

Abstract
Streptococcus pneumoniae is responsible for the majority of pneumonia, motivating ongoing searches for insights into its physiology that could enable new treatments. S. pneumoniae responds to exogenous fatty acids by suppressing its de novo biosynthetic pathway and exclusively utilizing extracellular fatty acids for membrane phospholipid synthesis. The first step in exogenous fatty acid assimilation is phosphorylation by fatty acid kinase (FakA), whereas bound by a fatty acid-binding protein (FakB). Staphylococcus aureus has two binding proteins, whereas S. pneumoniae expresses three. The functions of these binding proteins were not clear. We determined the SpFakB1- and SpFakB2-binding proteins were bioinformatically related to the two binding proteins of Staphylococcus aureus, and biochemical and X-ray crystallographic analysis showed that SpFakB1 selectively bound saturates, whereas SpFakB2 allows the activation of monounsaturates akin to their S. aureus counterparts. The distinct SpFakB3 enables the utilization of polyunsaturates. The SpFakB3 crystal structure in complex with linoleic acid reveals an expanded fatty acid-binding pocket within the hydrophobic interior of SpFakB3 that explains its ability to accommodate multiple cis double bonds. SpFakB3 also utilizes a different hydrogen bond network than other FakBs to anchor the fatty acid carbonyl and stabilize the protein. S. pneumoniae strain JMG1 (ΔfakB3) was deficient in incorporation of linoleate from human serum verifying the role of FakB3 in this process. Thus, the multiple FakBs of S. pneumoniae permit the utilization of the entire spectrum of mammalian fatty acid structures to construct its membrane.
Keywords: fatty acid-binding protein, X-ray crystallography, glycerophospholipid, bacterial metabolism, Streptococcus, acyl-phosphate, fatty acid kinase, nutrient acquisition
Introduction
The ability to acquire exogenous fatty acids (FA)2 for membrane phospholipid synthesis is a universal feature of lipid metabolism in Firmicutes, a phylum of Gram-positive bacteria that contains many important human pathogens. The first step in FA incorporation is activation by FA kinase, an enzyme system consisting of a kinase domain protein (FakA) that phosphorylates a FA carried by the FA-binding protein component (FakB) (1). The resulting acyl-phosphate (FA∼P) bound to FakB is either transferred to the glycerol phosphate acyltransferase (PlsY) to initiate phospholipid synthesis or transferred to acyl carrier protein (ACP) by PlsX (2). The resulting acyl-ACP is either utilized by PlsC to acylate the 2-position of 1-acyl-glycerolphosphate or it may enter the FASII cycle and be elongated. The elongated acyl-ACP is then metabolized via PlsX/PlsY or PlsC like the acyl-ACP derived from de novo biosynthesis. Two major orders of the Firmicutes have distinctly different physiological responses to the presence of extracellular FA. The Bacillales, exemplified by Staphylococcus aureus, do not genetically suppress the genes of type II FA biosynthesis (FASII) in the presence of exogenous FA (3). Rather, FASII continues to produce primarily anteiso15:0 that is placed in the 2-position, and the exogenous long-chain saturated and monounsaturated FA are activated by FA kinase and placed into the 1-position (4, 5). The Lactobacillales, exemplified by Streptococcus pneumoniae, have a different response. These organisms strongly suppress the expression of the genes encoding the FASII enzymes in response to exogenous FA. This response is mediated by the FabT transcriptional repressor (6–9). FabT bound to acyl-ACP tightly binds to promoters within the FASII gene cluster to potently suppress FASII gene expression (10). Thus, these organisms construct their membrane phospholipids almost exclusively using FA obtained from the environment to acylate both positions of the glycerol-phosphate backbone (3, 11). Elongation is not a significant fate for exogenous FA in the Lactobacillales because the FabT system suppresses the transcription of the entire gene set responsible for the FASII elongation cycle.
The ability of FA kinase to activate FA encountered in the environment is conferred by the FA selectivities of the FakB component. S. aureus expresses two FakBs (1, 5). SaFakB1 is specific for saturated FA (16:0), whereas SaFakB2 selectively binds monounsaturated FA (18:1) (1, 5, 12). S. aureus does not synthesize unsaturated FA, so SaFakB1 is the housekeeping protein and SaFakB2 is responsible for the uptake of host monounsaturated FA (1, 5, 12). S. pneumoniae encodes three FakB-binding proteins suggesting an expanded repertoire of FA-binding capabilities to enable the acquisition of environmental FA to replace de novo biosynthesis. Here, we report that two of these binding proteins, SpFakB1 and SpFakB2, are bioinformatically related to the two FakBs previously characterized in S. aureus (5, 12). Heterologous expression, biochemical analyses, and X-ray crystallography confirmed that SpFakB1 specifically bound saturated FA (16:0) and SpFakB2 selectively bound monounsaturated FA (18:1) but also bound saturated FA. Fatty acids are designated by number of carbons:number of double bonds. SpFakB3 is more distantly related to the S. aureus homologs, and heterologous expression and biochemical analyses showed that SpFakB3 expanded the repertoire of substrates to polyunsaturated FA (18:2). The SpFakB3(18:2) structure revealed a different hydrogen bond network anchoring the FA carbonyl and an expanded FA-binding pocket within the hydrophobic interior of the protein that explains its ability to bind FA with multiple cis double bonds. S. pneumoniae strain JMG1 (ΔfakB3) was deficient in 18:2 incorporation verifying the role of SpFakB3 in the incorporation of polyunsaturated FA into S. pneumoniae phospholipids. Thus, the multiple FakBs of S. pneumoniae permit the acquisition and utilization of the entire spectrum of mammalian FA structures to facilitate the acquisition of host FA to construct its membrane at the infection site.
Results
Incorporation of FA from human serum
The distinct difference between S. aureus and S. pneumoniae utilization of exogenous FA is illustrated by the effect of human serum supplementation on the molecular species composition of PG, a major membrane phospholipid in both organisms (Fig. 1). S. pneumoniae FASII produced both saturated and monounsaturated FA when grown in C+Y medium without FA supplement (3) (Fig. 1A). The two most abundant PG molecular species had 16:0 paired with either 16:1 or 18:1. S. pneumoniae is like most organisms in that it places saturated FA in the 1-position and unsaturated FA in the 2-position of its membrane phospholipids (13). S. aureus places anteiso15:0 in the 2-position of the glycerol backbone (3) giving rise to S. aureus PG molecular species with only saturated, straight, and branched-chain FA paired with anteiso15:0 when grown in media without exogenous FA (Fig. 1B). These data illustrate the difference in membrane phospholipid structures produced by de novo biosynthesis in these two Gram-positive pathogens.
Figure 1.

PG molecular species in the presence and absence of human serum. S. pneumoniae strain TIGR4 and S. aureus strain AH1263 were grown in their respective medium or in medium:human serum (1/1, v/v). The PG molecular species were determined under each of the four culture conditions in triplicate, and representative spectra are shown. The molecular species that contain an 18:2 or 18:2-derived FA are highlighted in red. A, S. pneumoniae grown in C+Y media. B, S. aureus grown in Luria broth. C, S. pneumoniae grown with 50% human serum, 50% C+Y medium. The presence of 18:2 in the m/z = 745 peak was verified by fragmentation (inset). D, S. aureus grown with 50% human serum, 50% Luria broth. E, major FA of human serum. Lipids were extracted from triplicate samples of human serum and the total FA composition determined by GC of the derived methyl esters. The weight percent of each FA was calculated. FA less than 1% of the total are not shown. F, triplicate biological replicates were obtained, the areas under each peak in the spectra were summed, and the PG molecular species containing 18:2 as a percent of the total area were calculated to provide an estimate of 18:2 incorporation. Peaks containing elongation products, like 20:2 derived from 18:2, were included in the calculation for the contribution of the parent FA.
Both S. pneumoniae and S. aureus incorporated FA derived from serum lipids when cultured in the presence of human serum. The S. pneumoniae PG molecular species pattern when grown in human serum was different from the pattern obtained in the absence of an exogenous lipid supplement (Fig. 1C). The most abundant peak, m/z = 747, corresponded to a 16:0/18:1 in both growth conditions, and in the presence of human serum the 16:0/16:1 peak was less prominent. This pattern is consistent with the observation that S. pneumoniae shuts down de novo lipid synthesis when supplied with an exogenous source of FA (3). Peaks in the spectrum arising from the synthesis of molecular species containing 18:2 are highlighted in red (Fig. 1C). A major new peak (m/z = 745) was fragmented to verify that 16:0/18:2 was the most abundant molecular species at this mass position (Fig. 1C, inset). S. pneumoniae cannot make 18:2, thus this FA must be derived from the human serum. The origin of the saturated and monounsaturated FA in PG cannot be unambiguously determined in this experiment because these FA are synthesized by S. pneumoniae and are present in human serum. The analysis of S. aureus grown with human serum showed that most PG molecular species still contained 15:0, but that 18:1 along with its elongation product 20:1 were incorporated from the serum (Fig. 1D). Human serum contains nonesterified FA plus an abundance of glycerolipids, and both S. aureus (geh and lip) (14) and S. pneumoniae (lipA) (15) express genes capable of hydrolyzing these lipids to release FA. The three most abundant FA in the human serum sample used in our study were 16:0, 18:1, and 18:2 (Fig. 1E). Although 18:1 and 18:2 were about equal in abundance, S. aureus incorporated significantly more 18:1 than 18:2 (Fig. 1D). The incorporation of 18:2 into PG was significantly less in S. aureus compared with the more robust incorporation of 18:2 into the S. pneumoniae lipidome (Fig. 1F). These data indicated that S. pneumoniae had a higher capacity to extract polyunsaturated FA from human serum than S. aureus.
Three fakB genes in S. pneumoniae
S. aureus expresses two FakB proteins, one specific for saturated FA and the other specific for monounsaturated FA (5). The fact that S. pneumoniae incorporates 18:2 into its phospholipids suggested that it had a FA-binding protein capable of binding polyunsaturated FA. We identified a single fakA gene (locus tag: Sp0443) and 3 fakB genes (locus tags: Sp1557, Sp1112, Sp0742) in the S. pneumoniae TIGR4 genome. The protein sequences were compared using BLASTP alignment software using the KEGG database sequence entries. The Sp1557 gene was named fakB1 because it encoded a protein that was 35% identical to SaFakB1 and 29% identical to SaFakB2. The Sp1112 gene was named fakB2 because it encoded a protein with 29% identity to SaFakB2 and 28% identity to SaFakB1. The Sp0742 gene encoded a protein that was the least similar to the S. aureus FakBs (18% to SaFakB1 and 26% to SaFakB2), and it was designated fakB3. Both S. pneumoniae and S. aureus are in the phylum Firmicutes and class Bacilli, but S. pneumoniae is in the Order Lactobacillales and S. aureus in the Order Bacillales. Analysis of both Orders using the KEGG database showed that the individual members of both Orders encode between 2 and 4 fakB genes.
FA selectivity of SpFakBs in cells
An experimental system to evaluate the FA selectivities of FA-binding proteins was developed using S. aureus strain JLB31 (ΔfakB1 ΔfakB2) that was unable to incorporate exogenous FA because it lacks FA-binding proteins but expresses the SaFakA kinase domain protein. A series of plasmids were constructed that expressed the individual FakBs governed by the sarA promoter (Table 1). The series of JLB31 strains each harboring a plasmid expressing a particular FakB were grown to an A600 of 0.5 and then a mixture of [d4]16:0, -18:1, and -18:2 (10 μm each) was added to the cultures. After a 30-min incubation, the extent of each FA incorporated into PG was determined by MS (Fig. 2A). The assay was validated using SaFakBs of known FA specificity. The expression of SaFakB1 resulted in robust incorporation of [d4]16:0, whereas the incorporation of the unsaturated FA in the mixture was not detected. In the strain expressing SaFakB2, 18:1 was the most highly incorporated FA, although lower amounts of both [d4]16:0 and -18:2 were detected. Strain JLB31/pCS119 (empty vector) did not express a FakB and did not incorporate any exogenous FA.
Table 1.
Strains, plasmids, and primers
| Description | Source | |
|---|---|---|
| Strains | ||
| AH1263 | S. aureus strain USA300–0114, Erm-sensitive | 46 |
| JLB31 | fakB1::ΦΝΣ ΔfakB2 of strain AH1263 | 1 |
| Sa178RI | Derived from S. aureus strain RN4220 | 47 |
| TIGR4 | Wildtype encapsulated S. pneumoniae | www.tigr.org |
| JMG1 | TIGR4 with fakB3 replaced with Kan cassette | This study |
| Plasmids | ||
| pG164 | Isopropyl 1-thio-β-d-galactopyranoside-inducible vector | 47 |
| pCS119 | pCM28SarAP1promoter | 48 |
| pPJ480 | pCS119 vector with RBS site | This study |
| pET28a | Expression vector | Novagen |
| pET28SpA | His tag SpFakA expression vector | This study |
| pET28SpB1 | His tag SpFakB1 expression vector | This study |
| pET28SpB2 | His tag SpFakB2 expression vector | This study |
| pET28SpB3 | His tag SpFakB3 expression vector | This study |
| pSaB1 | pCS119 expressing SaFakB1 | This study |
| pSaB2 | pCS119 expressing SaFakB2 | This study |
| pSpB1 | pPJ480 expressing SpFakB1 | This study |
| pSpB2 | pPJ480 expressing SpFakB2 | This study |
| pSpB3 | pPJ480 expressing SpFakB3 | This study |
| pDL278 | E. coli, Gram-positive shuttle vector | 49 |
| pSpFakB3 | SpFakB3 S. pneumoniae expression plasmid | This study |
| pABG5 | E. coli, Gram-positive shuttle vector | 34 |
| Primers | Sequence (5′ to 3′) | |
| Primer 1 | CTCAAGTGGTAAATGGGTCAA | |
| Primer 2 | AAATGGTTCGCTGGGTTTATCAGAAATTCTCCATCTTTGTCA | |
| Primer 3 | GATAAACCCAGCGAACCATTT | |
| Primer 4 | ATACAAATTCCTCGTAGGCGC | |
| Primer 5 | GCGCCTACGAGGAATTTGTATTTGCATTCTTGACAAGAGGTG | |
| Primer 6 | ATCATAATGCAGACTTCCCGC | |
| Primer 7 | TGGAGAATTTCTATGACTTGG | |
| Primer 8 | ATGCAATTAATCAATTTC | |
| pABGB3BamH1 | CTTGGATCCATTTTCTCTCCTCTCAAA | |
| pABGB3Sac1 | GATGAGCTCTTAATCAATTTCATAGCC |
Figure 2.
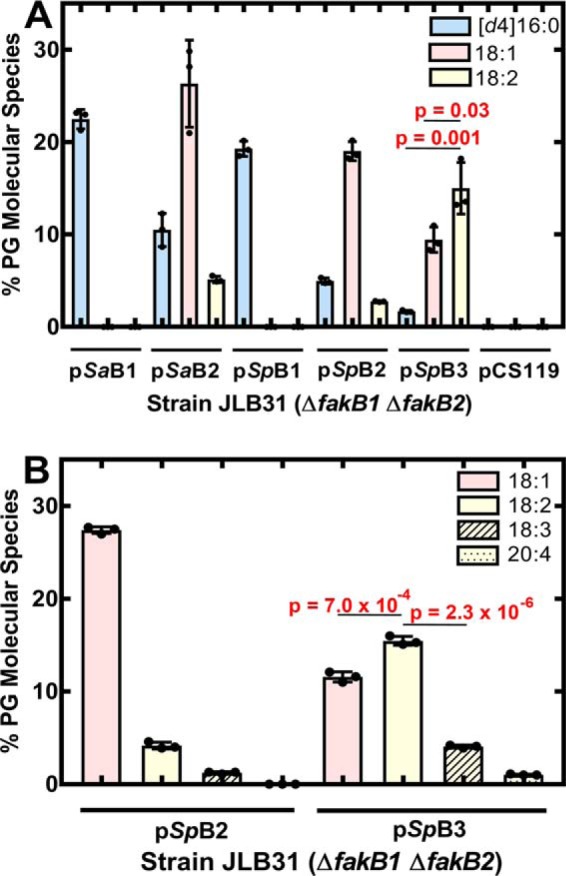
FA selectivity of S. pneumoniae FakB proteins in vivo. S. aureus strain JLB31 (ΔfakB1 ΔfakB2) contains no functional fakB genes. Plasmids derived from pCS119 (control empty vector) were constructed to drive the expression of each of the S. aureus and S. pneumoniae fakB genes using the sarA promoter and were introduced into strain JLB31. A, the strain set was labeled with an equimolar mixture of [d4]16:0, 18:1, and 18:2 (10 μm each) for 30 min and the contribution of each FA in the PG molecular species was determined by MS. B, strain JLB31 expressing SpFakB2 or SpFakB3 was labeled with an equimolar mixture of 18:1, 18:2, 18:3, and 20:4 (7.5 μm each) for 30 min, and the contribution of each unsaturated FA to the PG molecular species was determined by MS. Triplicate biological replicates were obtained, the areas under each peak in the spectra were summed and reported as a percent of the total area, and mean ± S.D. was plotted. Peaks containing elongation products, like 20:1 derived from 18:1, were included in the calculation for the contribution of the parent FA. Data are the mean ± S.D. of 3 individual data sets. Statistical differences between the FA incorporated in each strain was determined using Student's t test.
S. pneumoniae FakBs were interrogated in the same manner. SpFakB1 supported the incorporation of [d4]16:0, but not 18:1 or 18:2 (Fig. 2A). Oleate (18:1) was the highest incorporated FA in the strain expressing SpFakB2, and more 18:2 was incorporated in the strain expressing SpFakB3 as compared with 18:1 (p = 0.03) or [d4]16:0 (p = 0.001) (Fig. 2A). These data point to SpFakB3 as a binding protein for polyunsaturated FA, and the substrate preferences of SpFakB2 and SpFakB3 were further examined using a mixture of 3 polyunsaturated FA (Fig. 2B). SpFakB2 clearly preferred 18:1, whereas SpFakB3 was less specific in this assay utilizing 18:1, 18:2, and 18:3 although more 18:2 was incorporated as compared with 18:1 (p = 7.0 × 10−4) or 18:3 (p = 2.3 × 10−6) (Fig. 2B). These data suggested that SpFakB3 extends the range of environmental FA that can be incorporated by the FA kinase system to polyunsaturated FA.
Biochemical analysis of SpFakB selectivity
One potential caveat to the cellular FA selectivity experiment was that incorporation also depended on the action of PlsX and PlsY, which themselves may have acyl chain selectivity that would impact the results. Therefore, SpFakA and the three SpFakBs were expressed and purified to evaluate FA preferences in vitro (Fig. 3A). All four proteins expressed well and were >90% pure judged by gel electrophoresis. Next, the apparent Km for each of the SpFakBs was determined to establish the foundation for the biochemical competition assay (Fig. 3B). SpFakB2 and SpFakB3 had the same apparent Km (0.4 ± 0.09 and 0.5 ± 0.08 μm), whereas the SpFakB1 Km was 2.1 μm ± 0.6.
Figure 3.
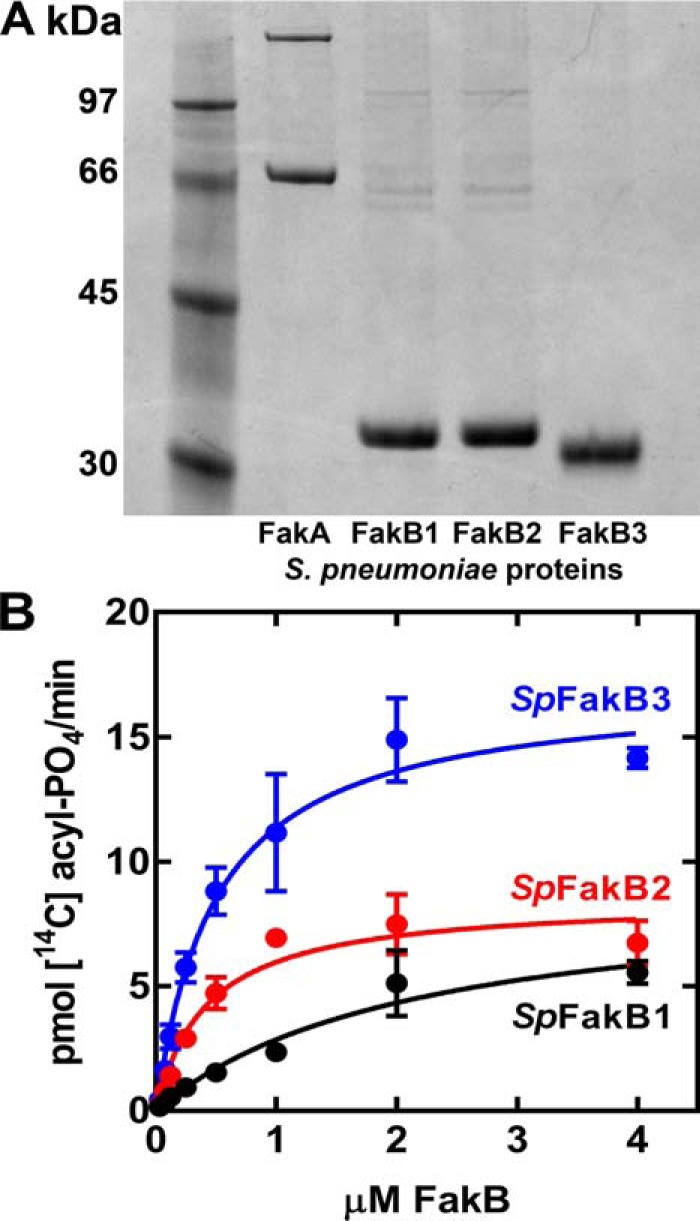
Biochemical validation and analysis of S. pneumoniae FakB proteins. A, purity of SpFakA and the three FakB proteins assessed by gel electrophoresis and Coomassie-staining. B, dependence of FA kinase activity on FakB concentration in the biochemical assays using 20 μm [14C]FA (SpFakB1, [14C]16:0; SpFakB2, [14C]18:1; and SpFakB3, [14C]18:2) and 0.2 μm SpFakA.
We used a competition assay to evaluate the utilization of FA by the individual SpFakBs (Fig. 4). This assay used a 14C-labeled FA substrate that matched the preferred substrate for each SpFakB: [14C]16:0 for SpFakB1; [14C]18:1 for SpFakB2; and [14C]18:2 for SpFakB3. Unlabeled FA competitors were 80 μm. If the added FA was a substrate for the FA kinase reaction, then the amount of labeled product is reduced. SpFakB1 activity measured with [14C]16:0 was robust, and an excess of cold 16:0 added to the reaction significantly diminished labeled [14C]16:0-phosphate formation. When an excess of either 18:1 or 18:2 was added, there was no change in the utilization of [14C]16:0 indicating that these FA are poor substrates for SpFakB1 (Fig. 4A). The activity of SpFakB2 measured with [14C]18:1 was effectively competed with 18:1, but not with 16:0 (Fig. 4B). The addition of 18:2 decreased the conversion of [14C]18:1-phosphate indicating that 18:2 exchanged onto SpFakB2, but the amount of [14C]18:1-phosphate was not significantly different from the control (Fig. 4B). FA kinase activity with SpFakB3 using [14C]18:2 as a substrate was not decreased by 16:0. Both 18:1 and 18:2 were effective competitors but statistically more [14C]18:2-phosphate remained when cold 18:1 was used as compared with when cold 18:2 was used (p = 0.002) meaning that 18:2 was a better substrate for SpFakB3 (Fig. 4C). The range of FA competitors was extended to other polyunsaturated FA (18:3 and 20:4) (Fig. 4D). These experiments showed that SpFakB3 utilized a broad spectrum of unsaturated FA but showed a statistically significant preference for 18:2 over 18:1 (p = 0.02), 18:3 (p = 0.01), or 20:4 (p = 2.7 × 10−4) (Fig. 4D).
Figure 4.

Analysis of FakB selectivity in vitro. FA kinase assays were used to investigate the selectivites of the SpFakBs. Each SpFakB was assayed in the presence of 0.2 μm SpFakA using 20 μm of an individual radiolabeled FA that corresponds to its substrate preference. [14C]16:0 was the substrate for SpFakB1, [14C]18:1 was the substrate with SpFakB2, and [14C]18:2 was the substrate for SpFakB3. Then to each assay 80 μm cold FA competitor was added. A, SpFakB1. B, SpFakB2. C, SpFakB3, and D, SpFakB3. Experiments were performed in triplicate. Data are the mean ± S.D. of 3 individual data sets. Statistical differences between the FA incorporated in each strain was determined using Student's t test.
FA utilization in a ΔfakB3 mutant
The physiological role of fakB3 in S. pneumoniae FA uptake was evaluated by constructing a ΔfakB3 knockout strain. Strain JMG1 (ΔfakB3) was constructed from strain TIGR4 by allelic replacement with a cassette encoding kanamycin resistance (Fig. 5A). PCR analysis showed the presence of the knockout allele and the absence of the fakB3 gene in strain JMG1. The PG molecular species of strain JMG1 (ΔfakB3) was indistinguishable from TIGR4 (WT) when grown in C+Y (Figs. 1A and 5B) but TIGR4 incorporated more 18:2 than strain JMG1 (ΔfakB3) when grown in human serum (Figs. 1C and 5C). The incorporation of 18:2 was restored to WT levels in the complemented strain JMG1/pSpFakB3 (Fig. 5D). These data validate a role for SpFakB3 in the utilization of exogenous 18:2 for membrane lipid synthesis in S. pneumoniae.
Figure 5.

FA acquisition in a S. pneumoniae ΔfakB3 deletion strain. A, schematic diagram illustrating the construction of strain JMG1 (ΔfakB3). The final construct has the fakB3 gene replaced with a cassette expressing kanamycin resistance. Primers 1 and 6 showed the exchange of the fakB3 gene for the kanamycin cassette, and primers 7 and 8 confirmed that fakB3 was absent from the genome. Construction details are provided under “Experimental procedures” and primer sequences are listed in Table 1. B, representative spectra of S. pneumoniae strain JMG1 (ΔfakB3) grown in C+Y media (C) and grown in 50% human serum, 50% C+Y medium human serum. D, TIGR4, JMG1 (ΔfakB3), and JMG1/pSpFakB3 were grown in 50% human serum, 50% C+Y medium, PG mass spectra from triplicate biological replicates were obtained, the areas under each peak in the spectra were summed, the molecular species containing 18:2 as a percent of the total area were calculated, and the mean ± S.D. was plotted.
Crystal structures of SpFakBs
The basis for the FA preferences of the SpFakB proteins was revealed by the crystal structures of the three S. pneumoniae FA-binding proteins in complex with their respective FA ligands (Table 2). The SpFakBs all have a two-domain protein-fold very similar to the other FakB protein family members that have been described (5, 12, 16, 17) (Fig. 6). The N-terminal domain consists of an EDD-fold fused to a 3-stranded antiparallel β-sheet and one α-helix. The carboxyl-terminal domain is a six-stranded β-sheet flanked by two α-helices on one side and three on the other. The surface arginine residue highlighted in Fig. 6 is conserved and required for high affinity binding to the FakA component of FA kinase (12). Its presence in all FakB structures explains why SpFakBs function interchangeably with SaFakA (Fig. 2A), and identifies the FakA-FakB binding locale. Furthermore, the overall surface charge distribution is similar in all three SpFakBs (Fig. 6). The conservation of the FakB structure across isoforms and species accounts for their ability to interchangeably interact with their three protein partners: FakA, PlsX, and PlsY (5).
Table 2.
Data collection statistics for SpFakB fatty acid complexes
| Protein | SpFakB1 | SpFakB1 | SpFakB2 | SpFakB2 | SpFakB3 |
|---|---|---|---|---|---|
| Fatty acid | Myristate (14:0) | Palmitate (16:0) | Oleate (18:1Δ9) | Vaccinate (18:1Δ11) | Linoleate (18:2Δ9,12) |
| PDB ID | 6NOK | 6DKE | 6DJ6 | 6NR1 | 6CNG |
| Precipitant | PEG2/D7 | PEG2/D7 | Classics suite/A5 | LMB/D12 | JCSG+/B7 |
| Cryoprotectant | Paraffin/paratone-N (1:1) | 25% Glycerol | 25% Glycerol | 25% Glycerol | 25% Glycerol |
| Data collection | |||||
| Beamline | SER-CAT 22-BM | SER-CAT 22-ID | SER-CAT 22-ID | SER-CAT 22-BM | SER-CAT 22-ID |
| Detector | MarCCD 300HS | Dectris Eiger X 16 m | Dectris Eiger X 16 m | MarCCD 300HS | MarCCD 300HS |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 |
| Wavelength (Å) | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.0000 |
| Space group | P21 | P21 | P212121 | P212121 | P21 |
| Unit cell parameters (Å) | |||||
| a, b, c | 37.56,66.32,46.95 | 46.83,62.34,50.43 | 59.72,103.85,108.17 | 59.65,103.41,107.93 | 52.78,103.41,67.47 |
| α, β, γ | 90.00,95.75,90.00 | 90.00,95.94,90.00 | 90.00,90.00,90.00 | 90.00,90.00,90.00 | 90.00,112.99,90.00 |
| Resolution range (Å) | 66.32–1.69 | 62.34–1.76 | 74.92–1.90 | 74.67–2.10 | 62.11–1.47 |
| Rmerge | 0.056 (0.669)a | 0.059 (0.497) | 0.109 (0.827) | 0.101 (0.616) | 0.036 (0.544) |
| Rr.i.m. or Rmeas | 0.066 (0.790) | 0.072 (0.613) | 0.129 (0.978) | 0.113 (0.693) | 0.044 (0.669) |
| Rp.i.m. | 0.34 (0.415) | 0.042 (0.355) | 0.067 (0.510) | 0.050 (0.311) | 0.025 (0.386) |
| Number observations | 93,246 (4,636) | 76,979 (4,403) | 197,121 (12,541) | 190,517 (15,213) | 323,223 (15,293) |
| Number unique reflections | 25,680 (1,310) | 28,063 (1,592) | 52,631 (3,366) | 39,625 (3,194) | 108,472 (5,188) |
| Multiplicity | 3.6 (3.5) | 2.7 (2.8) | 3.7 (3.7) | 4.8 (4.8) | 3.0 (2.9) |
| Mn I/σ(I) | 16.2 (2.0) | 10.6 (2.0) | 9.4 (1.9) | 9.0 (2.0) | 15.0 (2.0) |
| Mn(I) half-set corr. | 0.998 (0.667) | 0.995 (0.746) | 0.993 (0.479) | 0.998 (0.799) | 0.999 (0.574) |
| Completeness (%) | 99.8 (99.8) | 97.9 (98.7) | 98.3 (99.6) | 99.7 (99.7) | 96.2 (94.1) |
| Wilson B-factor (Å−2) | 17.3 | 22.0 | 17.03 | 21.7 | 17.3 |
| Model quality | |||||
| Twinning (operator, %) | NAb | NA | NA | NA | -h,-k,h+l, 49.8 |
| Monomers/asymetric unit | 1 | 1 | 2 | 2 | 2 |
| Rwork/Rfree value (%) | 15.2/20.0 | 17.0/20.6 | 17.1/22.7 | 18.4/24.1 | 13.7/16.5 |
| R.m.s. deviations | |||||
| Bonds (Å) | 0.017 | 0.005 | 0.010 | 0.009 | 0.014 |
| Angles (°) | 1.482 | 0.751 | 1.361 | 0.915 | 1.134 |
| Coordinates error (ML, Å)c | 0.17 | 0.22 | 0.12 | 0.24 | n/a |
| Protein residues | 285 | 283 | 555 | 557 | 562 |
| Average B factor (Å2) | |||||
| All atoms | 25.1 | 29.9 | 25.4 | 36.5 | 21.2 |
| Protein atoms | 23.4 | 28.4 | 23.8 | 36.4 | 19.1 |
| Fatty acid atoms | 17.5 | 19.8 | 31.2 | 33.9 | 30.3 |
| Solvent atoms | 38.9 | 41.9 | 37.7 | 37.8 | 30.6 |
| Ramachandran plot | |||||
| Favored (%) | 96.8 | 98.2 | 98.5 | 98.6 | 96.0 |
| Allowed (%) | 2.5 | 1.8 | 1.3 | 1.5 | 3.8 |
| Outliers (%) | 0.7 | 0 | 0.2 | 0.8 | 0.2 |
| Clashscore | 2.8 | 2.2 | 2.3 | 4.3 | 8.6 |
a Values in parentheses are for the highest resolution shell. Rmerge = Σ(I−〈I〉)/Σ(I); where I is the intensity measured for a given reflection, 〈I〉 is the average intensity for multiple measurements of this reflection. Rp.i.m. = (Σ[1/(n − 1)]∧1/2. Σ|I − 〈I〉|)/Σ(I). Rwork = Σ‖Fobs| − |Fcalc‖/Σ|Fobs|; where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively. Rfree is Rwork for the 5% excluded reflections during refinement.
b NA, not applicable.
c ML is maximum likelihood.
Figure 6.
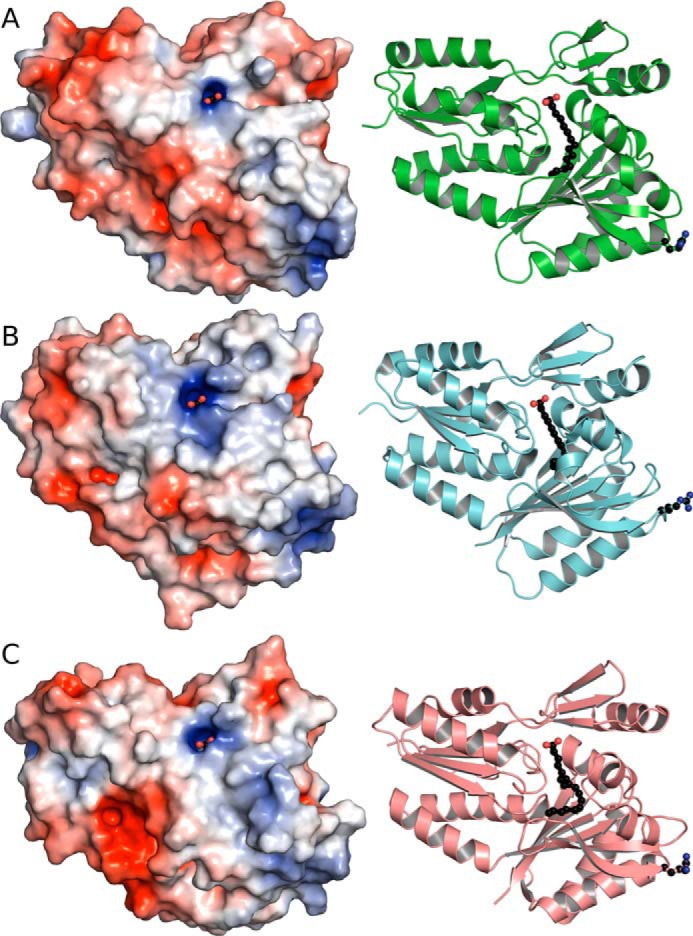
The overall structures of the three SpFakBs. The structures of the 3 SpFakBs were determined and are shown in ribbon diagram adjacent to the surface rendering. The surface Arg residue that is critical for FakA binding is located at the bottom right of each molecule and are shown as balls and sticks in the ribbon diagrams. The surface potential of each SpFakB show similar patterns of charge distribution. A, SpFakB1. B, SpFakB2. C, SpFakB3. The ribbon structures were rendered with PyMOL and the surface potentials were calculated with APBS in PyMOL.
The SpFakB FA-binding tunnels
The unique distinguishing feature of the FakBs is the size and shape of the hydrophobic FA-binding tunnel located in the protein's interior that defines the FA binding selectivity. The SpFakB1(16:0) and SaFakB1(16:0) complexes are structurally very similar (Fig. 7A), and there is an overall root mean square deviation of 1.45 Å between the two aligned structures. The gently curved shapes of the two FA-binding pockets and the residues used to construct the pocket are similar in the two proteins, and in both cases, the FA carboxyl is fixed by a hydrogen bond network at the mouth of the tunnel. However, a notable difference in SpFakB1 is a kink in the FA chain at carbon-2 that is not present in SaFakB1, which displaces the acyl chains by 1.2 Å at this position. The equivalent residues SpIle28–SaLeu28 and SpVal264–SaVal266 are displaced by 1.6 and 1.7 Å, respectively, to accommodate this kink at carbon-2 in the SpFakB1(16:0) structure. The kink generates additional changes in the conformation of the FA in SpFakB1(16:0) compared with SaFakB1(16:0), and these are accommodated by other displacements between aligned equivalent residues in the tunnels: SaGly277–SpGly277, 0.9 Å; SaIle124–SpPro123, 0.7 Å; SaLeu120–SpIle119, 1.2 Å; and SaAla158–SpAla157, 0.6 Å.
Figure 7.
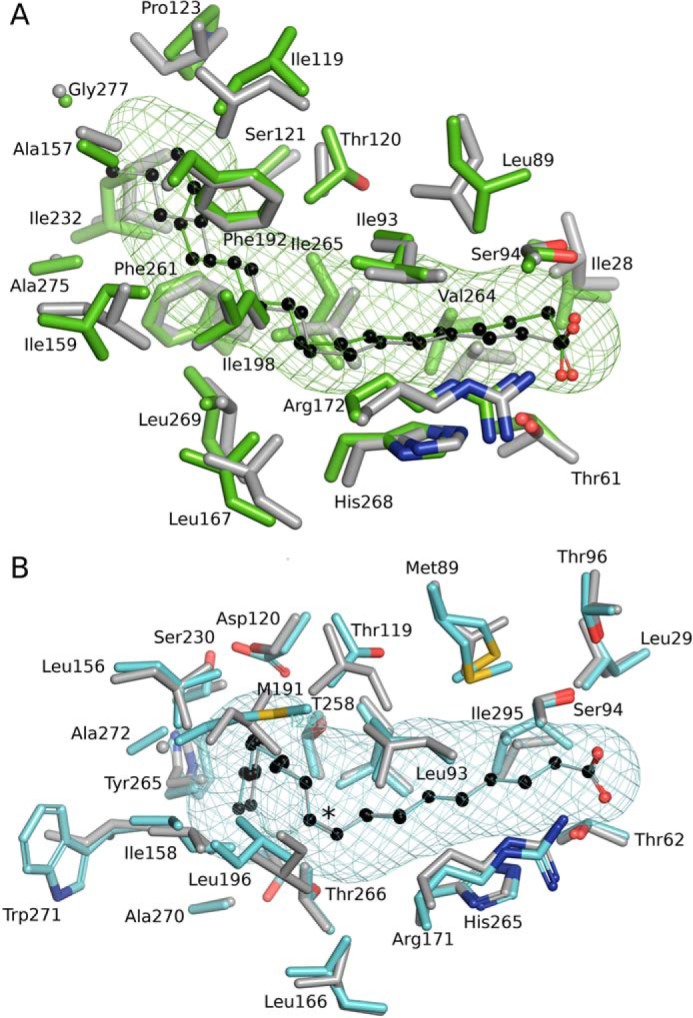
Conservation of the FA-binding pocket shapes in saturated and monounsaturated FA-binding proteins of S. pneumoniae and S. aureus. A, structural alignment of SpFakB1(16:0) (green, PDB ID, 6DKE) and SaFakB1(16:0) (gray, PDB ID, 5UTO) FA-binding tunnels and surrounding residues as aligned with PyMOL. B, SpFakB2(18:1Δ9) (cyan, PDB ID 6DJ6) and SaFakB2(18:1Δ9) (gray, PDB ID, 4X9X). The coordinate alignment was based on the aligned tunnel residues in PyMOL. Asterisk indicates sp2 hybridization of the double bond. The bound FA are depicted as colored sticks with black carbon spheres. The meshes that delineate the hydrophobic cavity within the proteins were computed with CAVER/PyMOL.
The SpFakB2(18:1Δ9) structure is also very similar to the SaFakB2(18:1Δ9) structure (Fig. 7B) with an overall root mean square deviation of 1.27 Å between the two aligned structures. Despite the low level of sequence identity between the two proteins (29%), the amino acids that create their FA tunnels both generate the same distinctive kinked cavities that are required to accommodate the cis double bond in the middle of the acyl chain. Similar to FakB1, the FA carboxyl is fixed by a hydrogen bond network at the entrance of the tunnel that comprises highly conserved and closely aligned residues in SpFakB2 and SaFakB2; SpLeu29–SaLeu23 (0.2 Å), SpSer94–SaSer93 (0.3 Å), SpArg171–SaArg170 (0.4 Å), and SpHis265–SaHis266 (0.5 Å). The bound 18:1 adopts very similar conformations in SpFakB2 and SaFakB2 but begins to diverge at carbon-13 and is separated by 0.5 Å at the terminal methyl. Accordingly, equivalent residues that surround the distal end of the FA are more displaced than those at the proximal end: SpSer230–SaAla230, 1.0 Å; SpTrp271–SaIle272, 0.5 Å; SpAla272–SaGly273, 1.3 Å.
The SpFakB3(18:2(Δ9Δ12)) complex structure reveals a distinctive gourd-like FA-binding pocket that highlights how the interiors of these proteins have evolved to perfectly accommodate FA of different structures (Fig. 8A). Like FakB1 and FakB2, the proximal end of the FakB3 tunnel is relatively narrow and straight to accommodate the first 7 carbons of the acyl chain. However, at this point, the tunnel expands into a wide cavern that allows FA with multiple cis double bonds to curl up inside the protein. The volume of the tunnel is 110.4 Å3, which is larger than the linear FakB1 tunnel (67.5 Å3) and slightly larger than the kinked FakB2 tunnel (102 Å3). Another distinguishing feature of SpFakB3 compared with FakB1 and FakB2 is that it uses a different hydrogen bond network to lock the FA carboxyl in place (Fig. 8B). The typical FakB configuration places Ser on one carbonyl oxygen and a His-Thr dyad on the other side to form a Ser-FA carbonyl-Thr-His network (12). In contrast, in SpFakB3, a tyrosine (Tyr-268) plays the role of histidine and a serine (Ser-63) replaces threonine to form a Ser-FA carbonyl-Ser-Tyr network. There are also two structured water molecules in SpFakB3 hydrogen bonded to the FA carbonyl that mediate hydrogen bond connections between the FA carbonyl and Arg-173, Gly-92, and Ser-63. Arg-173 also forms a hydrogen bond with Tyr-268 and a water-mediated connection with Asn-171.
Figure 8.
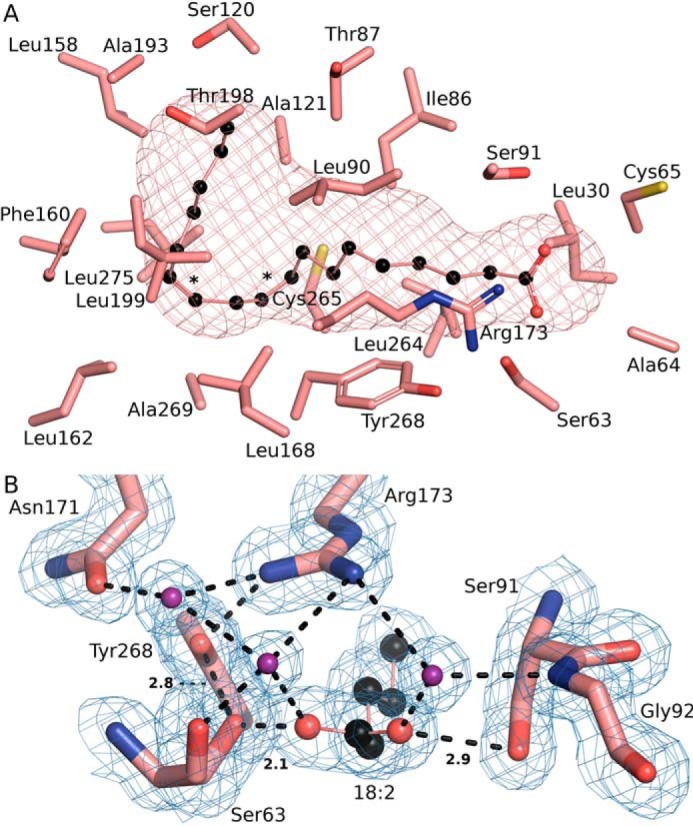
Unique aspects of the SpFakB3 structure. A, view of the SpFakB3 FA-binding tunnel and associated residues as visualized using CAVER to outline the interior volume of FakB3(18:2(Δ9Δ12)) (PDB ID 6CNG) (salmon). FA carbons are black. B, the SpFakB3 Ser-91–FA–Ser-63–Tyr-268 hydrogen bond network that fixes the position of the FA carbonyl in the protein. FA carbons are black, FA and amino acid oxygens are red, and water molecules are colored purple. The blue mesh represents the experimental electron density contoured at 1 σ. Dashed lines indicate hydrogen bonds and the lengths in Å of the three key interactions are shown.
Further insights into the SpFakB1 and SpFakB2 tunnel specificities
In SpFakB1(16:0), the terminal methyl of the 16:0 chain is packed against the protein at the end of the pocket suggesting that the bend at carbon-2 may be required for the 16:0 chain to fit into the pocket. This idea was tested by determining the structure of SpFakB1 in complex with the shorter 14:0 FA with the prediction that the carbon-2 bend would not be present. An overlay of the SpFakB1(16:0) and SpFakB1(14:0) structures shows that the two FA overlay along the entire length of the binding tunnel and that the bend at carbon-2 is also present in the 14:0 structure (Fig. 9A). SaIle232 acts as a swinging gate to increase or decrease the tunnel length in SaFakB1, but residue SpIle232 did not change orientation or position in the SpFakB1 structures loaded with either 16:0 or 14:0. In the SpFakB1(16:0) structure, the terminal FA methyl carbon is 3.7 Å away from SpIle232. There were small changes in Phe-192 side chain that slightly close the end of the tunnel in the SpFakB1(14:0) structure, but otherwise the two structures are almost identical except for the length of the acyl chain. Thus, the carbon-2 bend is an integral feature of the SpFakB1 FA complex.
Figure 9.
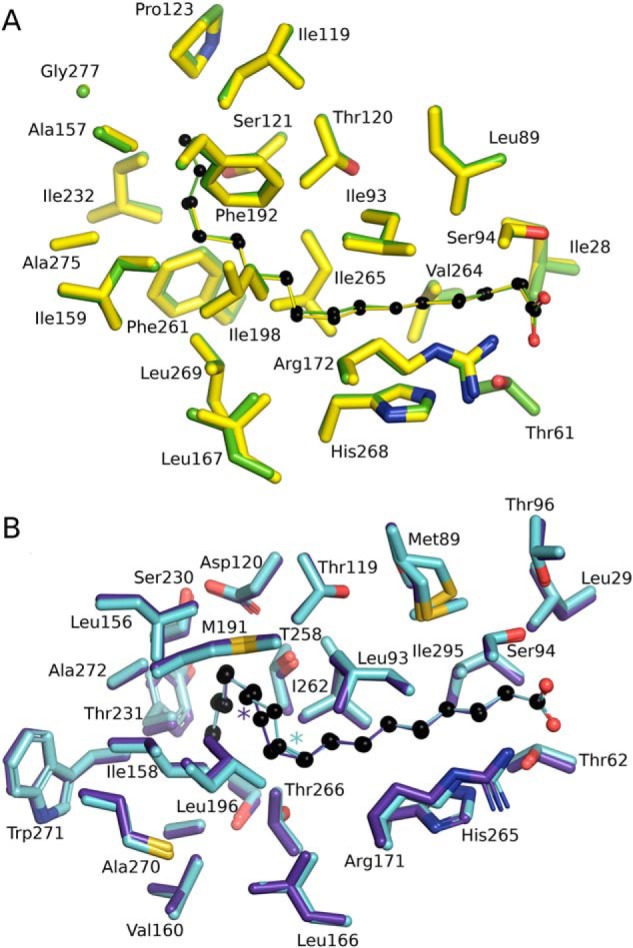
Versatility of FA binding by SpFakB1 and SpFakB2. A, structural alignment of SpFakB1(14:0) (yellow, PDB ID 6NOK) and SpFakB1(16:0) (green, PDB ID 6DKE). The fatty acids are presented as colored sticks with carbon spheres in black. B, structural alignment of SpFakB2 with C18:1Δ9, (cyan, PDB ID 6DJ6) and SpFakB2 with C18:1Δ11 (purple blue, PDB ID, 6NR1). Colored asterisks indicate locations of the sp2 hybridized double bonds. The coordinate alignments were based on the FA using MCSALIGN plugin for PyMOL.
Another question is how the substrate selectivity of SpFakB2 is related to its biological function(s). S. aureus does not produce unsaturated FA and thus SaFakB2 does not have a specific role in lipid homeostasis in the organism (5). However, S. pneumoniae does produce monounsaturated FA suggesting that SpFakB2 may have a role in lipid metabolism in this organism. S. pneumoniae de novo biosynthesis produces 18:1, although the primary location of the double bond is at carbon-11 instead of carbon-9 as found in mammals (6). The flexibility of SpFakB2 to bind these two monounsaturated FA was investigated by determining the structure of the SpFakB2(18:1Δ11) complex and comparing it to the structure of SpFakB2(18:1Δ9) (Fig. 9B). The binding pocket has enough space between the 9 and 12 carbons to accommodate either of the sp2-hybridized double bonds. The overlay of these two structures shows that they are essentially identical with regard to the size and shape of the FA-binding tunnel and the positioning of the residues that form the binding pocket. The different locations of the sp2 double bond conformations are clearly seen in the electron density map, and the maximum divergence of the two acyl chains is only 0.7 Å in the immediate area between the locations of the double bonds. These data illustrate that SpFakB2 participates in the activation of both endogenous cis-vaccinate and exogenous oleate derived from the host.
Discussion
The biochemical and structural data lead to a model for FA metabolism in S. pneumoniae outlined in Fig. 10. The biosynthetic pathway in the absence of exogenous FA proceeds via the FASII collection of enzymes to long-chain acyl-ACP (18), which is then distributed to PlsY (via PlsX) or PlsC to produce phosphatidic acid, the precursor to all membrane glycerophospholipids (19). In the presence of exogenous FA, transcription of the FASII genes is potently suppressed by binding of the FabT–acyl-ACP repressor complex to promoters within the FASII gene cluster (6, 10). The repression of FASII gene expression by exogenous FA is mediated by acyl-ACP2 (9) (the acpP2 gene that is in an operon with plsX). This regulatory loop shuts off de novo FA synthesis, and only exogenous FA are used for phospholipid synthesis (3). These exogenous FA are not significantly elongated even though they are converted to acyl-ACP because the FASII elongation system is suppressed. The three FakBs of S. pneumoniae characterized in this study participate in this FA sensing pathway by providing a mechanism to activate a spectrum of saturated, monounsaturated, and polyunsaturated FA found at the infection site for FA∼P and acyl-ACP formation. SpFakB1 has the most restricted FA binding preference and effectively excludes unsaturated FA with kinked acyl chains due to the straight shape of its FA-binding tunnel. SpFakB2 prefers monounsaturated FA. SpFakB2 has a kinked FA-binding tunnel that accommodates both 18:1Δ11, the monounsaturated fatty acid produced by S. pneumoniae, and 18:1Δ9, a prevalent mammalian FA. Although SpFakB2 prefers to bind monounsaturated FA, it does not exclude saturated FA, which are flexible and capable of fitting into the kinked binding pocket. SpFakB2 also has a weak capacity to bind 18:2. SpFakB3 has a wider FA-binding pocket that allows the binding of polyunsaturated FA, like 18:2, with multiple cis double bonds. The gourd-shaped pocket within SpFakB3 means that it can present a variety of FA structures to FakA. The multiple FakBs of the FA kinase activation system coupled with the repression of de novo synthesis means that S. pneumoniae can efficiently and exclusively utilize the entire spectrum of mammalian FA found at the infection site for phosphatidic acid synthesis (Fig. 10).
Figure 10.
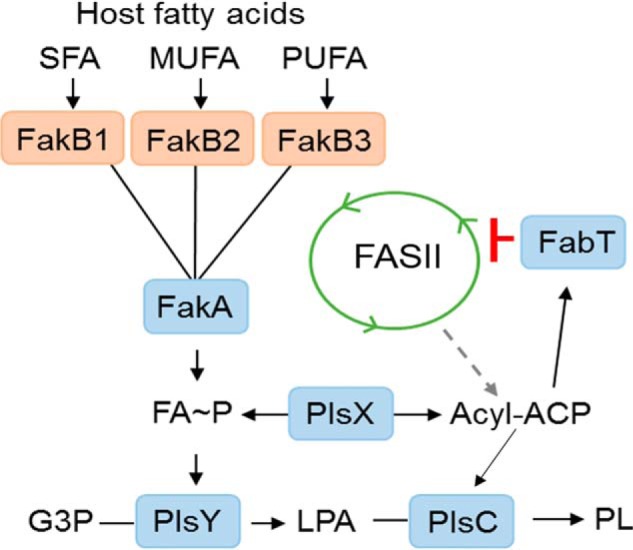
Model for the function of FakB proteins in S. pneumoniae. The de novo phospholipid biosynthetic pathway in S. pneumoniae delivers acyl-ACP generated by the FASII system to PlsX to provide FA∼P to the glycerol phosphate (G3P) acyltransferase (PlsY) to form lysophosphatidic acid (LPA) and initiate phospholipid synthesis. Acyl-ACP is also a substrate for PlsC that completes the formation of phosphatidic acid, the universal precursor to membrane glycerolipids (PL). At the infection site the host provides an environment containing a spectrum of saturated (SFA), monounsaturated (MUFA), and polyunsaturated (PUFA) FA. Each of these types of FA are the preferred ligand for binding to unique FakB-binding proteins that carry the FA to FakA, where they are phosphorylated. The resulting FA∼P is a substrate for PlsY or is converted to acyl-ACP by PlsX to act as substrate for PlsC. The long-chain acyl-ACP binds to the FabT transcriptional regulator, and the complex binds tightly to the promoters within the FASII gene cluster to potently suppresses the expression of the entire biosynthetic gene set and shut off acyl-ACP formation from FASII. The acyl-ACP may also enter FASII and be elongated, although this is a minor pathway due to the strong suppression of the elongation cycle enzymes by FabT–acyl-ACP complex.
SpFakB3 is differentiated from most FakBs by having a different hydrogen bond network that interacts with the FA carboxyl group (Fig. 9B). The majority of bacterial FA-binding proteins use a Ser-FA carboxyl-Thr-His hydrogen bond network to fix the FA carboxyl at the entrance of the FakB FA-binding tunnel (12), but in about 5% of the FakB sequences, a tyrosine is substituted at the position of the histidine residue. SpFakB3 is one of the FA-binding proteins with a Ser-FA carboxyl-Ser-Tyr hydrogen bond network. Disruption of this hydrogen bond network by mutagenesis shows that it is critical to the stability of the FA-binding protein. For example, the SaFakB2(S93A) mutant severs the connection to one side of the FA carbonyl and results in a 17 °C decrease in the denaturation temperature of the protein (12). Whether the hydrogen bond network is associated with polyunsaturated FA binding or has some other mechanistic significance is unclear.
The bacterial FA-binding proteins are very different from their mammalian counterparts (20, 21). The mammalian proteins are about half the size of the bacterial proteins and have a β-barrel structure consisting of 10 antiparallel β-sheets with 2 α-helices between the first and second β-strands (22–24). The carboxyl group of the FA is oriented inward and electrostatically bound to Arg-106 in the intestinal FA-binding protein (25), whereas the FA carboxyl in the bacterial binding proteins is exposed as the phosphorylation site in the FakB(FA) complexes. Another key difference is that the mammalian FA-binding proteins lack acyl chain selectivity (20, 21, 26) and have large cavities (PDB ID 4TJZ = 245 Å3; PDB ID 5CE4, 291 Å3; calculated using CASTp (27)) that in some cases bind two FAs (20, 22). The bacterial FA-binding proteins have smaller, narrow tunnels that impart substrate selectivity to these binding proteins. The expanded hydrophobic binding pocket of SpFakB3 is most reminiscent of the large, nonselective hydrophobic pockets that occur in mammalian FA-binding proteins, although the 110 Å3 volume of SpFakB3 is half the size of the larger mammalian pockets. Mammalian FA-binding proteins are isolated in both their ligand-free and FA-bound forms, and there is no clear relationship between FA binding and protein stability (28). The FA-free FakBs are not stable and only FA-bound FakBs have been purified (5, 12). Mutations in the hydrogen bond network that fixes the FA carboxyl significantly destabilize SaFakB2 (12). The mammalian FA-binding proteins pick up and deposit FA into phospholipid bilayers, and the 2-helix motif is thought to act as the entry and exit flap for the FA (22–24, 29, 30). The bacterial FA-binding proteins also exchange FA with FA in phospholipid bilayers (1) and transfer the phosphorylated FA to two enzyme partners, PlsX and PlsY (5). The protein movements in FakB that allow the entry and exit of the FA and acyl-phosphate remain to be defined.
Experimental procedures
Materials
Sources of supplies were: PerkinElmer Life Sciences, [14C]16:0 (specific activity, 56.1 mCi/mol), [14C]18:1 (specific activity, 59 mCi/mol), [14C]18:2 (specific activity, 54.5 mCi/mol); Millipore-Sigma, all FA and human serum (lot number SLBX0350); Cambridge Isotope Laboratories, Inc., 7,7,8,8-tetradeuteriohexadcanoic acid ([d4]16:0); Sigma, all reagents for buffers; New England Biolabs (Ipswich, MA), restriction enzymes; Invitrogen, Anza alkaline phosphatase, T4 DNA ligase master mix, and DNA blunt end kit for blunt end ligation. Proteins were expressed, purified, and FA exchange was accomplished as described (1, 5, 12).
Bacterial strains
DNA sequences were synthesized for each of the S. aureus and S. pneumoniae FakB genes (Invitrogen). The DNA sequences for S. aureus fakB1 (SaUSA300_0733) and fakB2 (SaUSA300_1318) included a ribosomal binding site and a His6 tag on the N terminus. Each S. aureus fakB sequence was cloned into plasmid pCS119, which has a pCM28 backbone (31), no ribosomal binding site, and uses a sarA P1 promoter to drive expression. DNA sequences for S. pneumoniae strain TIGR4 fakB1 (SP_1557), fakB2 (SP_1112), and fakB3 (SP_0742) genes did not contain a ribosomal binding site or a His6 tag and were cloned into pET28a (Novagen), to add an N-terminal His6 tag, and ligated into pPJ480 (32), which is a pCS119 plasmid derivative containing the sarA P1 promoter as well as a ribosomal binding site. Restriction followed by ligation reactions were used to insert the fakB gene of interest into either pCS119 (S. aureus expression) or pPJ480 (S. pneumoniae expression). The ligation mixture was transformed into Top10 Escherichia coli-competent cells (Invitrogen) and selected on Luria broth with carbenicillin (100 μg/ml). Purified plasmids were passaged through strain Sa178R1 to acquire the S. aureus DNA methylation pattern, re-isolated, and electroporated into strain JLB31.
Strain JMG1 (ΔfakB3) was constructed by gene splicing by overlap extension PCR, which allowed for the creation of a gene deletion with a kanamycin resistance cassette in its place (33). Briefly, primers 1 and 2 were used to amplify the upstream region (∼1 kb) and primers 5 and 6 were used to amplify the downstream region (∼1 kb) of the fakB3 (locus tag, SP_0742) gene from TIGR4 genomic DNA (Fig. 5A; Table 1). Primers 3 and 4 were used to amplify the kanamycin cassette (∼1.5 kb) from the pABG5 plasmid. These three PCR products underwent another PCR amplification using primers 1 and 6 to generate a linearized hybrid construct with the fakB3 gene replaced by the kanamycin cassette (Fig. 5A). The linear DNA was run on an agarose gel and purified before being transformed. To transform 1 ml of TIGR4 culture in C+Y media (A600 = 0.07), 3 μl of competence pheromone were incubated at 37 °C for 13 min before 5 μl of the linearized DNA was added and incubated at 37 °C for 4 h. Cells were plated on 3% blood agar plates with kanamycin (400 μg/ml) and neomycin (20 μg/ml) and incubated overnight at 37 °C. The colonies that arose were purified and genomic DNA was extracted. PCR was used to confirm the deletion using primers 1 and 6 and 7 and 8 (Fig. 5A; Table 1).
The SpFakB3 expression plasmid was made by engineering a gene sequence Genewiz) with an EcoRI site on the 5′ end followed by a ribosomal binding site taken from the pABG5 vector, a His6 tag, the TIGR4 S. pneumoniae fakB3 nucleotide sequence, and ClaI restriction site at the 3′ end of the gene. The assembled expression construct was inserted into pUC57. Purified plasmid was passaged through INV110 E. coli-competent cells as the ClaI site was sensitive to Dam methylation. The pUC57 containing the fakB3 insert and pABG5 vector were cut using EcoRI and ClaI, ligated, and transformed into Top10 E. coli-competent cells. The resulting plasmid was purified and amplified using PCR with primers pABGB3BamHI and pABGB3SacI to amplify the fakB3 insert as well as the upstream region of the pABG5 plasmid containing the rofA promoter (34). The pABGB3SacI primer changed the 3′ restriction site to SacI so that the resulting amplicon could be inserted into pDL278. A restriction digest was performed on the PCR product using BamHI and SacI and the product was run on an agarose gel before the band was extracted and purified. The fakB3 fragment was inserted into linearized pDL278 via blunt end ligation and subsequently transformed into Top10 E. coli-competent cells. The resulting plasmid was purified and sequenced before being transformed into strain JMG1 (ΔfakB3) and plated on 3% blood agar plates with kanamycin (400 μg/ml), spectinomycin (150 μg/ml), and neomycin (20 μg/ml). TIGR4/pDL278 and JMG1/pDL278 were made using the above-mentioned transformation protocol and plated on the same plates used to select for strain JMG1.
FA incorporation experiments
S. aureus and S. pneumoniae strains AH1263 and TIGR4, respectively, were inoculated into 5 ml of growth medium (S. aureus, Luria broth; S. pneumoniae, C+Y) at an A600 of 0.05, and grown at 37 °C to an A600 of 0.5, harvested, and resuspended in 5 ml of 50% human serum/LB (S. aureus) or 50% human serum/C+Y (S. pneumoniae). Cultures were incubated 4 h before cells were harvested, washed twice in growth medium, and once in PBS, and lipids were extracted (35). Plasmids containing S. pneumoniae or S. aureus genes in strain JLB31 were grown in Luria broth at 37 °C to an A600 of 0.5. A FA mixture (10 μm each) of [d4]16:0, -18:1, and -18:2, along with Brij-58 (0.1%) was added to the culture, incubated at 37 °C for 30 min, and the lipids were extracted. Protocol was the same for polyunsaturated FA incorporation with the exception that a mixture (7.5 μm each) of 18:1, 18:2, 18:3, and 20:4 was added to the growth medium.
Lipid extracts were resuspended in chloroform:methanol (1:1). PG was analyzed using a Shimadzu Prominence UFLC attached to a QTrap 4500 equipped with a Turbo V ion source (Sciex). Samples were injected onto an Acquity UPLC BEH HILIC, 1.7 μm, 2.1 × 150-mm column (Waters) at 45 °C with a flow rate of 0.2 ml/min. Solvent A was acetonitrile, and solvent B was 15 mm ammonium formate, pH 3. The HPLC program was the following: starting solvent mixture of 96% A/4% B, 0 to 2 min isocratic with 4% B; 2 to 20 min linear gradient to 80% B; 20 to 23 min isocratic with 80% B; 23 to 25 min linear gradient to 4% B; 25 to 30 min isocratic with 4% B. The QTrap 4500 was operated in the Q1 negative mode. The ion source parameters for Q1 were: ion spray voltage, −4500 V; curtain gas, 25 psi; temperature, 350 °C; ion source gas 1, 40 psi; ion source gas 2, 60 psi; and declustering potential, −40 V. The system was controlled by the Analyst® software (Sciex). The sum of the areas under each peak in the mass spectra was calculated and the percent of each molecular species present was calculated with LipidView software (Sciex). Incorporation of [d4]16:0 was calculated by combining the values of [d4]16:0, [d4]18:0, and [d4]20:0 molecular species, the values of 18:1 and 20:1 were combined for 18:1 incorporation; the values of 18:2 and 20:2 were combined for 18:2 incorporation; the values of 18:3 and 20:3 were combined for 18:3 incorporation; and the incorporation of 20:4 was a stand-alone value.
The samples were introduced to the QTrap 4500 by direct injection to perform product scans to verify the fatty acids present in a particular molecular species. The ion source parameters for the negative mode product scan were: ion spray voltage, −4500 V; curtain gas, 10 psi; collision gas, medium; temperature, 270 °C; ion source gas 1, 10 psi; ion source gas 2, 15 psi; declustering potential, −40 V; and collision energy −50 V.
Fatty acid composition of human serum
Lipids were extracted from human serum (Millipore-Sigma) by the method of Bligh and Dyer (35), and fatty acid methyl esters were prepared using methanol/hydrochloric acid. The fatty acid methyl esters were analyzed by a Hewlett-Packard model 5890 gas chromatograph equipped with a flame ionization detector and separated on 30 m × 0.536 mm × 0.50-μm DB-225 capillary column. The injector was set at 250 °C, and the detector was at 300 °C. The temperature program was as followed: initial temperature of 70 °C for 2 min, rate of 20 °C/min for 5 min (final 170 °C), rate of 2 °C/min for 10 min (final 190 °C), hold at 190 °C for 5 min, rate of 2 °C/min for 15 min (final 220 °C), hold at 220 °C for 5 min. The identity of fatty acid methyl esters was determined by comparing their retention times with fatty acid methyl ester standards (Matreya). The composition was expressed as weight percentages.
FA kinase assay
FA kinase assays contained 0.1 m Tris-HCl (pH 7.5), 10 mm ATP, 20 mm MgCl2, 0.1% Brij-58, 20 μm [14C]16:0, [14C]18:1, or [14C]18:2, 0.2 μm FakA, and the indicated concentrations of purified FakB proteins in a total volume of 60 μl. Tubes were incubated at 37 °C for 5 min before acetic acid (0.6%) was added and 40 μl was pipetted onto a DE81 Whatman filter paper disc and discs were washed three times, 20 min each, in ethanol containing acetic acid (1%). Discs were dried and counted by scintillation counting. Apparent Km values were determined performing a nonlinear regression and using the Michaelis-Menten equation.
FA competition assays contained 10 μm FakB1, 1 μm FakB2, or 1 μm FakB3, 20 μm of the preferred radiolabeled substrate ([14C]16:0, [14C]18:1, or [14C]18:2) plus 80 μm of one unlabeled FA (16:0, 18:1, 18:2, 18:3, or 20:4). The reaction conditions were the same as above with the exception that the total volume of the reaction was 100 μl. Eighty microliters was spotted onto DE81 Whatman filter paper discs. Discs were washed three times with an ethanol/acetic acid (1%) mixture for 20 min each wash. Discs were dried and counted by scintillation counting. Experiments were each performed three times in duplicate.
Protein crystallization and structure determination
All crystals were grown using the hanging drop method from pre-formed SpFakB-FA complexes that were prepared as previously described (5). Specific crystallization conditions are as follows: SpFakB1(16:0) and SpFakB1(14:0), 0.1 m MES (pH 6.5) and 30% (w/v) PEG4000; SpFakB2(C18:1Δ9), 0.1 m sodium HEPES (pH 7.5), 10% (v/v) isopropyl alcohol, 20% (w/v) PEG 4000; SpFakB2(18:1Δ11), 18% (w/v) PEG 3350, 4.8% (v/v) 2-propanol, 0.1 m CAPSO, 17% (v/v) PEG400; SpFakB3(18:2), 0.1 m sodium acetate (pH 4.6), and 8% (w/v) PEG4000. For data collection, crystals were cryoprotected in mother liquor containing 25% glycerol prior to flash freezing in liquid nitrogen. Glycerol could not be used as cryoprotectant with SpFakB1(14:0) crystals because they immediately cracked and dissolved. Instead, crystals were briefly immersed in a 1:1 mixture of paraffin oil/Paratone N prior to freezing. X-ray diffraction data (180°) were collected at SER-CAT beamlines 22-ID and 22-BM at the APS (Argonne National Laboratory). Data were integrated with XDS (36, 37) and scaled and merged using AIMLESS/CCP4 (38). The SpFakB1 and SpFakB2 structures were determined by molecular replacement using the SaFakB1 and SaFakB2 structures as search models. For SpFakB3, the structure was eventually determined using a structurally related protein from Eubacterium eligens (PDB ID 3FDJ) as the search model. Molecular replacement procedures were performed using MOLREP (39), MODBASE (40), MrBUMP/CCP4 (41), and PHASER (42). The FA 3-dimensional coordinates were designed using AVOGADRO (28) and the .cif dictionaries of restraints were generated with PHENIX.ELBOW (43). Multiple cycles of manual and real space refinement were performed using COOT (44), REFMAC (39), and PHENIX.REFINE (45). The final models of SpFakB1, SpFakB2, and SpFakB3 contained 1, 2, and 2 monomers per asymmetric unit, respectively. 5% of the reflections was excluded from the refinement process for calculation of Rfree. Structural statistics are presented in Table 2.
Author contributions
J. M. G., S. W. W., and C. O. R. conceptualization; J. M. G., M. G. C., M. W. F., S. W. W., and C. O. R. formal analysis; J. M. G., M. G. C., and M. W. F. investigation; J. M. G., M. G. C., and M. W. F. methodology; J. M. G. and C. O. R. writing-original draft; J. M. G., M. G. C., M. W. F., S. W. W., and C. O. R. writing-review and editing; M. G. C. data curation; M. G. C. visualization; S. W. W. validation; C. O. R. supervision; C. O. R. funding acquisition; C. O. R. project administration.
Acknowledgments
We thank Pam Jackson for assistance with the molecular biology, Karen Miller for protein purification, and Hannah Rowe and Jason Rosch for advice and assistance in creating strain JMG1. Use of the Advanced Photon Source was supported by the U. S. Dept. of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
This work was supported by National Institutes of Health Grant GM034496, Cancer Center Support Grant CA21765, and the American Lebanese Syrian Associated Charities. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- FA
- fatty acid
- FA∼P
- acyl-phosphate
- ACP
- acyl carrier protein
- FASII
- bacterial type II fatty acid biosynthesis pathway
- a15:0
- anteiso pentadecanoic acid
- LPA
- 1-acyl-sn-glycerol-3-phosphate
- PlsX
- acyl-phosphate:ACP transacylase
- PlsY
- acyl-phosphate-dependent glycerol-phosphate acyltransferase
- PG
- phosphatidylglycerol
- PDB
- Protein Data Bank
- CAPSO
- 3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic acid.
References
- 1. Parsons J. B., Broussard T. C., Bose J. L., Rosch J. W., Jackson P., Subramanian C., and Rock C. O. (2014) Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 111, 10532–10537 10.1073/pnas.1408797111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yao J., and Rock C. O. (2015) How bacterial pathogens eat host lipids: Implications for the development of fatty acid synthesis therapeutics. J. Biol. Chem. 290, 5940–5946 10.1074/jbc.R114.636241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parsons J. B., Frank M. W., Subramanian C., Saenkham P., and Rock C. O. (2011) Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc. Natl. Acad. Sci. U.S.A. 108, 15378–15383 10.1073/pnas.1109208108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parsons J. B., Frank M. W., Jackson P., Subramanian C., and Rock C. O. (2014) Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol. Microbiol. 92, 234–245 10.1111/mmi.12556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cuypers M. G., Subramanian C., Gullett J. M., Frank M. W., White S. W., and Rock C. O. (2019) Acyl chain selectivity and physiological roles of Staphylococcus aureus fatty acid-binding proteins. J. Biol. Chem. 294, 38–49 10.1074/jbc.RA118.006160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu Y. J., and Rock C. O. (2006) Transcriptional regulation of fatty acid biosynthesis in Streptococcus pneumoniae. Mol. Microbiol. 59, 551–566 10.1111/j.1365-2958.2005.04951.x [DOI] [PubMed] [Google Scholar]
- 7. Eckhardt T. H., Skotnicka D., Kok J., and Kuipers O. P. (2013) Transcriptional regulation of fatty acid biosynthesis in Lactococcus lactis. J. Bacteriol. 195, 1081–1089 10.1128/JB.02043-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Faustoferri R. C., Hubbard C. J., Santiago B., Buckley A. A., Seifert T. B., and Quivey R. G. Jr. (2015) Regulation of fatty acid biosynthesis by the global regulator CcpA and the local regulator FabT in Streptococcus mutans. Mol. Oral. Microbiol. 30, 128–146 10.1111/omi.12076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu L., Zou Q., Cao X., and Cronan J. E. (2019) Enterococcus faecalis encodes an atypical auxiliary acyl carrier protein required for efficient regulation of fatty acid synthesis by exogenous fatty acids. MBio 10, e00577–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jerga A., and Rock C. O. (2009) Acyl-acyl carrier protein regulates transcription of fatty acid biosynthetic genes via the FabT repressor in Streptococcus pneumoniae. J. Biol. Chem. 284, 15364–15368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brinster S., Lamberet G., Staels B., Trieu-Cuot P., Gruss A., and Poyart C. (2009) Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 458, 83–86 10.1038/nature07772 [DOI] [PubMed] [Google Scholar]
- 12. Broussard T. C., Miller D. J., Jackson P., Nourse A., White S. W., and Rock C. O. (2016) Biochemical roles for conserved residues in the bacterial fatty acid-binding protein family. J. Biol. Chem. 291, 6292–6303 10.1074/jbc.M115.706820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tatituri R. V., Brenner M. B., Turk J., and Hsu F. F. (2012) Structural elucidation of diglycosyl diacylglycerol and monoglycosyl diacylglycerol from Streptococcus pneumoniae by multiple-stage linear ion-trap mass spectrometry with electrospray ionization. J. Mass Spectrom. 47, 115–123 10.1002/jms.2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Delekta P. C., Shook J. C., Lydic T. A., Mulks M. H., and Hammer N. D. (2018) Staphylococcus aureus utilizes host-derived lipoprotein particles as sources of exogenous fatty acids. J. Bacteriol. 200, e00728–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim G. L., Luong T. T., Park S. S., Lee S., Ha J. A., Nguyen C. T., Ahn J. H., Park K. T., Paik M. J., Suhkneung, Pyo, Briles D. E., and Rhee D. K. (2017) Inhibition of autolysis by lipase LipA in Streptococcus pneumoniae sepsis. Mol. Cells 40, 935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schulze-Gahmen U., Pelaschier J., Yokota H., Kim R., and Kim S. H. (2003) Crystal structure of a hypothetical protein, TM841 of Thermotoga maritima, reveals its function as a fatty acid-binding protein. Proteins 50, 526–530 10.1002/prot.10305 [DOI] [PubMed] [Google Scholar]
- 17. Nan J., Zhou Y., Yang C., Brostromer E., Kristensen O., and Su X. D. (2009) Structure of a fatty acid-binding protein from Bacillus subtilis determined by sulfur-SAD phasing using in-house chromium radiation. Acta Crystallogr. D Biol. Crystallogr. 65, 440–448 10.1107/S0907444909007756 [DOI] [PubMed] [Google Scholar]
- 18. Parsons J. B., and Rock C. O. (2013) Bacterial lipids: metabolism and membrane homeostasis. Prog. Lipid Res. 52, 249–276 10.1016/j.plipres.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yao J., and Rock C. O. (2013) Phosphatidic acid synthesis in bacteria. Biochim. Biophys. Acta 1831, 495–502 10.1016/j.bbalip.2012.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Storch J., and Corsico B. (2008) The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu. Rev. Nutr. 28, 73–95 10.1146/annurev.nutr.27.061406.093710 [DOI] [PubMed] [Google Scholar]
- 21. Storch J., and McDermott L. (2009) Structural and functional analysis of fatty acid-binding proteins. J. Lipid Res. 50, S126–131 10.1194/jlr.R800084-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He Y., Yang X., Wang H., Estephan R., Francis F., Kodukula S., Storch J., and Stark R. E. (2007) Solution-state molecular structure of apo and oleate-liganded liver fatty acid-binding protein. Biochemistry 46, 12543–12556 10.1021/bi701092r [DOI] [PubMed] [Google Scholar]
- 23. Hodsdon M. E., and Cistola D. P. (1997) Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by 15N NMR relaxation and 1H exchange. Biochemistry 36, 2278–2290 10.1021/bi962018l [DOI] [PubMed] [Google Scholar]
- 24. Hodsdon M. E., and Cistola D. P. (1997) Discrete backbone disorder in the nuclear magnetic resonance structure of apo intestinal fatty acid-binding protein: implications for the mechanism of ligand entry. Biochemistry 36, 1450–1460 10.1021/bi961890r [DOI] [PubMed] [Google Scholar]
- 25. Sacchettini J. C., Gordon J. I., and Banaszak L. J. (1989) Refined apoprotein structure of rat intestinal fatty acid binding protein produced in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 86, 7736–7740 10.1073/pnas.86.20.7736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Richieri G. V., Ogata R. T., Zimmerman A. W., Veerkamp J. H., and Kleinfeld A. M. (2000) Fatty acid binding proteins from different tissues show distinct patterns of fatty acid interactions. Biochemistry 39, 7197–7204 10.1021/bi000314z [DOI] [PubMed] [Google Scholar]
- 27. Tian W., Chen C., Lei X., Zhao J., and Liang J. (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 46, W363–W367 10.1093/nar/gky473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prinsen C. F., and Veerkamp J. H. (1996) Fatty acid binding and conformational stability of mutants of human muscle fatty acid-binding protein. Biochem. J. 314, 253–260 10.1042/bj3140253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Corsico B., Cistola D. P., Frieden C., and Storch J. (1998) The helical domain of intestinal fatty acid binding protein is critical for collisional transfer of fatty acids to phospholipid membranes. Proc. Natl. Acad. Sci. U.S.A. 95, 12174–12178 10.1073/pnas.95.21.12174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Falomir-Lockhart L. J., Laborde L., Kahn P. C., Storch J., and Córsico B. (2006) Protein-membrane interaction and fatty acid transfer from intestinal fatty acid-binding protein to membranes: support for a multistep process. J. Biol. Chem. 281, 13979–13989 10.1074/jbc.M511943200 [DOI] [PubMed] [Google Scholar]
- 31. Pang Y. Y., Schwartz J., Thoendel M., Ackermann L. W., Horswill A. R., and Nauseef W. M. (2010) agr-Dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J. Innate. Immun. 2, 546–559 10.1159/000319855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subramanian C., Frank M. W., Batte J. L., Whaley S. G., and Rock C. O. (2019) Oleate hydratase from Staphylococcus aureus protects against palmitoleic acid, the major antimicrobial fatty acid produced by mammalian skin. J. Biol. Chem. 294, 9285–9294 10.1074/jbc.RA119.008439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wurch T., Lestienne F., and Pauwels P. J. (1998) A modified overlap extension PCR method to create chimeric genes in the absence of restriction enzymes. Biotechnol. Tech. 12, 653–657 10.1023/A:1008848517221 [DOI] [Google Scholar]
- 34. Granok A. B., Parsonage D., Ross R. P., and Caparon M. G. (2000) The RofA binding site in Streptococcus pyogenes is utilized in multiple transcriptional pathways. J. Bacteriol. 182, 1529–1540 10.1128/JB.182.6.1529-1540.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 36. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 10.1107/S0108767310004551,10.1107/S0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 10.1107/S0907444913000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 40. Pieper U., Webb B. M., Dong G. Q., Schneidman-Duhovny D., Fan H., Kim S. J., Khuri N., Spill Y. G., Weinkam P., Hammel M., Tainer J. A., Nilges M., and Sali A. (2014) ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 42, D336–D346 10.1093/nar/gkt1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keegan R. M., and Winn M. D. (2007) Automated search-model discovery and preparation for structure solution by molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 63, 447–457 10.1107/S0907444907002661 [DOI] [PubMed] [Google Scholar]
- 42. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boles B. R., Thoendel M., Roth A. J., and Horswill A. R. (2010) Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS ONE 5, e10146 10.1371/journal.pone.0010146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. D'Elia M. A., Pereira M. P., Chung Y. S., Zhao W., Chau A., Kenney T. J., Sulavik M. C., Black T. A., and Brown E. D. (2006) Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J. Bacteriol. 188, 4183–4189 10.1128/JB.00197-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ericson M. E., Subramanian C., Frank M. W., and Rock C. O. (2017) Role of fatty acid kinase in cellular lipid homeostasis and SaeRS-dependent virulence factor expression in Staphylococcus aureus. mBio 8, e00988–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. LeBlanc D. J., Lee L. N., and Abu-Al-Jaibat A. (1992) Molecular, genetic, and functional-analysis of the basic replicon of pva380–1, a plasmid of oral streptococcal origin. Plasmid 28, 130–145 10.1016/0147-619X(92)90044-B [DOI] [PubMed] [Google Scholar]