

Abstract
Background
Clostridioides difficile ribotype (RT) 019/sequence type (ST) 67 strains belong to a hypervirulent lineage closely related to RT027/ST1; however, limited data are available for hypervirulent clade 2 lineages in Japan. Herein, we report the draft genome of a C. difficile strain B18-123 belonging to clade 2, RT019/ST67 for the first time in Japan.
Results
The pathogenicity locus carried by B18-123 (19.6 kb) showed higher homology (97.29% nucleotide identity) with strain R20291 (RT027/ST1) than the reference strain 630 (RT012/ST54), and B18-123 harbored 8-nucleotide substitutions in tcdC. However, it did not contain an 18-base pair (bp) deletion or a single-bp deletion at position 117 in tcdC, which was identified in the previous strain R20291. A cytotoxicity assay revealed similar cytotoxicity levels between strains B18-123 and ATCC BAA-1870 (RT027/ST1). The B18-123 strain was found to be susceptible to metronidazole and vancomycin.
Conclusion
Our findings contribute to the further understanding of the characteristics of hypervirulent clade 2 including RT019/ST67 lineages.
Keywords: Clostridioides difficile, Hypervirulent clade 2, Ribotype 019, Sequence type 67, Toxin A, Toxin B, Binary toxin, Pathogenicity locus, Cdt locus
Introduction
Clostridioides difficile infection (CDI) is a well-known cause of healthcare-associated infections, ranging from mild diarrhea to severe pseudomembranous colitis and even death. It may be related to disruption of the indigenous intestinal microbiota by prolonged treatment with drugs such as antibiotics [1]. The incidence of CDI has increased over the past decade and clinical manifestations are typically triggered by toxin A (TcdA) and toxin B (TcdB) [2].
Epidemic hypervirulent strains with binary toxin (Cdt), North American pulsed-field type one (NAP1)/ribotype (RT) 027/sequence type (ST) 1 belonging to clade 2 and RT078/ST11 belonging to clade 5 have been identified as major lineages responsible for outbreaks in North America and Europe because of their high sporulation capacity and toxin production and resistance to fluoroquinolones [3–6]. However, these strains are rarely reported in Asia including in Japan, where other lineages such as RT014, RT017, and RT018 have been known to be predominant [7, 8]. RT019/ST67, which is closely related to NAP1/RT027/ST1 among clade 2 [9], produces high levels of toxins upon tcdC repression caused by an 18-base pair (bp) deletion and a single-bp deletion at position 117 in tcdC [10]. Although the previously reported RT019/ST67 strains [10–12] have not shown any resistance to vancomycin and metronidazole, additional data are needed to understand the characteristics of the RT019/ST67 lineage to implement more effective treatments and methods of infection control.
In Japan, NAP1/RT027/ST1 strains are occasionally isolated from patients with fulminant colitis [13, 14]. However, limited data, including genome sequences, are available for hypervirulent clade 2 because of its extremely low prevalence among CDI incidences in Japan [15]. We reported the detection of a C. difficile strain B18-123 belonging to hypervirulent clade 2, RT019/ST67, in Japan isolated from a patient with multiple recurrent episodes of severe diarrhea.
Methods
Bacterial isolates and growth conditions
Clostridioides difficile strain B18-123 was obtained from a stool sample of a 81-year-old elderly male patient who underwent chemotherapy for hepatocellular carcinoma at Medical Hospital of Tokyo Medical and Dental University in 2018. Bacterial identification was performed using the C. DIFF QUIK CHEK COMPLETE test (Abbott Diagnostics Medical, Tokyo, Japan) and confirmed by 16S rRNA gene sequencing [16] for quality assurance of the following data. C. difficile strains American Type Culture Collection (ATCC) 700057 (RT038/ST48; TcdA−, TcdB−, Cdt−) and ATCC BAA-1870 (NAP1/RT027/ST1; TcdA+, TcdB+, Cdt+) were also used in the study. The C. difficile strains were grown at 37 °C under anaerobic conditions using brain heart infusion (BHI; Oxoid, Basingstoke, UK) broth or agar plate supplemented with 5 g/L yeast extract and 0.1% (w/v) l-cysteine (BHIS).
Multilocus sequence typing, ribotyping, and toxin profiles
Multilocus sequence typing (MLST) and ribotyping were performed as previously described [16, 17]. tcdA, tcdB, cdtA, and cdtB were detected by multiplex PCR [18], followed by analysis of the length of tcdA as described [19]. A cytotoxicity assay was performed for B18-123 together with the ATCC BAA-1870 and ATCC 700057 strains as described previously [16].
Genome sequencing, de novo assembly, and annotation
Genomic DNA was extracted with a NucleoSpin Tissue kit (Takara Bio, Shiga, Japan) and subjected to quality control by agarose gel electrophoresis. The DNA obtained was quantified with a Synergy H1 (BioTek, Winooski, VT, USA) and QuantiFluor dsDNA System (Promega, Madison, WI, USA), and sequenced using a NextSeq platform (Illumina, Inc., San Diego, CA, USA) using a 2 × 151-bp paired-end approach. After quality control on raw reads was performed using Sickle (https://github.com/najoshi/sickle), the reads were assembled de novo using SPAdes version 3.10.1. Gene annotation was determined with Prokka version 1.11. The draft genome was subjected to in silico analyses, such as MLST using MLST version 2.0 [20] and detection of antibiotic resistance mechanisms using ResFinder version 3.1 [21], ARG-ANNOT [22], and CARD [23].
Comparative genomics
Phylogenetic analysis was performed with a pathogenicity locus (PaLoc) and tcdC sequences of 12 complete and draft genomes, including nine ST67 strains deposited in the GenBank, by the maximum likelihood method based on the Tamura-Nei model [24] using MEGA X software [25]. Sequence comparisons of PaLoc and Cdt locus (CdtLoc) were conducted using ClustalW (https://clustalw.ddbj.nig.ac.jp/) and Easyfig version 2.2.2 [26]. The presence of insertion sequences and tRNAs were identified on the PaLoc and its flanking region of B18-123 using ISfinder [27] and tRNAscan-SE [28], respectively. The presence of phage-related sequences was investigated in the whole genome of B18-123 using PHASTER [29].
Antimicrobial susceptibility testing
The minimum inhibitory concentrations (MICs) of six antibiotics (metronidazole, vancomycin, rifampin, erythromycin, clindamycin, and moxifloxacin) were determined using an Etest (bioMérieux, Marcy-l’Étoile, France) and the breakpoints were based on EUCAST epidemiological cut-off values (http://www.eucast.org/clinical_breakpoints/).
Statistical analysis
One-way analysis of variance was performed using GraphPad Prism version 7 software to determine the statistical significance of the differences for comparison. p < 0.05 was considered to indicate significance.
Results and discussion
Following draft genome sequencing of B18-123, a total of 4,284,537 reads were assembled into 98 contigs larger than 200 bp (largest scaffold = 526,102 bp; N50 = 277, 155 bp; L50 = 5; average GC = 28.39%) with an estimated genome size of 4,221,572 bp and average coverage of 50 ×. MLST analysis in silico along with conventional PCR assigned C. difficile strain B18-123 to ST67, which belongs to hypervirulent clade 2. B18-123 strain was classified into RT019 by ribotyping. Although in Japan, only two ST67 strains without ribotype data have been deposited in the GenBank in 2019 (last accessed on September 22, 2019), no report of CDI incidences with RT019/ST67 in Japan has been published so far in English literature. Therefore, our results indicate that a hypervirulent strain with RT019/ST67 responsible for severe diarrhea has been reported for the first time in Japan.
Strain B18-123 possesses tcdA and tcdB, and their presence was supported by in silico analysis. PaLoc carried by B18-123 was located between two conserved genes, cdu1 and cdd1 (Fig. 1a). BLAST results indicated that the PaLoc in B18-123 (19.6 kb) was homologous with the reference genome 630 (accession number AM180355; RT012/ST54; 99% query cover and 94.88% identity), R20291 (accession number FN545816; NAP1/RT027/ST1; 99% query cover and 97.29% identity), and M120 (accession number NC_017174; classified as clade 5, RT078/ST11; 99% query cover and 94.02% identity) strains. This indicates that the PaLoc carried by B18-123 is genetically closely related to that of R20291. These findings were supported by phylogenetic tree analysis of PaLoc carried by 630, R20291, M120, B18-123, and ST67 strains with complete PaLoc deposited in the GenBank (last accessed on September 22, 2019) (Fig. 1b).
Fig. 1.

Genetic characteristics of PaLoc in Clostridioides difficile strain B18-123. (a) Comparative genomic structure analysis of PaLoc among C. difficile strains used in this study. (b) Phylogenetic tree of PaLoc among 12 strains deposited in the GenBank by maximum likelihood method based on Tamura-Nei model [24] using MEGA X software [25]. Strains surrounded by dotted line belong to ST67. (c) tcdC sequence alignment for C. difficile strains used in this study. Identical nucleotides among their strains are indicated with an asterisk. (d) Phylogenetic tree of tcdC sequences among 12 strains deposited in the GenBank by maximum likelihood method based on Tamura-Nei model [24] using MEGA X software [25]. Strains indicated with an asterisk belong to ST67
In this study, no intact insertion sequences were found 10 kb upstream and downstream from PaLoc; however, truncated sequences (≤ 20 bp) of many insertion sequence families matched with our sequence. These results agree with a previous finding that C. difficile PaLoc does not fit the genetically accepted definition of pathogenicity islands containing direct repeats and insertion sequences [30]. However, the transfer of a chromosomal fragment containing the PaLoc proved to be caused by a chromosomally integrated mobile genetic element such as conjugative transposon [31, 32]. The detailed mechanism of this transfer has not yet been clarified.
Although no tRNA was identified on the scaffold containing PaLoc (location on scaffold of NODE013; 119,372 bp), 76 tRNAs representing all 20 amino acids were identified in the whole genome of B18-123. As the reference strain 630 and strain R20291 possess 87 tRNAs and 65 tRNAs, respectively, further investigations considering the entire cellular population of tRNAs may reveal the functional role of tRNAs against cellular physiological process, such as modulation of toxin production.
Additionally, although strain B18-123 harbored an 8-nucleotide substitution in tcdC, a negative regulator of tcdA and tcdB [33] compared to the reference genome 630 (Fig. 1c), only a single amino acid substitution of Asp7Glu (caused by a nucleotide T21G substitution), which was located outside of the three predicted domains [34], was found in 232 amino acids among both strains. Moreover, we did not detect an 18-bp deletion or a single-bp deletion at position 117, or a 39-bp deletion in tcdC as previously reported [3, 10, 11, 33, 35, 36]. On the other hand, we found that the tcdC sequences of ST67 strains (20100502, Lei034, F314, 5550, 51680, FD094, and FD109), except for strain LIBA5757 [10], were 100% identical to that of B18-123 (Fig. 1d). However, we did not find any information indicating that these strains were associated with severe CDI episodes. The amino acid sequence of B18-123 TcdC showed 95.83% identity with strain R20291 but showed very low identity with strain M120 because of the 39-bp deletion in tcdC. Collectively, our results suggest that TcdC carried by B18-123 causes different regulation of TcdA and TcdB production compared to previously identified hypervirulent lineages such as in the NAP1/RT027/ST1, RT019/ST67 (LIBA5757), and RT078/ST11 stains. These findings also indicate that it may be difficult to directly determine the TcdC characteristics of B18-123 by nucleic acid amplification tests available in clinical settings [37].
Although conflicting findings have been reported regarding the functional role of TcdC [38–40], the regulation of TcdA and TcdB production in B18-123, which possesses an Asp7Glu substitution in TcdC, may differ from that in previously identified hypervirulent clade 2 strains. Therefore, we further investigated the relationship between tcdC mutation and toxin activity in a cytotoxicity assay [16]. Vero cell viability in B18-123 was similar to that in strain ATCC BAA-1870 (NAP1/RT027/ST1) with an 18-bp deletion and a single-bp deletion at position 117 in tcdC (P = 0.7859) (Fig. 2a). This result was related to the morphological changes shown by cell rounding of Vero cells observed under a light microscope (Fig. 2b). These findings suggest that TcdC carried by B18-123 allows the cell to increase toxin production, leading to high virulence. However, we could not directly quantify both TcdA and TcdB or clarify the mechanism of this protein in toxin regulation in B18-123 in this study.
Fig. 2.
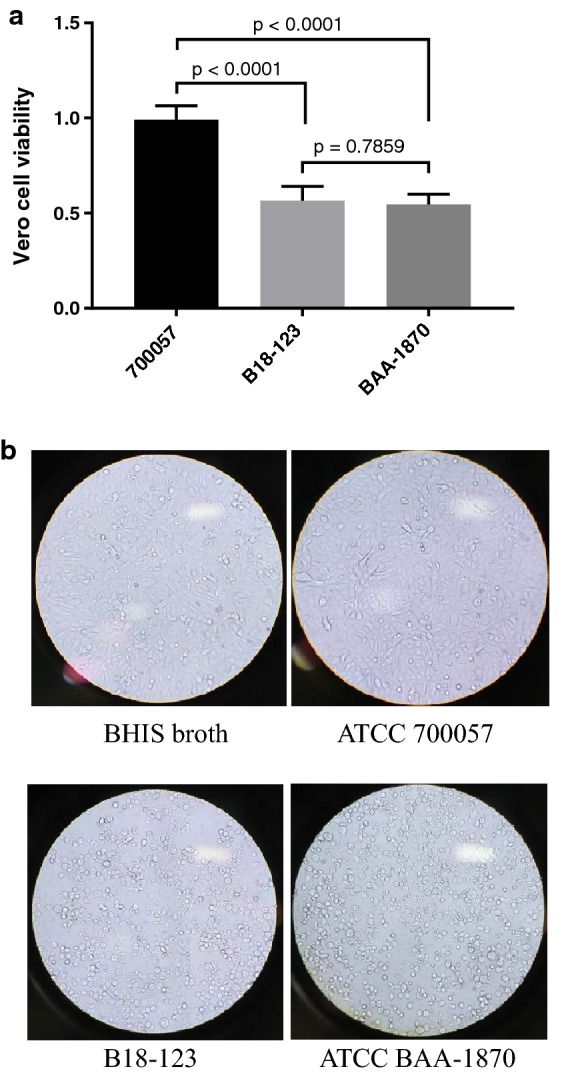
Cytotoxicity of Clostridioides difficile strain B18-123 on Vero cells. (a) Vero cell viability in B18-123 was evaluated together with positive control ATCC BAA-1870 (NAP1/RT027/ST1; TcdA+, TcdB+, Cdt+) and negative control ATCC 700057 (RT038/ST48; TcdA−, TcdB−, Cdt−) strains, as described previously [16]. The results are presented as means ± standard deviations of cell viability percentages. (b) Cytotoxic effect of C. difficile strains studied under a light microscope (original magnification × 200)
As strain B18-123 also possesses cdtA and cdtB, CdtLoc (5.2 kb) carried by B18-123 was compared to those of the R20291 and M120 strains. The BLAST results indicated that CdtLoc in B18-123 showed homology with the R20291 (100% query cover and 99.90% identity) and M120 (100% query cover and 97.99% identity) strains. The binary toxin regulator gene cdtR (747 bp) in B18-123 was completely conserved with that in strain R20291 (100% query cover and 100% identity) and highly conserved with that in strain M120 (100% query cover and 97.46% identity), indicating that CdtR carried by B18-123 modulates binary toxin production similar to in NAP1/RT027/ST1 strains.
PHASTER identified three complete prophage sequences (scores > 90 and over; Table 1) and showed that strain B18-123 possessed the C. difficile bacteriophages phiMMP02, phiCDHM19 (belong to Myoviridae family), and phiCD111 (belong to Siphoviridae family). Although the B18-123 genome also contained one questionable and three incomplete prophage sequences, further studies are needed to understand the role of the phages in the evolution of RT019/ST67 lineages.
Table 1.
Summary of phage-related regions identified by PHASTER in the B18-123 genome
| Region | Length of prophage (kb) | Completeness | PHASTER score | No. of total proteins | Region position | Most common phage | GC content (%) |
Location on scaffolds |
|---|---|---|---|---|---|---|---|---|
| 1 | 69.2 | Intact | 150 | 93 | 294078–363300 | PHAGE_Clostr_phiMMP02_NC_019421 (26) | 28.18 | NODE001 |
| 6 | 44.8 | Intact | 140 | 66 | 1–44849 | PHAGE_Clostr_phiCDHM19_NC_028996 (21) | 28.25 | NODE021 |
| 7 | 40.4 | Intact | 125 | 48 | 48–40472 | PHAGE_Clostr_phiCD111_NC_028908 (46) | 30.88 | NODE022 |
All contigs were merged into a single continuous sequence on PHASTER (http://phaster.ca/). B18-123 genome also contained three incomplete and one questionable prophage sequences (scores < 90)
Strain B18-123 was susceptible to metronidazole (MIC = 0.19 mg/L), vancomycin (MIC = 0.75 mg/L), rifampin (MIC ≤ 0.002 mg/L), and moxifloxacin (MIC = 1.0 mg/L). Additionally, B18-123 showed low-level MICs to erythromycin (1.5 mg/L) and clindamycin (0.75 mg/L). We then performed in silico analysis to investigate the presence of antibiotic resistance genes using ResFinder version 3.1 [21], ARG-ANNOT [22], and CARD [23]. We detected only a C656T nucleotide substitution within the 23S rRNA copy on the genome with CARD. However, as a previous study suggested, the substitution has been not associated with conferring resistance to macrolides and lincosamides in C. difficile [41]. Our findings obtained from the resistome analysis seem to be sound and coincide with phenotypic results of antibiotic MICs. As evidenced by our MIC results with vancomycin on B18-123, treatment with the same was very effective for treating the CDI in this patient. Additionally, no amino acid substitutions in GyrA or GyrB were identified between B18-123 and fluoroquinolone-susceptible C. difficile strain 630 [42]. These results agree with those of a previous study of RT019/ST67 strain [10].
Conclusion
In conclusion, we reported for the first time that C. difficile strain B18-123 in Japan belongs to hypervirulent clade 2, RT019/ST67. Our findings enhance our understanding of the characteristics of hypervirulent clade 2 strains and further emphasize the need for continuous monitoring of C. difficile isolates to assess the spread of clade 2 strains in Japan.
Acknowledgements
The manuscript was edited by Editage, a language editing company.
Abbreviations
- RT
ribotype
- MLST
multilocus sequence typing
- ST
sequence type
- TcdA
toxin A
- TcdB
toxin B
- Cdt
binary toxin
- PaLoc
pathogenicity locus
- CdtLoc
Cdt locus
- MIC
minimum inhibitory concentration
Authors’ contributions
All authors contributed toward bacterial isolation, data analysis, drafting, and critically revising the paper, and agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Funding
This work was partially supported by JSPS KAKENHI Grant Number JP17K08822 (R.S.).
Availability of data and materials
This whole-genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the Bioproject number PRJNA530817.
Ethics approval and consent to participate
As this study did not involve human participants or animals, informed consent and ethical approval were not required in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ryoichi Saito, Email: r-saito.mi@tmd.ac.jp.
Yukino Usui, Email: ma180005@tmd.ac.jp.
Alafate Ayibieke, Email: a.ayibieke@gmail.com.
Jun Nakajima, Email: nakamlab@tmd.ac.jp.
Isaac Prah, Email: pawa7@yahoo.com.
Kazunari Sonobe, Email: sonomlab@tmd.ac.jp.
Yoshibumi Aiso, Email: aisompha@tmd.ac.jp.
Shiori Ito, Email: nobuyo.i5963@gmail.com.
Yasuhiro Itsui, Email: yitsui.gast@tmd.ac.jp.
Yoshiro Hadano, Email: y-hadano.infe@tmd.ac.jp.
Yoko Nukui, Email: nucku-tky@umin.ac.jp.
Ryuji Koike, Email: koike.rheu@tmd.ac.jp.
Shuji Tohda, Email: tohda.mlab@tmd.ac.jp.
References
- 1.McDonald LC, Owings M, Jernigan DB. Clostridium difficile infection in patients discharged from US short-stay hospitals, 1996-2003. Emerg Infect Dis. 2006;12(3):409–415. doi: 10.3201/eid1205.051064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aktories K, Schwan C, Jank T. Clostridium difficile toxin biology. Annu Rev Microbiol. 2017;71:281–307. doi: 10.1146/annurev-micro-090816-093458. [DOI] [PubMed] [Google Scholar]
- 3.Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, et al. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366(9491):1079–1084. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 4.Goorhuis A, Bakker D, Corver J, Debast SB, Harmanus C, Notermans DW, et al. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin Infect Dis. 2008;47(9):1162–1170. doi: 10.1086/592257. [DOI] [PubMed] [Google Scholar]
- 5.Merrigan M, Venugopal A, Mallozzi M, Roxas B, Viswanathan VK, Johnson S, et al. Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol. 2010;192(19):4904–4911. doi: 10.1128/JB.00445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drudy D, Quinn T, O’Mahony R, Kyne L, O’Gaora P, Fanning S. High-level resistance to moxifloxacin and gatifloxacin associated with a novel mutation in gyrB in toxin-A-negative, toxin-B-positive Clostridium difficile. J Antimicrob Chemother. 2006;58(6):1264–1267. doi: 10.1093/jac/dkl398. [DOI] [PubMed] [Google Scholar]
- 7.Jia HB, Du PC, Yang H, Zhang YY, Wang J, Zhang W, et al. Nosocomial transmission of Clostridium difficile ribotype 027 in a Chinese hospital, 2012–2014, traced by whole genome sequencing. BMC Genomics. 2016;17:405. doi: 10.1186/s12864-016-2708-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins DA, Hawkey PM, Riley TV. Epidemiology of Clostridium difficile infection in Asia. Antimicrob Resist Infect Control. 2013;2:21. doi: 10.1186/2047-2994-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffiths D, Fawley W, Kachrimanidou M, Bowden R, Crook DW, Fung R, et al. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol. 2010;48(3):770–778. doi: 10.1128/JCM.01796-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quesada-Gomez C, Lopez-Urena D, Chumbler N, Kroh HK, Castro-Pena C, Rodriguez C, et al. Analysis of TcdB proteins within the hypervirulent clade 2 reveals an impact of RhoA glucosylation on Clostridium difficile proinflammatory activities. Infect Immun. 2016;84(3):856–865. doi: 10.1128/IAI.01291-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pires RN, Monteiro AA, Saldanha GZ, Falci DR, Caurio CFB, Sukiennik TCT, et al. Hypervirulent Clostridium difficile strain has arrived in Brazil. Infect Control Hosp Epidemiol. 2018;39(3):371–373. doi: 10.1017/ice.2017.280. [DOI] [PubMed] [Google Scholar]
- 12.Cao H, Wong SC, Yam WC, Liu MC, Chow KH, Wu AK, et al. Genomic investigation of a sequence type 67 Clostridium difficile causing community-acquired fulminant colitis in Hong Kong. Int J Med Microbiol. 2019;309(5):270–273. doi: 10.1016/j.ijmm.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura I, Yamaguchi T, Tsukimori A, Sato A, Fukushima S, Mizuno Y, et al. Fulminant colitis from Clostridium difficile infection, the epidemic strain ribotype 027, in Japan. J Infect Chemother. 2014;20(5–6):380–383. doi: 10.1016/j.jiac.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Nishimura S, Kou T, Kato H, Watanabe M, Uno S, Senoh M, et al. Fulminant pseudomembranous colitis caused by Clostridium difficile PCR ribotype 027 in a healthy young woman in Japan. J Infect Chemother. 2014;20(11):729–731. doi: 10.1016/j.jiac.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Kato H, Senoh M, Honda H, Fukuda T, Tagashira Y, Horiuchi H, et al. Clostridioides (Clostridium) difficile infection burden in Japan: a multicenter prospective study. Anaerobe. 2019;12(19):30046. doi: 10.1016/j.anaerobe.2019.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Kuwata Y, Tanimoto S, Sawabe E, Shima M, Takahashi Y, Ushizawa H, et al. Molecular epidemiology and antimicrobial susceptibility of Clostridium difficile isolated from a University teaching hospital in Japan. Eur J Clin Microbiol Infect Dis. 2015;34(4):763–772. doi: 10.1007/s10096-014-2290-9. [DOI] [PubMed] [Google Scholar]
- 17.Kato H, Ito Y, Akahane T, Izumida S, Yokoyama T, Kaji C, et al. Typing of Clostridium difficile isolates endemic in Japan by sequencing of slpA and its application to direct typing. J Med Microbiol. 2010;59(5):556–562. doi: 10.1099/jmm.0.016147-0. [DOI] [PubMed] [Google Scholar]
- 18.Persson S, Torpdahl M, Olsen KEP. New multiplex PCR method for the detection of Clostridium difficile toxin A (tcdA) and toxin B (tcdB) and the binary toxin (cdtA/cdtB) genes applied to a Danish strain collection. Clin Microbiol Infect. 2008;14(11):1057–1064. doi: 10.1111/j.1469-0691.2008.02092.x. [DOI] [PubMed] [Google Scholar]
- 19.Kato H, Kato N, Watanabe K, Iwai N, Nakamura H, Yamamoto T, et al. Identification of toxin A-negative, toxin B-positive Clostridium difficile by PCR. J Clin Microbiol. 1998;36(8):2178–2182. doi: 10.1128/jcm.36.8.2178-2182.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50(4):1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta SK, Padmanabhan BR, Diene SM, Lopez-Rojas R, Kempf M, Landraud L, et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. 2014;58(1):212–220. doi: 10.1128/AAC.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia BF, Raphenya AR, Alcock B, Waglechner N, Guo PY, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 25.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowe TM, Chan PP. tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44(W1):W54–W57. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang YJ, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44(W1):W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rupnik M, Dupuy B, Fairweather NF, Gerding DN, Johnson S, Just I, et al. Revised nomenclature of Clostridium difficile toxins and associated genes. J Med Microbiol. 2005;54(2):113–117. doi: 10.1099/jmm.0.45810-0. [DOI] [PubMed] [Google Scholar]
- 31.Mullany P, Allan E, Roberts AP. Mobile genetic elements in Clostridium difficile and their role in genome function. Res Microbiol. 2015;166(4):361–367. doi: 10.1016/j.resmic.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brouwer MSM, Roberts AP, Hussain H, Williams RJ, Allan E, Mullany P. Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nature Commun. 2013;4:2601. doi: 10.1038/ncomms3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matamouros S, England P, Dupuy B. Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol Microbiol. 2007;64(5):1274–1288. doi: 10.1111/j.1365-2958.2007.05739.x. [DOI] [PubMed] [Google Scholar]
- 34.van Leeuwen HC, Bakker D, Steindel P, Kuijper EJ, Corver J. Clostridium difficile TcdC protein binds four-stranded G-quadruplex structures. Nucleic Acids Res. 2013;41(4):2382–2393. doi: 10.1093/nar/gks1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dupuy B, Govind R, Antunes A, Matamouros S. Clostridium difficile toxin synthesis is negatively regulated by TcdC. J Med Microbiol. 2008;57(6):685–689. doi: 10.1099/jmm.0.47775-0. [DOI] [PubMed] [Google Scholar]
- 36.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci USA. 2010;107(16):7527–7532. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crobach MJT, Planche T, Eckert C, Barbut F, Terveer EM, Dekkers OM, et al. European society of clinical microbiology and infectious diseases: update of the diagnostic guidance document for Clostridium difficile infection. Clin Microbiol Infect. 2016;22:S63–S81. doi: 10.1016/j.cmi.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 38.Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol. 2012;78(13):4683–4690. doi: 10.1128/AEM.00249-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carter GP, Douce GR, Govind R, Howarth PM, Mackin KE, Spencer J, et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. Plos Pathogens. 2011;7(10):e1002317. doi: 10.1371/journal.ppat.1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray R, Boyd D, Levett PN, Mulvey MR, Alfa MJ. Truncation in the tcdC region of the Clostridium difficile pathLoc of clinical isolates does not predict increased biological activity of toxin B or toxin A. BMC Infect Dis. 2009;9:103. doi: 10.1186/1471-2334-9-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang BY, Lv Z, Zhang PP, Su JR. Molecular epidemiology and antimicrobial susceptibility of human Clostridium difficile isolates from a single institution in Northern. Medicine. 2018;97(25):e11219. doi: 10.1097/MD.0000000000011219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wasels F, Kuehne SA, Cartman ST, Spigaglia P, Barbanti F, Minton NP, et al. Fluoroquinolone resistance does not impose a cost on the fitness of Clostridium difficile in vitro. Antimicrob Agents Chemother. 2015;59(3):1794–1796. doi: 10.1128/AAC.04503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This whole-genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the Bioproject number PRJNA530817.