

Abstract
Glucagon plays an essential role in robust feedback regulation between the liver and α‐cells, and exerts suppressive or static effects on the plasma concentration of amino acids, especially glutamine. Thereby, “glutaminostatin” might be an alternative name in recognition of another facet of glucagon as a suppressor of plasma glutamine levels.
Glucagon was first described in 1923 as a hyperglycemic substance found in aqueous pancreas extracts. Since then, it has become well accepted that the major physiological role of glucagon is to increase blood glucose levels. Consequently, the suppression of glucagon activity has been considered as a potential avenue to treat diabetes mellitus1. A glucagonocentric view of diabetes mellitus was proposed in 2012 by Unger et al.2 that might have further reinforced the concept of glucagon as a hyperglycemic substance and, thereby, an aggravating factor of diabetes mellitus. The glucagonocentric view of diabetes is mainly based on the observation that mice deficient in the glucagon receptor gene (Gcgr −/−) showed lower blood glucose levels and resistance to developing diabetes after the streptozotocin‐induced destruction of islet β‐cells. However, it should be noted that in addition to glucagon, glucagon‐like peptide‐1 (GLP‐1) is also overproduced in Gcgr −/− mice, as both glucagon and GLP‐1 are derived from a common precursor, proglucagon1. As GLP‐1, a major incretin, harbors protective effects on islet β‐cells in addition to its well‐known insulinotropic effects, an increase in GLP‐1 activity, in combination with the absence of glucagon activity, might contribute to the resistance to diabetes observed in Gcgr −/− mice. The answer to the question of whether GLP‐1 is involved in the antidiabetic phenotype of Gcgr −/− mice was provided by studies in which a glucagon gene knockout (KO) animal model deficient in both glucagon and GLP‐1 (GCGKO: Gcg −/− or Gcg gfp/gfp) or a model that lacked receptors for both glucagon and GLP‐1 (Gcgr −/− Glp1r −/−) were analyzed. Both GCGKO and Gcgr −/− Glp1r −/− mice developed diabetes after streptozotocin injection, indicating that GLP‐1 played a pivotally important role in the resistance to diabetes observed in the Gcgr −/− mice1.
Intriguingly, both GCGKO and Gcgr −/− Glp1r −/− mice are virtually normoglycemic under normal, non‐diabetic conditions. Therefore, the absence of glucagon activity per se does not reduce blood glucose levels. As is the case for resistance to diabetes, GLP‐1 activity is a prerequisite for a decrease in blood glucose levels in the absence of glucagon activity. In contrast, serum amino acid levels are increased in both GCGKO and Gcgr −/− mice1. Therefore, the absence of glucagon activity by itself is sufficient to increase serum amino acid levels regardless of the presence or absence of GLP‐1. Hyperaminoacidemia has also been documented in mice and monkeys given a blocking antibody against the glucagon receptor3. In addition to hyperaminoacidemia, defects in glucagon activity induce an increase in islet α‐cell mass. Studies in mice showed that liver‐specific ablation of the glucagon receptor was enough to induce the proliferation of islet α‐cells1. Such data show that glucagon has little, if any, potential to directly suppress the proliferation of α‐cells, and that liver‐derived factors under the control of glucagon might regulate α‐cell proliferation. Whereas efforts to identify genes encoding specific regulators of α‐cell proliferation have been unsuccessful, data showing that amino acids, especially glutamine, are involved in the regulation of α‐cell proliferation has been accumulating1, 4.
The regulatory mechanisms that control the plasma concentration of amino acids remain largely to be elucidated. However, it is clear that glucagon does play a major role in this regulation. As discussed above, animal models deficient in glucagon activity do not always show lower blood glucose levels, but are characterized by hyperaminoacidemia. Furthermore, in the 1980s, it was found that the administration of glucagon decreased the plasma amino acid concentration5 and that hypoaminoacidemia was a major symptom in glucagonoma syndrome6. Therefore, glucagon excess and deficiency results in hypoaminoacidemia and hyperaminoacidemia, respectively. Of the various amino acids, glutamine is present in plasma at the highest concentration, with the plasma concentration being able to reach a level in the order of millimolars, comparable to that of glucose, in animals with defective glucagon activity1. Amino acids, including glutamine, can serve as substrates for gluconeogenesis. To function as a glucogenic amino acid, glutamine is first converted to glutamate through deamination by glutaminase. Recently, Miller et al.7 showed that glucagon activates hepatic glutaminolysis. They also showed that mice deficient in hepatic glutaminase (Gls2 −/−) had lower blood glucose and increased glutamine levels. Furthermore, Gls2 −/− mice showed an increase in islet α‐cell area and fasting glucagon levels7. The data by Miller et al.7 clearly show that impaired glutaminolysis in the liver is sufficient to induce α‐cell proliferation and an elevation in circulating glucagon levels, underscoring the importance of glutamine as a mediator of feedback regulation between the liver and islet α‐cells.
As a historical side note, the name “glucagon” is a combination of glucose and agonist, and was given in 1923 to a 29‐amino acid peptide secreted by islet α‐cells because of its activity in increasing blood glucose levels. Yet, “glucagon” plays an essential role in robust feedback regulation between the liver and α‐cells, and exerts suppressive or static effects on the plasma concentration of amino acids, especially glutamine (Figure 1). Thereby, I would like to propose here an alternative name “glutaminostatin” in recognition of another facet of glucagon as a suppressor of plasma glutamine levels.
Figure 1.
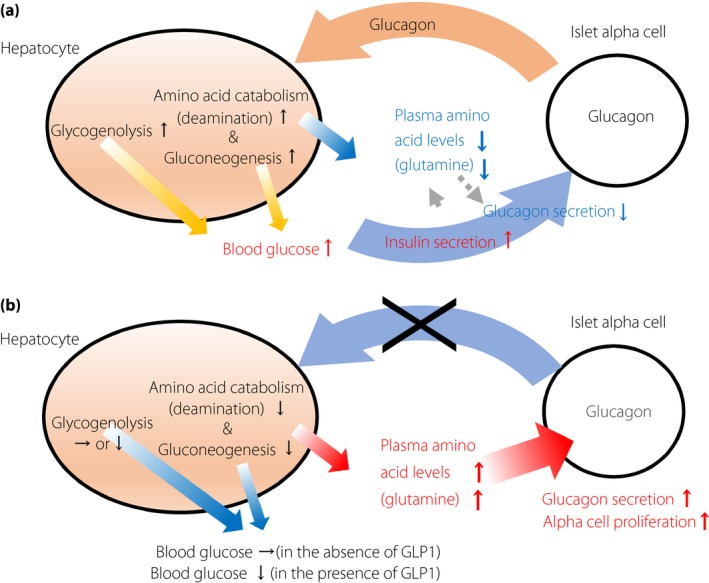
Feedback regulation between the liver and islet α‐cells mediated by glucagon and amino acids (a) under normal conditions or (b) under absent or blocked glucagon activity. Glucagon increases glycogenolysis and gluconeogenesis in hepatocytes, thereby increasing blood glucose levels. Elevated blood glucose levels stimulate the secretion of insulin, which in turn suppresses glucagon secretion. Glucagon also converts amino acids into substrates available for gluconeogenesis and reduces plasma amino acid levels. The absence of glucagon activity per se is insufficient to reduce the blood glucose level. Whether blood glucose levels decrease is dependent on glucagon‐like peptide‐1 (GLP‐1). In the absence of glucagon activity, plasma amino acid levels increase regardless of the presence or absence of GLP‐1. The elevation in plasma amino acid levels induces glucagon secretion, as well as α‐cell proliferation.
Analyses of the plasma concentrations of amino acids, especially glutamine, together with those of glucagon, glucose and insulin, are expected to shed light on unknown roles of glucagon/glutaminostatin under physiological and/or pathological conditions. Indeed, the association between plasma amino acid levels and the risk of type 2 diabetes has been analyzed in a number of studies. In particular, Chen et al.8 reported that a high glutamine concentration and glutamine‐to‐glutamate ratio were associated with a decreased risk of the incidence of type 2 diabetes in a nested case–control study in non‐diabetic Japanese individuals. As glucagon levels were not analyzed in the study, the association between glutamine concentration and glucagon levels in plasma remains a matter of conjecture. Interestingly, Wewer Albrechtsen et al.9 reported that both glutamine and glucagon levels are increased in Danish individuals with insulin resistance, and hypothesized that glucagon resistance, together with insulin resistance, plays some role in dysregulated metabolism. If glucagon resistance is the cause of an increase in plasma glucagon levels, therapeutic intervention should be directed to ameliorate the resistance, but not to suppress glucagon activity.
Collectively, glucagon/glutaminostatin plays an essential role in the homeostatic regulation of plasma amino acid levels. Therefore, glucagon/glutaminostatin cannot simply be regarded as a villain in the metabolic regulation of diabetes. To explore novel therapeutic maneuvers in order to modify glucagon activity in the treatment of diabetes, it is essential to consider another facet of glucagon, as a regulator of plasma amino acid concentrations.
Disclosure
The author received lecture fees from Astellas Pharma, AstraZeneca, Boehringer Ingelheim, Cosmic Corporation, Daiichi Sankyo, Kissei Pharmaceutical, Kyowa Hakko Kirin, MSD, Novartis Pharma, Novo Nordisk and Sumitomo Dainippon Pharma, and research support from Cosmic Corporation and Yamasa Corporation. The sponsors had no role in research design, data collection, data analysis, data interpretation or report preparation.
Acknowledgment
This work was supported in part by Grants‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (18H03176) and Novartis Research Grant 2018.
References
- 1. Hayashi Y, Seino Y. Regulation of amino acid metabolism and alpha‐cell proliferation by glucagon. J Diabetes Investig 2018; 9: 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 2012; 122: 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okamoto H, Kim J, Aglione J, et al Glucagon receptor blockade with a human antibody normalizes blood glucose in diabetic mice and monkeys. Endocrinology 2015; 156: 2781–2794. [DOI] [PubMed] [Google Scholar]
- 4. Dean ED, Li M, Prasad N, et al Interrupted glucagon signaling reveals hepatic alpha cell axis and role for L‐glutamine in alpha cell proliferation. Cell Metab 2017; 25: 1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boden G, Rezvani I, Owen OE. Effects of glucagon on plasma amino acids. J Clin Invest 1984; 73: 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parker CM, Hanke CW, Madura JA, et al Glucagonoma syndrome: case report and literature review. J Dermatol Surg Oncol 1987; 10: 884–889. [DOI] [PubMed] [Google Scholar]
- 7. Miller RA, Shi Y, Lu W, et al Targeting hepatic glutaminase activity to ameliorate hyperglycemia. Nat Med 2018; 24: 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen S, Akter S, Kuwahara K, et al Serum amino acid profiles and risk of type 2 diabetes among Japanese adults in the Hitachi Health Study. Sci Rep 2019; 9: 7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wewer Albrechtsen NJ, Færch K, Jensen TM, et al Evidence of a liver‐alpha cell axis in humans: hepatic insulin resistance attenuates relationship between fasting plasma glucagon and glucagonotropic amino acids. Diabetologia 2018; 61: 671–680. [DOI] [PubMed] [Google Scholar]