

Abstract
A generic Transcriptomics Reporting Framework (TRF) is presented that lists parameters that should be reported in ‘omics studies used in a regulatory context. The TRF encompasses the processes from transcriptome profiling from data generation to a processed list of differentially expressed genes (DEGs) ready for interpretation. Included within the TRF is a reference baseline analysis (RBA) that encompasses raw data selection; data normalisation; recognition of outliers; and statistical analysis. The TRF itself does not dictate the methodology for data processing, but deals with what should be reported. Its principles are also applicable to sequencing data and other ‘omics. In contrast, the RBA specifies a simple data processing and analysis methodology that is designed to provide a comparison point for other approaches and is exemplified here by a case study. By providing transparency on the steps applied during ‘omics data processing and analysis, the TRF will increase confidence processing of ‘omics data, and regulatory use. Applicability of the TRF is ensured by its simplicity and generality. The TRF can be applied to all types of regulatory ‘omics studies, and it can be executed using different commonly available software tools.
Keywords: Regulatory toxicology, normalisation of ‘omics data, statistical analysis, differentially expressed genes, reproducibility, bioinformatics, gene expression
1. Introduction
The evolution of ‘omics (e.g., transcriptomics, proteomics, metabolomics) occurred in parallel with developments in computational technology that allowed the storing, processing and analysis of large data sets (Peng, 2011). Analysis of these large sets of ‘omics data was made possible by advances in bioinformatics that incorporate the established principles of statistical data interpretation specifically for application to ‘omics studies.
‘Omics studies encompass the derivation of a measurement (the generation, curation and storage of raw data) using, e.g., a tissue or blood sample from an animal toxicity study; data processing and statistical analysis; and data interpretation to conclude on the outcome of the study (Figure 1). For each of the steps of an ‘omics study, multiple methodologies and approaches are available, many of which are incorporated into commercially or freely available software tools. However, the different data processing methodologies are inconsistently applied by different users and data analysts. Some inconsistencies arise from the uninformed application of the available software tools, but more often the inconsistencies are caused by differences in the study designs and bioinformatic approaches. Often the selection of the given methodology is justifiable, but the use of different analytical approaches makes it difficult to compare data from different studies, or to compare different evaluations of the same set of raw data. Due to differences in approaches to the processing, analysis and interpretation of ‘omics data, different results and conclusions can be derived from identical starting data sets (OECD, 2005; CATTPTRA – NRC, 2007).
Figure 1: Overview of the work streams considered at the ECETOC workshop Applying ‘omics technologies in chemical risk assessment (Buesen et al., 2017).
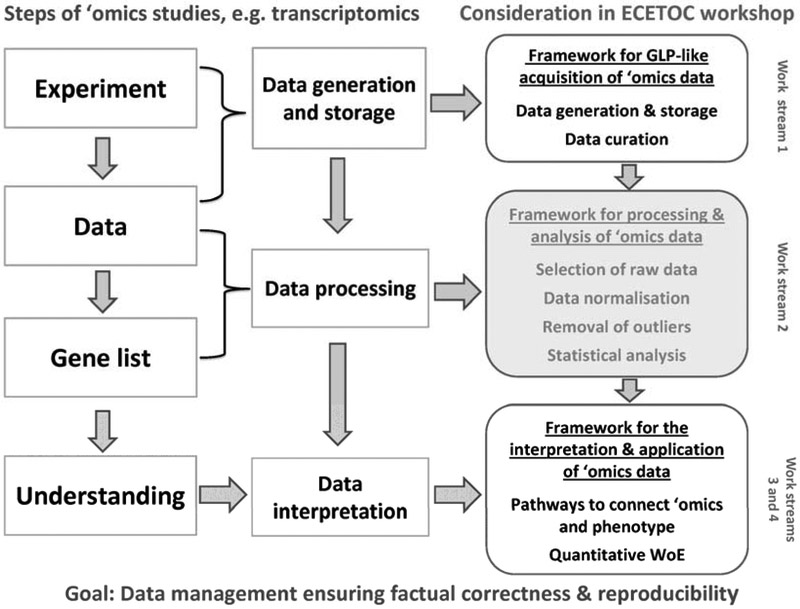
Footnote to Figure 1: The present article ‘A generic Transcriptomics Reporting Framework (TRF) for ‘Omics Data Processing and Analysis’ considers discussions from the ECETOC workshop work stream 2 (highlighted in grey). This box briefly lists bioinformatic processes involved in the use of transcriptomics data. The TRF incorporates a reference baseline analysis (RBA) method for the identification of differential gene expression against which other approaches can be benchmarked. While designed around microarray data analysis, the TRF and the embedded RBA are compatible with high throughput sequencing gene counting data.
The lack of standardisation and validation of ‘omics studies is a major obstacle preventing their wider regulatory adoption and use. In regulatory settings, ‘omics data can potentially contribute to the classification and labelling of substances; mode-of-action (MoA) analysis; the substantiation of chemical similarity for read-across; the determination of points-of-departure during hazard assessment; and the demonstration of species-specific effects and human health relevance (or absence thereof) (cf. Sauer et al. (2017) in this journal Supplement). However, the lack of standardisation is hindering delivery of these desirable outcomes.
This overview highlights that the standardisation of ‘omics studies is urgently required to foster the regulatory acceptance of ‘omics data. Against this background, the establishment of a framework describing steps that are relevant for the processing and analysis of ‘omics data was one of the key objectives of the European Centre for Ecotoxicology and Toxicology of Chemicals (ECETOC) workshop Applying ‘omics technologies in chemical risk assessment, that took place on 10-12 October 2016 in Madrid, Spain (Figure 1). The report of this workshop is provided in Buesen et al. (2017) in this journal Supplement. Ahead of the ECETOC workshop, a first draft framework on the processing and analysis of transcriptomics data was compiled, that was presented and further discussed during work stream 2 of the workshop (Figure 1). Based upon the recommendations from the ECETOC workshop, the drafted framework was updated, yielding this article that presents a generic Transcriptomics Reporting Framework (TRF). The TRF lists study elements and parameters that should be taken into consideration in designing ‘omics studies for regulatory use and in reporting such studies, e.g., in unpublished study reports or peer review publications. The TRF was drawn up with a focus on microarray studies for transcriptome profiling. Notwithstanding, it can easily be adapted to cover other sources of ‘omics data.
As a result of the difficulties presented above of comparing even the same data when subject to differing bioinformatics procedures, the TRF includes a Reference Baseline Analysis (RBA) method that indicates specific steps for data processing and statistical analysis from raw data to differentially expressed genes (DEGs). While the TRF, as such, merely provides a list of study elements and parameters that should be reported without being prescriptive, the embedded RBA stipulates one specific approach to convert the experimental measures (the raw data) to the processed data ready for interpretation (e.g., a list of differentially expressed genes (DEGs) for transcriptomics). Thereby, the TRF RBA serves to provide a benchmark reference list (common denominator) of DEGs for the comparative assessment of different studies. However, the TRF RBA can also be taken forward for interpretation of ‘omics data in its own right.
The TRF does not currently include parameters associated with the production of tissue, blood or cell samples (e.g., the animal or in vitro experimental design) or the generation, curation and storage of ‘omics data from these samples. The quality control (QC) of the generation of gene expression data has been dealt with previously by the U.S. Food and Drug Administration (FDA)-led MicroArray Quality Control (MAQC) consortium (MAQC Consortium et al., 2006; Shi et al., 2005, 2008, 2010; Xu et al., 2016). Therefore, the TRF does not make specific recommendations relating to the generation of ‘omics data, but it begins with data handling (i.e. the first manipulation of the raw data produced from the microarray platform). Data variance resulting from data generation is taken into account in the TRF as part of the analytical process, but the underlying experimental reasons for the variance are not addressed further.
Finally, the TRF does not cover the interpretation of ‘omics data. There is a greater level of latitude that can be applied in the interpretation of ‘omics data than in their generation and processing. With respect to data interpretation, the important issue for the TRF is to ensure that the interpretation is not based upon ‘omics data that are misleading due to the bioinformatic processing and statistical analysis. Aspects related to meta data (e.g., animal species, gender, animal numbers, diet, etc.) also do not form part of the TRF. In this respect, the Minimum Information About Microarray Experiments (MIAME) guidelines are to be followed (Brazma et al., 2011). Details on the work of the MAQC consortium and on the contents of the MIAME guidelines are summarised in Sauer et al. (2017) in this journal Supplement.
Since the TRF only covers two bioinformatic aspects of ‘omics studies, i.e. data processing and statistical analysis (Figure 1), it considers the use of ‘omics data from an overarching, methodological perspective for application in regulatory science and stipulates what should be reported. The RBA part of the TRF exemplarily uses standard methodologies that can be executed using commonly available software tools. The TRF RBA is not designed to be exclusive, and it does not preclude the use of any specific data processing or data analysis methodology; it simply allows simple within-study and between-study reproducibility and comparability of ‘omics data sets leaving room for the application of individual expertise.
Section 2 below presents the five steps of the TRF, i.e. (1) description of the raw data; (2) data normalisation; (3) data filtering; (4) identification and removal of bad or outlying data sets; and (5) statistical analysis of data.
Section 3 presents a case study in which the TRF RBA was submitted to an initial evaluation. The raw data for this case study were generated during a rat oral pre- and postnatal toxicity study using three endocrine-active compounds (flutamide, vinclozalin and prochloraz) that was conducted within the European Chemical Industry Council (CEFIC) Long-range Research Initiative (LRI) project EMSG56 Combined low-dose exposures to anti-androgenic substances (http://cefic-lri.org/projects/emsg56-basf-combined-low-dose-exposures-to-anti-androgenic-substances/).
Section 4 provides a discussion of and an outlook on the applicability of the TRF and the embedded TRF RBA method.
2. The Transcriptomics Reporting Framework (TRF) and the embedded Reference Baseline Analysis (RBA) method
Step 1 – Description of the raw data (including initial pre-processing)
Minimum core study elements and parameters to describe the raw data (including initial pre-processing)
- The experimental design (sample quality assessment metrics; sample labelling; assignment of samples to slides, flow cells, etc.)
- The type and version of the platform used and the (manufacturer’s) name
- Specification of the raw data (cf. Table 1 and Supplementary Information)
Table 1:
Specification of the raw data and the normalisation method taking the examples of Agilent and Affymetrix microarray platforms
| Agilent microarray platforms | Affymetrix microarray platforms | |
|---|---|---|
| Specification of the raw data | ||
| Images of signal intensities | If applicable | Yes |
| Raw data | Specification if median, mean or processed signal intensity was used as the raw data | |
| Quantification | Specification of feature extraction tool | Generally: Affymetrix software |
| Data files | TXT | CEL |
| Background subtraction | If applicable; and including background measured | |
| Specification of the normalisation method | ||
| Two colour (locally weighted scatterplot smoothing (LOWESS) normalisation; options include span, degree, etc.) or one colour (quantile, cyclic LOWESS, variance stabilisation normalisation (VSN), etc.) | e.g., robust multi-array average (RMA), MicroArray Suite (MAS), version 5.0, etc. Specification if data were adjusted for confounders before or after normalisation |
|
- The signal used from raw data output, including a specification whether background correction to the signal was performed, or not
-Technical and experimental replicates
- Spot quality assurance / quality control metrics reviewed on raw data; if bad quality and/or low and high/saturated intensity spots are pre-filtered prior to statistical analysis, such spots should be clearly defined and a justification for performing the pre-filtering should be provided (cf. also Step 4 of the TRF: Removal of bad or outlying data sets)
- The handling of replicate probes
- The handling of technical replicates (preferably, technical replicates should be combined into one data set by calculating the average of the measures for each probe or sequence; including an indication if this includes a filtering step). Multiple probes for the same target provide a particular issue that needs to be considered and reported by the bioinformatician. These probes are technical replicates and therefore cannot contribute to the degrees of freedom in the statistical analysis. If they are identical, they should be averaged into one result, but if targeting different regions of the gene sequence there is a case for keeping them separate. The choice made should be decided by the bioinformatician and reported as part of the TRF.
Quality of the source material
- For RNA, this can include a report of the RNA Integrity Number (RIN) reported for the RNA used (a low RIN indicates a loss of RNA integrity); if the RIN number is not available, e.g., visual inspection can be applied to assess RNA integrity; also, A230/280 and A260/280 ratio values and yield (ng/µL) are helpful to assess RNA quality and potential contamination.
- An indication whether any sample pooling has occurred (samples being combined into one hybridisation or sequencing analysis; Zhang and Gant, 2005).
When applying the TRF RBA, data collection should proceed according to the manufacturers’ guidelines. The median signal data should be used for further processing. The raw data should not be background-corrected or pre-filtered, except for the removal of spiked-in standards. All data sets generated during the experiment should be examined to ensure consistency of quality.
Explanatory notes
Raw data represent the magnitude of the expression of the genes under investigation in the selected biological samples (e.g. cells, tissue, blood). An unambiguous description of the raw data following the study elements and parameters listed above is indispensable for ensuring transparency on the design of any given ‘omics study. Generally, the software tools available for different microarray platforms provide the option of deriving different types of raw data (e.g., median or mean signal intensities) and of conducting (or not conducting) an initial processing of signal intensities. Further, manufacturers may alter data types when they develop a new platform. Thus, differences in the types of raw data can arise either from the manner in which pixel data are condensed into one number (e.g. mean or median) and/or from the initial processing of the data.
The specific type of raw data used as a starting point for the subsequent analysis may affect the outcome of the study (i.e. the composition of DEG lists). This issue is further explored taking the example of two commonly used, commercially available microarray platforms, i.e. the Agilent and the Affymetrix® microarray platforms. The Agilent microarray platform and the accompanying Agilent software tools were applied during the CEFIC LRI EMSG56 project whose data were used for the case study presented in Section 3 Of note, the Agilent and the Affymetrix microarray platforms were selected exemplarily, and this selection does not imply a preference over the microarray test kits or software tools that are available from other manufacturers (cf. Supplementary Information for further information on the data processing modules of the corresponding software tools as presented in the respective user manuals (Affymetrix, 2013, 2014; Agilent, 2014)).
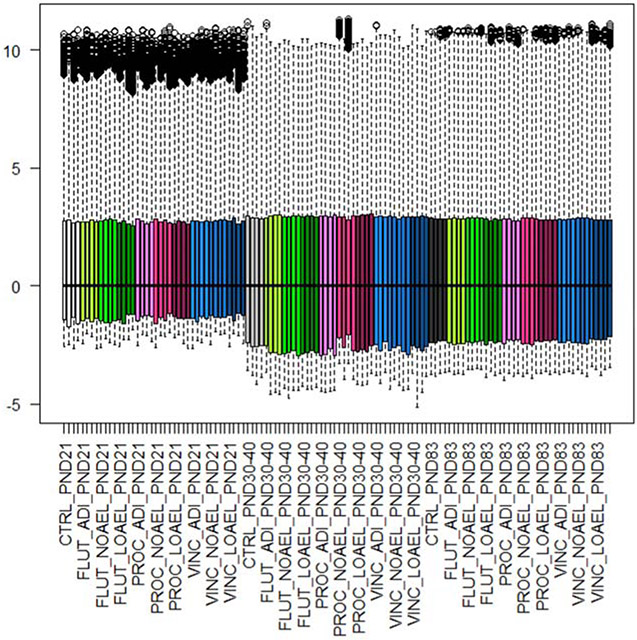
The Agilent software tools generally provide two numbers to quantify gene expression, i.e. the gMedianSignal and the gProcessedSignal (with the letter g referring to the green fluorescence labelling). The gProcessedSignal is specific for Agilent Technologies (Box 1) and thus not available to other technologies. Therefore, use of the gProcessedSignal could cause difficulties in the cross-comparison of data, because the gMedianSignal and the gProcessedSignal can produce different lists of DEGs even when analysed by the same statistical procedure. This effect of the processing on the distribution of signal intensities is visible in the boxplots in Figure 2 by the intense signals at the upper end of the distributions. This example highlights that the type of measure that is used as a starting point for the subsequent analysis should be specified, ideally together with a justification.
Box 1: The gProcessedSignal (Agilent Technologies).
The gProcessedSignal is generated by first subtracting an average background signal (yielding the BSubSignal) and then dividing this BSubSignal by the MultiplicativeDetrendingSignal (a factor derived by using the replicate probes on the array and correcting for their variation). Accordingly, the gProcessedSignal can include inherent background correction and multiple probe information. However, there are several options for the derivation of the gProcessedSignal, and it can also be presented without background subtraction. Therefore, the options used in deriving this signal measure need to be reported in the TRF.
Figure 2: Boxplots of the non-normalised log2 data from the EMSG56 study comparing data that are based upon the Agilent Technologies gProcessedSignal (left) with those that are based upon the gMedianSignal (right).

Footnote to Figure 2: The data are derived from 120 microarrays across three time points (postnatal days (PND) 21, 30-40, 83). At each time point, a total of 10 samples were measured, i.e. the control and three concentrations each for the three substances flutamide (FLUT), prochloraz (PROC) and vincolzalin (VINC). The three concentrations were set to represent each substance’s acceptable daily intake level (ADI; the low dose), no observed adverse effect level (NOAEL; the mid-dose) and lowest observed adverse effect level (LOAEL; the high dose). Each dose/time combination is indicated by a colour, and there are four experimental sets of data at each combination. The data are either the gProcessedSignal data or the gMedianSignal data and have been transformed to the log2. For each measurement, the coloured boxes represent the first to third quartile, the dotted lines the minimum to maximum values, and the black circles (for the gMedianData) outliers that have exceeded the intensity threshold for the scanner.
If the given study aims to derive ‘omics data that can be cross-compared between different data sets, it may be advisable to use a signal that is available on all major platforms. This is the median signal, i.e. the gMedianSignal in case of the Agilent technology. For this reason, the TRF RBA uses the median signal as type of raw data for further data processing and analysis. There is no reason why the gProcessedSignal should not be used to mine data if the responsible bioinformatician considers this most relevant (e.g., because background corrections better reflect the actual biological targets). Processing of the gProcessedSignal alongside use of the TRF RBA enables a cross comparison between the DEG lists obtained following either procedure. It is important though that the options used in the derivation of the gProcessedSignal are reported as part of the TRF.
Further work is advisable to determine how DEG lists acquired using the Agilent gMedianSignal compare to DEG lists acquired using the median signals generated using other microarray platforms and software tools. Nevertheless, in spite of possible inherent differences in the technology used (e.g., location of probes, signal capture, etc.), cross-comparisons between datasets obtained using the platforms from different manufacturers may be enhanced by using median signals as raw data, as compared to the use of pre-processed signals.
Step 2 – Data normalisation
Minimum core study elements and parameters to report data normalisation
- Data transformation: ln, log10, log2, etc.; variance stabilisation normalisation (VSN)
- The method of normalisation, including a reference, if applicable, and justifying its relevance for the type of data; generally, data normalisation should be restricted to the minimum necessary (cf. Table 1)
- Selection of probes (all, invariant subset, house-keeping genes, spike-in controls)
- Quality assurance of normalisation assessment
When applying the TRF RBA, data should be logged to the base 2 to ensure an equal distribution about 0 for the expression of each of the genes in the set prior to statistical analysis. This can be done before or after the normalisation. Median centring normalisation is performed. Normalisation should be performed within-sample only, and not across the entire experiment (i.e. as between-sample normalisation).
Explanatory notes
Data normalisation is generally considered a minimum prerequisite to correct for experimental and technical variables (cf. Sauer et al. (2017) in this journal Supplement). Generally, the normalisation methods should be chosen depending on data and sample properties and the goal of the analysis (Fundel et al., 2008). In all ‘omics studies, the applied normalisation method should be properly reported taking into account data supporting the choice of normalisation methods (e.g. analysing variance pre- and post-normalisation).
The TRF RBA includes a within-data set median centring normalisation to ensure that distributions are organised to lie about the same axis (Zhang and Gant, 2004). This one of the simplest non-proprietary normalisation methods that can be applied to all data types. It is used for this reason. Single data set normalisation methods prevent data sets from affecting each other (by altering existing (normalised) expression values from the addition of new sample sets (Welsh et al., 2013).
Step 3 – Data filtering
Minimum core study elements and parameters for data filtering
Recognition and removal of data below the detection limit, including specification (1) how poorly expressed and measured genes contribute to the uncertainty; and (2) how poorly expressed and measured genes were removed (in a consistent manner)
- Recognition and removal of low abundance genes (including specification of threshold)
- Recognition and removal of absent probes (i.e. empty spots; including definition how identified)
- Any non-specific filtering
- Process used to identify and remove poor quality probes (e.g., poor areas of hybridisation or high noise)
The TRF RBA does not include any specific filtering of data below detection limits or non-specific filtering of data and relies on statistical analysis to remove high variance data.
Explanatory notes
‘Omics data that are below the detection limits of the respective analysis methods contribute to the variance in the data sets and ultimately to the occurrence of false positives and negatives in the data. However, removal of data below the detection limit can interfere with the calculation of fold changes in gene expression. For example, when substance treatment induces an increase in the expression of a gene starting from a very low level, then removal of the very low level of expression can result in a division by zero error when calculating the ratio of fold change. The same applies when the expression of a gene is reduced from a high level to a very low one. For any genes with expression levels close to the detection limit, there will likely be a high level of variance. Thus, with the exception of genes that exhibit large fold change in expression (e.g., one group having low expression at the detection limit and another group having detectable expression), genes with high variance should be removed by the statistical limits. Thus, it is necessary to record how such data points are identified and handled in an experiment.
Step 4 – Identification and removal of bad or outlying data sets
Minimum core study elements and parameters for the identification and removal of bad or outlying data sets
Bad signal detection; describe how samples yielding poor signal were identified for removal; this may be based on:
- Low or biased dye incorporation
- Failure of the manufacturer’s QC
- Low signal to noise ratio
- Failure of spiked-in controls, if present, to pass the manufacturer’s QC
- Data set does not conform to statistically assessed normality
- Failure of biological or technical replicates to cluster together, e.g., on a principal component analysis (PCA) plot
- Other sample coherence quality assurance metrics
Note: Biological replicates should only be removed if the presence of the outlier can be explained both statistically and biologically.
Identification and removal of outliers
- Removal of outliers before normalisation? (if so: provide justification and describe applied algorithm)
- Removal of additional outliers after normalisation? (if so: provide justification and describe applied algorithm)
When applying the TRF RBA, outliers are identified via PCA distribution and clustering; any obvious outlying data sets are removed, describing how outliers were identified and justifying the removal.
Explanatory notes
The identification and removal of outlying data can decrease the variance in the data, but it needs to be borne in mind that each removed data set takes out a degree of freedom for the subsequent statistical analysis. Outlying data can be identified, e.g., by reviewing (1) boxplots for each filtered data set; (2) principal component analysis (PCA), plots; (3) clustering analysis, in which biological replicates should cluster together and, (4) failure of the internal spiked in standards. On the basis of these plots, a justification should be written for both the retention and removal of data sets from the further evaluation.
When applying the TRF RBA, outlying data are only removed if they are a result of a traceable technical problem (e.g., failed microarray QC, low counts, spiked standards, etc.), or fail to cluster with replicates. Outliers are not removed based on PCA, hierarchal clustering, etc. alone. Unless technical problems can be identified there is a danger that a sample is in fact reflecting real biological variation.
Step 5 - Statistical analysis of data
Minimum core study elements and parameters regarding the statistical analysis of data
- Statistical software applied to identify DEGs (including name and version of the software tool, algorithms, etc.)
-Number of technical and experimental replicates and treatment of these in the statistical analysis.
- Application of fold change and/or p-value filters?
- Application of additional filters (e.g., false discovery rate (FDR))?
- Relation of statistical significance (e.g., to the control samples or to a specific data set or global mean)
For application of the TRF RBA, the Welch’s t-test should be used for the calculation of statistical significance (i.e. the p-value) of the data (i.e. as pre-processed in Steps 1-4 of the TRF RBA). A fold change of 1.5 and a p-value < 0.05 should be used as cut-off values to identify DEGs (cf. explanatory note). Additional filters, such as FDR, are not used in the TRF RBA. In all 'omics studies, the raw data should be provided alongside the analysed data. Depending on the scope of the study, Analysis of Variance (ANOVA) and Linear Models for Microarray And RNA-Seq Data (LIMMA) may be equally well suited for the analysis and can be used, but should always be compared back to the TRF RBA analysis to provide the common denominator between experiments and analysis methods.
Explanatory notes
For the data remaining in the experiment upon completion of the first four steps of the TRF, all of the steps used in identifying DEGs should be transparently documented. Within the TRF RBA, significant changes in gene expression as compared to the control samples are calculated using Welch’s t-test and the statistical cut-offs indicated below. This test is more robust to unequal variance and sample size than the Student’s t-test and is included in the TRF RBA for this reason. The TRF RBA uses an empirically set fold change threshold of 1.5 and a p-value < 0.05 as cut-off values to identify DEGs.
This approach may not be optimal for all uses, e.g., when the ‘omics study uses (invertebrate) species whose tissues are not well differentiated, or that are so small that sampling of the entire animal is inevitable. In such cases, the most appropriate approach to determine the statistical (and biological) relevance of the data may have to be established on a case-by-case basis, and the TRF RBA should be used as the benchmark reference.
Following the statistical analysis, DEGs can then be calculated as the ratio of the gene expression of the treated and the control samples (subtracting the log data). The standard deviation (SD) for significant changes from the control can be calculated by analysis of the log ratio of gene expression for each of the replicate experiments against the mean of the control values. This approach was selected for the TRF RBA since it is universally applicable, non-propriety, and can be undertaken using various computer packages. Technical replicates should not contribute to the degrees of freedom used and should be averaged together into one data set for the sample they represent. It may not be appropriate to average together probes targeting the same gene but in different regions, though these are still technical replicates and should not contribute to the degrees of freedom. The bioinformatician should report the method used for these as part of the TRF. For two colour microarrays (now not very common) the use of reverse hybridisation is strongly encouraged and should be reported (Zhang and Gant, 2004).
Depending on the objective of the study, other statistical tests and formats for identifying the DEGs than those specified for the TRF RBA may be selected (and conducted in parallel with the TRF RBA, if applicable). In all ‘omics studies, the data analysis protocols should be provided together with the study results. Care should be taken that the use of specific fold change or p-value thresholds does not impose statistical restrictions that preclude the subsequent biological interpretation of ‘omics data (that is not addressed in this article), especially when also considering false discovery rate (FDR) corrections. For this reason, the embedded TRF RBA has specific a requirement for the p and fold change cut off values thus forming a benchmark reference analysis that is fairly permissive against which the results of other statistical filtering limits can be compared.
Of note, statistical tests that employ permutation or bootstrap analysis (Fang and Ma, 2017) are generally considered less reproducible than methods with parametric assumptions.
3. Application of the TRF RBA to a sample data set
Exemplarily, the TRF RBA was applied to a data set that was generated during the CEFIC LRI EMSG56 project Combined low-dose exposures to anti-androgenic substances. ‘Omics data were generated using blood samples from rats submitted to an extended one generation-like toxicity study investigating the pre- and postnatal effects of three endocrine-active compounds, i.e. flutamide, vinclozalin and prochloraz (Fussell et al., 2012). For each test compound, the ‘omics data were generated at three dose levels that were selected to mimic the respective compound’s (i) acceptable daily intake level; (ii) no observed adverse effect level; and (iii) lowest observed adverse effect level. Three time points were analysed for each test compound and dose level (postnatal days 21, 30-40, and 83), and 4 replicates were tested at each time point and dose level. The control groups encompassed 4 animals per time point. Agilent microarray platforms were used to quantify gene expression levels. The study design yielded a total of 120 sets of microarray data. Further details on the study design are presented in Fussell et al. (2012).
Data processing and statistical analysis were carried out using the R Statistical Environment (R Core Team, 2013). This environment provides a relatively simple envelope in which to make complex calculations with large data sets. It has the advantage that the scripts used can be saved as a record of the bioinformatics process undertaken and reported. Thereby, the records can be reviewed by anyone with a basic knowledge of the package. Furthermore, the R package is non-proprietary and publicly available, and it can be used without costly computer equipment.
The main steps of the TRF RBA are summarised in Figure 3.
Figure 3: Overview of the Transcriptomics Reporting Framework (TRF) Reference Baseline Analysis (RBA) method.
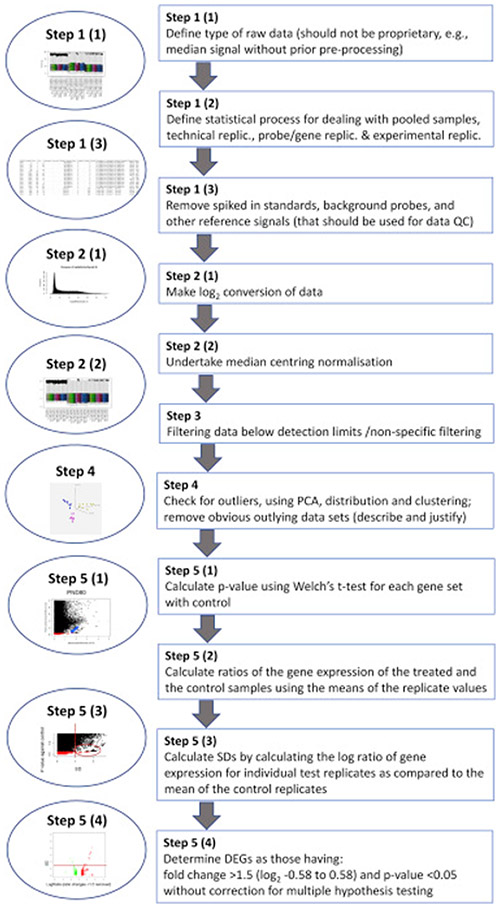
Footnote to Figure 3: Abbreviations: DEG: Differentially expressed gene, PCA: Principal component analysis; Replic.: Replicates; SD: Standard deviation.
3.1. Definition of the type of raw data
As discussed in Step 1 of the TRF (cf. Section 2), the Agilent software tools provide two types of output data, the gProcessedSignal and the gMedianSignal. As specified in the TRF RBA and to facilitate future use of ‘omics data sets (including, possibly, cross-platform comparability), the gMedianSignal data were used as raw data. Microarray markers and internal standards were removed by analysis of the accession numbers. It was noted that all such markers and spiked in standards did not have associated accession numbers. Therefore, this provided the easiest means to remove the corresponding data by taking out all those expression values that did not have an associated accession number. The means of achieving this will be different for different platforms and should be reported as part of the TRF.
3.2. Log2 conversion and median centring normalisation
Step 2 of the TRF (and the TRF RBA) specifies data transformation and normalisation. All data were converted to log2 values. As compared to the non-logged data, the log2-converted data followed a bimodal distribution with a tight distribution of the low intensity data and a much wider distribution of the more highly expressed genes (Figure 4). Background subtraction was not performed. Background is usually measured in the area surrounding the probe and is not always reflective of the actual background in the area of the probe that cannot be measured directly. This can result in values that are less than zero if the background is removed. For these reasons, the TRF RBA does not include background subtractions. Median centring normalisation was conducted on the gMedianSignal data (Figure 5). By comparison with Figure 2, the boxplot presented in Figure 5 reveals that the normalisation served to centre the medians, but that it did not alter the distribution on the data (as expected).
Figure 4: Transforming the data by log2 results in a bimodal distribution of data.

Footnote to Figure 4: Rat data for all gene expressions at one experimental point (A) are log transformed (B) spreading the data. The bimodal distribution results from the majority of genes being expressed at a similar level with a small set having a distribution over a much greater expression level. Individual genes show a normal distribution across the experiment (C).
Figure 5: Effect of median centring normalization on the total data set.
Footnote to Figure 5: The log transformed gMedianSignal data is transformed by centring to the median. This ensures that the data distributions in each experiment lie over each other allowing calculation of the ratios of differential gene expression for each experiment. For each measurement, the coloured boxes represent the first to third quartile, the dotted lines the minimum to maximum values, and the black circles outliers that have exceeded the intensity threshold for the scanner.
3.3. Data filtering
Step 3 of the TRF includes testing for bad data and removal thereof. The TRF RBA does not specify any data filtering, but instead relies on the statistical analysis in Step 5. Thereby, data are not removed that might be required for cross-comparison with other data processing methods.
3.4. Identification and removal of bad and outlying data sets
Step 4 of the TRF calls for an assessment of bad and outlying data sets. PCA analysis did not show any data sets that required removing from the analysis (data not shown) and therefore a justification for the removal of datasets was not required.
3.5. Statistical analysis
Step 5 of the TRF encompasses the statistical analysis of the data. Variance in the data is dependent on signal strength (Figure 6), and the statistical test needs to be sufficiently discriminatory to remove high variance data and sufficiently permissive to ensure there is no data loss that would prevent the comparisons for which the TRF RBA has been designed. Applying TRF RBA in the case study, the statistical analysis was conducted using the Welch’s t-test comparing the experimental average expression of each gene expression value at each dose and time point with the relevant control and applying the cut-off value of p-value < 0.05 alone (Figure 7A) and together with the 1.5-fold change cut-off value (Figure 7B). Application of the p-value < 0.05 cut-off alone is permissive. Nevertheless, as stated above, the purpose of the TRF RBA is to provide a common denominator data set against with other analyses of the same data set or TRF RBA analyses from other ‘omics studies can be compared. While application of the p-value < 0.05 cut-off alone proved sufficiently discriminatory for high variance data, this results in some DEGs being recognised that are very small but have small variance between experiments. For this reason, TRF RBA includes the p-value < 0.05 cut-off and the fold change cut off of 1.5-fold (Figure 7).
Figure 6: Log signal plotted against the SD of that signal showing that more variance occurs at the lower expression levels.

Footnote to Figure 6: Greater intensity of fluorescence is easier and more reproducible to measure leading to a decrease in variance at the higher levels of expression. Circled region: The most pronounced variance occurs in the measurement of lower expressed genes that produce lower signals on the microarray.
Figure 7: Statistical analysis applying both the 1.5-fold change and p <0.05 cut-off values.

Footnote to Figure 7: p <0.05 cut-off applied to the data. This removes most of the more variable data though some still remain as indicated by the circle (A). These data could result though from a high level of gene expression variance between the control and test groups with one measure being of low intensity and the other high. This would result in some variance from the low expression sample but would still be significant due to the high level of differential gene expression. Panel B indicates that the 1.5-fold change cut off value removes those genes of low variance in the measure of expression but also low differential expression that could still be significant (B).
4. Discussion and outlook
The TRF presented in this article has been developed as a generic framework to enhance transparency on two key steps of ‘omics studies, i.e. the processing of raw data and the statistical analysis of the processed data. It builds on previous work published Zhang and Gant (2004) and Gant and Zhang (2005). The TRF has been conceived generally to accommodate the different purposes of regulatory use of ‘omics data (e.g., hazard identification or MoA analysis) as well as the complexity and diversity of ’omics technologies.
Differences in the processing and analysis of ‘omics data can result in the derivation of different results and conclusions from identical starting data sets (OECD, 2005; CATTPTRA – NRC, 2007). Therefore, the RBA has been embedded within the TRF to allow easy comparison of different bioinformatics approaches and to serve as a benchmark reference analysis, or common denominator, against which to compare other approaches. Use of the TRF RBA fosters the within-study and between-study comparability of the outcomes of ‘omics studies. Such comparability is essential to obtain a profound biological understanding of the implications of gene expression changes.
In the long term, the transparency and comparability that the TRF and the embedded RBA yield can also foster the establishment of best practices in the processing and statistical analysis of ‘omics data when performing ‘omics studies for different regulatory applications thereby facilitating the standardisation and regulatory applicability of ‘omics studies. The transparency and reproducibility of ‘omics studies are further enhanced when data analysis scripts (including the entire source code) and standards are made available with the ‘omics study reports. Preferentially, such data analysis scripts should follow reproducible research paradigms, e.g. by using literate programming for statistical analysis and methods that are open to scrutiny (Peng, 2011). In the TRF RBA, this is achieved by the use of publicly available software tools, such as the R Statistical Environment.
While currently focusing on the processing of data from transcriptomics microarray studies, the TRF may eventually provide the basis for the development of further technology-specific data processing and analysis frameworks (e.g., RNA-Seq, etc.) and/or for the development of such frameworks for further ‘omics (e.g., proteomics, metabolomics, etc.). The manufacturers and vendors of the technologies should be involved in developing data processing and analysis frameworks to ensure that the selected study elements and parameters to cover their product readouts. The likely future development of further data processing and analysis methods provides justification for the generic TRF RBA that allows for cross-comparison between different ‘omics studies. This is exemplified by the ongoing developments to collect high-throughput functional genomics data such as the Gene Expression Omnibus (GEO) database repository of the U.S. National Center for Biotechnology Information (available at: https://www.ncbi.nlm.nih.gov/geo/).
The case study presented in this article confirms the general usefulness of the TRF RBA. Further case studies are recommended to enhance the regulatory applicability and use of the TRF and the embedded RBA (and possibly to provide a starting point for validation studies). Such work should address if and how the TRF and the embedded RBA should be adapted to comply with the structure of different commercially available (manufacturer-specific) or publicly available software tools. Such evidence will also be useful for the standardisation of approaches to process and statistically analyse ‘omics data. The current TRF RBA is restricted to one specific study design, and it will need to be adapted or expanded to take account of other study designs. In this respect, further work is also merited to investigate the most appropriate design for ‘omics studies (e.g., with respect to the time points for cell, tissue or blood sampling), and the outcome of such investigations should be used to expand the TRF to include parameters that are relevant for the experimental design of ‘omics studies. Similarly, the TRF should eventually be expanded to also accommodate the bioinformatics applied for data interpretation, an issue beyond the scope of this work.
The flexible organisation of the TRF allows for the inclusion of new evidence as it becomes available. Its focus is not prescriptive, but performance-based. Transparency on the steps applied in processing ‘omics data allows the identification of sources of error and variance in these studies. The RBA embedded within the TRF also allows this evolution while facilitating within-study and between-study comparison as new evidence becomes available. To enable wide-spread and straight-forward application of (technology-specific) TRFs, it is advisable to make them publicly available as common resources, possibly combined with web-based reporting forms that include export functions to allow adding documentary files to study reports (similar to the web-based reporting and evaluation resource Science in Risk Assessment and Policy (SciRAP; available at: http://www.scirap.org/).
Finally, apart from the goals to standardise approaches for the processing and statistical analysis of ‘omics data and to ensure transparency and reproducibility of such data, the formal establishment of a set of performance standards and their widespread use in ‘omics studies conducted for regulatory purposes (as well as their inclusion in the TRF as essential study element) can form an important pillar for the quality control of ‘omics studies and enhance the reproducibility and comparability of ‘omics data. When ‘omics studies are performed for regulatory purposes, such reference samples could also be used for benchmark dose modelling. An example for such performance standards are the MAQC reference standards (Wen et al., 2010; Zheng et al., 2015). Finally, during ‘omics data interpretation (outside the scope of the present article), the biological relevance of ‘omics data needs to be assessed by comparison with the other outcome measures of the given study.
Supplementary Material
5. Acknowledgements
Emma Marczylo (PHE, UK) and Tewes Tralau (German Federal Institute for Risk Assessment, Germany) are thanked for valuable discussions during the preparation of the manuscript. Ian Cummings, ECETOC, Belgium, is thanked for support in editing the figures.
Abbreviations
- ADI:
Acceptable daily intake level
- ANOVA
Analysis of variance
- CEFIC
European Chemical Industry Council
- DEG
Differentially expressed gene
- ECETOC
European Centre for Ecotoxicology and Toxicology of Chemicals
- FDA
Food and Drug Administration
- FDR
False discovery rate
- GEO
Gene Expression Omnibus (database)
- LIMMA
Linear models for microarray and RNA-Seq data
- LOWESS
locally weighted scatterplot smoothing
- LRI
Long-Range Research Initiative
- LOAEL
Lowest observed adverse effect level
- MAQC
MicroArray Quality Control
- MIAME
Minimum Information About Microarray Experiments
- MoA
Mode-of-action
- NOAEL
No observed adverse effect level
- PCA
Principal component analysis
- PND
Postnatal day
- QC
Quality control
- RBA
Reference Baseline Analysis (method)
- Replic
Replicate
- RIN
RNA Integrity Number
- RMA
Robust multi-array (multi-chip) average
- SciRAP
Science in Risk Assessment and Policy
- SD
Standard deviation
- TRF
Transcriptomics Reporting Framework
- VSN
Variance stabilisation normalisation
Footnotes
Conflict of interest
UGS was hired by ECETOC and the CEFIC LRI to assist in the preparation of the manuscript. The other authors were engaged in the course of their normal employment. The authors alone are responsible for the content and writing of the paper.
Disclaimer
The content described in this article has been reviewed by the National Health and Environmental Research Laboratory of the U.S. Environmental Protection Agency and approved for publication. Approval does not signify that the contents necessarily reflect the views and the policies of the Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
8. References
Of note: All websites were accessed in May 2017.
- Affymetrix, 2013. Affymetrix® GeneChip® Command Console® (AGCC) 4.0 User Manual, available at: http://www.affymetrix.com/support/technical/byproduct.affx?product=commandconsole.
- Affymetrix, 2014. Transcriptome Analysis Console (TAC) 3.0. User Guide, available at: http://www.affymetrix.com/estore/browse/level_seven_software_products_only.jsp?productId=prod760001#1_1
- Agilent, 2014. Agilent GeneSpring. User manual, available at: http://www.agilent.com/cs/library/usermanuals/public/GeneSpring-manual.pdf.
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FC, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, Stewart J, Taylor R, Vilo J, Vingron M, 2001. Minimum information about a microarray experiment (MIAME) - toward standards for microarray data. Nat. Genet 29, 365–371. [DOI] [PubMed] [Google Scholar]
- Buesen R, Chorley BN, da Silva Lima B, Daston G, Deferme L, Ebbels T, Gant TW, Goetz A, Greally J, Gribaldo L, Hackermüller J, Hubesch B, Jennen D, Johnson K, Kanno J, Kauffmann H-M, Laffont M, Meehan R, Pemberton M, Perdichizzi S, Piersma AH, Sauer UG, Schmidt K, Seitz H, Sumida K, Tollefsen KE, Tong W, Tralau T, van Ravenzwaay B, Weber R, Worth A, Yauk C, Poole A, 2017. Applying ‘omics technologies in chemicals risk assessment: Report of an ECETOC workshop Regulat. Toxicol. Pharmacol. epub ahead of print 25 September 2017, doi: 10.1016/j.yrtph.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CATTPTRA – NRC, 2007. Committee on Applications of Toxicogenomic Technologies to Predictive Toxicology and Risk Assessment, National Research Council. Applications of Toxicogenomic Technologies to Predictive Toxicology and Risk Assessment. ISBN: 0-309-11299-0, 300 pages, available at: http://www.nap.edu/catalog/12037.html. [Google Scholar]
- Fang K, Ma S, 2017. Analyzing large datasets with bootstrap penalization. Biom. J 59(2), 358–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fundel K, Küffner R, Aigner T, Zimmer R, 2008. Normalization and gene p-value estimation: issues in microarray data processing. Bioinform. Biol. Insights 2, 291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fussell KC, Melching-Kollmuss S, Groeters S, Strauss V, Siddeek B, Benahmed M, Frericks M, van Ravenzwaay B, Schneider S, 2012. Endocrine exposure at environmentally relevant concentrations. Poster presented at the 14th Cefic-LRI Annual Workshop 2012, available at: http://cefic-lri.org/projects/emsg56-basf-combined-low-dose-exposures-to-anti-androgenic-substances/. [Google Scholar]
- Gant TW, Zhang SD, 2017. In pursuit of effective toxicogenomics. Mut. Res 575(1-2), 4–16. [DOI] [PubMed] [Google Scholar]
- MAQC Consortium; Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, Luo Y, Sun YA, Willey JC, Setterquist RA, Fischer GM, Tong W, Dragan YP, Dix DJ, Frueh FW, Goodsaid FM, Herman D, Jensen RV, Johnson CD, Lobenhofer EK, Puri RK, Scherf U, Thierry-Mieg J, Wang C, Wilson M, Wolber PK, Zhang L, Amur S, Bao W, Barbacioru CC, Lucas AB, Bertholet V, Boysen C, Bromley B, Brown D, Brunner A, Canales R, Cao XM, Cebula TA, Chen JJ, Cheng J, Chu TM, Chudin E, Corson J, Corton JC, Croner LJ, Davies C, Davison TS, Delenstarr G, Deng X, Dorris D, Eklund AC, Fan XH, Fang H, Fulmer-Smentek S, Fuscoe JC, Gallagher K, Ge W, Guo L, Guo X, Hager J, Haje PK, Han J, Han T, Harbottle HC, Harris SC, Hatchwell E, Hauser CA, Hester S, Hong H, Hurban P, Jackson SA, Ji H, Knight CR, Kuo WP, LeClerc JE, Levy S, Q.Z. Li, Liu C Liu Y, Lombardi MJ, Ma Y, Magnuson SR, Maqsodi B, McDaniel T, Mei N, Myklebost O, Ning B, Novoradovskaya N, Orr MS, Osborn TW, Papallo A, Patterson TA, Perkins RG, Peters EH, Peterson R, Philips KL, Pine PS, Pusztai L, Qian F, Ren H, Rosen M, Rosenzweig BA, Samaha RR, Schena M, Schroth GP, Shchegrova S, Smith, DD, Staedtler F, Su Z, Sun H, Szallasi Z, Tezak Z, Thierry-Mieg D, Thompson KL, Tikhonova I, Turpaz Y, Vallanat B, Van C, Walker SJ, Wang SJ, Wang Y, Wolfinger R, Wong A, Wu J, Xiao C, Xie Q, Xu J, Yang W, Zhang L, Zhong S, Zong Y, Slikker W Jr, 2006. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol 24, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OECD, 2005. Organisation for Economic Co-operation and Development. Series on Testing and Assessment No. 50. Report of the OECD/IPCS workshop on toxicogenomics. ENV/JM/MONO(2005)10. 29.04.2005. [Google Scholar]
- Peng RD, 2011. Reproducible research in computational science. Sci. 334(6060), 1226–12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team, 2013. A language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria, available at: https://www.r-project.org. [Google Scholar]
- Sauer UG, Deferme L, Gribaldo L, Hackermüller J, Tralau T, van Ravenzwaay B, Yauk C, Poole A, Tong W, Gant TW, 2017. The challenge of the application of ‘omics technologies in chemicals risk assessment: background and outlook. Regulat. Toxicol. Pharmacol epub ahead of print, 18 September 2017, doi: 10.1016/j.yrtph.2017.09.020. [DOI] [PubMed] [Google Scholar]
- Shi L, Tong W, Fang H, Scherf U, Han J, Puri RK, Frueh FW, Goodsaid FM, Guo L, Su Z, Han T, Fuscoe JC, Xu ZA, Patterson TA, Hong H, Xie Q, Perkins RG, Chen JJ, Casciano DA, 2005. Cross-platform comparability of microarray technology: intra-platform consistency and appropriate data analysis procedures are essential. BMC Bioinformatics 6(Suppl 2), S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Jones WD, Jensen RV, Harris SC, Perkins RG, Goodsaid FM, Guo L, Croner LJ, Boysen C, Fang H, Qian F, Amur S, Bao W, Barbacioru CC, Bertholet V, Cao XM, Chu TM, Collins PJ, Fan XH, Frueh FW, Fuscoe JC, Guo X, Han J, Herman D, Hong H, Kawasaki ES, Li QZ, Luo Y, Ma Y, Mei N, Peterson RL, Puri RK, Shippy R, Su Z, Sun YA, Sun H, Thorn B, Turpaz Y, Wang C, Wang SJ, Warrington JA, Willey JC, Wu J, Xie Q, Zhang L, Zhang L, Zhong S, Wolfinger RD, Tong W, 2008. The balance of reproducibility, sensitivity, and specificity of lists of differentially expressed genes in microarray studies. BMC Bioinformatics 9(Suppl 9), S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, Su Z, Chu TM, Goodsaid FM, Pusztai L, Shaughnessy JD Jr., Oberthuer A, Thomas RS, Paules RS, Fielden M, Barlogie B, Chen W, Du P, Fischer M, Furlanello C, Gallas BD, Ge X, Megherbi DB, Symmans WF, Wang MD, Zhang J, Bitter H, Brors B, Bushel PR, Bylesjo M, Chen M, Cheng J, Cheng J, Chou J, Davison TS, Delorenzi M, Deng Y, Devanarayan V, Dix DJ, Dopazo J, Dorff KC, Elloumi F, Fan J, Fan S, Fan X, Fang H, Gonzaludo N, Hess KR, Hong H, Huan J, Irizarry RA, Judson R, Juraeva D, Lababidi S, Lambert CG, Li L, Li Y, Li Z, Lin SM, Liu G, Lobenhofer EK, Luo J, Luo W, McCall MN, Nikolsky Y, Pennello GA, Perkins RG, Philip R, Popovici V, Price ND, Qian F, Scherer A, Shi T, Shi W, Sung J, Thierry-Mieg D, Thierry-Mieg J, Thodima V, Trygg J, Vishnuvajjala L, Wang SJ, Wu J, Wu Y, Xie Q, Yousef WA, Zhang L, Zhang X, Zhong S, Zhou Y, Zhu S, Arasappan D, Bao W, Lucas AB, Berthold F, Brennan RJ, Buness A, Catalano JG, Chang C, Chen R, Cheng Y, Cui J, Czika W, Demichelis F, Deng X, Dosymbekov D, Eils R, Feng Y, Fostel J, Fulmer-Smentek S, Fuscoe JC, Gatto L, Ge W, Goldstein DR, Guo L, Halbert DN, Han J, Harris SC, Hatzis C, Herman D, Huang J, Jensen RV, Jiang R, Johnson CD, Jurman G, Kahlert Y, Khuder SA, Kohl M, Li J, Li L, Li M, Li QZ, Li S, Li Z, Liu J, Liu Y, Liu Z, Meng L, Madera M, Martinez-Murillo F, Medina I, Meehan J, Miclaus K, Moffitt RA, Montaner D, Mukherjee P, Mulligan GJ, Neville P, Nikolskaya T, Ning B, Page GP, Parker J, Parry RM, Peng X, Peterson RL, Phan JH, Quanz B, Ren Y, Riccadonna S, Roter AH, Samuelson FW, Schumacher MM, Shambaugh JD, Shi Q, Shippy R, Si S, Smalter A, Sotiriou C, Soukup M, Staedtler F, Steiner G, Stokes TH, Sun Q, Tan PY, Tang R, Tezak Z, Thorn B, Tsyganova M, Turpaz Y, Vega SC, Visintainer R, von Frese J, Wang C, Wang E, Wang J, Wang W, Westermann F, Willey JC, Woods M, Wu S, Xiao N, Xu J, Xu L, Yang L, Zeng X, Zhang J, Zhang L, Zhang M, Zhao C, Puri RK, Scherf U, Tong W, Wolfinger RD; MAQC Consortium, 2010. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat. Biotechnol 28, 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh EA, Eschrich SA, Berglund AE, Fenstermacher DA, 2013. Iterative rank-order normalization of gene expression microarray data. BMC Bioinformatics 14, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Wang C, Shi Q, Huang Y, Su Z, Hong H, Tong W, Shi L, 2010. Evaluation of gene expression data generated from expired Affymetrix GeneChip® microarrays using MAQC reference RNA samples. BMC Bioinformatics 11(Suppl 6), S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Thakkar S, Gong B, Tong W, 2016. The FDA's experience with emerging genomics technologies - past, present, and future. A.A.P.S. J 18, 814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SD, Gant TW, 2004. A statistical framework for the design of microarray experiments and effective detection of differential gene expression. Bioinformatics 20(16), 2821–2828. [DOI] [PubMed] [Google Scholar]
- Zhang SD, Gant TW, 2005. Effect of pooling samples on the efficiency of comparative studies using microarrays. Bioinformatics 21(24), 4378–4383. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Qing T, Song Y, Zhu J, Yu Y, Shi W, Pusztai L, Shi L, 2015. Standardization efforts enabling next-generation sequencing and microarray based biomarkers for precision medicine. Biomark Med. 9(11), 1265–1272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.