

Abstract
One-bead-one-compound (OBOC) libraries constructed by solid-phase split-and-pool synthesis are a valuable source of protein ligands. Most OBOC libraries are comprised of oligoamides, particularly peptides, peptoids and peptoid-inspired molecules. Further diversification of the chemical space covered by OBOC libraries is desirable. Towards this end, we report here that the proline-catalyzed asymmetric aldol reaction, developed by List and Barbas for solution-phase synthesis, also works well for coupling immobilized aldehydes and soluble ketones. These reaction conditions do not compromise the amplification of DNA by the polymerase chain reaction. Thus, this chemistry should be useful for the construction of novel DNA-encoded OBOC libraries by solid-phase synthesis.
Keywords: Aldol reaction, solid-phase synthesis, organocatalysis, one bead one compound libraries, DNA-encoded libraries
Graphical Abstract

Introduction
The creation of one bead one compound (OBOC) libraries by split and pool solid-phase synthesis1, 2 is a powerful method for the rapid and inexpensive synthesis of large libraries of potential protein ligands.3–5 When created on a suitable resin, such as TentaGel, these libraries can be screened by incubation with a labeled protein and manually picking beads that retain a high level of fluorescence.6 The compound is released from the bead and its structure characterized, usually by mass spectrometry (MS).7, 8 While the utility of this approach to the discovery of protein ligands had previously been limited by a number of nagging technical problems, most of these issue have now been addressed. For example, bead screening has a notoriously high false positive rate, but it has been shown that if one screens redundant libraries, hits that are isolated on multiple beads are almost always bona fide ligands.9
An important advance in OBOC technology was the application of encoding10 tags to the beads.11, 12 This eliminates the need for direct MS-based characterization of hit compounds, which had largely limited OBOC libraries to oligomers that fragment in a logical fashion in the tandem MS or can be sequenced by Edman degradation, such as peptides and peptoids.3, 13–15 DNA-encoded OBOC libraries, in particular, confer the additional advantage that the library can be constructed and screened on 10 μm TentaGel beads, which are small enough to pass through a flow cytometer. Isolation of hit beads by using a fluorescent activated cell sorter (FACS) is far more convenient than manual picking and facilitates two-color screens that demand selectivity for a target over one or more off-targets.16 The encoding tags are then deep sequenced.
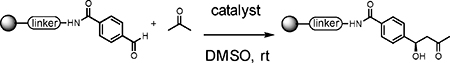
With this powerful technology now established, it is important to expand the repertoire of chemical reactions that can be used to make OBOC libraries. Reactions that create chiral centers are of particular interest, since there is a general feeling in the screening community that molecules with a high level of “three-dimensionality” and natural product-like character would tend to bind their targets with higher selectivity than the flat, largely aromatic compounds that dominate most current screening collections.17–20 Towards this end, we report here conditions for the efficient solid-phase synthesis of β-hydroxy ketones via the organocatalytic aldol reaction21–28 of immobilized aldehydes and soluble ketones (Scheme 1C). Another important consideration in developing reactions for the synthesis of DNA-encoded libraries is to evaluate the level of chemical damage suffered by the encoding DNA that would compromise it amplifiability. For example, routine amino acid or peptoid couplings are known to degrade encoding tags to some degree (Scheme 1 A and B).29 The mild conditions for the aldol reaction are shown to be quite compatible with the presence of the DNA tags. Thus, this chemistry should be useful in the synthesis of novel DNA-encoded OBOC libraries.12
Scheme 1.

Chemistry suitable for the construction of one bead one compound libraries by solid-phase synthesis. The red stars on the DNA indicate chemical modifications that would impede amplification of the encoding tag.
Results and Discussion
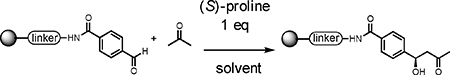
TentaGel HL NH2 resin (110 μm, 0.46 mmol/g) was modified with a disulfide-containing linker to allow the release of compounds from the solid support under mild reductive conditions (treatment with TCEP) to avoid potential dehydration following the aldol addition product under the usual acidic cleavage conditions. The amino terminal end of the linker was acylated with 4-formyl benzoic acid using Oxyma coupling conditions30 (see supporting information). Various solvents and conditions were then explored for the (S)-proline-catalyzed addition of acetone to this immobilized aldehyde. We included in all of the reactions beads that contained an inactive control substrate made by coupling benzoic acid (with no formyl group) to the linker. This inert compound served as an internal standard to aid in the quantification of the conversion of the formyl-containing substrate.
We found using DMSO as the solvent and incubating for 16 hours at room temperature in the presence of one equivalent of proline (with respect to the aldehyde on the resin) provided the highest yield of the desired aldol product (Table 1, entry 6), as determined by liquid chromatography-mass spectrometry (LC-MS). This result was obtained using a large excess of ketone with respect to the immobilized aldehyde (100:1 molar ratio). Reactions employing lower ketone levels resulted in poor conversion. Poor results were also obtained when the amount of proline was reduced. Note that proline was used as a suspension due to its modest solubility in all of the solvents examined. We also examined various proline derivatives such as tetrazole substituted-proline31 2-(methoxymethyl)-pyrrolidine23 and prolineamide,23 but found that (S)-proline was the best choice (Table 2), which was also true for the solution-phase organocatalytic aldol reaction.23
Table 1.
Establishment of optimal conditions.
 | |||
|---|---|---|---|
| Entry | Solvent | Temp (°C) | Conversion (%) |
| 1 | acetonitrile | 4 | 0 |
| 2 | acetonitrile | 20 | 2 |
| 3 | NMP | 4 | 11 |
| 4 | NMP | 20 | 42 |
| 5 | DMSO | 4 | 75 |
| 6 | DMSO | 20 | 95 |
| 7 | DMF | 4 | 4 |
| 8 | DMF | 20 | 35 |
Reaction conditions: Reactions were carried out using 1 eq. of (S)-proline (relative to the immobilized aldehyde (0.006 mmol)) and acetone (100 eq, 44 μL) in 120 μL DMSO. Conversion was determined from cleaved crude mixture by monitoring LC-MS analysis of the crude product after reductive cleavage from the beads., This was calculated by measuring the ratio of the remaining aldehyde peak and that of an internal standard (the equivalent molecule lacking a formyl group) (see supporting info).
Table 2.
Comparison of various proline-derived catalysts.
 | ||||
|---|---|---|---|---|
| Entry | catalyst | amount of catalyst (eq) | Conversion (%) | ee (%) |
| 1 | 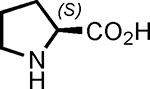 |
1 | 99 | 73 |
| 2a |  |
1 | 99 | 70 |
| 3 | 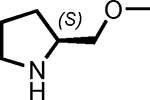 |
5 | 98 | 8 |
| 4 | 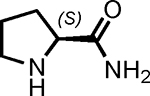 |
3 | 99 | 1 |
Reaction conditions: Reactions were carried out using organocatalyst and acetone (100 eq, 44 μL) to resin loading (0.006 mmol) in 120 μL of DMSO for 16 hours. Conversion was determined from cleaved crude mixture by monitoring LC-MS, calculated by two peak ratio between starting aldehyde and inactive benzoic ring which is used for internal standard. (see supporting info)
Reaction was completed in 4h.
The aldol product was formed in a modest 73% e.e. The use of a variety of additives such as chiral diols,32 thioureas33 and guanidinium salts34 or lowering the temperature (4°C and −10°C) did not improve the enantioselectivity significantly (data not shown). Hence, the conditions described above were used to explore the scope of the reaction with respect to the aldehyde and ketone components.
Table 3 shows the results of the reaction of acetone with a number of immobilized aldehydes. All of the reactions proceeded with almost quantitative conversion, even when an ortho-substituent was present (1g, 1h). However, when the formyl group was ortho to the amide no reaction was observed (data not shown). A heteroaromatic aldehyde also worked well (1i). In general, the enantioselectivities were modest, with e.e. ranging from 54%−79%. A notable exception was 1f, which was produced as a single stereoisomer. This is typical for the solution phase aldol reaction of benzaldehydes with electron-withdrawing substituents ortho to the aldehyde, as noted by List and co-workers.35
Table 3.
Substrate scope of acceptor aldehyde.
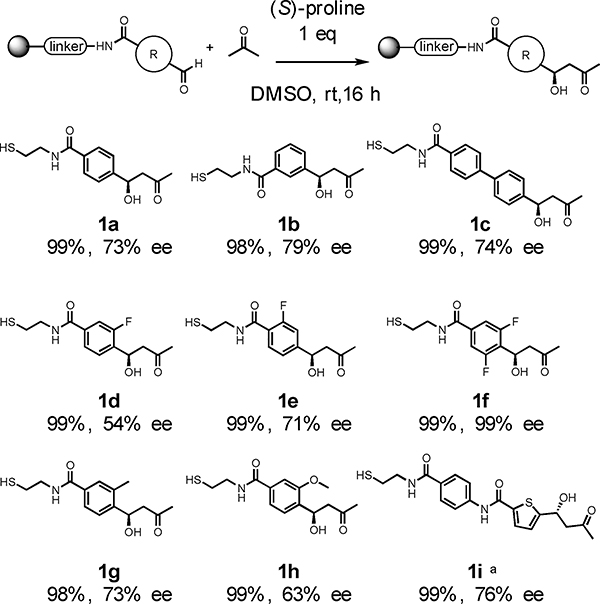 |
Reaction conditions: Reactions were carried out using 1 eq (S)-proline and acetone (100 eq, 44 ∝L) to resin loading (0.006 mmol) in 120 ∝L DMSO. Conversion was determined from cleaved crude mixture by monitoring LC-MS, calculated by two peak ratio between starting aldehyde and inactive benzoic ring which is used for internal standard. Ee were determined by chiral HPLC (see supporting info).
To monitor products by UV length of 254 nm, Fmoc-4-Abz-OH was coupled before coupling 5-formylthiophen-2-carboxylic acid.
Next, direct aldol reactions between the p-formyl benzoic acid-derived immobilized substrate and various soluble ketones were investigated (Table 4). The reactivity of most soluble ketones was excellent (90%−99% conversion), with the sole exception of dimethoxyacetone, which was a respectable 78%. In all cases, the anti product was favored, generally providing drs of 3:1 – 4:1. Hydroxyacetone was exceptional in that no detectable syn product was formed using this ketone. The enantioselectivity was improved over the reactions employing acetone. With the exception of cyclopentanone (54% e.e.), the major anti product was produced in 84%−98% e.e. The use of nonsymmetrical ketones produced mixtures of regioisomers. For 2-butanone, there was no preference, with both regioisomers produced in equal amounts. However, for hydroxyacetone and methoxyacetone, the regioisomer resulting from the more substituted carbon acting as the nucleophile (called the non-terminal product in Table 4) was strongly favored (>20:1). These results largely mirror those obtained in the solution phase aldol reaction.36
Table 4.
Substrate scope of donor ketone
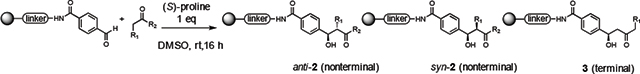 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ketone | Conversion (anti-2 + syn-2 + 3) (%) | 2 |
rr (2:3) | 3 |
|
| dr (anti:syn) | ee (%) anti; (syn) | ee (%) | ||||
| 1 | cyclopentanone | 99 (anti-2a + syn-2a) | 4:1 | 57 (69) | – | – |
| 2 | cyclohexanone | 90 (anti-2b + syn-2b) | 1:1 | 85 (80) | – | – |
| 3 | dimethoxyacetone | 78 (anti-2c + syn-2c) | 4:1 | 84 (n.d.)a | – | – |
| 4 | methoxyacetone | 90 (anti-2d + syn-2d + trace3d) | 3:1 | 98 (n.d.)b | > 20:1 | n.d.b |
| 5 | 2-butanone | 99 (anti-2e + syn-2e + 3e) | 4:1 | 96 (80) | 1:1 | 94 |
| 6 | hydroxyacetone | 99 (anti-2f + syn-2f + trace3f) | > 20:1 | 84 (n.d.)b | > 20:1 | n.d.b |
Reaction conditions: Reactions were carried out using 1 eq (S)-proline and 100 eq of ketones to resin loading (0.006 mmol) in 120 μL DMSO. Conversion were determined from cleaved crude mixture by monitoring LC-MS, calculated by two peak ratio between starting aldehyde and inactive benzoic ring which is used for internal standard. ee were determined by chiral HPLC. Diastereoisomeric and regioisomeric ratios were obtained by 1H NMR of crude mixture.
Not determined because chiral separation was failured.
Not determied because we could not obtain enough amount of compound for analysis.
With an eye towards incorporating useful functionality into the aldol products, Fmoc-protected 1-amino-2-propanone and Fmoc-protected 4-piperidinone were examined as substrates, but these reactions failed to produce the desired aldol products. This is probably due to the lower solubility of these ketones in DMSO. The same high concentration that could be achieved for the ketones shown in Table 4 was not possible with these substrates. This is likely to be a problem using any ketone containing an Fmoc-protected amine. Therefore, we turned our attention to trying to incorporate an azido moiety into the aldol product. An azide can be further elaborated using copper-catalyzed Huisgen cycloadditions, aza-Wittg reactions or can be reduced to an amine with a phosphine. Unfortunately, under the standard conditions described above, azidoacetone provided a poor yield of the desired aldol product. However, we found that the addition of a guanidinium-type ionic liquid (see SI for a detailed procedure), as reported by Martinez-Casteneda, et al.,37 provided the desired α-azido-β-hydroxy ketone in excellent yield as well as with high stereo-, regio-, and enantioselectivity (Fig 1A). However, a two-day incubation at 4°C was required to obtain this result. Thus, we also examined the possibility of displacing the alcohol in the aldol product with azide under Mitsunobu conditions (Fig 1B). After several attempts, we found that using a large excess (50 equivalents) of all the soluble reagents required for the Mitsunobu reaction provided a high yield of the desired product (see SI for a detailed procedure). Thus, using either of these protocols, a versatile azido group can be incorporated into the aldol product, allowing further elaboration of the molecule.
Figure 1.

Incorporation of azide group to aldol product (A) aldol reaction using azido acetone. Diastereoisomeric and regioisomeric ratios were obtained by 1H NMR of crude mixture. ee were determined by chiral HPLC. (B) Convert hydroxy group to azide using Mitsunobu reaction.
We hypothesized that the mild conditions employed for the proline-catalyzed aldol reaction would be compatible with encoding DNA. To address this point, we employed a quantitative polymerase chain reaction (qPCR) assay described by Malone and Paegel29 in which a 200 bp double-stranded DNA molecule is subjected to the reaction conditions and the amount of DNA that remains amplifiable by the polymerase chain reaction (PCR) is quantified. Thus, the immobilized DNA was incubated for 16 hours at room temperature in the presence of acetone, immobilized aldehyde and proline. The qPCR data showed that at least 90% of the DNA could be amplified relative to a control reaction in which the DNA was not exposed to any chemical reagents (Fig. 2).
Figure 2.

Quantification of amplifiable DNA remaining ratio by DNA damage assay. (A) Caliburation curve made by DNA standard samples of a known concentration. R=0.96(B) Remaining DNA damage ratio of aldol reaction and mitsunobu reaction. The remaing DNA ratio of aldol reaction values were 91±14%. That of mitsunobu reaction were 93±8%. Results were generated from triplicate experiments.
In conclusion, the proline-catalyzed aldol reaction has been adapted to bead-based synthesis. To a large degree, the trends observed in the solid-phase reaction parallel those observed when the reaction is carried out in solution, the major difference being the need for a large excess of ketone and an equivalent of proline. As anticipated, the reaction conditions have little or no effect on the amplifiability of encoding DNA. Therefore, this process should be a valuable tool for the construction of DNA-encoded OBOC libraries. Finally, we have employed in this study a disulfide linker that can be cleaved reductively38 to ensure that our analysis of the intrinsic stereoselectivity of the aldol reaction was not compromised by the chemistry used to release the compound from the bead, as might be the case for acidic cleavage, for example. In subsequent work we have found that cleavage of compounds from resins with RAM linkers by brief treatment with dilute trifluoroacetic acid does not result in detectable racemization (data not shown), so there is no need to use the disulfide linker if this is not convenient.
Supplementary Material
Acknowledgements
We thank Ms. Paige Dickson, Dr. Hongchan An, Dr. Hao Wu and Mr. Scott Simanski for technical assistance.
Funding
This research was supported by a grant form the NIH (AG054892).
Footnotes
The authors have no conflicts of interest to declare.
References
- (1).Lam KS; Salmon SE; Hersh EM; Hruby VJ; Kazmierski WM; Knapp RJ, A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- (2).Houghten RA; Pinilla C; Blondelle SE; Appel JR; Dooley CT; Cuervo JH, Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 1991, 354, 84–6. [DOI] [PubMed] [Google Scholar]
- (3).Alluri PG; Reddy MM; Bachhawat-Sikder K; Olivos HJ; Kodadek T, Isolation of protein ligands from large peptoid libraries. J. Amer. Chem. Soc 2003, 125, 13995–4004. [DOI] [PubMed] [Google Scholar]
- (4).Lian W; Upadhyaya P; Rhodes CA; Liu Y; Pei D, Screening Bicyclic Peptide Libraries for Protein-Protein Interaction Inhibitors: Discovery of a Tumor Necrosis Factor-alpha Antagonist. J. Amer. Chem. Soc 2013, 135, 11990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Moon H; Lim HS, Synthesis and screening of small-molecule alpha-helix mimetic libraries targeting protein-protein interactions. Curr. Opin. Chem. Biol 2015, 24, 38–47. [DOI] [PubMed] [Google Scholar]
- (6).Olivos HJ; Baccawat-Sikder K; Kodadek T, Quantum dots as a visual aid for screening bead-bound combinatorial libraries. ChemBiochem 2003, 4, 1242–1245. [DOI] [PubMed] [Google Scholar]
- (7).Paulick MG; Hart KM; Brinner KM; Tjandra M; Charych DH; Zuckermann RN, Cleavable hydrophilic linker for one-bead-one-compound sequencing of oligomer libraries by tandem mass spectrometry. J. Comb. Chem 2006, 8, 417–426. [DOI] [PubMed] [Google Scholar]
- (8).Thakkar A; Cohen AS; Connolly MD; Zuckermann RN; Pei D, High-throughput sequencing of peptoids and peptide-peptoid hybrids by partial Edman degradation and mass spectrometry. J. Comb. Chem 2009, 11, 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Doran TM; Gao Y; Mendes K; Dean S; Simanski S; Kodadek T, The utility of redundant combinatorial libraries in distinguishing high and low quality screening hits. ACS Comb. Sci 2014, 16, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ohlmeyer MH; Swanson RN; Dillard LW; Reader JC; Asouline G; Kobayashi R; Wigler M; Still WC, Complex synthetic chemical libraries indexed with molecular tags. Proc. Natl. Acad. Sci. USA 1993, 90, 10922–10926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Liu R; Marik J; Lam KS, A novel peptide-based encoding system for “one-bead one-compound” peptidomimetic and small molecule combinatorial libraries. J. Amer. Chem. Soc 2002, 124, 7678–7680. [DOI] [PubMed] [Google Scholar]
- (12).MacConnell AB; McEnaney PJ; Cavett VJ; Paegel BM, DNA-Encoded Solid-Phase Synthesis: Encoding Language Design and Complex Oligomer Library Synthesis. ACS Comb Sci 2015, 17, 518–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lam KS; Lake D; Salmon SE; Smith JD; Chen M-L; Wade S; Abdul-Latif F; Knapp RJ; Leblova Z; Ferguson RD; Krchnak V; Sepetov NF; Lebl M, A one-bead one-peptide combinatorial library method for B-cell epitope mapping. Methods 1996, 9, 482–493. [DOI] [PubMed] [Google Scholar]
- (14).Figliozzi GM; Goldsmith R; Ng SC; Banville SC; Zuckermann RN, Synthesis of N-substituted glycine peptoid libraries. Methods Enzymol. 1996, 267, 437–447. [DOI] [PubMed] [Google Scholar]
- (15).Zuckermann RN; Martin EJ; Spellmeyer DC; Stauber GB; Shoemaker KR; Kerr JM; Figliozzi GM; Goff DA; Siani MA; Simon RJ; al., e., Discovery of nanomolar ligands for 7-transmembrane G-protein-coupled receptors from a diverse N-(substituted)glycine peptoid library. J. Med. Chem 1994, 37, 2678–2685. [DOI] [PubMed] [Google Scholar]
- (16).Mendes K; Malone ML; Ndungu JM; Suponitsky-Kroyter I; Cavett VJ; McEnaney PJ; MacConnell AB; Doran TM; Ronacher K; Stanley K; Utset O; Walzl G; Paegel BM; Kodadek T, High-throughput identification of DNA-encoded IgG ligands that distinguish active and latent mycobacterium tuberculosis infections. ACS Chem. Biol 2017, 19, 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lovering F; Bikker J; Humblet C, Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem 2009, 52, 6752–6. [DOI] [PubMed] [Google Scholar]
- (18).Ritchie TJ; Macdonald SJ, The impact of aromatic ring count on compound developability--are too many aromatic rings a liability in drug design? Drug Discov Today 2009, 14, 1011–20. [DOI] [PubMed] [Google Scholar]
- (19).Ritchie TJ; Macdonald SJ; Young RJ; Pickett SD, The impact of aromatic ring count on compound developability: further insights by examining carbo- and hetero-aromatic and -aliphatic ring types. Drug Discov Today 2011, 16, 164–71. [DOI] [PubMed] [Google Scholar]
- (20).Bauer RA; Wurst JM; Tan DS, Expanding the range of “druggable” targets with natural product-based libraries: an academic perspective. Curr. Op. Chem. Biol 2010, 14, 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cordova A; Notz W; Barbas CF 3rd, Direct organocatalytic aldol reactions in buffered aqueous media. Chem. Comm 2002, 3024–5. [DOI] [PubMed] [Google Scholar]
- (22).Mase N; Nakai Y; Ohara N; Yoda H; Takabe K; Tanaka F; Barbas CF 3rd, Organocatalytic direct asymmetric aldol reactions in water. J. Amer. Chem. Soc 2006, 128, 734–5. [DOI] [PubMed] [Google Scholar]
- (23).Mase N; Tanaka F; Barbas CF 3rd, Synthesis of beta-hydroxyaldehydes with stereogenic quaternary carbon centers by direct organocatalytic asymmetric aldol reactions. Angew. Chem. (Int. ed.) 2004, 43, 2420–3. [DOI] [PubMed] [Google Scholar]
- (24).Notz W; Tanaka F; Barbas CF 3rd, Enamine-based organocatalysis with proline and diamines: the development of direct catalytic asymmetric Aldol, Mannich, Michael, and Diels-alder reactions. Acc Chem Res 2004, 37, 580–91. [DOI] [PubMed] [Google Scholar]
- (25).Sakthivel K; Notz W; Bui T; Barbas CF 3rd, Amino acid catalyzed direct asymmetric aldol reactions: a bioorganic approach to catalytic asymmetric carbon-carbon bond-forming reactions. J. Amer. Chem. Soc 2001, 123, 5260–7. [DOI] [PubMed] [Google Scholar]
- (26).Hoang L; Bahmanyar S; Houk KN; List B, Kinetic and stereochemical evidence for the involvement of only one proline molecule in the transition states of proline-catalyzed intra- and intermolecular aldol reactions. J. Amer. Chem. Soc 2003, 125, 16–7. [DOI] [PubMed] [Google Scholar]
- (27).List B; Hoang L; Martin HJ, New mechanistic studies on the proline-catalyzed aldol reaction. Proc Natl Acad Sci U S A 2004, 101, 5839–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).List B; Pojarliev P; Castello C, Proline-catalyzed asymmetric aldol reactions between ketones and alpha-unsubstituted aldehydes. Org. Lett 2001, 3, 573–5. [DOI] [PubMed] [Google Scholar]
- (29).Malone ML; Paegel BM, What is a “DNA-compatible” reaction? ACS Comb Sci 2016, 18, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Subiros-Funosas R; Prohens R; Barbas R; El-Faham A; Albericio F, Oxyma: an efficient additive for peptide synthesis to replace the benzotriazole-based HOBt and HOAt with a lower risk of explosion. Chemistry 2009, 15, 9394–403. [DOI] [PubMed] [Google Scholar]
- (31).Hartikka A; Arvidsson PI, Rational design of asymmetric organocatalysts––increased reactivity and solvent scope with a tetrazolic acid. Tetrahedron Asymmetry 2004, 15, 1831–1834. [Google Scholar]
- (32).Zhou Y; Shan Z, Chiral diols: a new class of additives for direct aldol reaction catalyzed by L-proline. J Org Chem 2006, 71, 9510–2. [DOI] [PubMed] [Google Scholar]
- (33).Reis O; Eymur S; Reis B; Demir AS, Direct enantioselective aldol reactions catalyzed by a proline-thiourea host-guest complex. Chemical communications (Cambridge, England) 2009, 1088–90. [DOI] [PubMed] [Google Scholar]
- (34).Martinez-Castaneda A; Poladura B; Rodriguez-Solla H; Concellon C; del Amo V, Direct aldol reactions catalyzed by a heterogeneous guanidinium salt/proline system under solvent-free conditions. Org. Lett 2011, 13, 3032–5. [DOI] [PubMed] [Google Scholar]
- (35).Matrtinez A; van Gemmeren M; List B, Unexpected beneficial effect of ortho-substituents on the (S)-proline-catalyzed asymmetric aldol reaction of acetone with aromatic aldehydes. Synlett 2014, 25, 961–964. [Google Scholar]
- (36).List B, Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar]
- (37).Martinez-Castaneda A; Kedziora K; Lavandera I; Rodriguez-Solla H; Concellon C; del Amo V, Highly enantioselective synthesis of alpha-azido-beta-hydroxy methyl ketones catalyzed by a cooperative proline-guanidinium salt system. Chem Comm 2014, 50, 2598–600. [DOI] [PubMed] [Google Scholar]
- (38).Fisher KJ; Turkett JA; Corson AE; Bicker KL, Peptoid Library Agar Diffusion (PLAD) Assay for the High-Throughput Identification of Antimicrobial Peptoids. ACS Comb Sci 2016, 18, 287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.