

Abstract
α-Linked GalNAc (α-GalNAc) is most notably found at the nonreducing terminus of the blood type–determining A-antigen and as the initial point of attachment to the peptide backbone in mucin-type O-glycans. However, despite their ubiquity in saccharolytic microbe-rich environments such as the human gut, relatively few α-N-acetylgalactosaminidases are known. Here, to discover and characterize novel microbial enzymes that hydrolyze α-GalNAc, we screened small-insert libraries containing metagenomic DNA from the human gut microbiome. Using a simple fluorogenic glycoside substrate, we identified and characterized a glycoside hydrolase 109 (GH109) that is active on blood type A-antigen, along with a new subfamily of glycoside hydrolase 31 (GH31) that specifically cleaves the initial α-GalNAc from mucin-type O-glycans. This represents a new activity in this GH family and a potentially useful new enzyme class for analysis or modification of O-glycans on protein or cell surfaces.
Keywords: glycosidase, glycobiology, metagenomics, glycoprotein, blood, glycopeptide cleavage, glycoside hydrolase 31 (GH31), human gut microbiome, mucin, O-glycoprotein, metagenomic analysis, glycoside hydrolase 109 (GH109), O-glycanase, N-acetylgalactosamine (alpha-GalNAc)
Introduction
GalNAc is found relatively sparingly in nature but plays two particularly important roles. One of these is as the first sugar attached to proteins within mucin-type O-glycans, linked to a serine or threonine residue. Installation of this sugar is achieved by one of the family of polypeptide N-acetyl-galactosaminyltransferases (1) with specificity toward an, as yet unclear, amino acid sequence. Attachment of galactose, GlcNAc, or GalNAc to this initial GalNAc creates the other O-linked glycan core structures commonly found on secreted proteins (Fig. 1) (2). These core structures can then be terminated by attachment of sialic acid or fucose residues or further elaborated on with N-acetyllactosamine repeats before termination with various oligosaccharide structures. The other important role of α-GalNAc, at least from a human health perspective, is as the terminal sugar attached α-1,3 to the galactose of H-antigen ((fucose-α-1,2)-galactose-), thereby forming the A-antigen (GalNAc-α-1,3-(fucose-α-1,2-)galactose-), an important part of the human blood immune system (3). α-GalNAc residues also appear in a number of contexts in other life forms. For example, the outer coat of Mycobacterium tuberculosis contains both α-GalNAc residues and their deacetylated versions (4). Enzymes that cleave these α-GalNAc residues, α-N-acetyl-galactosaminidases (α-GalNAcases)7 (EC 3.2.1.49), are of interest for two particular applications. First, α-GalNAcases that can efficiently cleave the core α-GalNAc from peptides or proteins are of interest, because these could be valuable analytical tools for the analysis of O-glycan structures and their attachment points to peptides. Relatively few glycoside hydrolase (GH) families, as defined by the Carbohydrate-Active enZyme (CAZy) database (http://www.cazy.org/)8 (5), are known to have the requisite activity. Currently, the best-characterized enzymes that cleave this bond include the GH101 endo-α-GalNAcases, which primarily cleave the T-antigen (also known as core 1; Galβ1–3GalNAcα1-Ser/Thr) from the peptide (6); fungal and mammalian GH27 exo-α-GalNAcases (such as human α-NAGAL), which cleave the initial GalNAc of mucin-type O-glycans as well as the terminal GalNAc of blood type A-antigens (7, 8); and GH129s, which are found primarily in Bifidibacteria and are capable exo/endo-α-GalNAcases (9). An additional class of enzymes capable of cleaving α-GalNAc are those of the less well-characterized GH135 enzymes, which appear to cleave α-GalNAc from fungal galactosaminogalactans, but not from proteins (10). Access to additional enzymes that can cleave α-GalNAc from peptides or proteins would increase the scope and utility of this enzyme class and may open up the possibility of engineering such enzymes into glycosynthases or glycoligases that are capable of attaching α-GalNAc to peptides or proteins (11–13). This would provide an alternative to the use of polypeptide N-acetylgalactosaminyltransferase in the generation of O-linked glycoproteins.
Figure 1.

O-Linked glycans and blood antigens. a, scheme of mucin-type O-linked glycosylation. O-Linked glycans are shown bound to either serine or threonine on the surface of a protein. Structures shown on the right are a selection of known mucin-type O-glycans. b, structures of ABO blood antigens present on red blood cells. All antigens are shown as bound to the surface of red blood cells though a Type 1 linkage. Sugars are shown using the Consortium for Functional Glycomics notation (76).
As well, access to α-GalNAcases that can efficiently cleave α-GalNAc from the A-antigen on red blood cells at pH 7.4 could allow the practical and economic conversion of A-type red blood cells to O-type (14). Whereas several glycosidases that remove A-antigen from RBCs had been discovered (15–18), most worked under nonideal conditions of pH and buffer for this application until the study of Liu et al. (19). This study led to the discovery of GH family 109, a new family of α-GalNAcases (5). GH109 α-GalNAcases use the same NAD-dependent mechanism reported previously for GH4 family members (19, 20). More recently, Rahfeld et al. (21) used functional metagenomic screening to identify a pair of enzymes, a deacetylase and a GH36 α-galactosaminidase, which together were capable of even more efficient conversion of A-type to O-type blood. These previous studies show the potential for identification of highly efficient enzymes (and enzymatic pathways) for A-type blood antigen cleavage from microbial genomes.
The mucous membranes of the human gut harbor large glycoproteins known as mucins. These glycoproteins present a wide variety of O-glycan structures on their surface, including A, B, and H blood antigens (22) as well as shorter mucin-type O-glycan chains (23). Gut microbes are known to digest these glycans as a carbon source within the competitive environment of the gastro-intestinal tract (2). Due to the importance of host glycans as a carbon source and the relatively few characterized microbial α-GalNAcases, it seems likely that gut microbes produce more enzymes capable of glycan degradation than are currently known. Therefore, screening of the gut microbiome for α-GalNAcase activity should expand the pool of available enzymes. The identification of these activities using classic cultivation-based screening of the human gut microbiome would neglect the majority of the bacteria, because many microbial organisms therein are thought to be uncultivable under normal laboratory conditions (24, 25). Over the past decade, metagenomic techniques have been developed as a method for the investigation of genetic material in an environmental sample without the need to culture individual organisms (26). Using a functional metagenomic screening approach, new glycoside hydrolases have already been discovered in microbiomes from decaying wood, soil, and bioremediation systems (27–29) as well as human and other gut microbiomes (21, 30–32).
In this paper, we describe the generation of two small-insert (<10 kb) metagenomic libraries derived from the gut microbiomes (fecal material) of human volunteers and their screening using fluorogenic α-N-acetyl-galactosaminide substrates. This approach has allowed us to identify and characterize a GH109 family member that cleaves A-type blood antigens, as well as, most interestingly, two α-GalNAcases from GH31, a family not previously known to harbor this activity. These GH31 α-GalNAcases efficiently cleave α-GalNAc from glycopeptides and may play a role in the intracellular harvesting of host-derived glycans by gut microorganisms.
Results
Functional metagenomic screening
Two small-insert metagenomic libraries were constructed using extracted genomic DNA from the feces of two healthy human participants with blood type O and A. The resulting libraries consisted of 10,750 and 23,000 clones for O- and A-type, respectively, arrayed in 384-well plates. In the first screening round, an activated aryl monosaccharide glycoside substrate that releases the highly fluorescent 4-methylumbelliferone (MU) reporter upon cleavage was used to screen for α-GalNAcase activity (Fig. 2). Screening the metagenomic libraries against the GalNAc-α-MU substrate identified six hits (three from each library) that produced substantial (>10 S.D.) fluorescent signal following extended incubation (Fig. 3). Of those six clones, one did not retain activity following secondary validation assays, and after sequencing, two turned out to be duplicates. Plasmids from the three unique hits, a single hit from the type O gut metagenomic library (O1) and two hits from the type A gut metagenomic library (A1 and A2) were isolated for validation, sequencing, and further characterization.
Figure 2.

Overview of the substrates used in this study for screening or for kinetic analysis. The fluorophore used was either MU, pNP, or BODIPY. The sequence of the peptide BODIPY-IL29a is represented in single-letter format. Sugars are shown using the Consortium for Functional Glycomics notation (76).
Figure 3.

Z-scores of the screens of the Type O and A libraries using 100 μm GalNAc-α-MU substrate. The dotted line shows the chosen cut-off at Z-score > 10. Z-score = (fluorescence − mean value)/S.D.
For each plasmid sequence, only a single ORF containing the proposed glycoside hydrolase domains was identified in silico. The hits O1 and A2 both matched to putative GH31 α-xylosidases, from Bacteroides plebeius (GenBankTM accession no. WP_007559952.1) and Bacteroides caccae (WP_005682123.1), respectively (33) (Table S1). Based on the GH classification and annotated species, O1 will be hereafter named BpGH31 and A2 BcGH31. The genes present in our small-insert library were in fact truncated relative to the full-length genes seen in the genome. BpGH31 and BcGH31 were missing 337 and 329 aa, respectively, at their C termini. These truncated versions are hereafter referred to as tBpGH31 and tBcGH31 and are shown in Fig. 4. Both GH31s included signal peptides, identified by SignalP (34), with a length of 26 aa for BcGH31 and 33 aa for BpGH31. The other hit, A1, matched a putative GH109 α-N-acetylgalactosaminidase from Bacteroides vulgatus (WP_005848863.1), called BvGH109_1. This too possessed a 27-amino acid signal peptide and was missing 56 aa at its C terminus. Full-length genes for all three were thus generated by cloning in the missing C-terminal amino acids based on the top BLAST hit for each and then expressed in Escherichia coli and purified. BvGH109_1 has recently also been identified in a separate functional screen of a blood type AB human gut metagenomic library carried out by our group (21). That publication describes the characterization of BvGH109_1 activity on blood group antigen substrates.
Figure 4.

Protein domain structure of GH31 enzymes in this study. The protein domain structures are presented based on the domains identified based on InterPro prediction (77) and the NCBI conserved domain search (78) and are visualized using DOG2.0 (79). Signal peptides are shown in yellow, whereas the identity and length of all other domains are indicated by color and text within the figure. The truncations of the proteins employed in the present study are displayed below each structure. DUF, domain of unknown function; FN3, fibronectin type 3; F5/8, F5/F8 Type C domain.
BvGH109_1 characterization
The specificity of BvGH109_1 was tested using a range of substrates previously found to be cleaved by enzymes within the GH31 and GH109 families (Table 1). Only α-GalNAc–containing substrates were cleaved, consistent with expectations of the GH109 family. Kinetic parameters for the cleavage of GalNAc-α-pNP are shown in Table 2 (Table S2 shows GalNAc-α-MU data).
Table 1.
Substrate specificity of the GH31s and GH109 examined in this study
0.1 mm substrates were incubated at 37 °C with 0.5 mg/ml of the enzymes in 50 mm NaH2PO4, pH 7.5, for at least 30 min. NT, not tested.
| Substrate | tBpGH31/BpGH31 | tBcGH31/BcGH31 | tCpGH31 | BvGH109_1 |
|---|---|---|---|---|
| GalNAc-α-MU | Yes | Yes | Yes | Yes |
| GalNAc-α-pNP | Yes | Yes | NT | Yes |
| Gal-α-MU | No | No | No | No |
| Glc-α-MU | No | No | No | No |
| Man-α-MU | No | No | NT | No |
| Xyl-α-pNP | No | No | NT | No |
| A-antigen Type2tetra-MU | No | No | NT | Yes |
Table 2.
Kinetic parameters for the hydrolysis of GalNAc-α-pNP by truncated and full-length BcGH31 and BpGH31, truncated BpGH31 mutants, and BvGH109_1
Values indicate means ± S.E. (n = 3). Reactions were carried out in 50 mm HEPES, pH 7.0, at 37 °C and observed via absorbance at 405 nm. Kinetic parameters were obtained by fitting initial velocities obtained from three replicate assays to the Michaelis–Menten equation using GraFit. Truncated BpGH31 mutants D413A/G/S were inactive.
| Enzyme | kcat | Km | kcat/Km |
|---|---|---|---|
| s−1 | μm | s−1 mm−1 | |
| BcGH31 | 3.6 ± 0.2 | 110 ± 10 | 32 ± 4 |
| tBcGH31 | 2.1 ± 0.1 | 140 ± 20 | 16 ± 2 |
| BpGH31 | 7.2 ± 0.3 | 270 ± 20 | 27 ± 2 |
| tBpGH31 | 2.22 ± 0.09 | 140 ± 20 | 16 ± 2 |
| tBpGH31 D465A | 5.0 ± 0.3 × 10−3 | 42 ± 9 | 0.12 ± 0.02 |
| tBpGH31 D465G | 5.7 ± 0.3 × 10−3 | 340 ± 40 | 0.017 ± 0.002 |
| tBpGH31 D465S | 4.3 ± 0.1 × 10−3 | 46 ± 5 | 0.09 ± 0.01 |
| BvGH109_1 | 0.25 ± 0.01 | 11.5 ± 0.3 | 22.3 ± 0.6 |
GH31 characterization
The activities of BcGH31, tBcGH31, BpGH31, and tBpGH31 were tested against a range of substrates, and only α-GalNAc glycosides were cleaved (Table 1). This was interesting because no such α-GalNAcase activity has been observed previously with any GH31 enzymes. Michaelis–Menten kinetic parameters were thus determined for cleavage of the p-nitrophenyl α-GalNAc glycosides by both the truncated and the full-length versions of each protein, and similar values of Km, kcat, and kcat/Km (Table 2 and Table S2) were observed in each case. Likewise, very similar pH profiles for hydrolysis of GalNAc-α-pNP with an optimum at pH 6–6.5 were seen for both versions of the two enzymes, consistent with their similar function and origin (Fig. S1).
GH31 mechanism and catalytic residues
The initial classification as a member of the GH31 family was based on sequence similarity toward the closest BLAST hit, an α-xylosidase. Because the substrate specificity was distinct from known activities within the GH31 family, a more detailed mechanistic investigation, including stereochemical outcome and catalytic residue identification, was performed to ensure that these enzymes were correctly classified within the GH31 family. The stereochemical outcome of cleavage of GalNAc-α-MU by tBpGH31 and tBcGH31 was monitored by 1H NMR, and the first-formed product was shown to be α-GalNAc (Fig. 5). Thus, hydrolysis indeed occurred with net retention of stereochemistry as seen for other GH31 enzymes (35) and is consistent with a double-displacement mechanism involving a covalent glycosyl-enzyme intermediate.
Figure 5.

Determination of the stereochemical outcome of enzymatic hydrolysis at the anomeric center by 1H NMR. The hydrolyses of GalNAc-α-DNP (5 mm) by tBpGH31 and tBcGH31 at room temperature were monitored by 1H NMR. A 1H NMR spectrum of the reaction was obtained after 5 min, and the anomeric stereochemistry of the hydrolysis product was determined. Stereochemistry was determined by observation of the product anomeric hydrogen peak appearing at either 5.2 ppm (α-GalNAc) or 4.6 ppm (β-GalNAc). Only α-GalNAc product was observed following rapid hydrolysis of the starting material, indicating net retention of stereochemistry.
In most GH31 enzymes, the catalytic nucleophile and the general acid/base are highly conserved aspartic acids (36, 37). Alignment of BpGH31 and BcGH31 with other GH31 sequences identified the potential catalytic aspartic acids for BpGH31 as Asp-413 and Asp-465 and that of BcGH31 to be Asp-427 and Asp-479 (Fig. 6). To confirm these assignments for tBpGH31, the alanine, glycine, and serine mutants at each of these positions were constructed and purified. Far-UV CD of the resulting nucleophile D413A/G/S and general acid/base D465A/G/S mutants (Fig. S2) revealed that the mutants adopted the same/similar secondary structure as WT tBpGH31 and thus were likely folded correctly. Assay of these mutants using GalNAc-α-pNP as substrate revealed rate reductions consistent with their predicted roles (38) (Table 1). Whereas no activity could be detected for the nucleophile mutants, kcat values for the acid/base mutants are ∼450-fold lower than those of the WT enzyme. Km was also reduced by ∼3-fold for the D465A and D465S mutants, whereas an increased Km was seen for the D465G mutant.
Figure 6.
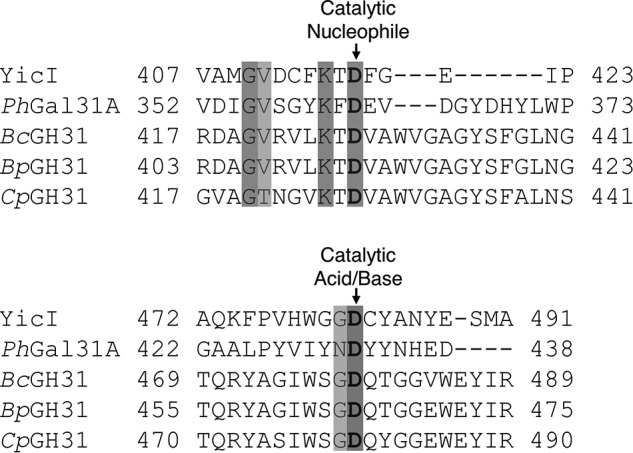
Alignment of the amino acid sequences around the catalytic nucleophile and general acid/base. GH31s with α-GalNAcase activity examined in this work were aligned against the well-characterized GH31 α-xylosidase YicI from E. coli (GenBankTM accession no. AAC76680.1) (37), as well as a representative GH31 α-galactosidase PhGal31A from P. heparinus (ACU04898.1) (45) using Clustal Omega (80).
GH31 specificity for glycopeptide substrates
None of the GH31 enzymes tested were able to cleave A-antigen–based substrates (Table 1). It therefore seemed possible that the natural function of these enzymes was the cleavage of the GalNAc residues present in O-glycans on glycopeptide or glycoprotein substrates. To test this, fetuin, a glycoprotein, was chosen as substrate because it has a well-defined glycosylation pattern, primarily featuring short mucin-type O-glycans (39). None of the enzymes showed any cleavage activity on fetuin, probably due to inaccessibility of the GalNAc residue. To expose the GalNAc residues present within the sialyl-T-antigen glycans, the fetuin substrate was treated with a sialidase (NedA) and a 1,3-β-galactosidase (BgaC) prior to GH31 enzyme exposure. Both BpGH31 and BcGH31, as well as their truncated versions, were able to cleave the exposed α-GalNAc, as shown by Dionex HPLC analysis of reaction mixtures (Fig. 7 and Fig. S3). GalNAc was not released when the fetuin was not treated with the GH31s in tandem with NedA and BgaC (data not shown), indicating that these GH31 α-GalNAcases do not release oligosaccharides via an endo-mechanism but rather are exo-α-GalNAcases. Moreover, the enzymes were active on both denatured and native fetuin, provided that the α-GalNAc was accessible (Fig. 7 and Figs. S3 and S4). Reactions with truncated GH31s proceed more slowly than with the full-length GH31s, both on denatured and native fetuin (Fig. S5). However, after overnight incubation, equal amounts of α-GalNAc were eventually released.
Figure 7.
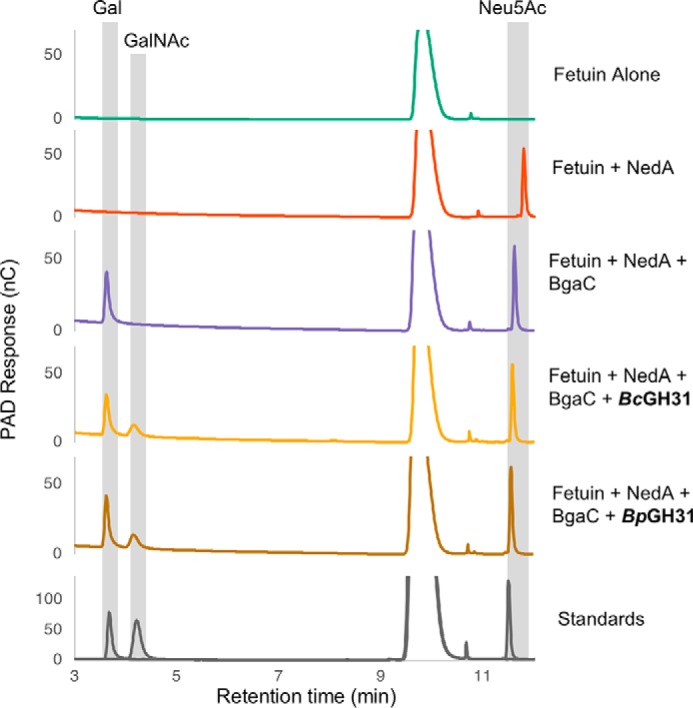
α-GalNAcase GH31s show exo-cleavage of O-glycans from fetuin. Native fetuin was subjected to overnight enzymatic cleavage of glycans by sequential activity of a neuraminidase (NedA), β-1,3-galactosidase (BgaC), and α-GalNAcases (BcGH31 or BpGH31). Cleavage products were then separated and analyzed via HPAEC-PAD. All GH31 enzymes tested were able to cleave GalNAc from the fetuin. Sugars were identified based on buffer (20 mm HEPES, pH 7.0, 150 mm NaCl) spiked with free Neu5Ac, Gal, and GalNAc. Some variations in the elution time accrued during the HPAEC-PAD runs, so the released sugars have been assigned with allowance for these variations.
The activity of the GH31 enzymes toward a glycopeptide substrate was verified using a fluorescently labeled peptide modified with α-GalNAc (BODIPY-IL29 seq: QPQPT-(α-GalNAc)AGPV) (Fig. 2). Both the full-length enzymes and their truncations readily removed α-GalNAc from the O-GalNAcylated peptide substrate, as evidenced by the formation of free GalNAc following incubation (Fig. 8). These results further indicate that the enzymes are true protein/peptide exo-α-GalNAcases.
Figure 8.

α-GalNAcase GH31s cleave α-GalNAc from the peptide backbone. a, a peptide containing a GalNAc-α-threonine residue (BODIPY-IL29) was subjected to overnight enzymatic cleavage by the GH31 exo-α-GalNAcases from B. caccae and B. plebeius identified in this study. Cleavage products were then separated and analyzed via HPAEC-PAD. b, the cleavage of GalNAcylated BODIPY-IL29 was additionally followed via TLC at 5 and 45 min.
Phylogeny
A phylogeny was generated based on all known GH31 sequences present in the CAZy database (5), as of June 2018, to gain a better understanding of the GH31 family and the different activities present within it. Both BpGH31 and BcGH31 are located in a clade containing no previously characterized members (Fig. 9). However, a GH31 from Clostridium perfringens (CpGH31) lay in close proximity to α-GalNAcase GH31 enzymes identified in our screen. Grondin et al. (40) had previously determined that the three carbohydrate-binding module 32 (CBM32) domains in CpGH31 bind to GalNAc, consistent with a possible α-GalNAcase activity. However, they did not test for α-GalNAcase activity within the GH31 catalytic domain. To see if this was the case and whether the principal activity within the clade is that of an α-GalNAcase, the GH domain (aa 33–933) of CpGH31 was cloned from the genomic DNA of C. perfringens DSM 798 (tCpGH31) (Fig. 2), and the protein was expressed and purified. Substrate specificity testing with tCpGH31 showed specific cleavage of GalNAc-α-MU (Table 1). Likewise, GalNAc was cleaved from treated fetuin in the same manner as seen for BpGH31 and BcGH31 (Fig. S4).
Figure 9.

Phylogeny of GH31 family. All available GH31 members from the CAZy database are presented in a phylogeny. Red dots, locations of sequences of characterized GH31 enzymes. Black dot, location of the BcGH31, BpGH31, and CpGH31 within the phylogeny. Colored areas, major enzymatic activities within the different clades of the GH31 family. Lighter-colored turquoise, putative α-GalNAcases that lack the consensus motif present in the GH31s characterized in this study.
Discussion
Our use of a small-insert library construction approach in conjunction with the use of highly sensitive fluorescent glycoside substrates for screening led to the identification of two GH31 enzymes with a novel substrate specificity and one GH109. These enzymes were found to have distinct specificities toward the two known α-GalNAc moieties seen in the gut: mucin-type O-glycans and blood type A-antigens. Whereas GH109s are well-known α-GalNAcases, the GH31 family was not previously known to contain α-GalNAcase activity. Our identified α-GalNAcase GH31s act solely upon the GalNAc present at the linkage of mucin-type O-glycans with no activity toward blood type A-antigens. Characterization of these GH31s suggests that they are indeed bona fide GH31s and that the α-GalNAcase activity is conserved within related GH31s.
The use of small-insert libraries, rather than the larger fosmid libraries used in our previous studies (21, 29, 30, 32), has both advantages and disadvantages. Use of the fosmid approach, in which large fragments of metagenomic DNA (typically 35–45 kb) are ligated into fosmid vectors, packaged in phage, and transduced into E. coli has the advantage of introducing more genes per host bacterium. Whereas this allows screening of larger numbers of genes in a single clone and allows possible expression of more complex metabolic pathways, the probability of expression per gene is lower than for small-insert libraries with active and inducible promoters in closer proximity to environmental DNA. Genomic analysis of various bacteria suggests that ∼40% of environmental genes can be heterologously expressed in E. coli without external promoters, but this can vary widely, depending upon the microbe from which the DNA is isolated (41). Fosmid libraries also come with higher production costs, not only through the DNA extraction process and phage-based delivery method, but also through the larger amounts of sequencing necessary to assemble active clones (42). The use of small-insert libraries largely removes the possibility of cloning of pathways and reduces the number of genes that are screened in a single clone. However, the probability of expression per gene is higher, costs are considerably lower, and the process is operationally simpler.
By screening with a simple, sensitive and reasonably chemically activated (MU pKa ∼8) methylumbelliferyl GalNAc derivative, we were able to search for α-GalNAcases of all types, including enzymes with activity toward complex oligosaccharide and glycopeptide substrates. The substrate could be synthesized on a scale that allows screening of a reasonably large library (∼20,000 clones), something that would not be feasible with more complex substrates using our current plate-based screen.
Our discovery of a GH109 enzyme in this screen is consistent with previously observed substrate specificities (19, 43). Four members of this family have been investigated to date, but only one, that from Elizabethkingia meningosepticum (EmGH109), has been characterized in depth. Indeed, only EmGH109 showed a useful ability to cleave the A-antigen from red blood cells, thereby allowing the production of universal donor blood (21). GH109 from Bacteroides vulgatus also has exo-α-GalNAcase activity, cleaving GalNAc-α-pNP with a kcat/Km value around 6 times lower than that of EmGH109 (11).
The surprising finding was that the other two hits from the screen were identified as GH31 members, because this family was previously only known to harbor α-glucosidase, α-galactosidase, α-mannosidase, α-xylosidase, α-sulfoquinovosidase, and α-glucan lyase activities. Substrates for all of these enzymes have a 2-hydroxyl, whereas GalNAc has a bulky N-acetamide (36, 37, 44, 45). As noted in the Introduction, α-GalNAcase activity has so far only been reported in the GH27, GH36, GH101, GH109, GH129, and GH135 families. Members of GH31 share similar structural characteristics with members of GH27 and GH36 and indeed have been assigned to the same Clan D of glycoside hydrolases on this basis; thus, crossover of substrate specificity is conceivable (46).
More insights are obtained by looking in greater detail at the phylogeny of the GH31 members. As shown in Fig. 9, BpGH31 and BcGH31 are both located in a cluster of the GH31 family that previously contained no characterized members. Both of these Bacteroides sp. GH31s share conserved aspartic acid residues that have been assigned in other family members as the catalytic nucleophile and general acid/base (Fig. 6). Kinetic analysis of mutants of the key catalytic residues showed a complete loss of detectable activity of the nucleophile mutants and large changes in the kinetic parameters of the acid/base mutants. The decreased kcat seen for all acid/base mutants and decreased Km of the tBpGH31 D465A/S mutants is consistent with slowed turnover of the covalent glycosyl-enzyme intermediate due to a lack of general base catalysis (47). The increased Km of the D465G mutant is perhaps due to fine changes in the active site resulting in a loss of affinity for the substrate. The kinetic behavior of these mutants along with the “retaining” stereochemical outcome (Fig. 5) and similar pH profiles all suggest that, despite their different substrate specificity, the enzymes from this clade use the same double-displacement catalytic mechanism as other characterized GH31 members (37, 44, 45, 48, 49).
The characterized members of GH31 that are closest in sequence to BcGH31 and BpGH31 are PsGH31A and PhGH31 from Pseudopedobacter saltans and heparinus (45). These GH31s were identified as α-galactosidases, with no activity on α-N-acetylgalactosaminides. Therefore, enzymes within these two clades contain elements of the same specificity determinants that allow binding of galacto- substrates with an axial 4-hydroxyl.
To determine whether the α-GalNAcase activity is conserved, we investigated the activity of another member of this clade. Grondin et al. (40) had carried out investigations on the CBM32 domains of what was noted as a potential α-glucosidase CpGH31. During our study, we noted that their enzyme is located in the same cluster as BpGH31 and BcGH31 and that their investigations revealed GalNAc as the best CBM ligand. However, the hydrolytic domain of CpGH31 had never been characterized. To determine whether CpGH31 is an α-GalNAcase, and that this activity is conserved within the clade, we generated a truncated version of CpGH31, containing the GH31 domain (Fig. 4). This protein, tCpGH31, was then assayed and indeed found to be a specific α-GalNAcase (Table 1 and Fig. S4). Thus, BpGH31, BcGH31, and CpGH31 constitute a new α-GalNAcase cluster within the GH31 family. Notably, this cluster does not contain the KiDEvD catalytic nucleophile sequence motif that was noted by Miyazaki et al. (45) as being present in the Gal31 subfamily (Figs. 6 and 8). The absence of this motif is thus consistent with the assignment of these newly discovered enzymes to a novel GH31 subfamily. Sequence alignment of the putative GalNAc31 subfamily suggests a potential motif of kTDVAWVGaGYSFgL. This motif contains residues that would likely interact with the 2-acetamide, based on comparison with the YicI crystal structure (37). It should be noted that an extended branch within the putative GalNAc31 subfamily (shown in light turquoise in Fig. 9) lacks this motif despite having likely descended from an α-GalNAcase ancestor.
Both tBpGH31 and tBcGH31 contained the putative GH domain plus an unknown N-terminal domain (DUF 5110) and a C-terminal fibronectin type III (FN3) domain. However, these genes were missing C-terminal regions of the putative full genes in the screened library plasmids (Fig. 4). These missing portions include an F5/F8 Type C domain, as well as a dockerin and cohesin domains. As noted by Briliūtė et al. (50), F5/F8 Type C domains are similar in structure to family 32 CBMs and could play a role in glycan binding. Dockerin and cohesin domains are generally involved in the assembly of multiprotein complexes on the cell surface in the degradation of carbohydrates (51). These missing domains were not necessary for activity, as evidenced by the only minor differences seen in kinetic parameters (Table 2) and pH optima (Fig. S1) for cleavage of GalNAc-α-pNP by full-length and truncated forms. Both truncated and full-length forms are active on fetuin, provided that the initial α-GalNAc is accessible (Fig. 7 and Figs. S3 and S4), but the truncated GH31s proceed more slowly than the full-length (Fig. S5). Given their structural similarity to CBM32s, it seems possible then that these F5/F8 Type C domains are responsible for interactions with the target glycan and surrounding environment in the targeting of the enzyme to the glycoprotein, as shown previously for other CBMs (2). Further studies could show potential roles of the dockerin and cohesin domains in the interaction with other enzymes for polysaccharide degradation as has been observed in cellulosome assembly (51).
The context in which the enzymes BpGH31 and BcGH31 naturally function is unknown at this stage. Because the epithelial cells that line the entire intestinal tract (52) are coated in heavily O-glycosylated (O-GalNAc) mucins, the gut is a very O-GalNAc–rich environment for microorganisms and a likely target. Based on the inactivity of our GH31 enzymes on A-type blood antigen substrates, yet efficient release of the core α-GalNAc from O-glycoproteins such as fetuin after enzymatic removal of other sugar residues, it is clear that it is the core GalNAc that is cleaved and not the terminal one. This was confirmed through the activity of the GH31s tested toward a synthetic glycopeptide containing a GalNAc-α-1-Thr residue (Fig. 8).
The presence of signal peptides on both BpGH31 and BcGH31 suggests that they are secreted into the gut lumen as part of a set of O-glycan–degrading enzymes dedicated to nutritional scavenging. Analysis of the genes surrounding BcGH31 within the sequenced genome of B. caccae ATCC 43185 supports this hypothesis. The close proximity of SusD and SusC/RagA proteins indicates the presence of a potential polysaccharide utilization locus (PUL) (Fig. 10). PULs are sets of physically linked genes organized around a susCD gene pair that are prevalent in the Bacteroidetes phylum and are responsible for the degradation of large carbohydrate chains (53, 54).
Figure 10.

BcGH31 PUL. Overview of the genes located within the putative PUL containing BcGH31. Genes are annotated based upon domains that could potentially be involved in polysaccharide utilization. Gene loci are noted for the beginning and end of the putative PUL, BcGH31 in B. caccae ATCC 43185, and for the SusC/D pair that is central to PUL organization.
PULs often encode all of the genes necessary for carbohydrate degradation and uptake by bacteria. Generally, sugars are released from the polysaccharide substrate via the action of CAZymes present within the PUL, with subsequent binding and uptake being carried out by the SusC/D proteins and further carbohydrate degradation occurring after uptake (54). Upstream of the BcGH31 gene, there appears to be GH2 and GH42 β-galactosidases and one GH29 α-fucosidase (Fig. 10). These enzymes could conceivably act in concert to remove other residues present in the O-GalNAc chains to expose α-GalNAc at the glycoprotein backbone for cleavage by BcGH31.
Other genes of interest in the BcGH31 PUL include an M60 peptidase and a GH88. Enzymes from the M60 peptidase family seem to specialize in the degradation of glycoproteins (55). Notably, the most closely related characterized M60 peptidase (28.1% sequence identity) to that within our putative PUL in BcGH31 is a glycoprotein-specific peptidase (BT4244) which requires the TN-antigen to be present for activity (55). It should be noted that on certain substrates at least, the GH31s described in this study are capable of acting on intact glycoproteins and do not require the activity of a glycopeptidase (Fig. 7 and Fig. S4). The putative GH88 neighboring BcGH31 may play a role in degrading glycosaminoglycans present on the intestinal mucosal lining by cleaving at the unsaturated glucuronic acid units formed by polysaccharide lyases (56, 57). Analysis of this gene set in the CAZy Polysaccharide Utilization Loci Database (PULDB) reveals (54) similarity to other predicted PULS from Bacteroides sp. 2_2_4, Bacteroides sp. D20, and Bacteroides uniformis ATCC 8492 containing a combination of GH31, GH29, and GH2 enzymes.
The region surrounding BpGH31 in the genome of B. plebeius DSM 17135 was less informative and does not seem to form a PUL. However, an M23 peptidase is located upstream of the BpGH31 gene. The Gly-Gly endopeptidase (58) encoded may well cleave the peptide into smaller fragments after removal of the O-GalNAc chain.
The putative PULs in which the described α-GalNAcase GH31s are located had no similarity to known mucin-degrading PULs, so more work will be required to conclusively determine the specific roles of BpGH31 and BcGH31 in a coordinated glycan-scavenging strategy. Despite the well-known adaptations of certain Bacteroides spp. for degradation of mucosal glycans (59), they lack the GH101 or GH129 family enzymes that are used by other gut bacteria in host glycan degradation (60). Thus, the GH31s discovered in this study, with homologues found in PULs within other Bacteroides, highlight a potential means for more complete degradation of mucosal glycans by Bacteroides spp.
Conclusion
In conclusion, two small-insert (<10-kb) metagenomic libraries derived from the gut microbiomes (fecal material) of human volunteers with blood type O and A were successfully screened for α-GalNAcase activity. Three bacterial α-GalNAcases were identified in the screen, including two novel GH31 enzymes and a GH109. BpGH31 and BcGH31 were isolated, characterized, and shown to be the first GH31 enzymes with α-GalNAcase activity (EC 3.2.1.49), thus forming the new GH31 α-GalNAcase clade. These enzymes were shown to specifically cleave the core α-GalNAc from mucin-type O-glycans. The discovery of this enzymatic activity highlights a new means for host glycan utilization by certain gut microorganisms. However, further experiments will be required to determine the specific role of these enzymes in the concerted degradation of host glycans by gut microbiota.
Experimental procedures
Materials and reagents
Substrates used in this study were purchased from Sigma-Aldrich, if not otherwise indicated. TLC was performed using TLC Silica Gel 60 F254 TLC plates (EMD Millipore Corp., Billerica, MA).
Sampling and metagenomic DNA extraction
The collection of human fecal samples was approved by the Clinical Research Ethics Board of the University of British Columbia (ID: H15-02967). Human fecal samples were collected in a small sterile plastic receptacle, from which ∼3-g aliquots were transferred to individual 50-ml Falcon tubes (×6) and either stored at −70 °C or immediately subjected to DNA extraction. DNA extraction was performed using a modified chemical lysis procedure. For each 3-g sample, 1.5 ml of denaturing buffer (10 mm Tris/HCl, pH 7.0, 4 m guanidinium isothiocyanate, 1 mm EDTA, 1% β-mercaptoethanol) and 9 ml of extraction buffer (100 mm sodium phosphate at pH 7.0, 100 mm Tris/HCl at pH 7.0, 100 mm EDTA at pH 8.0, 1.5 m NaCl, 1% CTAB, 2% SDS) was added to the sample. Two small glass beads (3 mm) were added to each sample tube to aid in homogenization, and then each sample was mixed vigorously by vortexing. Samples were incubated at 60 °C for 40 min with light rotation. Following incubation, each sample was spun down (1800 rpm, 10 min), and the supernatant was transferred to a new 50-ml Falcon tube. A second extraction step was then performed by adding 5 ml of extraction buffer to the sample, homogenizing, incubating (60 °C, 30 min), spinning the sample down, and transferring the additional supernatant to the new tube. An equivalent volume of ice-cold chloroform was added to the supernatant, and the tube was shaken lightly for 10 min. The sample was then centrifuged (1800 rpm, 10 min) and the clear, aqueous layer was transferred to 2-ml ultracentrifuge tubes in aliquots of 1 ml. Isopropyl alcohol (0.6 eq to sample volume) and ammonium acetate ( total volume of 3 m stock) were added to ultracentrifuge tubes to a final concentration of 0.3 m (pH 5.2). Tubes were mixed and then centrifuged (15,000 rpm, 20 min, 4 °C) and decanted (carefully so as not to disturb the DNA pellet). Each sample was washed with 1 ml of fresh 70% ethanol and then spun down and decanted. Each pellet was allowed to air-dry for 10 min prior to the addition of 150 μl of resuspension buffer (TE buffer, pH 8.0). DNA pellets were resuspended overnight at 4 °C, and then DNA concentration and yields were determined by absorbance at 260 nm. Approximately 200 μg of DNA was recovered from each 3-g sample.
Library construction
Purified metagenomic DNA (1 μg) was partially digested with the restriction enzyme Sau3AI (0.5 units) at 37 °C for 30 min. The resulting DNA was separated on a 1% agarose gel by electrophoresis (100 V, 40 min). DNA of the size 3–10 kb was excised from the gel and purified using the Qiagen DNA Gel Extraction Kit. This insert DNA was then ligated (T4 DNA ligase) with the vector pHSG396, which had been digested previously with BamHI and dephosphorylated with the alkaline phosphatase FastAP (Thermo Fisher Scientific). The insert DNA (3–10 kb) was ligated with the vector (pHSG396) at a vector/insert ratio of ∼1:3. The resulting ligation product was purified using the Qiagen PCR Purification Kit, concentrated by eluting (EB Buffer) into a smaller volume (20 μl), and stored at −20 °C prior to transformation. An aliquot of the ligation product (5 μl) was transformed into ReplicatorFOS cells (80 μl) by electroporation (1-mm cuvette, output voltage = 1.6 kV, capacitance = 25 microfarads, pulse time = 4–5 ms) and immediately recovered in SOC medium (1 ml) at 37 °C for 60 min. A very small amount (2 μl) of the cells was used to plate 1×, 100×, and 10,000× dilutions on LB (12.5 μg/ml chloramphenicol) and determine library titers. DMSO ( volume) was added to the library, and the library was stored at −80 °C. Dilutions of the libraries were plated and grown on solid LB (chloramphenicol 25 μg/ml) overnight at 37 °C such that each plate contained 150–400 colonies. Colonies were picked using the QPix II (Genetix) colony picker and transferred to 384-well plates containing LB (chloramphenicol 25 μg/ml + 10% glycerol (v/v)) in each well (80 μl). The plates were incubated at 37 °C for 20 h and then stored at −70 °C.
Plate screening
Library plates were thawed at room temperature and then replicated to 384-well plates containing growth/induction medium (LB + 25 μg/ml chloramphenicol + 0.2 mm isopropyl 1-thio-β-d-galactopyranoside) in each well (40 μl). Plates were incubated at 37 °C for 20 h (in a humid chamber to minimize evaporation). Following growth/induction, 40 μl of a lysis/assay mixture (50 mm NaPO4, pH 7.5, 1% Triton X-100, 200 μm GalNAc-α-MU) was added to each well, and the plate was incubated at 37 °C. All plates were filled with medium or lysis/assay mixture using a QFill instrument (Genetix). Following 22 h of incubation, the fluorescence (excitation, 365; emission, 440 nm) of each plate was determined by a plate reader (Opticon). All data were compiled and analyzed in Microsoft Excel. Hits were identified as those wells fluorescing at least 10 S.D. values above the mean. Wells in the library storage plates corresponding to hits (>10 S.D. above mean) were picked and retested in duplicate on 384-well plates to validate α-N-acetyl-galactosaminidase activity.
Hit identification, cloning, and gene extension
DNA from each hit was isolated (Qiagen DNA Miniprep Kit) and sequenced (Sanger) using primers flanking each metagenomic insert (M13F and M13R). For inserts over 1 kb in length, primer walking was conducted until the entire insert sequence had been identified. Sequences were analyzed using the default setting of the ORFfinder program (NCBI) to identify protein-coding regions. ORFs were analyzed using BLAST (NCBI) to identify any sequence similarity to known proteins (Table S1). All hits found in the sequenced plasmids were missing parts of the sequence, relative to the BLAST hits on NCBI.
For the first hit, A1, called BvGH109_1, the ORF was extended to include 57 amino acids missing from its C terminus relative to its full putative protein sequence. This was achieved by ordering the remaining DNA sequence (150 bp) as a codon-optimized DNA fragment (Genewiz, Geneblocks). Overlap-extension PCR was performed with the BvGH109_1 ORF fragment along with the additional fragment. The resulting BvGH109_1 sequence was amplified, digested using the XhoI and NdeI restriction enzymes, and then ligated into the expression vector pET16b.
The other hits, O1 and A2, called BcGH31 and BpGH31, were both missing 342 and 337 amino acids, respectively, here called tBcGH31 and tBpGH31. They were cloned into pET28 via Golden Gate cloning (61). The full length was archived by ordering the missing C-terminal parts as Gene Strings (Thermo Fisher Scientific) and using PIPE cloning (62) for extension of pET28a-tBcGH31 and pET28a-tBpGH31 to the full-length constructs. tCpGH31 was cloned out of the genomic DNA of C. perfringens DSM 798, generating pET28a-tCpGH31.
All proteins contained signal peptides that were identified by SignalP (34) and were not included in the final constructs. All primers used are noted in Table S3.
Site-directed mutagenesis
All single aa substitution mutants were generated using the QuikChange protocol for site-directed mutagenesis (63) and the primers noted in Table S3.
Protein expression
The production of proteins for characterization was performed in BL21(DE3) cells, cultured in ZY5052 autoinduction medium (64) for 20 h at 30 °C with shaking at 220 rpm inoculated from an overnight LB culture. Cells were harvested by centrifugation (4000 rcf, 4 °C, 10 min) and resuspended in 10 ml of lysis buffer/2 g of cell pellet (wet mass) (50 mm Tris/HCl, 150 mm NaCl, 1% (v/v) glycerol, 20 mm imidazole, pH 7.4, 1× protease inhibitor EDTA-free (Pierce), 2 units of benzonase (Novagen), 0.3 mg/ml lysozyme, 10 mm MgCl2), followed by sonication (3-min pulse time; 5-s pulse, 10-s pause, 35% amplitude) on ice. After removal of cell debris by centrifugation (14,000 rcf, 4 °C, 30 min), supernatant was collected and loaded onto a nickel-nitrilotriacetic acid chromatography column (5-ml HisTrap HP column (GE Healthcare)) using a peristaltic pump. The elution was performed and monitored on an ÄKTA purifier system (GE Healthcare) with a 10–75% gradient of 50 mm Tris/HCl, 400 mm imidazole, pH 7.4. Fractions containing the highest concentrations of protein were identified via SDS-PAGE and then pooled. Buffer exchange into 50 mm Tris/HCl, 150 mm NaCl, pH 7.4, and concentration were performed in Amicon Ultra-15 centrifugal filter units, MWCO 10 kDa (Merck Millipore).
tBcGH31, BcGH31, tBpGH31, and BpGH31 were purified additionally on a size-exclusion chromatography column (Superdex 200 (GE Healthcare)), utilizing the following buffer conditions: 50 mm Tris/HCl, 150 mm NaCl, 1% (v/v) glycerol, pH 7.4. Fractions containing the protein were identified via SDS-PAGE and then pooled. They were then concentrated using Amicon Ultra-15 centrifugal filter units MWCO 30 kDa (Millipore) in 50 mm Tris/HCl, 150 mm NaCl, 1% glycerol, pH 7.4.
Substrate specificity assay
All enzymes were initially assayed against a number of substrates in a 384-well plate (clear) (NUNC). Each well contained 50 μl of an assay mixture consisting of 50 mm NaH2PO4, pH 7.5 (40 μl); 0.1 mm (final concentration) of either GalNAc-α-MU, GalNAc-α-pNP, Gal-α-MU, Glc-α-MU, Man-α-MU, Xyl-α-pNP, A-antigen Type2tetra-MU; and 20 μg of purified candidate enzyme. Upon the addition of enzyme to each well, the plate was incubated at 37 °C and monitored continuously at 405 nm by a plate reader (Biotek Synergy) for release of oNP or pNP. For GalNAc-a-MU, the fluorescence signal (excitation, 365; emission, 435 nm) resulting from MU release by hydrolysis was monitored using a Synergy H1 plate reader (BioTek) for at least 30 min. For A-antigen Type2tetra-MU, a coupled assay was employed as described previously (21).
Kinetic characterization
Michaelis–Menten kinetic parameters were determined for the substrate GalNAc-α-pNP for all enzymes. Kinetic assays were carried out at 37 °C in 50 mm HEPES, pH 7.0 (total volume of 100 μl). These were performed in multiwell plates (96-well plate (NUNC)) monitoring the increased absorbance (405 nm) or fluorescence (excitation, 360 nm; emission, 450 nm) resulting from pNP or MU release, respectively, using a plate reader (Biotek Synergy). Reactions were performed in triplicate, with varying concentrations of substrate and enzymes (36.2–45.8 nm tBpGH31, 49.9 nm BpGH31, 16.3–22.1 nm tBcGH31, 38.5 nm BcGH31, 54.6–109 nm BvGH109, 863 nm tBpGH31 D465A, 400 nm tBpGH31 D465G, and 532 nm tBpGH31 D465S). Initial rates (milli-optical density × s−1 or milli-relative fluorescence units × s−1) were determined within the plate reader Synergy software and converted from absorbance to concentration (mm × s−1) using pNP or MU standard concentration curves determined under identical reaction conditions. All measurements were made within the linear range of the standard curves. Kinetic parameters were then determined via fitting to the Michaelis–Menten equation using nonlinear regression in GraFit version 7.0.
pH optima of GH31 enzymes
pH optima were determined using the substrate depletion method in a manner similar to that described previously (65). In brief, at low concentrations of substrate, where [substrate] < Km, the kcat/Km value can be approximated upon nonlinear fitting of the reaction time course to a first-order curve. Enzymes were brought to appropriate concentrations in 5 mm NaH2PO4, pH 7.0, before 8-fold dilution into the assay mixture (10 μm GalNAc-α-pNP, 50 mm citrate, 50 mm NaH2PO4, and 50 mm glycine at pH values from 5 to 8) for up to 25 min. Reaction progress was followed by the absorbance at 348 nm (pH 5–6) and 405 nm (pH 6.5–8). Control experiments, in which enzymes were incubated in the appropriate buffers (pH 4–8) before 15-fold dilution and assaying in 200 μm GalNAc-α-pNP, 50 mm citrate, 50 mm NaH2PO4, and 50 mm glycine, pH 7.0, were performed to determine conditions under which the enzymes did not lose substantial activity (>15%) due to denaturation over the course of 25 min. Data on activity versus pH were then analyzed using GraFit version 7.0 to determine pKa values and pH optima.
High-performance anion-exchange chromatography coupled with pulsed amperometric detection (HPAEC-PAD)
The ability of the candidate enzymes to digest biological substrates was determined through digestion tests on native and denatured fetuin. In brief, fetuin from fetal calf serum was denatured and reduced over a period of 3 h at 37 °C (18.9 mm DTT, 6 m guanidinium hydrochloride, 9.4 mm EDTA, 94 mm Tris/HCl, pH 8.2). Reduced Cys residues were then alkylated through the addition of iodoacetamide (final concentration of 23 mm) for 90 min at 37 °C while protected from light. The buffer was then exchanged to 20 mm HEPES, pH 7.0, 150 mm NaCl, 10 mm CaCl2 using the Amicon Ultra-15 centrifugal filter with a 10-kDa molecular weight cut-off (Merck Millipore). The fetuin substrate (3 mg/ml) was then incubated at 37 °C with different combinations of sialidase NedA (0.1 mg/ml), β-1,3-galactosidase BgaC (0.2 mg/ml), and purified candidate enzyme (0.1 mg/ml). To stop the reactions and remove proteins prior to HPLC analysis, the samples were applied to a prewashed Amicon Ultra-0.5 centrifugal filter with a 10-kDa molecular weight cut-off (Merck Millipore). The permeates were then analyzed via HPAEC-PAD. Assays using native fetuin proceeded in a similar manner, albeit without the denaturation steps.
The ability of the enzymes tBpGH31, BpGH31, tBcGH31, and BcGH31 to cleave GalNAc-α-O-threonine on a peptide substrate was determined using a glycopeptide substrate containing an N-terminal BODIPY residue (BODIPY-QPQP(GalNAc-α-)TAGPV). 20 μg of glycopeptide substrate and ∼50 ng of purified enzyme were incubated overnight at room temperature in 50 mm HEPES, pH 7.0. 10 μl was diluted in H2O (100 μl) and submitted to analysis on a Dionex HPAEC-PAD instrument.
Separation was obtained on a CarboPAC PA200 (150-mm) column with guard column, and detection was achieved using a disposable gold on polytetrafluoroethylene electrode and a four-potential waveform. The separation conditions were as follows: 100 mm sodium hydroxide and a sodium acetate gradient from 70 to 300 mm over the first 10 min of the separation. The eluent was held at the final gradient conditions for 1 min and then returned to the starting conditions over the next minute. The flow rate was 1.0 ml/min, and an injection was made every 27 min. A standard of the free sugars GalNAc, Gal, and Neu5Ac (50 μm) was also applied to HPAEC-PAD to determine the peak elution times for reference.
Far-UV CD spectroscopy for secondary structure prediction
Enzymes were diluted into 5 mm NaH2PO4, pH 7, to a final concentration of 0.075 mg/ml. Data were acquired in a Jasco J-815 CD spectrophotometer using a 1-mm quartz cuvette (VWR) with the following parameters: 180–250-nm measuring range, speed 50 nm/min, pitch 0.5, scans 20, room temperature. Raw data were input into a web server that analyzed the data using the method described by Raussens et al. (66) for secondary structure prediction.
Stereochemistry of GalNAc cleavage
α-N-Acetylgalactosaminidase reactions (500 μl) were set up in NMR tubes (Sigma) prior to analysis. Each 500-μl reaction contained 50 mm HEPES buffer (pH 7.5), GalNAc-α-DNP (5 mm final concentration), and either 100 or 8 μg of purified tBpGH31 and tBcGH31, respectively. All buffer salts and substrates were lyophilized and resuspended in D2O. The enzyme was buffer-exchanged into D2O prior to addition to the reaction mixture. Upon enzyme addition and mixing, the reaction-containing NMR tube was immediately submitted to 1H NMR analysis for monitoring. 1H NMR spectra were recorded following 5 min of reaction (D2O, 298 K, 400-MHz Bruker INV) to determine stereochemistry of the initial product formed (prior to formation of both α- and β-products at equilibrium).
GH31 phylogenetic mapping
Reference sequences of GH31 were downloaded from the CAZy database using SACCHARIS' cazy_extract.pl script (67). A phylogenetic-based protein-profiling software, TreeSAPP (available on GitHub), was used to both build the reference trees and map the sequences to these trees. Briefly, HMMs from dbCAN were used to extract protein family domains from all full-length sequences downloaded from CAZy (68). These sequences were then clustered at 70% sequence similarity using UCLUST to remove redundant sequence space and decrease the size of the tree (69). RAxML version 8.2.0 was used to build the reference trees with the “- -autoMRE” to decide when to quit bootstrapping before 1000 replicates have been performed and PROTGAMMAAUTO to select the optimal protein model (70, 71). TreeSAPP was then used to map the query sequences onto these reference trees. Briefly, protein sequences were aligned to HMMs using hmmsearch, and the aligned regions were extracted (72). hmmalign was used to include the new query sequences in the reference multiple alignment, and then TrimAl removed the unconserved positions from the alignment file (73). RAxML was used to classify the query sequences in the reference tree through insertions. Placements of each query sequence were filtered and concatenated into a single .Jplace file before being visualized in iTOL (74, 75).
Ethics
The collection of human fecal samples was approved by the Clinical Research Ethics Board of the University of British Columbia (ID: H15-02967).
Data availability
The data sets generated and/or analyzed during this study are available from the corresponding author on reasonable request.
Author contributions
P. R., K. M., D. H., and S. G. W. conceptualization; P. R., S. J. H., and S. G. W. resources; P. R., J. F. W., and K. M. data curation; P. R., J. F. W., K. M., D. H., C. M.-L., H.-M. C., and S. G. W. formal analysis; P. R., J. F. W., S. J. H., and S. G. W. supervision; P. R., S. J. H., and S. G. W. funding acquisition; P. R., J. F. W., and K. M. validation; P. R., J. F. W., K. M., D. H., C. M.-L., H.-M. C., and S. G. W. investigation; P. R., J. F. W., K. M., C. M.-L., and H.-M. C. visualization; P. R., J. F. W., K. M., D. H., and S. G. W. writing-original draft; P. R. and S. G. W. project administration; P. R., J. F. W., K. M., and S. G. W. writing-review and editing; J. F. W., K. M., D. H., H.-M. C., and S. G. W. methodology; C. M.-L., H.-M. C., and S. J. H. software.
Supplementary Material
Acknowledgments
We thank Dr. Harry Brumer for the use of his Dionex HPLC, Dr. Warren Wakarchuk for the BODIPY-labeled glycopeptide sample, and Dr. David Kwan for the A-antigen substrate.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S4 and Figs. S1–S10.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- α-GalNAcase
- α-N-acetyl-galactosaminidase
- GH
- glycoside hydrolase
- pNP and oNP
- para- and ortho-nitrophenol, respectively
- Xyl
- xylose
- Man
- mannose
- MU
- 4-methylumbelliferone
- aa
- amino acids
- CBM32
- carbohydrate-binding module 32
- PUL
- polysaccharide utilization locus
- LB
- lysogeny broth
- rcf
- relative centrifugal force
- MWCO
- molecular weight cutoff
- HPAEC-PAD
- high-performance anion-exchange chromatography coupled with pulsed amperometric detection.
References
- 1. Ten Hagen K. G., Fritz T. A., and Tabak L. A. (2003) All in the family: the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 13, 1R–16R 10.1093/glycob/cwg007 [DOI] [PubMed] [Google Scholar]
- 2. Tailford L. E., Crost E. H., Kavanaugh D., and Juge N. (2015) Mucin glycan foraging in the human gut microbiome. Front. Genet. 6, 81 10.3389/fgene.2015.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clausen H., and Hakomori S. (1989) ABH and related histo-blood group antigens; immunochemical differences in carrier isotypes and their distribution. Vox Sang. 56, 1–20 10.1111/j.1423-0410.1989.tb03040.x [DOI] [PubMed] [Google Scholar]
- 4. Skovierová H, Larrouy-Maumus G., Pham H., Belanová M., Barilone N., Dasgupta A., Mikusová K., Gicquel B., Gilleron M., Brennan P. J., Puzo G., Nigou J., and Jackson M. (2010) Biosynthetic origin of the galactosamine substituent of arabinogalactan in Mycobacterium tuberculosis. J. Biol. Chem. 285, 41348–41355 10.1074/jbc.M110.188110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fujita K., Oura F., Nagamine N., Katayama T., Hiratake J., Sakata K., Kumagai H., and Yamamoto K. (2005) Identification and molecular cloning of a novel glycoside hydrolase family of core 1 type O-glycan-specific endo-α-N-acetylgalactosaminidase from Bifidobacterium longum. J. Biol. Chem. 280, 37415–37422 10.1074/jbc.M506874200 [DOI] [PubMed] [Google Scholar]
- 7. Clark N. E., and Garman S. C. (2009) The 1.9 Å structure of human α-N-acetylgalactosaminidase. J. Mol. Biol. 393, 435–447 10.1016/j.jmb.2009.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ashida H., Tamaki H., Fujimoto T., Yamamoto K., and Kumagai H. (2000) Molecular cloning of cDNA encoding α-N-acetylgalactosaminidase from Acremonium sp. and its expression in yeast. Arch. Biochem. Biophys. 384, 305–310 10.1006/abbi.2000.2114 [DOI] [PubMed] [Google Scholar]
- 9. Kiyohara M., Nakatomi T., Kurihara S., Fushinobu S., Suzuki H., Tanaka T., Shoda S., Kitaoka M., Katayama T., Yamamoto K., and Ashida H. (2012) α-N-Acetylgalactosaminidase from infant-associated Bifidobacteria belonging to novel glycoside hydrolase family 129 is implicated in alternative mucin degradation pathway. J. Biol. Chem. 287, 693–700 10.1074/jbc.M111.277384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bamford N. C., Snarr B. D., Gravelat F. N., Little D. J., Lee M. J., Zacharias C. A., Chabot J. C., Geller A. M., Baptista S. D., Baker P., Robinson H., Howell P. L., and Sheppard D. C. (2015) Sph3 is a glycoside hydrolase required for the biosynthesis of galactosaminogalactan in Aspergillus fumigatus. J. Biol. Chem. 290, 27438–27450 10.1074/jbc.M115.679050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Danby P. M., and Withers S. G. (2016) Advances in enzymatic glycoside synthesis. ACS Chem. Biol. 11, 1784–1794 10.1021/acschembio.6b00340 [DOI] [PubMed] [Google Scholar]
- 12. Mackenzie L. F., Wang Q., Warren R. A. J., and Withers S. G. (1998) Glycosynthases: mutant glycosidases for oligosaccharide synthesis. J. Am. Chem. Soc. 120, 5583–5584 10.1021/ja980833d [DOI] [Google Scholar]
- 13. Jahn M., Marles J., Warren R. A. J., and Withers S. G. (2003) Thioglycoligases: mutant glycosidases for thioglycoside synthesis. Angew. Chem. Int. Ed. Engl. 42, 352–354 10.1002/anie.200390114 [DOI] [PubMed] [Google Scholar]
- 14. Goldstein J., Siviglia G., Hurst R., Lenny L., and Reich L. (1982) Group B erythrocytes enzymatically converted to group O survive normally in A, B, and O individuals. Science 215, 168–170 10.1126/science.6274021 [DOI] [PubMed] [Google Scholar]
- 15. Levy G. N., and Aminoff D. (1980) Purification and properties of α-N-acetylgalactosaminidase from Clostridium perfringens. J. Biol. Chem. 255, 11737–11742 [PubMed] [Google Scholar]
- 16. Hsieh H.-Y., Calcutt M. J., Chapman L. F., Mitra M., and Smith D. S. (2003) Purification and characterization of a recombinant α-N-acetylgalactosaminidase from Clostridium perfringens. Protein Expr. Purif. 32, 309–316 10.1016/j.pep.2003.08.007 [DOI] [PubMed] [Google Scholar]
- 17. Hsieh H.-Y., Mitra M., Wells D. C., and Smith D. (2000) Purification and characterization of α-N-acetylgalactosaminidase from Clostridium perfringens. IUBMB Life 50, 91–97 10.1080/713803702 [DOI] [PubMed] [Google Scholar]
- 18. Calcutt M. J., Hsieh H. Y., Chapman L. F., and Smith D. S. (2002) Identification, molecular cloning and expression of an α-N-acetylgalactosaminidase gene from Clostridium perfringens. FEMS Microbiol. Lett. 214, 77–80 10.1111/j.1574-6968.2002.tb11327.x [DOI] [PubMed] [Google Scholar]
- 19. Liu Q. P. Y. P., Sulzenbacher G., Yuan H., Bennett E. P., Pietz G., Saunders K., Spence J., Nudelman E., Levery S. B., White T., Neveu J. M., Lane W. S., Bourne Y., Olsson M. L., Henrissat B., and Clausen H. (2007) Bacterial glycosidases for the production of universal red blood cells. Nat. Biotechnol. 25, 454–464 10.1038/nbt1298 [DOI] [PubMed] [Google Scholar]
- 20. Yip V. L. Y., Varrot A., Davies G. J., Rajan S. S., Yang X., Thompson J., Anderson W. F., and Withers S. G. (2004) An unusual mechanism of glycoside hydrolysis involving redox and elimination steps by a family 4 β-glycosidase from Thermotoga maritima. J. Am. Chem. Soc. 126, 8354–8355 10.1021/ja047632w [DOI] [PubMed] [Google Scholar]
- 21. Rahfeld P., Sim L., Moon H., Constantinescu I., Morgan-Lang C., Hallam S. J., Kizhakkedathu J. N., and Withers S. G. (2019) An enzymatic pathway in the human gut microbiome that converts A to universal O type blood. Nat. Microbiol. 4, 1475–1485 10.1038/s41564-019-0469-7 [DOI] [PubMed] [Google Scholar]
- 22. Vitiazeva V., Kattla J. J., Flowers S. A., Lindén S. K., Premaratne P., Weijdegård B., Sundfeldt K., and Karlsson N. G. (2015) The O-linked glycome and blood group antigens ABO on mucin-type glycoproteins in mucinous and serous epithelial ovarian tumors. PLoS One 10, e0130197 10.1371/journal.pone.0130197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rossez Y., Maes E., Lefebvre Darroman T., Gosset P., Ecobichon C., Joncquel Chevalier Curt M., Boneca I. G., Michalski J. C., and Robbe-Masselot C. (2012) Almost all human gastric mucin O-glycans harbor blood Group A, B or H antigens and are potential binding sites for Helicobacter pylori. Glycobiology 22, 1193–1206 10.1093/glycob/cws072 [DOI] [PubMed] [Google Scholar]
- 24. Riesenfeld C. S., Schloss P. D., and Handelsman J. (2004) Metagenomics: genomic analysis of microbial communities. Annu. Rev. Genet. 38, 525–552 10.1146/annurev.genet.38.072902.091216 [DOI] [PubMed] [Google Scholar]
- 25. Pasolli E., Asnicar F., Manara S., Zolfo M., Karcher N., Armanini F., Beghini F., Manghi P., Tett A., Ghensi P., Collado M. C., Rice B. L., DuLong C., Morgan X. C., Golden C. D., et al. (2019) Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 10.1016/j.cell.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Handelsman J. (2004) Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68, 669–685 10.1128/MMBR.68.4.669-685.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pope P. B., Denman S. E., Jones M., Tringe S. G., Barry K., Malfatti S. A., McHardy A. C., Cheng J.-F., Hugenholtz P., McSweeney C. S., and Morrison M. (2010) Adaptation to herbivory by the Tammar Wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc. Natl. Acad. Sci. U.S.A. 107, 14793–14798 10.1073/pnas.1005297107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nacke H., Engelhaupt M., Brady S., Fischer C., Tautzt J., and Daniel R. (2012) Identification and characterization of novel cellulolytic and hemicellulolytic genes and enzymes derived from German grassland soil metagenomes. Biotechnol. Lett. 34, 663–675 10.1007/s10529-011-0830-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mewis K., Armstrong Z., Song Y. C., Baldwin S. A., Withers S. G., and Hallam S. J. (2013) Biomining active cellulases from a mining bioremediation system. J. Biotechnol. 167, 462–471 10.1016/j.jbiotec.2013.07.015 [DOI] [PubMed] [Google Scholar]
- 30. Tasse L., Bercovici J., Pizzut-Serin S., Robe P., Tap J., Klopp C., Cantarel B. L., Coutinho P. M., Henrissat B., Leclerc M., Doré J., Monsan P., Remaud-Simeon M., and Potocki-Veronese G. (2010) Functional metagenomics to mine the human gut microbiome for dietary fiber catabolic enzymes. Genome Res. 20, 1605–1612 10.1101/gr.108332.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Culligan E. P., Sleator R. D., Marchesi J. R., and Hill C. (2013) Functional environmental screening of a metagenomic library identifies stlA; a unique salt tolerance locus from the human gut microbiome. PLoS One 8, e82985 10.1371/journal.pone.0082985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Armstrong Z., Mewis K., Liu F., Morgan-Lang C., Scofield M., Durno E., Chen H. M., Mehr K., Withers S. G., and Hallam S. J. (2018) Metagenomics reveals functional synergy and novel polysaccharide utilization loci in the Castor canadensis fecal microbiome. ISME J. 12, 2757–2769 10.1038/s41396-018-0215-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peterson J., Garges S., Giovanni M., McInnes P., Wang L., Schloss J. A., Bonazzi V., McEwen J. E., Wetterstrand K. A., Deal C., Baker C. C., Di Francesco V., Howcroft T. K., Karp R. W., Lunsford R. D., et al. (2009) The NIH human microbiome project. Genome Res. 19, 2317–2323 10.1101/gr.096651.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nielsen H. (2017) Predicting secretory proteins with SignalP. Methods Mol. Biol. 1611, 59–73 10.1007/978-1-4939-7015-5_6 [DOI] [PubMed] [Google Scholar]
- 35. Chiba S., Hiromi K., Minamiura N., Ohnishi M., Shimomura T., Suga K., Suganuma T., Tanaka A., Tomioka S., and Yamamoto T. (1979) Quantitative study on anomeric forms of glucose produced by α-glucosidases. J. Biochem. 85, 1135–1141 [PubMed] [Google Scholar]
- 36. Lee S. S., Yu S., and Withers S. G. (2003) Detailed dissection of a new mechanism for glycoside cleavage: α-1,4-glucan lyase. Biochemistry 42, 13081–13090 10.1021/bi035189g [DOI] [PubMed] [Google Scholar]
- 37. Lovering A. L., Lee S. S., Kim Y.-W., Withers S. G., and Strynadka N. C. J. (2005) Mechanistic and structural analysis of a family 31 α-glycosidase and its glycosyl-enzyme intermediate. J. Biol. Chem. 280, 2105–2115 10.1074/jbc.M410468200 [DOI] [PubMed] [Google Scholar]
- 38. Zechel D. L., and Withers S. G. (2001) Dissection of nucleophilic and acid-base catalysis in glycosidases. Curr. Opin. Chem. Biol. 5, 643–649 10.1016/S1367-5931(01)00260-5 [DOI] [PubMed] [Google Scholar]
- 39. Windwarder M., and Altmann F. (2014) Site-specific analysis of the O-glycosylation of bovine fetuin by electron-transfer dissociation mass spectrometry. J. Proteomics 108, 258–268 10.1016/j.jprot.2014.05.022 [DOI] [PubMed] [Google Scholar]
- 40. Grondin J. M., Duan D., Kirlin A. C., Abe K. T., Chitayat S., Spencer H. L., Spencer C., Campigotto A., Houliston S., Arrowsmith C. H., Allingham J. S., Boraston A. B., and Smith S. P. (2017) Diverse modes of galacto-specific carbohydrate recognition by a family 31 glycoside hydrolase from Clostridium perfringens. PLoS One 12, e0171606 10.1371/journal.pone.0171606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gabor E. M., de Vries E. J., and Janssen D. B. (2004) Construction, characterization, and use of small-insert gene banks of DNA isolated from soil and enrichment cultures for the recovery of novel amidases. Environ. Microbiol. 6, 948–958 10.1111/j.1462-2920.2004.00643.x [DOI] [PubMed] [Google Scholar]
- 42. Armstrong Z., Rahfeld P., and Withers S. G. (2017) Discovery of new glycosidases from metagenomic libraries. Methods Enzymol. 597, 3–23 10.1016/bs.mie.2017.06.001 [DOI] [PubMed] [Google Scholar]
- 43. Balabanova L. A., Golotin V. A., Bakunina I. Y., Slepchenko L. V., Isakov V. V., Podvolotskaya A. B., and Rasskazov V. A. (2015) Recombinant α-N-acetylgalactosaminidase from marine bacterium-modifying A erythrocyte antigens. Acta Naturae 7, 117–120 10.32607/20758251-2015-7-1-117-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Larsbrink J., Izumi A., Ibatullin F. M., Nakhai A., Gilbert H. J., Davies G. J., and Brumer H. (2011) Structural and enzymatic characterization of a glycoside hydrolase family 31 α-xylosidase from Cellvibrio japonicus involved in xyloglucan saccharification. Biochem. J. 436, 567–580 10.1042/BJ20110299 [DOI] [PubMed] [Google Scholar]
- 45. Miyazaki T., Ishizaki Y., Ichikawa M., Nishikawa A., and Tonozuka T. (2015) Structural and biochemical characterization of novel bacterial α-galactosidases belonging to glycoside hydrolase family 31. Biochem. J. 469, 145–158 10.1042/BJ20150261 [DOI] [PubMed] [Google Scholar]
- 46. Ernst H. A., Lo Leggio L., Willemoës M., Leonard G., Blum P., and Larsen S. (2006) Structure of the Sulfolobus solfataricus α-glucosidase: implications for domain conservation and substrate recognition in GH31. J. Mol. Biol. 358, 1106–1124 10.1016/j.jmb.2006.02.056 [DOI] [PubMed] [Google Scholar]
- 47. Zechel D. L., and Withers S. G. (2000) Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc. Chem. Res. 33, 11–18 10.1021/ar970172 [DOI] [PubMed] [Google Scholar]
- 48. Frandsen T. P., and Svensson B. (1998) Plant α-glucosidases of the glycoside hydrolase family 31: molecular properties, substrate specificity, reaction mechanism, and comparison with family members of different origin. Plant Mol. Biol. 37, 1–13 10.1023/A:1005925819741 [DOI] [PubMed] [Google Scholar]
- 49. Lee S. S., He S., and Withers S. G. (2001) Identification of the catalytic nucleophile of the family 31 α-glucosidase from Aspergillus niger via trapping of a 5-fluoroglycosyl-enzyme intermediate. Biochem. J. 359, 381–386 10.1042/0264-6021:3590381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Briliūtė J., Urbanowicz P. A., Luis A. S., Baslé A., Paterson N., Rebello O., Hendel J., Ndeh D. A., Lowe E. C., Martens E. C., Spencer D. I. R., Bolam D. N., and Crouch L. I. (2019) Complex N-glycan breakdown by gut Bacteroides involves an extensive enzymatic apparatus encoded by multiple co-regulated genetic loci. Nat. Microbiol. 4, 1571–1581 10.1038/s41564-019-0466-x [DOI] [PubMed] [Google Scholar]
- 51. Bayer E. A., Belaich J. P., Shoham Y., and Lamed R. (2004) The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu. Rev. Microbiol. 58, 521–554 10.1146/annurev.micro.57.030502.091022 [DOI] [PubMed] [Google Scholar]
- 52. Brockhausen I., Schachter H., and Stanley P. (2009) O-GalNAc glycans. in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., and Etzler M. E., eds) pp. 115–127, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 53. Martens E. C., Chiang H. C., and Gordon J. I. (2008) Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457 10.1016/j.chom.2008.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Terrapon N., Lombard V., Drula É., Lapébie P., Al-Masaudi S., Gilbert H. J., and Henrissat B. (2018) PULDB: the expanded database of polysaccharide utilization loci. Nucleic Acids Res. 46, D677–D683 10.1093/nar/gkx1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Noach I., Ficko-Blean E., Pluvinage B., Stuart C., Jenkins M. L., Brochu D., Buenbrazo N., Wakarchuk W., Burke J. E., Gilbert M., and Boraston A. B. (2017) Recognition of protein-linked glycans as a determinant of peptidase activity. Proc. Natl. Acad. Sci. U.S.A. 114, E679–E688 10.1073/pnas.1615141114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hobbs J. K., Pluvinage B., and Boraston A. B. (2018) Glycan-metabolizing enzymes in microbe-host interactions: the Streptococcus pneumoniae paradigm. FEBS Lett. 592, 3865–3897 10.1002/1873-3468.13045 [DOI] [PubMed] [Google Scholar]
- 57. Jongkees S. A. K., and Withers S. G. (2014) Unusual enzymatic glycoside cleavage mechanisms. Acc. Chem. Res. 47, 226–235 10.1021/ar4001313 [DOI] [PubMed] [Google Scholar]
- 58. Rawlings N. D., Morton F. R., Kok C. Y., Kong J., and Barrett A. J. (2008) MEROPS: the peptidase database. Nucleic Acids Res. 36, D320–D325 10.1093/nar/gkm954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pudlo N. A., Urs K., Kumar S. S., German J. B., Mills D. A., and Martens E. C. (2015) Symbiotic human gut bacteria with variable metabolic priorities for host mucosal glycans. MBio 6, e01282–15 10.1128/mBio.01282-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marcobal A., Southwick A. M., Earle K. A., and Sonnenburg J. L. (2013) A refined palate: bacterial consumption of host glycans in the gut. Glycobiology 23, 1038–1046 10.1093/glycob/cwt040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Engler C., Kandzia R., and Marillonnet S. (2008) A one pot, one step, precision cloning method with high throughput capability. PLoS One 3, e3647 10.1371/journal.pone.0003647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Klock H. E., Koesema E. J., Knuth M. W., and Lesley S. A. (2008) Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins 71, 982–994 10.1002/prot.21786 [DOI] [PubMed] [Google Scholar]
- 63. Zheng L., Baumann U., and Reymond J. L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Studier F. W. (2005) Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 65. Vocadlo D. J., Mayer C., He S., and Withers S. G. (2000) Mechanism of action and identification of Asp242 as the catalytic nucleophile of Vibrio furnisii N-acetyl-β-d-glucosaminidase using 2-acetamido-2-deoxy-5-fluoro-α-l-idopyranosyl fluoride. Biochemistry 39, 117–126 10.1021/bi991958d [DOI] [PubMed] [Google Scholar]
- 66. Raussens V., Ruysschaert J.-M., and Goormaghtigh E. (2003) Protein concentration is not an absolute prerequisite for the determination of secondary structure from circular dichroism spectra: a new scaling method. Anal. Biochem. 319, 114–121 10.1016/S0003-2697(03)00285-9 [DOI] [PubMed] [Google Scholar]
- 67. Jones D. R., Thomas D., Alger N., Ghavidel A., Inglis G. D., and Abbott D. W. (2018) SACCHARIS: an automated pipeline to streamline discovery of carbohydrate active enzyme activities within polyspecific families and de novo sequence datasets. Biotechnol. Biofuels 11, 27 10.1186/s13068-018-1027-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yin Y., Mao X., Yang J., Chen X., Mao F., and Xu Y. (2012) dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, W445–W451 10.1093/nar/gks479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Edgar R. C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 70. Stamatakis A., Hoover P., and Rougemont J. (2008) A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 71. Stamatakis A. (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 10.1093/bioinformatics/btl446 [DOI] [PubMed] [Google Scholar]
- 72. Eddy S. R. (1998) Profile hidden Markov models. Bioinformatics 14, 755–763 10.1093/bioinformatics/14.9.755 [DOI] [PubMed] [Google Scholar]
- 73. Capella-Gutiérrez S., Silla-Martínez J. M., and Gabaldón T. (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 10.1093/bioinformatics/btp348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Matsen F. A., Hoffman N. G., Gallagher A., and Stamatakis A. (2012) A format for phylogenetic placements. PLoS One 7, e31009 10.1371/journal.pone.0031009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Letunic I., and Bork P. (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 10.1093/nar/gkw290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Varki A., Cummings R. D., Aebi M., Packer N. H., Seeberger P. H., Esko J. D., Stanley P., Hart G., Darvill A., Kinoshita T., Prestegard J. J., Schnaar R. L., Freeze H. H., Marth J. D., Bertozzi C. R., et al. (2015) Symbol nomenclature for graphical representations of glycans. Glycobiology 25, 1323–1324 10.1093/glycob/cwv091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mitchell A. L., Attwood T. K., Babbitt P. C., Blum M., Bork P., Bridge A., Brown S. D., Chang H.-Y., El-Gebali S., Fraser M. I., Gough J., Haft D. R., Huang H., Letunic I., Lopez R., et al. (2019) InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360 10.1093/nar/gky1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Marchler-Bauer A., Bo Y., Han L., He J., Lanczycki C. J., Lu S., Chitsaz F., Derbyshire M. K., Geer R. C., Gonzales N. R., Gwadz M., Hurwitz D. I., Lu F., Marchler G. H., Song J. S., et al. (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 10.1093/nar/gkw1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ren J., Wen L., Gao X., Jin C., Xue Y., and Yao X. (2009) DOG 1.0: illustrator of protein domain structures. Cell Res. 19, 271–273 10.1038/cr.2009.6 [DOI] [PubMed] [Google Scholar]
- 80. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated and/or analyzed during this study are available from the corresponding author on reasonable request.