in cancer treatment. International Journal of Biological Macromolecules, 50, 796–814. 10.1016/j.ijbiomac.2011.11.021 [DOI] [PubMed] [Google Scholar]
- Raghav, P. K. , Verma, Y. K. , & Gangenahalli, G. U. (2012b). Molecular dynamics simulations of the Bcl‐2 protein to predict the structure of its unordered flexible loop domain. Journal of Molecular Modeling, 18, 1885–1906. 10.1007/s00894-011-1201-6 [DOI] [PubMed] [Google Scholar]
- Ramírez‐Aportela, E. , López‐Blanco, J. R. , & Chacón, P. (2016). FRODOCK 2.0: Fast protein–protein docking server. Bioinformatics, 32, 2386–2388. [DOI] [PubMed] [Google Scholar]
- Reed, J. C. , & Tanaka, S. (1993). Somatic point mutations in the translocated bcl‐2 genes of Non‐Hodgkin’s Lvmphomasrl and Lvmphocvtic Leukemias: Imblications for mechanisms of tumor progression. Leukaemia & Lymphoma, 10, 157–163. [DOI] [PubMed] [Google Scholar]
- Rupnarain, C. , Dlamini, Z. , Naicker, S. , & Bhoola, K. (2004). Colon cancer: Genomics and apoptotic events. Biological Chemistry, 385, 449–464. 10.1515/BC.2004.053 [DOI] [PubMed] [Google Scholar]
- Schmitt, M. W. , Loeb, L. A. , & Salk, J. J. (2016). The influence of subclonal resistance mutations on targeted cancer therapy. Nature Reviews Clinical Oncology, 13, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz, J. M. , Johnson, N. A. , Morin, R. D. , Scott, D. W. , Tan, K. , Ben‐Nierah, S. , … Gascoyne, R. D. (2012). BCL2 mutations in diffuse large B‐cell lymphoma. Leukemia, 26, 1383–1390. 10.1038/leu.2011.378 [DOI] [PubMed] [Google Scholar]
- Schwede, T. , Kopp, J. , Guex, N. , & Peitsch, M. C. (2003). SWISS‐MODEL: An automated protein homology‐modeling server. Nucleic Acids Research, 31, 3381–3385. 10.1093/nar/gkg520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, K. , & Briggs, J. M. (2016). Functional Implications of the spectrum of BCL2 mutations in Lymphoma. Mutation Research, Reviews in Mutation Research, 769, 1–18. 10.1016/j.mrrev.2016.06.001 [DOI] [PubMed] [Google Scholar]
- Sitbon, E. , & Pietrokovski, S. (2007). Occurrence of protein structure elements in conserved sequence regions. BMC Structural Biology, 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitziel, N. O. , Tseng, Y. Y. , Pervouchine, D. , Goddeau, D. , Kasif, S. , & Liang, J. (2003). Structural Location of Disease‐associated Single‐nucleotide Polymorphisms. Journal of Molecular Biology, 327(5), 1021–1030. 10.1016/S0022-2836(03)00240-7 [DOI] [PubMed] [Google Scholar]
- Sunyaev, S. , Ramensky, V. , & Bork, P. (2000). Towards a structural basis of human non‐synonymous single nucleotide polymorphisms. Trends in Genetics, 16, 198–200. 10.1016/S0168-9525(00)01988-0 [DOI] [PubMed] [Google Scholar]
- Talavera, D. , Taylor, M. S. , & Thornton, J. M. (2010). The (non)malignancy of cancerous amino acidic substitutions. Proteins, 78, 518–529. [DOI] [PubMed] [Google Scholar]
- Taylor, B. S. , Schultz, N. , Hieronymus, H. , Gopalan, A. , Xiao, Y. , Carver, B. S. , … Gerald, W. L. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell, 18, 11–22. 10.1016/j.ccr.2010.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, A. E. , Orengo, C. A. , & Thornton, J. M. (2002). Plasticity of enzyme active sites. Trends in Biochemical Sciences, 27, 419–426. 10.1016/S0968-0004(02)02158-8 [DOI] [PubMed] [Google Scholar]
- Valdar, W. S. J. (2002). Scoring residue conservation. Proteins: Structure, Function, and Genetics, 48, 227–241. 10.1002/prot.10146 [DOI] [PubMed] [Google Scholar]
- Vaser, R. , Adusumalli, S. , Leng, S. N. , Sikic, M. , & Ng, P. C. (2016). SIFT missense predictions for genomes. Nature Protocols, 11, 1–9. 10.1038/nprot.2015.123 [DOI] [PubMed] [Google Scholar]
- Vaux, D. L. , Cory, S. , & Adams, J. M. (1988). Bcl‐2 gene promotes haemopoietic cell survival and cooperates with c‐myc to immortalize pre‐B cells. Nature, 335, 440–442. 10.1038/335440a0 [DOI] [PubMed] [Google Scholar]
- Yan, Y. , Zhang, D. , Zhou, P. , Li, B. , & Huang, S.‐Y. (2017). HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Research, 45, W365–W373. 10.1093/nar/gkx407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yugandhar, K. , & Gromiha, M. M. (2014). Protein–protein binding affinity prediction from amino acid sequence. Bioinformatics, 30, 3583–3589. [DOI] [PubMed] [Google Scholar]
- Zacarías‐Lara, O. J. , Correa‐Basurto, J. , & Bello, M. (2016). Exploring the conformational and binding properties of unphosphorylated/phosphorylated monomeric and trimeric Bcl‐2 through docking and molecular dynamics simulations. Biopolymers, 105(7), 393–413. 10.1002/bip.22839 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials