

ABSTRACT
The use of fixed fibroblasts from familial and sporadic Alzheimer's disease patients has previously indicated an upregulation of mitochondria–ER contacts (MERCs) as a hallmark of Alzheimer's disease. Despite its potential significance, the relevance of these results is limited because they were not extended to live neurons. Here we performed a dynamic in vivo analysis of MERCs in hippocampal neurons from McGill-R-Thy1-APP transgenic rats, a model of Alzheimer's disease-like amyloid pathology. Live FRET imaging of neurons from transgenic rats revealed perturbed ‘lipid-MERCs’ (gap width <10 nm), while ‘Ca2+-MERCs’ (10–20 nm gap width) were unchanged. In situ TEM showed no significant differences in the lipid-MERCs:total MERCs or lipid-MERCs:mitochondria ratios; however, the average length of lipid-MERCs was significantly decreased in neurons from transgenic rats as compared to controls. In accordance with FRET results, untargeted lipidomics showed significant decreases in levels of 12 lipids and bioenergetic analysis revealed respiratory dysfunction of mitochondria from transgenic rats. Thus, our results reveal changes in MERC structures coupled with impaired mitochondrial functions in Alzheimer's disease-related neurons.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Alzheimer, Transgenic rats, Amyloid β/βCTF, Primary neuronal cultures, MERC, Mitochondrial lipids, FRET
Highlighted Article: Dynamic analysis of live neurons modelling Alzheimer's disease (AD)-like amyloid pathology shows loosened mitochondria-ER contacts (MERC), providing a novel perturbation of the MERC axis in AD.
INTRODUCTION
Despite a plethora of studies that date back more than two decades, the early mechanisms that associate accumulation of amyloid β (Aβ) with neuronal dysfunction in Alzheimer's disease have not been elucidated. However, bioenergetic failure has been suggested as an early event responsible for neuronal death in Alzheimer's disease (Leuner et al., 2007; Mancuso et al., 2007; Mosconi et al., 2009; Kapogiannis and Mattson, 2011). It has been shown that amyloid β precursor protein (APP) and Aβ colocalize with mitochondria (Devi and Ohno, 2012; Hansson Petersen et al., 2008), that Aβ inhibits respiratory chain function (reviewed in Swerdlow, 2012) and that mitochondrial function also changes APP processing increasing or decreasing the production of amyloidogenic derivatives (Gabuzda et al., 1994; Gasparini et al., 1997; Leuner et al., 2012; Pereira et al., 1998). Apart from their essential role in bioenergetics, mitochondria are also involved in a great variety of other cellular processes, such as Ca2+ homeostasis and lipid biosynthesis. These functions require a dynamic spatial organization that allows signaling from and to other organelles. In particular, mitochondria are associated with the endoplasmic reticulum (ER) – i.e. the mitochondria–ER contacts (MERCs) – form between the outer mitochondrial membrane (OMM) and specialized regions of the ER, in which membrane and luminal components can intermix and exchange (Shore and Tata, 1977; Vance, 1990). These membranes can run in juxtaposition for hundreds of nanometers with a gap width of 5–30 nm between them when mitochondria are associated with smooth ER. The number of contacts, the interface length and the gap width are parameters regulated by the cell and may vary depending on the metabolic needs and the presence of stress agents (Bravo et al., 2011; Csordás et al., 2006; Sood et al., 2014).
Although many studies have investigated MERCs in Alzheimer's disease, none has analyzed in detail their number, interface length and structure within primary neurons in vivo. Moreover, not much attention has been paid to the gap width between a mitochondrion and the ER, an important structural parameter that regulates the function of MERCs in the transfer of ions and molecules between organelles (reviewed in Csordás et al., 2018; Giacomello and Pellegrini, 2016). It has, however, been shown that mitochondria can capture and accumulate Ca2+ to regulate the activity of enzymes that are central in energetic metabolism (Rizzuto et al., 2012; Hajnóczky et al., 1995; Jouaville et al., 1999; McCormack et al., 1990). The uptake of Ca2+ by the mitochondrial Ca2+ uniporter (MCU) (Baughman et al., 2011; De Stefani et al., 2011) – although possible without the physical coupling of the ER to a mitochondrion – is favored when the ER membrane is facing the OMM with a gap width of ∼15 nm (Csordás et al., 2006, 2010). For efficient Ca2+ transfer, the organelles would have to be separated by 10–25 nm (Ca2+-MERC). In contrast, when lipids are being synthesized and exchanged in MERCs (lipid-MERCs), the organelles need their membranes to be in close proximity. Assuming that the density of the enzymes that synthesize lipids in these structures is homogeneous, any increase or decrease in the MERCs area should be accompanied by a similar change in the amount of lipids synthesized. Two models have been proposed to explain the transport of lipids between the two membranes. The first states that lipids are transported by hydrophobic protein complexes. This transport would be interrupted when the width of the mitochondria–ER gap increases. The second model suggests that lipids are coated by transporter proteins that diffuse from one membrane to the other. In this case, the increased in gap width would impact on the rate of delivery. In any case, the gap width required for lipid transfer is expected to be as narrow as ≤10 nm, given the proposed molecular mechanism implicated in the transfer of largely hydrophobic molecules between the organelles (Schauder et al., 2014).
In the last few years, studies in which cell lines or fibroblasts from Alzheimer's disease patients (Area-Gomez et al., 2012; Del Prete et al., 2017) or zebrafish neuronal progenitors (Newman et al., 2017) were used, have reported alterations in MERCs, with contradictory results (both increases and decreases). However, none had analyzed the gap width between organelles in live neurons, or used methods to evaluate proximity with the appropriate resolution. Moreover, the biochemical impact of altered MERCs in primary neurons expressing endogenous Aβ has not yet been reported. We performed dynamic and ultrastructural analysis of hippocampal neurons from wild-type (WT) and McGill-R-Thy1-APP transgenic rats, using multicolor live imaging, Förster resonance energy transfer (FRET) to generate drug-inducible interorganellar linkers (lateral resolution 8–10 nm) and in situ transmission electron microscopy (TEM).
RESULTS
Neuronal expression of human Aβ peptides leads to time-dependent morphological alterations in mitochondria and endoplasmic reticulum
Neurons from McGill-R-Thy1-APP transgenic rats express the entire coding region of the human APP751 cDNA, including the familial Alzheimer's disease Swedish double mutation (K670N, M671L) and the Indiana mutation (V717F) (Leon et al., 2010). We first determined in the conditioned medium of days in vitro (DIV) 7 and DIV14 cultures (Fig. S1) the time-course accumulation of the three human Aβ isoforms, and found that Aβ38, Aβ40 and Aβ42 were detectable (Fig. 1A). The mean value of the extracellular levels of Aβ40 and of Aβ42 was substantially lower at DIV7 (2.13±0.5 pM and 0.63±0.2 pM, respectively) and DIV14 (16.77±4.92 pM and 4.47±1.2 pM, respectively) than values previously reported in Swedish-type APP-HEK 293 transfected cells (∼400 pM for Aβ40 and ∼10 pM for Aβ42; Ohshima et al., 2018). This reinforces the use of this model to decipher cellular mechanisms involved in early stages of Aβ pathology. In agreement with what was described in cell culture experiments (Ohshima et al., 2018), Aβ monomers were not detected in cell lysates of neurons from transgenic rats by ELISA, suggesting an efficient clearance mechanism that precludes intraneuronal Aβ accumulation. To evaluate the impact of endogenous Aβ accumulation on neuronal ultrastructure, we performed TEM analysis. To avoid disturbances in the subcellular organization, TEM was carried out on neurons adhered to coverslips. At DIV7, the morphology of neurons from transgenic rats was similar to that of WT neurons; whereas at DIV14, when the highest Aβ concentration was detected in the conditioned medium, neurons from transgenic (Tg) rats (hereafter referred to as Tg neurons) exhibited mitochondria with fragmented cristae and ER swelling (Fig. 1B). Notice that, at DIV7, the number of mitochondria per soma (Fig. 1C) and the ER lumen area (Fig. 1D) were not significantly different between genotypes. However, at DIV14, an increase in the ER lumen area was detected compared to DIV7 neurons – regardless the presence of the human transgene.
Fig. 1.

Hippocampal primary neurons from transgenic rats produce human Aβ isoforms, and show mitochondria and endoplasmic reticulum swelling after DIV14. (A) Quantification of Aβ levels in the conditioned media of hippocampal primary neurons. Bar graphs show the concentration (in pM) of each Aβ isoform in Tg neurons at DIV7 or DIV14 from three different independent cultures and genotype. Unpaired two-tailed Student's t-test (Aβ38: P=0.610; Aβ40: P=0.040; Aβ42: P=0.035) was used to compare between DIV data. (B) Representative TEM images showing mitochondria (Mt) and ER ultrastructures in coverslip-attached embryonic primary hippocampal neurons from WT rats at DIV7 (top left panel) and DIV14 (bottom left panel), and in Tg neurons at DIV7 (top right panel) or DIV14 (bottom right panel). Scale bar: 250 nm. (C) Quantification of mitochondria (Mt) number in neuronal somata. For each genotype three independent hippocampal primary neuronal cultures were used, with 10–12 embryos per culture. Data were obtained after TEM analysis of coverslip-attached primary cultures from WT and Tg rats at DIV7 or DIV14. WT DIV7: n=9 cells; Tg DIV7: n=9 cells; WT DIV14: n=4 cells; Tg DIV14: n=8 cells. Two-way ANOVA test was used to make comparisons among the means of the four groups of data, where two independent variables (DIV and Genotype) were considered. The ANOVA result is reported as an F-statistic and its associated degrees of freedom and P-value. The effect of DIV (F1,26=0.1, P=0.749) or Genotype (F1,26=0.78, P=0.385) were not considered significant, neither was the interaction DIV×Genotype (F1,26=0.15, p=0.704). (D) Quantification of ER lumen area in the same neuronal somata as used in C. Kruskal–Wallis test (P<0.0001) followed by Dunn's post-hoc test. ***P<0.001. In the box-and-whisker plots in C,D, the box represents the 25–75th percentiles, the median is indicated with a line and the mean with '+'. The whiskers show the minimum and maximum values.
To then assess whether levels of soluble Aβ isoforms detected in DIV7 Tg neurons are associated with changes in the composition of endogenous free fatty acids (FFAs), which are known to modulate organelle membrane fluidity, oxidative stress and inflammation (Wang et al., 2012) we applied liquid chromatography-mass spectrometry (LC-MS) to DIV7 WT and Tg neurons. Six FFAs were detected across the neuron samples, with palmitic acid, oleic acid and stearic acid being by far the most abundant ones and roughly equal in concentration. A multiple t-test comparison was carried out between WT and Tg neurons, but no significant (P<0.05) differences of any of the FFAs were observed (Table S1).
Live Tg neurons exhibit decreased coupling in lipid-MERC
To perform an in vivo analysis of neuronal MERCs at early stages of Aβ pathology, without morphological alterations of mitochondria or ER and in the absence of disturbed lipid metabolism, we studied pre-existing physiological- MERCs in live hippocampal neurons from WT and Tg rats at DIV7. For this, we employed inducible interorganellar linkers tagged with CFP or YFP, which allow the measurement of mitochondria–ER coupling in living cells at a resolution of 8–10 nm. We transfected neurons with rapamycin-inducible synthetic interorganellar linkers and, upon expression of these constructs and following addition of rapamycin, a ‘bridge formation’ was induced at the areas where ER and OMM are naturally close. CFP and YFP formed a FRET pair, and the FRET signal confined to the mitochondria–ER interface was visualized by multicolor epifluorescence microscopy. Rapamycin was applied only for a short time (<8 min) to minimize the broad effects initiated through its endogenous receptor mTOR (Kim and Guan, 2015). Addition of rapamycin led to immediate increase of the FRET signal and to a decrease in CFP fluorescence in both WT and Tg neuronal somata. In WT neurons, the FRET:CFP ratio rapidly rose in a linear way until reaching a plateau at 200 s (short linkers) (Fig. 2A) or 150 s (long linkers) (Fig. 2B). Both WT and Tg neurons exhibited a similar FRET:CFP ratio with long linkers along the measurement (Fig. 2B) while with short linkers, the curve was linear during all its trajectory without reaching a plateau (Fig. 2A), indicating decreased mitochondria–ER coupling at a gap width of <10 nm.
Fig. 2.

Lipid-MERCs are decreased in live hippocampal primary neurons from transgenic rats. (A,B) FRET to CFP ratios (FRET:CFP) before and after linkage induction in response to rapamycin (indicated by an arrow) in WT and Tg neurons with short (A) or long (B) linkers, i.e. gap width <10 nm or ∼10–20 nm, respectively. Calculations were made for neuronal somata. Independent hippocampal primary neuronal cultures (n=3, WT; n=4, Tg) were performed using 10–12 embryos per culture. Data were obtained from 9–17 cells per group. Morphometric analysis of lipid-contact sites (<10 nm) in transmission electron microphotographs. (C,D) Box-and-whisker plots show percentage of lipid-MERCs (<10 nm) per total MERCs (10–30 nm; C) and percentage of lipid-MERCs (<10 nm) per number of mitochondria (Mt number) (D). Box represents the 25–75th percentiles, the median is indicated with a line and the mean with '+'. The whiskers show the minimum and maximum values.
Fixed Tg neurons display similar percentages of lipid-MERCs but decreased length of appositions
To further test the mitochondria–ER interface, TEM images were quantified using an in-house-developed macro for the image-processing package Fiji (Weaver et al., 2017). Following the same methodology as was used for the live FRET imaging, fixation was performed on cells attached to coverslips to conserve the characteristic morphology of the neurons. The analysis revealed that the percentage of lipid-MERCs versus total MERCs (Fig. 2C) and the percentage of lipid-MERCs versus total number of mitochondria (Fig. 2D) was similar between genotypes. However, we found that the length of ER-mitochondrial appositions (Sood et al., 2014) with a gap width of <10 nm (lipid-MERC) was significantly reduced in Tg as compared to WT neurons (Fig. 3B). Since the decreased length of appositions in Tg neurons could be the consequence of decreased mitochondria size, we quantified the perimeter of mitochondria from WT and Tg neurons, and found no significant differences between groups (Fig. 3C). Moreover, the decrease in the interface length of appositions in lipid-MERCs of Tg neurons persisted after normalizing it to the perimeter of mitochondria (Fig. 3D), suggesting that reduced length of appositions in lipid-MERCs of Tg rats is not a consequence of altered mitochondrial morphology. Representative TEM micrographs depicting the interface length in WT and Tg rats are shown in Fig. 3E. Thus, both the FRET and ultrastructure measurements support our finding that the tight lipid-MERCs are reduced in Tg neurons.
Fig. 3.
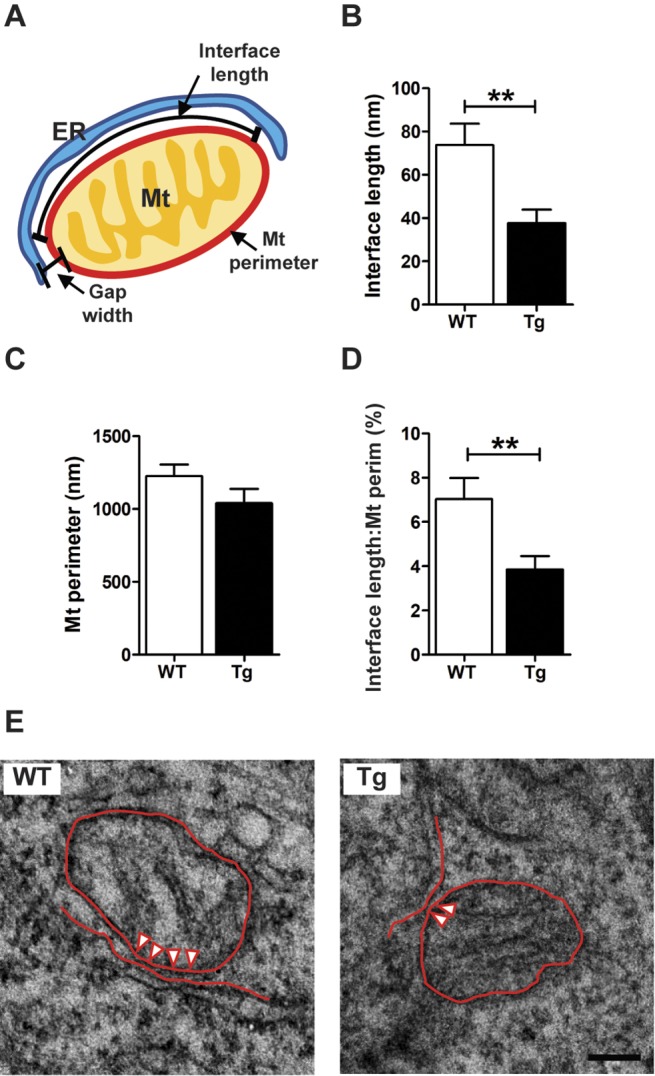
Tg neurons at DIV7 display decreased interface length in lipid-MERCs. (A) Scheme of the structural parameters of MERCs, i.e. gap width, mitochondrial (Mt) perimeter and interface length, which were analyzed in coverslip-attached neurons using TEM. (B) Bar graph shows the quantification of the interface length with a gap width <10 nm. Unpaired two-tailed Student's t-test with Welch's correction (t=3.112, P=0.003). (C) Bar graph showing the mitochondrial perimeter of the same mitochondria used in B. Data were analyzed by unpaired two-tailed Student's t-test (t=1.393, **P=0.168). (D) Bar graph showing the ratio of interface length to mitochondrial perimeter (Mt perim) using data shown in B and C, respectively. Unpaired two-tailed Student's t-test with Welch's correction (t=2.804, **P=0.007) was employed. (E) Representative microphotographs of MERCs in WT and Tg neurons. The interface where the gap width is <10 nm is indicated by arrowheads. All calculations were made for neuronal somata. Analysis was performed in WT (n=13) and Tg (n=16) cells. Neurons from each genotype came from three independent cultures using 10–12 embryos per culture. The number of evaluated mitochondria was n=47 for WT and n=21 for Tg neurons. Cells came from three different cultures or genotypes using 10–12 embryos per culture. ER, endoplasmic reticulum; Mt, mitochondria; perim, perimeter. Scale bar: 100 nm.
Decreased interface length in lipid-MERCs is associated with alterations in mitochondrial lipid profiles and bioenergetics functionality
To associate the decreased lipid-MERCs with a biochemical read-out, we isolated mitochondria from primary neuronal cells of WT and Tg rats (Fig. S2) and analyzed the lipid profile using high resolution liquid chromatography–tandem mass spectrometery (LC-MS/MS). Lipid peaks were identified in each sample using LipidBlast (Kind et al., 2013), and the resulting hits were quantified using in-house scripts (available upon request). The lipid profile of the mitochondrial extracts showed a significant deviation in the Tg group compared with WT (Fig. 4A). From the total number of lipids detected (n=106, Table S2), twelve lipids were significantly different in mitochondria of Tg rats when compared to those of WT rats (P≤0.015; Fig. 4B). Among those lipids, we found a 60% decrease in main mitochondrial phospholipids, i.e. cardiolipin and phosphatidylethanolamine (PtdEtn) in mitochondria preparations from Tg neurons as compared to WT, suggesting that alterations on lipid-MERCs directly impact on the biological relationship of membrane composition, structure and function in mitochondria, which can induce bioenergetic dysfunction.
Fig. 4.

Alterations of the mitochondrial lipid profile impact on mitochondrial bioenergetics functionality in Tg neurons at DIV7. (A) Base-peak chromatogram of detected lipids by positive (upper panel) and negative (lower panel) mode untargeted LC-MS/MS, scan range=350–2000 m/z. Lipid retention time is shown on the x-axis while peak intensity is shown on the y-axis. Each profile represents an overlay of the samples tested (n=3 per genotype). Numbers indicate the lipid peaks (listed below) with significant differences between WT and Tg neurons. (B) Bar graph showing the MS intensity in arbitrary units (A.U.) of the 12 lipids in Tg neurons that exhibited significant differences with a P-value of ≤0.015. ***P<0.001, **P<0.01, *P<0.05. Peak 4, Plasmenyl-PE 36:4; Peak 5, CerP 32:4; Peak 6, PE 37:6, Peak 7, PC 34:1; Peak 8, PC 20; 0; Peak 9, DGDG 15:0. Peak 10, PC 33:5; Peak 11, PC 31:4; Peak 12, DG 31:4; Peak 13, PI 38:4; Plasm-PE, plasmenyl-PE; PC, phosphatidylcholine; DG, diacylglycerol; PI, phosphatidylinositol; PE, phosphatidylethanolamine; CerP, ceramide-1-phosphate; DGDG, digalactosyldiacylglycerol; CL, cardiolipin. PE, phosphatidylethanolamine (PtdEtn); PI, phosphatidylinositol (PtdIns); PC, phosphatidylcholine (PtdCho. (C) Bioenergetic profile of DIV7 WT and Tg neurons assessed with an extracellular flux analyser. Oxygen consumption rate (OCR) was normalized to the mean value of the four baseline measurements (100%), before the addition of inhibitors or uncoupler, and after subtraction of non-mitochondrial respiration. (D) Bar graph shows the spare respiratory capacity calculated as the difference between maximal and baseline OCR from the same cells as used in A. Oligo, oligomycin; FCCP, carbonyl cyanide-4-phenylhydrazone. Three independent cultures per genotype were used. Paired two-tailed Student's t-test (t=6.479, *P=0.023) was used to compare genotypes.
In this regard, we assessed mitochondrial bioenergetics in intact neurons by extracellular flux analysis. We found an altered bioenergetics profile (Fig. 4C) and a significantly reduced spare respiratory capacity (Fig. 4D) in Tg versus WT neurons, suggesting a possible impairment in meeting a sudden increase in energy demand with increased ATP synthesis.
DISCUSSION
According to the Aβ levels detected in Tg neuron cultures, we speculate that our system represents a mild cellular model of Alzheimer's disease-like amyloid pathology. The fact that we were unable to detect intracellular Aβ accumulation suggests that, in embryonic primary neurons, the degradation of aggregation-prone proteins, which are retained in the ER, is efficiently mediated by proteasome (Kostova and Wolf, 2003) and by insulin-degrading enzyme (Schmitz et al., 2004) activities. However, we found morphological alterations of mitochondria and ER in Tg neurons at DIV14, similar to those reported in brains of Alzheimer's disease patients (Baloyannis, 2011), suggesting that extracellular Aβ peptides somehow impact on neuronal functionality. Mitochondrial swelling has been associated with the pathogenesis of Alzheimer's disease; however, mitochondrion alterations in neurons from brains of Alzheimer's disease patients were not related to accumulation of amyloid deposits (Baloyannis, 2011). Even though we did not determine any parameter of the unfolded protein response (UPR) response in WT and Tg neurons at DIV7 and DIV14, we speculate that Tg neurons at DIV14 show signs of activated UPR. The induction of artificial ER stress by overexpressing a membrane protein (as APP) must be considered as a possibility in our experimental setting, as previously reported (Hashimoto and Saido, 2018). However, the fact that the sub-cellular structure of mitochondria and ER is conserved in DIV7 Tg neurons and that no significant differences were observed between genotypes for any of the six FFAs detected, suggests that the genetic (human APP expression) and environmental (extracellular Aβ accumulation) insults in Tg neurons at DIV7 are not enough to disturb the function of the ER and induce ER enlargement. In this regard, it has previously been reported that exogenous palmitic acid decreases the expression of neurotrophic cytokines through induction of ER stress in SH-SY5Y human neuroblastoma cells, N2a mouse neuroblastoma cells, and cortices and hippocampi from WT C57BL/6J mice (Marwarha et al., 2016). Regarding our cellular model, additional experiments are required to assess whether the significant ER enlargement detected in Tg neurons at DIV414 is mediated by an increase of endogenous FFAs.
To our knowledge, this is the first report of MERCs in live neurons, using methods to evaluate the proximity of these organelles with a lateral resolution of 8–10 nm. In a similar way, other studies have employed the same synthetic linkers to evaluate the physical extension and integrity of MERCs in different cell lines, considering FRET signal as proportional to the extension of the mitochondria–ER apposition. Decreased mitochondria–ER coupling has also been reported in caveolin-1 knockout (Sala-Vila et al., 2016) and mitofusin-2-ablated mouse embryonic fibroblasts (MEFs) (Naon et al., 2016), reinforcing the validity of this genetic tool to assess altered mitochondria–ER interaction in live cells. However, any effects the expression of synthetic linkers per se might have, need to be taken into account in any quantification (Shi et al., 2018).
During the last five years, MERC analysis in Alzheimer's disease pathology was based on TEM – a ‘static’ approach – which suggested that an upregulated function of MERCs could induce mitochondrial bioenergetic dysfunction. However, this experimental evidence was never assessed in live primary neurons expressing endogenous Aβ peptides. MERCs were evaluated in different Alzheimer's disease-related models, such as PSEN1-knockout and/or PSEN2-knockout MEFs, and fibroblasts from patients suffering from familial or sporadic Alzheimer's disease (Area-Gomez et al., 2012), the neuroblastoma cell line SH-SY5Y overexpressing APP with the Swedish familial mutation (Del Prete et al., 2017), and mouse hippocampal primary neurons acutely exposed to synthetic Aβ peptides in the culture medium (Hedskog et al., 2013). In contrast to studies using these model systems, our dynamic and static results indicate that, at early stages of Aβ/APP fragment overproduction – i.e. without intraneuronal Aβ accumulation and conserved mitochondrion and ER ultrastructure – there are no changes in Ca2+-MERCs, whereas a loosening of lipid-MERC apposition is evident. In agreement with our observations, analysis of brain biopsies from patients with idiopathic normal pressure hydrocephalus, a neuropathology often associated with Alzheimer's disease, found that the presence of both amyloid plaques and neurofibrillary tangles (pathological hallmarks of Alzheimer's disease) correlates with a decreased stretch of contact between ER and mitochondria, analyzed by TEM (Leal et al., 2018). Even though Leal and colleagues did not take into account the gap width between the organelles or study any biochemical impact of this alteration, their results support our observation in Alzheimer's disease-like neurons of a decreased interface length in lipid-MERCs with a gap width <10 nm. In addition, fewer MERCs were reported in senescent cells (Janikiewicz et al., 2018), supporting the assumption that our cellular model represents not only early stages of neuronal dysfunction associated to Alzheimer's disease-like amyloid pathology, but also aging.
Reasons for the differences between our results of MERC analysis and those in previous reports using Alzheimer's disease-related in vitro models are not entirely clear but might be related to the cell type and methods used to quantify MERCs. Previously, MERCs were evaluated in fixed fibroblasts using confocal microscopy (lateral resolution of 250 nm) (Area-Gomez et al., 2012; Lidke and Lidke, 2012) or in fixed Aβ-exposed primary neurons employing a proximity ligation assay (lateral resolution of >30 nm) (Hedskog et al., 2013). These determinations were not performed in live cells and the techniques employed exclude an accurate quantification of the <10–30 nm distances that define MERCs. Even though Area-Gomez et al. analyzed the interface length in human fibroblasts by TEM, i.e. the gold-standard to evaluate inter-organelle distance, they did not assess the gap width between the ER and mitochondria (Area-Gomez et al., 2012). Finally, the results obtained by using cells lines do present – although they are informative – limitations compared with those from live neurons. In this regard, Newman and colleagues recently used TEM to analyze the interface length – as previously reported by Area-Gomez et al. (2012) in fibroblast from human subjects – in neuronal progenitors from zebrafish embryos injected with morpholinos against PSEN1 and/or PSEN2, and found no significant differences in PSEN1/2 knockdown cells as compared to controls (Newman et al., 2017). This reinforces the concept that MERC function and/or activity is cell dependent and that, although neurons share many features found in other cell types, they possess some special characteristics that cannot be reproduced in non-differentiated cells. Although Hedskog et al. employed primary mouse hippocampal neurons, the increase in MERCs detected by confocal microscopy after acute exposure of synthetic Aβ (Hedskog et al., 2013) might represent an artefact because Aβ affects intracellular trafficking of mitochondria, inducing accumulation of mitochondria in perinuclear regions – which are regions rich in ER (De Vos et al., 2008).
It has been already proposed that disturbances of MERCs having a gap width of <10 nm impair lipid exchange between the ER and mitochondria (Giacomello and Pellegrini, 2016). In our current report, we experimentally addressed this issue, using primary neuronal cultures. It is known that mitochondria depend on the import of certain proteins and lipids for mitochondrial membrane formation and for maintaining cell bioenergetics. Although phosphatidylglycerol, cardiolipin and PtdEtn are synthesized in mitochondria by specific enzymes, phosphatidylcholine (PtdCho), phosphatidylinositol (PtdIns), phosphatidylserine (PtdSer) and sterols need to be imported from other organelles. The origin of most lipids imported into mitochondria is the ER, which requires MERCs to be properly assembled and functionally operational (Flis and Daum, 2013). Functionally, cardiolipin and PtdEtn interact with many mitochondrial proteins (Osman et al., 2011) and stabilize their conformation (Joshi et al., 2012), playing a crucial role in maintaining mitochondrial morphology. Cardiolipin, the signature lipid of mitochondria, regulates mitochondrial bioenergetics owing to its fundamental role in functional membrane assembly of redox complexes. Notice that, in this report, we found that both cardiolipin levels and bioenergetics were altered in Tg neurons, reinforcing a direct link between mitochondrial lipid composition and functionality. Notably, mitochondria are the main supplier of PtdEtn to other organelles (Voelker, 1984), suggesting that a reduction of the mitochondrial PtdEtn content – as shown in Tg neuron samples, can impact on neuronal functionality. We also detected significantly lower levels of PtdCho and PtdIns in Tg as compared to WT neurons but, in contrast to cardiolipin and PtdEtn, the specific functions of PtdCho and PtdIns are not well defined; however, their role in the maintenance of mitochondrial functionality seems to be indispensable (van Meer and Hoetzl, 2010). PtdCho is the most abundant phospholipid in eukaryotic cells (van Meer et al., 2008) but, given that mitochondria lack PtdCho-synthesizing enzymes, it is imported from the ER. In a similar way, PtdIns is produced in the ER and imported into mitochondria. On the whole, our results suggest that the decreased levels of mitochondrial lipids are a consequence of perturbed MERC apposition length.
In our model, different cellular events might be responsible for the reduced apposition between organelles. Stoica and colleagues proposed previously that, in cells overexpressing Tar DNA-binding protein 43, the main pathology in amyotrophic lateral sclerosis with associated frontotemporal dementia – i.e. the loosening of MERCs – is a consequence of activation of glycogen synthase kinase-3β (GSK3β), which, in turn, disrupts binding of vesicle-associated membrane protein-associated protein B (located in the ER) to regulator of microtubule dynamics protein 3 (resident in the OMM) (Stoica et al., 2014). Increasing evidence suggests that dysregulation of GSK3β is implicated in Alzheimer's disease (DaRocha-Souto et al., 2012), and these findings support the idea that GSK3β is aberrantly activated by the presence of Aβ and contributes, at least in part, to the neuronal anatomical derangement associated with Aβ pathology itself. Another mechanism that, in our case, seems to be less probable, is steric hindrance that could cause accumulation of Aβ oligomers on the OMM. Although we could not detect the presence of soluble Aβ isoforms in cell homogenates of DIV7 Tg neurons by ELISA, we cannot rule out the presence of intracellular Aβ oligomers. Further experiments and the use of specific ELISA kits to detect Aβ oligomers will clarify this issue. Finally, it has been reported that, in addition to Aβ peptides, cleavage of APP by β-secretase generates a C-terminal APP fragment (βCTF), which was also markedly increased in cells overexpressing the Swedish mutant APP (eightfold increase as compared to WT APP) (Forman et al., 1997). Accumulation of βCTF impacts on brain microsomes and mitochondrial function (Kim et al., 1998; Chang and Suh, 2005), cellular events that may cause disturbances, ultimately leading to inappropriate organelle coupling. In addition, an alternative cause of an incompetent interaction between organelles may be the lipid composition of the membranes. In this regard, Grimm and colleagues reported previously that the control of cellular cholesterol and sphingomyelin metabolism involves APP processing, and that Aβ42 directly activates neutral sphingomyelinases and downregulates levels of sphingomyelin, whereas Aβ40 reduces de novo synthesis of cholesterol by inhibiting the activity of 3-hydroxy-methylglutaryl-CoA reductase (Grimm et al., 2005).
Collectively, our results advance the current knowledge about the organization of MERCs in neurons at early stages of Alzheimer's disease-like amyloid pathology, which will help to interpret functional roles of MERC axes in the context of Alzheimer's disease.
MATERIALS AND METHODS
Ethical statements
This study was carried out in strict accordance with Animal Research: Reporting of In Vivo Experiments (ARRIVE) and Office Laboratory Animal Welfare (OLAW) NIH guidelines. The protocols were approved by the local Animal Care Committee of the Fundación Instituto Leloir (Argentina), Assurance # A5168-01.
Reagents
Hank's buffered saline solution (HBSS), trypsin-EDTA, DMEM/F12, Neurobasal, B27 and Lipofectamine 3000 were purchased from Thermo Fisher Scientific. Oligomycin, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), antimycin A, rotenone and XF medium were purchased from Seahorse Bioscience (Agilent). Poly-l-lysine and rapamycin were purchased from Merck.
Hippocampal primary neuron cultures
Homozygous transgenic McGill-R-Thy1-APP (Tg+/+) rats, harboring the human APP751 transgene with the Swedish and Indiana mutation under the control of the murine Thy1.2 promoter, were generated using the HsdBrI:WH Wistar strain (Leon et al., 2010). Animals were provided to Fundación Instituto Leloir (FIL) by The Royal Institution for the Advancement of Learning/McGill University, Montreal, Canada, and an in-house colony was established at FIL (Galeano et al., 2014). Cultures were performed as previously described (de Tullio et al., 2013), with minor modifications. Briefly, 10–12 embryos at E18–19 were extracted from the uterus of pregnant WT or Tg+/+ rats, and hippocampi were isolated in HBSS buffer. Tissues were incubated with 2 ml of 0.25% trypsin-EDTA for 20 min at 37°C and then washed three times with DMEM/F12 supplemented with 10% FBS. Hippocampi were homogenized in DMEM/F12 mixture supplemented with 10% FBS by up- and down-pipetting; viability was assessed using Trypan Blue dye. Cells were plated in p poly-l-lysine-coated dishes and, after 2 h, medium was replaced by neuronal medium (Neurobasal, 2 mM L-glutamine, 2% B27, 100 U/ml penicillin, 100 µg/ml streptomycin). Three days after plating, i.e. at DIV3, cytosine β-D-arabinofuranoside (AraC) was added to conditioned medium to a final concentration of 1 µM to inhibit proliferation of glia cells, yielding neuronal cultures with 92% purity. Neurons were maintained at 37°C in a humidified incubator under 5% CO2 until DIV7–9 or DIV14–16.
Immunofluorescence
Neurons were cultured on 12 mm-diameter coverslips and fixed with 4% PFA at DIV7. Cells were then incubated with the primary antibodies against βIII tubulin (mouse, 1:1000, Promega) and Tom20 (rabbit,1:500, Santa Cruz Biotechnology), or glial fibrillary acidic protein (anti-GFAP; rabbit, 1:1000, Dako) overnight at 4°C, followed by incubation with secondary antibodies labelled with Alexa-Fluor 488 or Alexa-Fluor-546. Neurons were counterstained with DAPI to label nuclei.
Neuronal expression of Aβ isoforms
To quantify soluble Aβ 38/40/42, the V-PLEX PLUS Aβ Peptide Panel 1 ELISA kit (Meso Scale Diagnostics) was used following the manufacturer's instructions. Conditioned media and protein homogenates were loaded in duplicates onto MULTI-SPOT® microplates pre-coated with antibodies specific to the C-termini of Aβ38, Aβ40 and Aβ42, and were detected with SULFO-TAG™-labeled 6E10 antibody. Light emitted upon electrochemical stimulation was read using the MSD QuickPlex SQ120 instrument. Data were analyzed using MSD Workbench 4.0 software.
Liquid chromatography–mass spectrometry of free fatty acids
Samples were subjected to liquid chromatography–mass spectrometry LC-MS analysis to detect and quantify levels of free fatty acids in sample extracts. Fatty acid extraction was carried out on each sample using 100% methanol as the homogenization solvent. Cell pellets (2×106 cells/sample) were lysed with 500 μl of methanol and ∼100 μl of zircon beads (0.5 mm). Methanol extracts were centrifuged (21,000 g for 3 min) and transferred to glass LC-MS inserts for analysis. The LC column was a Waters TM BEH-C18 (2.1×100 mm, 1.7 μm) coupled to a Dionex Ultimate 3000™ system and the column oven temperature was set to 25°C for gradient elution. The flow rate was 0.1 ml/min and we used the following buffers: H2O with 0.1% formic acid (buffer A) and acetonitrile with 0.1% formic acid (buffer B). The gradient profile was as follows: 60–99% B from 0–6 min, hold at 99% B from 6–10 min, 99-60% B from 10–11 min, hold at 60% B from 11–15 min. Injection volume was set to 1 μl for all analyses (15 min total run time per injection).
MS analyses were carried out by coupling the LC system to a Thermo Q Exactive HFTM mass spectrometer operating in heated electrospray ionization mode (HESI). Data acquisition was over 10 min with a negative mode full-MS scan (profile mode) and one microscan, with an AGC target of 3×106 and a maximum ion time (IT) of 100 ms at 120,000 resolution, with a scan range from 160–400 m/z. Spray voltage was 3.5 kV and capillary temperature was set to 320°C with a sheath gas rate of 35, aux gas of 10, and max spray current of 100 μA. The acquisition order of samples and standard curve points was randomized, with blank matrix controls before and after each standard curve point to assess carry over (none detected). The resulting free fatty acid peaks were quantified by measuring the relative intensities (peak heights) of the high resolution extracted ion chromatogram (XIC) for each fatty acid across the samples and external standard curve samples. All fatty acids were detected as the negative-mode [M-H]-ion and retention times of the fatty acids were defined using a cocktail of authentic standards. For each XIC, the theoretical m/z of each fatty acid (±5 ppm) was used to extract the peak height (24 s retention time window, 12 s retention time tolerance) as follows: lauric acid (199.1704 m/z, 2.3 min), myristic acid (227.2017 m/z, 3.1 min), palmitoleic acid (253.2173 m/z, 3.4 min), palmitic acid (255.2330 m/z, 4.1 min), oleic acid (281.2486 m/z, 4.4 min), stearic acid (283.2643 m/z, 5.1 min), arachidic acid (311.2956 m/z, 6.0 min), nervonic acid (365.3425 m/z, 6.9 min), lignoceric acid (367.3582 m/z, 7.5 min). The resulting standard curve points (in duplicate) were fit to a linear regression (GraphPad Prism7), and this equation was used to interpolate the concentration of fatty acids in the sample extracts, as prepared.
Neuronal bioenergetics
Cells (6×104 per well) were plated in Seahorse XFp Cell Culture Miniplates (Agilent) and maintained in culture until DIV7 when bioenergetics was assessed. The day of the assay, conditioned medium was aspirated and replaced by XF Base Medium (Agilent) supplemented with 25 mM d-glucose, 1 mM sodium pyruvate and 2 mM l-glutamine (pH 7.4). Cells were incubated with this medium for 1 h at 37°C in a non-CO2 incubator, and then the microplate was loaded into Seahorse XFp Analyzer (Agilent) following manufacturer's guidelines. Oxygen consumption rate (OCR) was determined at the beginning of the assay (baseline OCR) and after the sequential addition of 2 µM oligomycin, 1 µM FCCP and 0.5 µM rotenone+antimycin A. In general, four baseline rates and three response rates (after the addition of a compound) were measured and the average of these rates used for data analysis. Neurons were titrated with 0.125–2 µM FCCP and 1 µM FCCP rendered the maximum OCR, so this concentration was used for the experiments. Non-mitochondrial OCR was determined after the addition of 0.5 µM rotenone+antimycin A and subtracted from all other values before calculation of the respiratory parameters as previously described (Martino Adami et al., 2017). Respiratory parameters were obtained as follows: basal respiration, baseline OCR; respiration driving proton leak, OCR after the addition of 2 mM oligomycin; respiration driving ATP synthesis, basal respiration – respiration driving proton leak; maximum respiration, OCR after the addition of 1 mM FCCP; respiratory control ratio (RCR), maximum respiration/respiration driving proton leak; spare respiratory capacity, maximum respiration – basal respiration; coupling efficiency, respiration driving ATP synthesis/basal respiration.
Plasmids and transfections
Neurons were plated on 25 mm-diameter coverslips and transfected at DIV5 using Lipofectamine 3000 (Thermo Fisher Scientific) with AKAP1-FKBP-YFP and CFP-FRB-ER (short linkers) or AKAP1-18×-FKBP-YFP and CFP-ER-9×-FRB (long linkers) plasmids. The mouse AKAP1 N-terminal sequence (residues 34–63: 5′-MAIQLRSLFPLALPGLLALLGWWWFFSRKK-3′) targets the outer mitochondrial membrane (OMM), and the mouse Sac1 C-terminal sequence (residues 260–326). 5′-FLALPIIMVVAFSMCIICLLMAGDTWTETLAYVLFWGVASIGTFFIILYNGKDFVDAPRLVQKEKID-3′) the ER. The rapamycin-inducible heterodimerization domains FKBP and FRB (Ho et al., 1996) were fused to the AKAP1 and Sac1 sequences, respectively, through repetitive sequences that act like arms, with a longer repetitive sequence for the long linkers and a shorter one for the short linkers. FKBP and FRB domains were also fused to fluorescent proteins YFP and CFP, respectively, which can form a FRET pair.
Multicolor live cell imaging
Forty-eight hours after transfection (DIV7), neuronal medium was replaced by imaging medium, composed of 50% fresh neuronal culture medium (Neurobasal, 2 mM L-glutamine, 2% B27, 100 U/ml penicillin, 100 µg/ml streptomycin) and 50% extracellular medium (ECM; 120 mM NaCl, 5 mM NaHCO3, 10 mM Na-HEPES, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 10 mM glucose 0.25% BSA, pH 7.4). Wide-field fluorescence was recorded at 37°C with a ProEM1024 EM-CCD camera (Princeton Instruments) coupled to an epi-fluorescence inverted Leica DMI 6000B microscope with a 40×/NA 1.35 oil immersion objective (Olympus). The following filters were used: YFP (excitation: 495/20, emission: 532/54), CFP (excitation: 415/30, emission: 502/38) and FRET (excitation: 415/30, emission: 532/54). Each triplet was obtained every 3 s (to avoid photobleaching) during 7 min 30 s (150 images). Rapamycin (100 nM) was added after the first 60 s to induce the increase in FRET signal. Fluorescence was expressed as the FRET:CFP ratio after background subtraction of each individual wavelength using Spectralyzer software (Thomas Jefferson University, Philadelphia,). An average of ten measurements before the addition of rapamycin was subtracted from all other values of each individual experiment to normalize to basal FRET fluorescence.
Transmission electron microscopy
Neurons were plated on 15 mm-diameter coverslips until DIV7 or DIV14 and were fixed in cold 2% glutaraldehyde overnight. The following day, they were fixed in 1% osmium tetroxide, contrasted with 1% uranyl acetate, dehydrated with increasing ethanol concentrations and embedded in Durcupan. Blocks were immersed in liquid nitrogen to release the coverslips and were then sliced into ultrathin sections. Sections were analyzed with a transmission electron microscope (Hitachi 7000). Gap width, interface length and mitochondrial perimeter analyses were performed using a macro for the image-processing package Fiji (Thomas Jefferson University, Philadelphia, PA; Weaver et al., 2017); the ER lumen area was analyzed with ImageJ.
Neuronal mitochondria isolation
Hippocampal primary neuronal cultures were prepared as described above. After DIV7, cells were detached and mitochondria isolated by differential centrifugation following standard procedures (Cadenas et al., 1980). Briefly, neurons were homogenized with a manual Teflon glass homogenizer in ice-cold MSTE buffer (0.23 M mannitol, 0.07 M sucrose, 10 mM Tris-HCl, 1 mM EDTA pH 7.4). Homogenates were centrifuged at 600 g for 10 min to discard cell debris and nuclei, and then supernatants were centrifuged at 8000 g to obtain crude mitochondrial pellets. Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific).
Western blotting
Samples (10 μg/lane) were run on SDS-tricine 12.5% gels and transferred to a PVDF membrane. The membrane was blocked with 5% skimmed milk in PBS-Tween 0.01% for 1 h at room temperature and incubated overnight at 4°C with rabbit anti-Tom20 (1:500, Santa Cruz Biotechnology). Peroxidase-conjugated secondary antibody and enhanced chemiluminescence detection system (ECL, Thermo Fisher Scientific) were used to detect immunoreactivity, which was quantitated with a STORM 840 Phosphor Imager (GE Healthcare).
Liquid Chromatography with tandem mass spectrometry in lipidomics
Mitochondrial samples (10 µg) were subjected to liquid chromatography–mass spectrometry (LC-MS) analysis to detect and identify phospholipid molecules, and quantify the relative levels of identified lipids. Lipid extraction was carried out on each sample based on the method by Vorkas et al. (2015a,b). The dried samples were resolubilized in 10 μl of a isopropanol:acetonitrile:H2O (4:3:1) mixture and analyzed by ultra-performance liquid chromatography–tandem mass spectrometery (UPLC-MS/MS) with a polarity switching method modified from Vorkas et al. (2015a,b). The LC column was a Waters TM CSH-C18 (2.1×100 mm, 1.7 μm) coupled to a Dionex Ultimate 3000™ system, the column oven temperature was set to 55°C for the gradient elution. A flow rate of 0.3 ml/min was used with, buffer A: acetonitrile:H2O (60:40), 10 mM ammonium formate, 0.1% formic acid and, buffer B: 90:10 isopropanol:acetonitrile, 10 mM ammonium formate, 0.1% formic acid. The gradient profile was as follows: 40–43% buffer B (0–1.25 min), 43–50% buffer B (1.25–2 min), 50–54% buffer B (2–11 min), 54–70% buffer B (11–12 min), 70–99% buffer B (12–18 min), 70–99% buffer B (18–32 min), 99–40% buffer B (23–24 min), hold 40% buffer B (1 min). Injection volume was set to 1 μl for all analyses (25 min total run time per injection). MS analyses were carried out by coupling the LC system to a Thermo Q Exactive HFTM mass spectrometer operating in heated electrospray ionization mode (HESI). Method duration was 20 min with a polarity switching data-dependent Top 10 method for both positive and negative modes. Spray voltage for both positive and negative modes was 3.5 kV and capillary temperature was set to 320°C with a sheath gas rate of 35, auxiliary gas of 10, and maximum spray current of 100 μA. The full MS scan for both polarities utilized 120,000 resolution with an AGC target of 3×106 and a maximum IT of 100 ms, and the scan range was from 350–2000 m/z. MS/MS spectra for both positive and negative mode used a resolution of 15,000, AGC target of 1×105, maximum IT of 50 ms, isolation window of 0.4 m/z, isolation offset of 0.1 m/z, fixed first mass of 50 m/z, and three-way multiplexed normalized collision energies (nCE) of 10, 35 and 80. The minimum AGC target was 5×104 with an intensity threshold of 1×106. All data were acquired in profile mode and normalized to protein levels.
Statistical analysis
Data were expressed as the mean±s.e.m. of at least three independent experiments. Mean differences were analyzed using unpaired or paired Student's t-test or two-way ANOVA. Assumptions of normality and homoscedasticity were assessed using the D'Agostino-Pearson omnibus normality test and the F-test of equality of variances, respectively. In some cases the assumption of homoscedasticity was violated and data were analyzed using Welch's unequal variances t-test. Significance level was set at P<0.05. Two-tailed probabilities were always reported. Statistical analysis was performed using GraphPad Prism 6 software (GraphPad software, Inc, La Jolla, CA).
Supplementary Material
Acknowledgements
We acknowledge the helpful assistance of the NYU Metabolomics Core Resource Laboratory at New York University, Langone Medical Center (New York, NY) in the lipidomics research. We thank María Cecilia Rotondaro and Pablo Galeano from Fundación Instituto Leloir (FIL) (Buenos Aires, Argentina) for assistance in immunocytochemistry, confocal microscopy and statistical analysis.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: P.V.M.A., G.H., L.M.; Methodology: P.V.M.A., Z.N., D.B.W., A.B., T.W., D.R.J., S.D.C., A.C.C., G.H.; Validation: Z.N., D.B.W., A.B.; Investigation: P.V.M.A., L.M.; Resources: T.W., D.R.J., G.H., L.M.; Writing - original draft: P.V.M.A., L.M.; Writing - review & editing: P.V.M.A., S.D.C., E.M.C., A.C.C., G.H., L.M.; Supervision: G.H., L.M.; Project administration: L.M.; Funding acquisition: T.W., E.M.C., A.C.C., G.H., L.M.
Funding
This study was supported by funding from the Agencia Nacional de Promoción Científica y Tecnológica (PICT-2015-0285, PICT-2016-4647 and PIBT/09-2013 to L.M.), International Society for Neurochemistry (CAEN Award 2016 to P.V.M.A.), International Union of Biochemistry and Molecular Biology (Wood-Whelan Fellowship 2016 to P.V.M.A.), Canadian Institutes of Health Research (201603PJT-364544 to A.C.C.) and National Institutes of Health grants (DK36609 and GM53407 to G.H.; AG008051, AG056850 and NS073502 to T.W.). P.V.M.A. was supported by Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) fellowship. L.M. and E.M.C. are members of the Research Career of CONICET. S.D.C. is the holder of the Charles E. Frosst/Merck Research Associate position. A.C.C. is member of the Canadian Consortium of Neurodegeneration in Aging (CCNA) and holder of the McGill University Charles E. Frosst/Merck Chair in Pharmacology. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.229906.supplemental
References
- Area-Gomez E., Del Carmen Lara Castillo M., Tambini M. D., Guardia-Laguarta C., de Groof A. J. C., Madra M., Ikenouchi J., Umeda M., Bird T. D., Sturley S. L. et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106-4123. 10.1038/emboj.2012.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloyannis S. J. (2011). Mitochondria are related to synaptic pathology in Alzheimer's disease. Int. J. Alzheimers Dis. 2011, 305395 10.4061/2011/305395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman J. M., Perocchi F., Girgis H. S., Plovanich M., Belcher-Timme C. A., Sancak Y., Bao X. R., Strittmatter L., Goldberger O., Bogorad R. L. et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341-345. 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R., Vicencio J. M., Parra V., Troncoso R., Munoz J. P., Bui M., Quiroga C., Rodriguez A. E., Verdejo H. E., Ferreira J. et al. (2011). Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 124, 2143-2152. 10.1242/jcs.080762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas E., Boveris A. and Chance B. (1980). Low-level chemiluminescence of bovine heart submitochondrial particles. Biochem. J. 186, 659-667. 10.1042/bj1860659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K.-A. and Suh Y.-H. (2005). Pathophysiological roles of amyloidogenic carboxy-terminal fragments of the beta-amyloid precursor protein in Alzheimer's disease. J. Pharmacol. Sci. 97, 461-471. 10.1254/jphs.CR0050014 [DOI] [PubMed] [Google Scholar]
- Csordás G., Renken C., Várnai P., Walter L., Weaver D., Buttle K. F., Balla T., Mannella C. A. and Hajnóczky G. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915-921. 10.1083/jcb.200604016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Várnai P., Golenár T., Roy S., Purkins G., Schneider T. G., Balla T. and Hajnóczky G. (2010). Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121-132. 10.1016/j.molcel.2010.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Weaver D. and Hajnóczky G. (2018). Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 28, 523-540. 10.1016/j.tcb.2018.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaRocha-Souto B., Coma M., Pérez-Nievas B. G., Scotton T. C., Siao M., Sánchez-Ferrer P., Hashimoto T., Fan Z., Hudry E., Barroeta I. et al. (2012). Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer's disease. Neurobiol. Dis. 45, 425-437. 10.1016/j.nbd.2011.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D., Raffaello A., Teardo E., Szabò I. and Rizzuto R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336-340. 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Tullio M. B., Castelletto V., Hamley I. W., Martino Adami P. V., Morelli L., Castaño E. M. (2013). Proteolytically inactive insulin-degrading enzyme inhibits amyloid formation yielding non-neurotoxic Aβ peptide aggregates. PLoS ONE 8, e59113 10.1371/journal.pone.0059113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos K. J., Grierson A. J., Ackerley S. and Miller C. C. J. (2008). Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 31, 151-173. 10.1146/annurev.neuro.31.061307.090711 [DOI] [PubMed] [Google Scholar]
- Del Prete D., Suski J. M., Oulès B., Debayle D., Gay A. S., Lacas-Gervais S., Bussiere R., Bauer C., Pinton P., Paterlini-Bréchot P. et al. (2017). Localization and processing of the amyloid-beta protein precursor in mitochondria-associated membranes. J. Alzheimers Dis. 55, 1549-1570. 10.3233/JAD-160953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L. and Ohno M. (2012). Mitochondrial dysfunction and accumulation of the beta-secretase-cleaved C-terminal fragment of APP in Alzheimer's disease transgenic mice. Neurobiol. Dis. 45, 417-424. 10.1016/j.nbd.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flis V. V. and Daum G. (2013). Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harbor Perspect. Biol. 5, a013235. 10.1101/cshperspect.a013235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman M. S., Cook D. G., Leight S., Doms R. W. and Lee V. M.-Y. (1997). Differential effects of the Swedish mutant amyloid precursor protein on beta-amyloid accumulation and secretion in neurons and nonneuronal cells. J. Biol. Chem. 272, 32247-32253. 10.1074/jbc.272.51.32247 [DOI] [PubMed] [Google Scholar]
- Gabuzda D., Busciglio J., Chen L. B., Matsudaira P. and Yankner B. A. (1994). Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 269, 13623-13628. [PubMed] [Google Scholar]
- Galeano P., Martino Adami P. V., Do Carmo S., Blanco E., Rotondaro C., Capani F., Castaño E. M., Cuello A. C. and Morelli L. (2014). Longitudinal analysis of the behavioral phenotype in a novel transgenic rat model of early stages of Alzheimer's disease. Front. Behav. Neurosci. 8, 321 10.3389/fnbeh.2014.00321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini L., Racchi M., Benussi L., Curti D., Binetti G., Bianchetti A., Trabucchi M. and Govoni S. (1997). Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci. Lett. 231, 113-117. 10.1016/S0304-3940(97)00536-3 [DOI] [PubMed] [Google Scholar]
- Giacomello M. and Pellegrini L. (2016). The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. 23, 1417-1427. 10.1038/cdd.2016.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M. O., Grimm H. S., Pätzold A. J., Zinser E. G., Halonen R., Duering M., Tschäpe J.-A., De Strooper B., Müller U., Shen J. et al. (2005). Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat. Cell Biol. 7, 1118-1123. 10.1038/ncb1313 [DOI] [PubMed] [Google Scholar]
- Hajnóczky G., Robb-Gaspers L. D., Seitz M. B. and Thomas A. P. (1995). Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415-424. 10.1016/0092-8674(95)90430-1 [DOI] [PubMed] [Google Scholar]
- Hansson Petersen C. A., Alikhani N., Behbahani H., Wiehager B., Pavlov P. F., Alafuzoff I., Leinonen V., Ito A., Winblad B., Glaser E. et al. (2008). The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 105, 13145-13150. 10.1073/pnas.0806192105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S. and Saido T. C. (2018). Critical review: involvement of endoplasmic reticulum stress in the aetiology of Alzheimer's disease. Open Biol. 8, 180024 10.1098/rsob.180024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedskog L., Pinho C. M., Filadi R., Ronnback A., Hertwig L., Wiehager B., Larssen P., Gellhaar S., Sandebring A., Westerlund M. et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer's disease and related models. Proc. Natl. Acad. Sci. USA 110, 7916-7921. 10.1073/pnas.1300677110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. N., Biggar S. R., Spencer D. M., Schreiber S. L., Crabtree G. R. (1996). Dimeric ligands define a role for transcriptional activation domains in reinitiation. Nature 382, 822-826. 10.1038/382822a0 [DOI] [PubMed] [Google Scholar]
- Janikiewicz J., Szymański J., Malinska D., Patalas-Krawczyk P., Michalska B., Duszyński J., Giorgi C., Bonora M., Dobrzyn A. and Wieckowski M. R. (2018). Mitochondria-associated membranes in aging and senescence: structure, function, and dynamics. Cell Death Dis. 9, 332 10.1038/s41419-017-0105-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A. S., Thompson M. N., Fei N., Hüttemann M. and Greenberg M. L. (2012). Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J. Biol. Chem. 287, 17589-17597. 10.1074/jbc.M111.330167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville L. S., Pinton P., Bastianutto C., Rutter G. A. and Rizzuto R. (1999). Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 96, 13807-13812. 10.1073/pnas.96.24.13807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D. and Mattson M. P. (2011). Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer's disease. Lancet Neurol. 10, 187-198. 10.1016/S1474-4422(10)70277-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. C. and Guan K.-L. (2015). mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest. 125, 25-32. 10.1172/JCI73939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-S., Park C.-H. and Suh Y.-H. (1998). C-terminal fragment of amyloid precursor protein inhibits calcium uptake into rat brain microsomes by Mg2+-Ca2+ ATPase. Neuroreport 9, 3875-3879. 10.1097/00001756-199812010-00020 [DOI] [PubMed] [Google Scholar]
- Kind T., Liu K.-H., Lee D. Y., DeFelice B., Meissen J. K. and Fiehn O. (2013). LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 10, 755-758. 10.1038/nmeth.2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostova Z. and Wolf D. H. (2003). For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 22, 2309-2317. 10.1093/emboj/cdg227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal N. S., Dentoni G., Schreiner B., Kämäräinen O.-P., Partanen N., Herukka S.-K., Koivisto A. M., Hiltunen M., Rauramaa T., Leinonen V. et al. (2018). Alterations in mitochondria-endoplasmic reticulum connectivity in human brain biopsies from idiopathic normal pressure hydrocephalus patients. Acta Neuropathol. Commun. 6, 102 10.1186/s40478-018-0605-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon W. C., Canneva F., Partridge V., Allard S., Ferretti M. T., DeWilde A., Vercauteren F., Atifeh R., Ducatenzeiler A., Klein W. et al. (2010). A novel transgenic rat model with a full Alzheimer's-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J. Alzheimers Dis. 20, 113-126. 10.3233/JAD-2010-1349 [DOI] [PubMed] [Google Scholar]
- Leuner K., Hauptmann S., Abdel-Kader R., Scherping I., Keil U., Strosznajder J. B., Eckert A. and Müller W. E. (2007). Mitochondrial dysfunction: the first domino in brain aging and Alzheimer's disease? Antioxid Redox Signal. 9, 1659-1676. 10.1089/ars.2007.1763 [DOI] [PubMed] [Google Scholar]
- Leuner K., Schütt T., Kurz C., Eckert S. H., Schiller C., Occhipinti A., Mai S., Jendrach M., Eckert G. P., Kruse S. E. et al. (2012). Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal. 16, 1421-1433. 10.1089/ars.2011.4173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidke D. S. and Lidke K. A. (2012). Advances in high-resolution imaging--techniques for three-dimensional imaging of cellular structures. J. Cell Sci. 125, 2571-2580. 10.1242/jcs.090027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso M., Coppede F., Murri L. and Siciliano G. (2007). Mitochondrial cascade hypothesis of Alzheimer's disease: myth or reality? Antioxid Redox Signal. 9, 1631-1646. 10.1089/ars.2007.1761 [DOI] [PubMed] [Google Scholar]
- Martino Adami P. V., Quijano C., Magnani N., Galeano P., Evelson P., Cassina A., Do Carmo S., Leal M. C., Castaño E. M., Cuello A. C., Morelli L. (2017). Synaptosomal bioenergetic defects are associated with cognitive impairment in a transgenic rat model of early Alzheimer's disease. J. Cereb. Blood Flow Metab. 37, 69-84. 10.1177/0271678X15615132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marwarha G., Claycombe K., Schommer J., Collins D. and Ghribi O. (2016). Palmitate-induced endoplasmic reticulum stress and subsequent C/EBPα homologous protein activation attenuates leptin and insulin-like growth factor 1 expression in the brain. Cell. Signal. 28, 1789-1805. 10.1016/j.cellsig.2016.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack J. G., Halestrap A. P. and Denton R. M. (1990). Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391-425. 10.1152/physrev.1990.70.2.391 [DOI] [PubMed] [Google Scholar]
- Mosconi L., Mistur R., Switalski R., Tsui W. H., Glodzik L., Li Y., Pirraglia E., De Santi S., Reisberg B., Wisniewski T. et al. (2009). FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging 36, 811-822. 10.1007/s00259-008-1039-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naon D., Zaninello M., Giacomello M., Varanita T., Grespi F., Lakshminaranayan S., Serafini A., Semenzato M., Herkenne S., Hernández-Alvarez M. I. et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 113, 11249-11254. 10.1073/pnas.1606786113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M., Halter L., Lim A. and Lardelli M. (2017). Mitochondrion to endoplasmic reticulum apposition length in zebrafish embryo spinal progenitors is unchanged in response to perturbations associated with Alzheimer's disease. PLoS ONE 12, e0179859 10.1371/journal.pone.0179859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima Y., Taguchi K., Mizuta I., Tanaka M., Tomiyama T., Kametani F., Yabe-Nishimura C., Mizuno T. and Tokuda T. (2018). Mutations in the beta-amyloid precursor protein in familial Alzheimer's disease increase Abeta oligomer production in cellular models. Heliyon 4, e00511 10.1016/j.heliyon.2018.e00511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C., Voelker D. R. and Langer T. (2011). Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 192, 7-16. 10.1083/jcb.201006159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira C., Santos M. S. and Oliveira C. (1998). Mitochondrial function impairment induced by amyloid beta-peptide on PC12 cells. Neuroreport 9, 1749-1755. 10.1097/00001756-199806010-00015 [DOI] [PubMed] [Google Scholar]
- Rizzuto R., De Stefani D., Raffaello A. and Mammucari C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566-578. 10.1038/nrm3412 [DOI] [PubMed] [Google Scholar]
- Sala-Vila A., Navarro-Lérida I., Sánchez-Alvarez M., Bosch M., Calvo C., López J. A., Calvo E., Ferguson C., Giacomello M., Serafini A. et al. (2016). Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci. Rep. 6, 27351 10.1038/srep27351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauder C. M., Wu X., Saheki Y., Narayanaswamy P., Torta F., Wenk M. R., De Camilli P. and Reinisch K. M. (2014). Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature 510, 552-555. 10.1038/nature13269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz A., Schneider A., Kummer M. P. and Herzog V. (2004). Endoplasmic reticulum-localized amyloid beta-peptide is degraded in the cytosol by two distinct degradation pathways. Traffic 5, 89-101. 10.1111/j.1600-0854.2004.00159.x [DOI] [PubMed] [Google Scholar]
- Shi F., Kawano F., Park S.-E., Komazaki S., Hirabayashi Y., Polleux F. and Yazawa M. (2018). Optogenetic control of endoplasmic reticulum-mitochondria tethering. ACS Synthetic Biol. 7, 2-9. 10.1021/acssynbio.7b00248 [DOI] [PubMed] [Google Scholar]
- Shore G. C. and Tata J. R. (1977). Two fractions of rough endoplasmic reticulum from rat liver. I. Recovery of rapidly sedimenting endoplasmic reticulum in association with mitochondria. J. Cell Biol. 72, 714-725. 10.1083/jcb.72.3.714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood A., Jeyaraju D. V., Prudent J., Caron A., Lemieux P., McBride H. M., Laplante M., Tóth K. and Pellegrini L. (2014). A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. USA 111, 16017-16022. 10.1073/pnas.1408061111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica R. De Vos K. J., Paillusson S. Mueller S. Sancho R. M., Lau K.-F., Vizcay-Barrena G., Lin W.-L., Xu Y.-F., Lewis J. et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5, 3996 10.1038/ncomms4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow R. H. (2012). Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer's disease. Antioxid Redox Signal. 16, 1434-1455. 10.1089/ars.2011.4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G. and Hoetzl S. (2010). Sphingolipid topology and the dynamic organization and function of membrane proteins. FEBS Lett. 584, 1800-1805. 10.1016/j.febslet.2009.10.020 [DOI] [PubMed] [Google Scholar]
- van Meer G., Voelker D. R. and Feigenson G. W. (2008). Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112-124. 10.1038/nrm2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J. E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248-7256. [PubMed] [Google Scholar]
- Voelker D. R. (1984). Phosphatidylserine functions as the major precursor of phosphatidylethanolamine in cultured BHK-21 cells. Proc. Natl. Acad. Sci. USA 81, 2669-2673. 10.1073/pnas.81.9.2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorkas P. A., Isaac G., Anwar M. A., Davies A. H., Want E. J., Nicholson J. K. and Holmes E. (2015a). Untargeted UPLC-MS profiling pipeline to expand tissue metabolome coverage: application to cardiovascular disease. Anal. Chem. 87, 4184-4193. 10.1021/ac503775m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorkas P. A., Shalhoub J., Isaac G., Want E. J., Nicholson J. K., Holmes E. and Davies A. H. (2015b). Metabolic phenotyping of atherosclerotic plaques reveals latent associations between free cholesterol and ceramide metabolism in atherogenesis. J. Proteome Res. 14, 1389-1399. 10.1021/pr5009898 [DOI] [PubMed] [Google Scholar]
- Wang D.-C., Sun C.-H., Liu L.-Y., Sun X.-H., Jin X.-W., Song W.-L., Liu X.-Q. and Wan X.-L. (2012). Serum fatty acid profiles using GC-MS and multivariate statistical analysis: potential biomarkers of Alzheimer's disease. Neurobiol. Aging 33, 1057-1066. 10.1016/j.neurobiolaging.2010.09.013 [DOI] [PubMed] [Google Scholar]
- Weaver D., Bartok A., Csordas G. and Hajnoczky G. (2017). A standardized method to quantify ER-mitochondrial interfaces in electron mircographs. Biophys. J. 112, 133A 10.1016/j.bpj.2016.11.73528076804 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.