

Abstract
The low-calorie sweetener erythritol is endogenously produced from glucose through the pentose phosphate pathway in humans. Erythritol is of medical interest because elevated plasma levels of this polyol are predictive for visceral adiposity gain and development of type 2 diabetes. However, the mechanisms behind these associations remain unknown because the erythritol biosynthesis pathway, particularly the enzyme catalyzing the final step of erythritol synthesis (reduction of erythrose to erythritol), is not characterized. In this study, we purified two enzymes from rabbit liver capable of catalyzing the conversion of erythrose to erythritol: alcohol dehydrogenase 1 (ADH1) and sorbitol dehydrogenase (SORD). Both recombinant human ADH1 and SORD reduce erythrose to erythritol, using NADPH as a co-factor, and cell culture studies indicate that this activity is primarily NADPH-dependent. We found that ADH1 variants vary markedly in both their affinity for erythrose and their catalytic capacity (turnover number). Interestingly, the recombinant protein produced from the ADH1B2 variant, common in Asian populations, is not active when NADPH is used as a co-factor in vitro. We also confirmed SORD contributes to intracellular erythritol production in human A549 lung cancer cells, where ADH1 is minimally expressed. In summary, human ADH1 and SORD catalyze the conversion of erythrose to erythritol, pointing to novel roles for two dehydrogenase proteins in human glucose metabolism that may contribute to individual responses to diet. Proteomics data are available via ProteomeXchange with identifier PXD015178.
Keywords: glucose metabolism, enzyme kinetics, enzyme catalysis, biomarker, alcohol dehydrogenase (ADH), erythritol, sorbitol dehydrogenase
Introduction
Erythritol is a four-carbon polyol that was approved as low calorie sweetener by the FDA in 2001 (1). It naturally occurs in various alcoholic beverages and fruits and has 70% of the sweetness of sucrose (2). In a large cohort of college freshmen, Hootman et al. (3) identified elevated plasma erythritol levels as a predictive marker for central adiposity gain and high glycemia. Plasma erythritol was also recently associated with onset of type 2 diabetes mellitus (T2DM)2 in adults followed over 20 years in the Atherosclerosis Risk in Communities cohort (4). Erythritol synthesis was not believed to occur in humans (2, 5). However, using stable isotope tracing, evidence was provided for the first time that human erythrocytes are capable of erythritol synthesis from glucose via erythrose-4-phosphate (E4P) in the pentose phosphate pathway (PPP) (Fig. 1) (3). In a recent case report, deficiency in the PPP enzyme transaldolase (TALDO) was accompanied by high concentrations of polyols including erythritol, ribitol, and sedoheptitol in the urine, supporting the link between erythritol production and the PPP (6). Although the metabolic pathway for erythritol synthesis is now recognized to be active in humans, the identity of the enzyme(s) catalyzing the conversion of erythrose to erythritol remains unknown.
Figure 1.
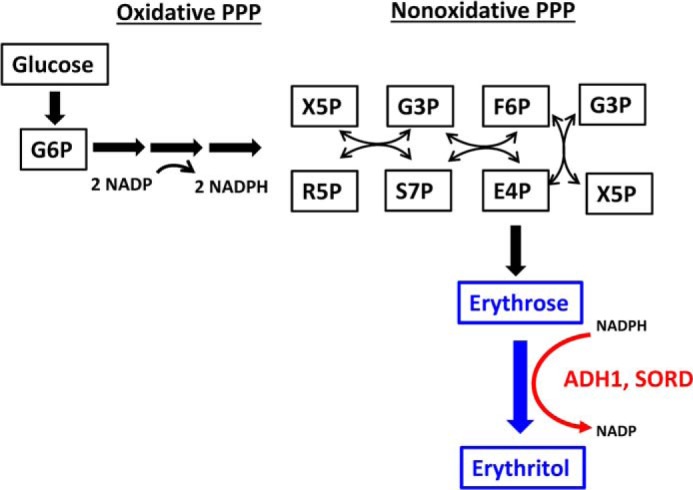
Conversion of glucose to erythritol via the pentose phosphate pathway. Stable isotope tracers indicated that glucose was converted to erythritol through the pentose phosphate pathway in human cells (3). Here we demonstrate that the substrate for erythritol synthesis is erythrose, not erythrose-4-phosphate and that the required cofactor in human cells is NADPH. In addition, this reaction is catalyzed by ADH1 and SORD. F6P, fructose 6-phosphate; G3P, glyceraldehyde 3-phosphate; G6P, glucose 6-phosphate; R5P, ribose 5-phosphate; S7P, sedoheptulose 7-phosphate; X5P, xylulose 5-phosphate.
Based on extensive toxicological investigations, erythritol is in general considered safe for human consumption (2). The low calorie sweetener is of interest to diabetic patients because blood glucose and insulin levels are not affected by erythritol consumption (7). Wen et al. (8) recently demonstrated that erythritol exhibits anti-postprandial hyperglycemic activities by competitively inhibiting α-glucosidase, which is beneficial for diabetes management and a recent pilot study correlated erythritol exposure with reduced central aortic stiffness and improved endothelial function (9). Interestingly, erythritol ingestion is toxic for the fruit fly Drosophila melanogaster and its insecticidal properties are currently under investigation (10, 11).
Currently, industrial erythritol production relies on biotechnological methods using bacterial, fungal, or yeast strains (1, 12), and the metabolic pathway using glucose as a substrate is well-characterized. In eukaryotes, glucose is phosphorylated yielding glucose 6-phosphate (G6P), which is subsequently converted to ribulose 5-phosphate in the oxidative part of the PPP. In the following, nonoxidative part of the PPP, carbon backbone rearrangements occur resulting in the formation of E4P. Prior to erythritol formation, E4P is dephosphorylated to erythrose and subsequently reduced in an NADPH-dependent manner to erythritol (1). In many eukaryotic erythritol-producing microorganisms, such as Candida magnoliae (13), Yarrowia lipolytica (14), and filamentous fungi (15), the enzyme erythrose reductase, catalyzing the final reduction step is well-characterized. However, in humans, to our knowledge, the exact metabolic conversions among E4P, erythrose, and erythritol, as well as the identity of the catalyzing enzyme, are not described and remain unknown. In this study, we isolated from rabbit liver two distinct protein fractions capable of catalyzing conversion of erythrose to erythritol. In addition, we confirmed that the human recombinant forms of these enzymes also exhibit this activity, effectively identifying novel roles for two dehydrogenase proteins in glucose metabolism (Fig. 1).
Results
Erythritol synthesis in A549 cells is NADPH-dependent
Hootman et al. (3) previously showed that erythritol can be synthesized endogenously from glucose, most probably via E4P through the pentose phosphate pathway (PPP) (Fig. 1). However, the enzyme responsible for the formation of erythritol in humans is not known. A colorimetric enzyme kinetics assay was used to determine the cofactor (NADH or NADPH) and the substrate (erythrose or E4P) required for erythritol synthesis in A549 lung cancer cells, in which erythritol production was previously detected (data not shown). Because the substrate must be reduced to form erythritol, the amount of reduced cofactor (NADH or NADPH, λmax = 340 nm) decreases as the reaction proceeds. Although the NADH level did not decrease during the time course of the assay, NADPH levels strongly decreased when erythrose and the cell lysate were present in the assay mixture (Fig. 2a, condition 1, black line). To ensure that the erythrose was converted to erythritol, we extracted the assay mixture at four different time points during the assay and measured the erythritol using GC-MS (Fig. 2b). Erythritol formation occurred only in the presence of NADPH and not in the presence of NADH, indicating that the enzyme of interest requires NADPH as a cofactor. A similar assay was used to determine the physiological substrate. These experiments revealed that erythrose, rather than E4P, is the substrate used by the target enzyme both with respect to NADPH consumption (Fig. 2a, black line) and erythritol formation in A549 cells (Fig. 2b, black line).
Figure 2.

The cofactor NADPH is required for erythritol synthesis activity in A549 lung cancer cell lysate. Enzyme activity assay for (a) cofactor (NADH or NADPH) and (c) substrate (erythrose or E4P) determination using A549 lung cancer cell lysate; b and d, GC-MS measurement of erythritol_4TMS extracted from the enzyme activity assay mixture of all tested conditions tested at specified time points to confirm the production of erythritol (n = 3, representative results).
Rabbit liver ADH1 and SORD catalyze erythritol synthesis
Two distinct, pure protein fractions (Fig. 3) were purified from rabbit liver with NADPH-dependent erythrose reduction activity (Table 1), as described under “Experimental procedures.” After purification and SDS-PAGE, three protein bands were excised (the most prominent 37-kDa band from purified fraction “A” and two bands belonging to purified fraction “B1” and “B2”) and subjected to tryptic digestion. Peptides from each protein were separated using LC-MS/MS and subsequently identified as belonging to alcohol dehydrogenase 1 (ADH1, fraction A, Table 2) and sorbitol dehydrogenase (SORD, fraction B1, Table 3) based on the coverage of the protein from identified peptides and the “Sequest HT” scores. Although the protein with the highest Sequest HT score for the smaller band excised from fraction B2 (Table 4) was identified as 33-kDa glycine N-methyltransferase, it is likely this band is a degradation product of the larger 38-kDa SORD protein, which exhibited the second-highest Sequest HT score. First, glycine N-methyltransferase comprises a significant proportion (1%) of total soluble liver protein in mammals (16). Second, the purification required several purification steps, all of which took place at room temperature over several days and included several freeze-thaw cycles of the liver fractions. Rabbit liver ADH1, fraction A, exhibited lower apparent Km values for erythrose than did rabbit liver SORD, fraction B1 (Table 1, Fig. S1). SORD, however, exhibited a much higher turnover number (kcat) than did ADH1, especially when NADPH was used as a cofactor (Table 1, Fig. S1).
Figure 3.

Two protein fractions (A and B) purified from rabbit liver exhibit erythrose reduction activity. Each pure protein fraction was loaded in duplicate (10 and 20 μg of total protein) on a 10% Tris glycine SDS-PAGE gel and visualized using Coomassie Brilliant Blue R-250. Molecular weights are defined by Precision Plus ProteinTM All Blue Prestained Protein Standards (Bio-Rad). The 37-kDa band from the 20 μg lane for protein A and the 10 μg lane for protein B were excised, digested, and analyzed using LC-MS/MS. In addition, the smaller band in “fraction B” was also analyzed and determined to be a degradation product of the 37-kDa protein due to the high degree of identity of identified peptides between the 37- and 35-kDa bands.
Table 1.
Kinetic characterization of protein fractions with erythrose reduction activity isolated from rabbit liver
Reaction buffer consisted of 50 mm sodium phosphate buffer, pH 6.8, 200 μm NADPH or NADH, and at least 5 concentrations of erythrose ranging from 0.5 to 100 mm. Reactions were initiated with addition of purified protein. Final concentrations of protein were 49 μm fraction A (ADH1) for NADPH, 25 μm fraction A (ADH1) for NADH, 0.7 μm fraction B (SORD) for NADPH, and 2.6 μm fraction B (SORD) for NADH. Kinetic constants were determined for the NAD-dependent conversion of ethanol to acetaldehyde for fraction A (ADH1) by monitoring production of NADH at 340 nm. Kinetic constants were also determined for the NAD-dependent conversion of sorbitol to fructose for fraction B (SORD) by monitoring production of NADH at 340 nm. Reactions were initiated with addition of purified protein (final concentration 1.3 μm for fraction A (ADH) and 0.13 μm for fraction B (SORD)). Measurements at each substrate concentration were performed in duplicate. Data are shown as average and standard deviation determined from fitting the data to Y = Vmax × X/(Km + X), where Y represents reaction velocity and X indicates substrate concentration using GraphPad Prism software. Km values are presented as “apparent Km” because these values were determined at only one saturating concentration of cofactor.
| Substrate | Cofactor | Apparent Km | kcat | kcat/Km | |
|---|---|---|---|---|---|
| mm | s−1 | m−1 s−1 | |||
| Fraction A, ADH1 | Erythrose | NADPH | 64.5 ± 7.7 | 0.0041 ± 0.0003 | 0.064 ± 0.009 |
| Erythrose | NADH | 80.1 ± 18.9 | 0.016 ± 0.002 | 0.205 ± 0.059 | |
| Ethanol (control) | NAD | 5.7 ± 0.5 | 0.106 ± 0.004 | 18.6 ± 1.8 | |
| Fraction B, SORD | Erythrose | NADPH | 340.3 ± 29 | 1.65 ± 0.11 | 4.84 ± 0.53 |
| Erythrose | NADH | 266 ± 120 | 0.13 ± 0.05 | 0.51 ± 0.29 | |
| Sorbitol (control) | NAD | 1.15 ± 0.16 | 0.26 ± 0.01 | 230 ± 33 |
Table 2.
Identification of rabbit liver ADH1
Database searches were performed with Proteome Discoverer (PD) 2.2 software and the Sequest HT algorithm. The database search was conducted against an NCBI database for O. cuniculus that contained 38,600 entries with two missed trypsin-cleavage sites allowed.
| Accession number | Protein description | Percentage of protein sequence covered by identified peptides | Number of distinct peptides in the protein group | Total number of identified protein sequences, including those redundantly identified | Number of protein groups in which peptide is found | Molecular mass of peptide | Score Sequest HT: Sequest HT |
|---|---|---|---|---|---|---|---|
| kDa | |||||||
| NP_001095174.1 | Alcohol dehydrogenase 1 | 92 | 34 | 574 | 1 | 39.6 | 1808.66 |
| NP_001171758.1 | Alcohol dehydrogenase class-2 isozyme 2 | 61 | 16 | 44 | 1 | 40.5 | 125.73 |
| XP_008250935.2 | Predicted: low quality protein: 3-ketoacyl-CoA thiolase, peroxisomal | 68 | 18 | 35 | 1 | 45.6 | 104.83 |
| XP_008248783.1 | Predicted: 4-hydroxyphenylpyruvate dioxygenase | 59 | 16 | 35 | 1 | 44.8 | 95.62 |
| XP_008267298.2 | Predicted: low quality protein: sorbitol dehydrogenase | 44 | 9 | 30 | 1 | 37.9 | 82.13 |
| XP_008261518.1 | Predicted: arginase-1 isoform X1 | 58 | 15 | 22 | 1 | 37.4 | 74.05 |
| NP_001075740.1 | Phosphoserine aminotransferase | 44 | 14 | 24 | 1 | 40.6 | 58.6 |
| NP_001164585.1 | Fructose-bisphosphate aldolase B | 41 | 9 | 17 | 1 | 39.6 | 51.23 |
| NP_001075778.1 | serine–pyruvate aminotransferase | 25 | 8 | 11 | 1 | 43.1 | 29.62 |
| XP_002719410.1 | Predicted: keratin, type I cytoskeletal 10 isoform X1 | 7 | 4 | 4 | 1 | 60.8 | 12.42 |
| XP_002711256.1 | Predicted: probable imidazolonepropionase | 13 | 4 | 4 | 1 | 46.5 | 10.31 |
| NP_001095187.1 | Serine hydroxymethyltransferase, cytosolic | 8 | 3 | 4 | 1 | 52.9 | 7.92 |
| XP_002714459.1 | Predicted: glycine N-methyltransferase | 11 | 2 | 2 | 1 | 32.8 | 6.35 |
| XP_002719859.1 | Predicted: short-chain specific acyl-CoA dehydrogenase, mitochondrial | 6 | 2 | 2 | 1 | 44.3 | 5.55 |
| XP_002711597.1 | Predicted: aspartate aminotransferase, mitochondrial | 6 | 2 | 2 | 1 | 47.4 | 5.47 |
| XP_008269673.1 | Predicted: keratin, type I cytoskeletal 15 | 4 | 2 | 2 | 1 | 49.9 | 4.68 |
| XP_017197922.1 | Predicted: fructose-bisphosphate aldolase A isoform X1 | 5 | 2 | 2 | 1 | 45.2 | 3.25 |
Table 3.
Identification of rabbit liver SORD, fraction B1
Database searches were performed with Proteome Discoverer (PD) 2.2 software and the Sequest HT algorithm. The database search was conducted against an NCBI database for O. cuniculus that contained 38,600 entries with two missed trypsin-cleavage sites allowed.
| Accession number | Protein description | Percentage of protein sequence covered by identified peptides | Number of distinct peptides in the protein group | Total number of identified protein sequences, including those redundantly identified | Number of protein groups in which peptide is found | Molecular mass of peptide | Score Sequest HT: Sequest HT |
|---|---|---|---|---|---|---|---|
| kDa | |||||||
| XP_008267298.2 | Predicted: low quality protein: sorbitol dehydrogenase | 84 | 27 | 496 | 1 | 37.9 | 1589.24 |
| XP_002714459.1 | Predicted: glycine N-methyltransferase | 48 | 8 | 41 | 1 | 32.8 | 124.26 |
| NP_001182607.1 | Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase | 30 | 9 | 15 | 1 | 36.5 | 44.71 |
| XP_002711050.1 | Predicted: keratin, type II cytoskeletal 1 | 4 | 2 | 2 | 1 | 63.6 | 6.51 |
Table 4.
Identification of rabbit liver SORD degradation product, fraction B2
Database searches were performed with Proteome Discoverer (PD) 2.2 software and the Sequest HT algorithm. The database search was conducted against an NCBI database for O. cuniculus that contained 38,600 entries with two-missed trypsin-cleavage sites allowed.
| Accession number | Protein description | Percentage of protein sequence covered by identified peptides | Number of distinct peptides in the protein group | Total number of identified protein sequences, including those redundantly identified | Number of protein groups in which peptide is found | Molecular weight of peptide | Score Sequest HT: Sequest HT |
|---|---|---|---|---|---|---|---|
| kDa | |||||||
| XP_002714459.1 | Predicted: glycine N-methyltransferase | 78 | 25 | 297 | 1 | 32.8 | 1058.34 |
| XP_008267298.2 | Predicted: low quality protein: sorbitol dehydrogenase | 69 | 19 | 42 | 1 | 37.9 | 126.08 |
| XP_002719410.1 | Predicted: keratin, type I cytoskeletal 10 isoform X1 | 9 | 6 | 7 | 1 | 60.8 | 19.41 |
| XP_002711050.1 | Predicted: keratin, type II cytoskeletal 1 | 9 | 5 | 6 | 1 | 63.6 | 14.37 |
| XP_008254715.1 | Predicted: keratin, type II cytoskeletal 1b | 7 | 4 | 8 | 1 | 63.1 | 13.38 |
| XP_017197646.1 | Predicted: keratin, type II cytoskeletal 2 epidermal | 6 | 3 | 3 | 1 | 64.2 | 9.32 |
| NP_001182607.1 | Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase | 11 | 3 | 3 | 1 | 36.5 | 8.74 |
| XP_002711048.1 | Predicted: keratin, type II cytoskeletal 5 isoform X1 | 4 | 2 | 2 | 1 | 62.2 | 6.47 |
Human recombinant ADH1B1, ADH1C2, and SORD catalyze NADPH-dependent erythritol synthesis
To determine whether human ADH1 and SORD catalyze erythrose reduction to erythritol, three common variants of human ADH1 and human SORD were cloned into pet28a(+) to make His6 fusions proteins, which were expressed in Escherichia coli and purified. More specifically, the ADH1B1 and ADH1C2 cDNAs were directly cloned into pet28a(+). ADH1B2 results from a SNP of ADH1B1 (rs 1229984), which consists of a Gly to Ala transition that causes a single amino acid change (Arg to His). Site-directed mutagenesis of Arg-48 in ADH1B1 to His-48 was used to create the ADH1B2 protein (17). Human recombinant ADH1B1, ADH1C2, and SORD proteins all catalyze the NADPH-dependent conversion of d-erythrose to erythritol, although apparent Km values are about 5-fold higher for ADH1 variant proteins than for SORD in an enzymatic assay (Table 5, Fig. S2). Unexpectedly, the recombinant ADH1B2 protein was not active with respect to d-erythrose reduction activity with its physiological cofactor NADPH (Table 5, Fig. S2), although the recombinant protein was capable of catalyzing ethanol oxidation (ethanol Km = 1.5 mm), indicating that an active protein was purified. In addition, the human ADH1 variant proteins and SORD were capable of reducing d-erythrose to erythritol when NADH was used as a cofactor (Table 5, Fig. S3), but this reaction may only occur in enzymatic assays in vitro, as ADH1 and SORD are cytosolic enzymes (18, 19) and NADH levels are relatively low in the cytosol (20). It is also worth noting that none of the ADH1 variants, or SORD exhibited erythrose reduction activity in vitro when the unnatural sugar isomer l-erythrose was used as a substrate, regardless of whether the cofactor was NADPH or NADH.
Table 5.
Kinetic characterization of erythrose reduction activity of human recombinant ADH1 variant proteins and SORD in vitro using NADPH as the cofactor
Reaction buffer consisted of 50 mm sodium phosphate, pH 7.2, 200 μm NADPH or NADH, and at least 5 concentrations of erythrose ranging from 17.5 to 1120 mm. Reactions were initiated with addition of 100 nm purified protein. The rate of NADPH or NADH consumption at 340 nm was monitored using a Shimadzu UV-2600 spectrophotometer. Measurements at each substrate concentration were performed in duplicate (two technical replicates). For ADH1B2, ADH1C2, and SORD, the Km and Vmax values from each independent experiment was determined by fitting the data to Y = Vmax × X/(Km + X), where Y represents reaction velocity and X indicates substrate concentration using GraphPad Prism software. For ADH1B1, the Km,Vmax, and Ki values from each independent experiment was determined by fitting the data to Y = Vmax × X/(Km + X × (1 + X/Ki)), where Y represents reaction velocity and X indicates substrate concentration using GraphPad Prism software. Data are shown as average and S.D. of two independent experiments. Km values are presented as “apparent Km” because these values were determined at only one saturating concentration of cofactor.
| Allele | Highest prevalence | Apparent Km | kcat | kcat/Km | Substrate inhibition |
|---|---|---|---|---|---|
| mm | s−1 | m−1 s−1 | |||
| NADPH used as cofactor | |||||
| ADH1B1 | Caucasian, African American | 322 ± 168 | 0.9 ± 0.3 | 1.4 ± 1.2 | Ki = 335 ± 237 mm |
| ADH1B2 | Asian | No activity detected | No activity detected | No activity detected | No activity detected |
| ADH1C2 | Caucasian | 518 ± 180 | 0.40 ± 0.07 | 0.9 ± 0.3 | None |
| SORD | 118 ± 11 | 1.58 ± 0.05 | 13.8 ± 1.4 | None | |
| NADH used as cofactor | |||||
| ADH1B1 | Caucasian, African American | 272 ± 70 | 1.04 ± 0.19 | 3.8 ± 1.2 | Ki = 403 ± 127 mm |
| ADH1B2 | Asian | 228 ± 36 | 0.70 ± 0.05 | 3.1 ± 0.6 | None |
| ADH1C2 | Caucasian | 443 ± 78 | 1.60 ± 0.17 | 3.6 ± 0.7 | None |
| SORD | 94 ± 2 | 4.10 ± 0.03 | 43.1 ± 2.0 | None |
A computational modeling analysis of the ADH1 active site containing a zinc atom, d-erythrose, and NADPH reveals that the Arg-47 residue of ADH1B1 makes three contacts with NADPH (Fig. 4A), including two contacts with the phosphate moiety that distinguishes NADPH from NADH. It is important to note that the primary structure of ADH1B1 in the protein data bank file lacks the start methionine residue, so Arg-47 is equivalent to Arg-48 in the cDNA of the recombinant protein assayed. His-47 of ADH1B2 makes only one contact with NADPH (Fig. 4B), and no contacts with the distinguishing phosphate moiety, suggesting that the lack of activity observed in ADH1B2 with erythrose and NADPH is the result of impaired cofactor binding by ADH1B2. SORD and ADH1 are structurally and kinetically homologous (21), but the binding site for NADH or NADPH is more open in SORD; several of the NADH/NADPH-protein contacts found in ADH, including those involving residue 47, are replaced with NADH/NADPH solvent contacts (Fig. 4, A–C). However, there appears to be a specific contact made between the phosphate moiety of NADPH and SORD Arg-208 (Fig. 4C); in ADH the corresponding residue is Lys-228, which is present in all the forms studied and also contacts NADPH. To interrogate the role of the SORD Arg-208 residue, recombinant protein was expressed in which Arg-208 of SORD was mutated to histidine. The SORD R208H mutation did not affect the apparent Km for d-erythrose (118 ± 11 for the WT compared with 141 ± 26 mm for R208H, p > 0.05, Fig. S4) when NADPH was used as the cofactor, but did reduce kcat to less than 20% of WT levels (1.6 ± 0.1 compared with 0.23 ± 0.02 s−1 for R208H, p < 0.05, Fig. S4). These observations suggest that arginine contributes to NADPH binding in these enzymes (22).
Figure 4.

Active site model of the ADH1B1, ADH1B2, SORD, erythrose, and NADPH complex. Modeled NADP(H) binding sites for (A) ADH1B1, (B) ADH1B2, and (C) SORD. In ADH1B1, residue Arg-47 makes 3 H-bonds with NADP(H); in the B2 variant the corresponding residue, His-47, makes only one. In SORD, the corresponding residue is Gly-45; it makes no contacts with NADP(H), but residue Arg-208 is in position for H-bonding. Modeled H-bonds are labeled with their lengths, in Ångstroms. Zinc (gray) and a modeled bound erythrose (brown) are included, with their coordinating residues. The main chain is shown as a green “worm” except for residues involved in coordinating ligands. Protein surrounding the active site is shown as a gray surface, with portions in front of the ligands omitted for clarity. The figure was prepared with the program CCP4MG.
SORD contributes to erythritol production in A549 cells
To confirm that the identified enzymes, ADH1 and SORD, contribute to endogenous erythritol formation in human cells, we performed silencing experiments using siRNA in A549 lung cancer cells and subsequently measured intracellular erythritol levels. Forty-eight hours after siRNA application to the cells, we determined the expression levels of the target genes by qPCR to verify knockdown (Fig. 5, a and b). This demonstrated that in A549 cells, ADH1B was not expressed at all (data not shown), whereas ADH1C was expressed at very low levels indicated by the consistently high CT values (CT > 34) (Fig. 5a). This is in line with the publicly available expression data reported by the Human Protein Atlas (www.proteinatlas.org) (43)3 for ADH1B and ADH1C in A549 cells. As a result, A549 cells cannot be used to determine the role of ADH1B and ADH1C in erythritol synthesis and further studies in suitable culture models are required. SORD-targeting siRNA, however, significantly reduced SORD mRNA levels to less than 15% of control levels (p < 0.01, Fig. 5b). Intracellular erythritol levels were reduced by 50% as a result of SORD knockdown (p < 0.001, Fig. 5c). Moreover, based on the obtained mass isotopomer distributions (MIDs) after labeling with [U-13C]glucose (Fig. 5d), the entire intracellular erythritol pool was labeled at all four carbon atoms (M4) suggesting that in A549 lung cancer cells, 100% of the intracellular erythritol is derived from glucose.
Figure 5.

SORD contributes to intracellular erythritol in A549 lung cancer cells. Validation of gene expression knockdown, intracellular erythritol levels, and erythritol labeling from [U-13C]glucose. mRNA levels were determined by quantitative real-time PCR for (a) ADH1C and (b) SORD, (c) resulting intracellular erythritol levels represented by the measurement of the derivative erythritol_4TMS: signal intensity was normalized to internal standard (IS) d6-pentanedioic acid and live cell number. d, MID of erythritol_4TMS (mean ± S.E.) after 30 h of [U-13C]glucose labeling in A549 lung cancer cells, extraction and derivatization; obtained p values compared with siCtrl using Welch's t test and Bonferroni correction (n = 5) to adjust for multiple comparison: *, p < 0.05; **, p < 0.01; ***, p < 0.001 (n = 4, representative results).
Discussion
Two recent publications revealed a strong correlation between plasma erythritol levels and incident central adiposity gain (3), increased glycaemia (3), and T2DM onset (4). In addition, one study uncovered the capacity of human metabolism to endogenously synthesize erythritol via E4P in the PPP from glucose (3). The human metabolic erythritol synthesis pathway is further characterized here. In humans, the final step of erythritol formation requires erythrose rather than E4P as the direct substrate and the enzymes that catalyze this reaction prefer NADPH over NADH as a cofactor (Fig. 2). These finding are analogous to erythritol-producing enzymes from eukaryotes including C. magnoliae, Trichoderma reesei, Aspergillis niger, and Fusarium graminearum, that also require NADPH as a cofactor (13, 15).
Two enzymes catalyzing the NADPH-dependent conversion of erythrose to erythritol, namely ADH1 and SORD, were purified from rabbit liver (Fig. 3). ADH1 constitutes one of six classes of mammalian alcohol dehydrogenases (23). One primary function of ADH1 is the NAD+-dependent oxidation of ethanol to acetaldehyde for alcohol detoxification (19). Different ethnic groups exhibit variable capacity to metabolize ethanol, reflecting the varying kinetic properties of the allelic variants of ADH1 that exist among populations (19, 24). SORD catalyzes the NAD+-dependent conversion of sorbitol to fructose, which constitutes the second step of the polyol pathway (21). The flux through the polyol pathway is increased under hyperglycemic conditions as occur in T2DM (18, 21).
The kinetic properties of three human recombinant variants of ADH1 including ADH1B1, ADH1B2, and ADH1C2 and human recombinant SORD were characterized with respect to their ability to catalyze the NADPH-dependent conversion of erythrose to erythritol. These represent the predominant variants in the Caucasian/African America, Asian, and Caucasian populations, respectively (19). SORD exhibited a lower Km and higher kcat value than the ADH1 proteins (Table 3), suggesting that SORD more efficiently catalyzes the conversion of erythrose to erythritol as compared with the ADH1 variants.
Interestingly, the ADH1B2 variant protein did not catalyze the conversion of erythrose to erythritol in vitro when the physiological cofactor, NADPH, was included in the reaction (Table 3). The lack of erythrose reduction activity of ADH1B2 may be explained in part by substitution of arginine with histidine at position 48, which eliminates a hydrogen bond to the phosphate moiety of NADPH (Fig. 4). The homology model suggests that this substitution reduces the potential for NADPH to bind to the enzyme, apparently to the point where catalysis does not occur. The importance of Arg-48 for ADH1B1 and Arg-208 for SORD has been recognized before in the context of NAD+ binding (25, 26). However, to our knowledge, the essentiality of the Arg-48 residue for NADPH-dependent function of ADH1B2 has not been described before. ADH1B2 exhibits a high catalytic efficiency for ethanol using NAD as a cofactor (24) and exhibits erythrose reductase activity when NADH is used as a cofactor. As described above, use of NADH as a cofactor for erythritol synthesis likely does not occur in vivo because ADH1 and SORD localize primarily to the cytosol, where NADH levels are low (20). In addition, our results clearly indicated that the enzymes catalyzing the reaction, ADH1 and SORD, distinguish between the stereoisomers d- and l-erythrose. Only the physiological isomer, d-erythrose, was enzymatically converted to erythritol. This is also consistent with the inability to model l-erythrose into the active site of ADH1B1 without creating unacceptably close interatomic contacts.
siRNA targeting SORD demonstrated that SORD catalyzes the formation of 50% of the intracellular erythritol pool in A549 cells (Fig. 5c). Moreover, based on stable isotope labeling using [U-13C]glucose as a tracer, intracellular erythritol is derived exclusively from glucose in A549 cells (Fig. 5d). These findings indicate that, in a controlled in vitro experiment, all possible precursors of erythritol, e.g. erythrose and E4P, are derived from glucose. In the experimental setup chosen by Hootman et al. (3) intracellular erythritol in blood cells was labeled to a lesser extent from [U-13C]glucose. This may be explained by a significantly shorter incubation time of the blood cells with the tracer. In that experiment, only 50% of the total glucose available was labeled after tracer addition. In addition, whole blood represents a very complex mixture containing a vast array of metabolites; additional, nonlabeled metabolites might be present that serve as potential substrates for erythritol synthesis.
Hootman et al. (3) demonstrated a strong association between elevated plasma erythritol and incident central adiposity gain and elevated glycemia. However, it is unclear whether increased dietary intake or increased endogenous synthesis of erythritol from glucose lead to these increased plasma erythritol levels (3). Our results revealed two enzymes capable of erythritol synthesis from the substrate erythrose. Therefore, enzymatic function of either SORD or ADH1 could result in altered erythrose metabolism and directly influence plasma erythritol levels. Based on the well-known association between hyperglycemia and increased polyol pathway flux (18, 27), increased plasma erythritol levels might represent a SORD-dependent, early-stage readout of hyperglycemia. Additionally, ADH1B is expressed in adipose tissue where expression strongly correlated with increased waist circumference, BMI, and insulin resistance (28). This suggests that increased central adiposity may lead to increased erythritol synthesis because of increased ADH1B expression.
The findings presented here have some important limitations that highlight the need for ongoing investigation. First, the plots of enzyme activity (Figs. S1–S4) used to determine kinetic parameters for erythritol synthesis do not fully saturate, which decreases the reliability of some of the resulting kinetic parameters, although it does not significantly impact the primary findings of the study. This is due largely to the lack of erythrose of sufficient purity and concentration from commercial sources required for the highest concentrations in kinetic assays. Second, the expression levels of ADH1 variants were too low to obtain meaningful conclusions on the contribution of ADH1 to intracellular erythritol synthesis in A549 lung cancer cells. Third, although erythritol formation was significantly reduced upon SORD knockdown, considerable amounts of erythritol were still produced, suggesting that there may be an additional enzyme capable of erythritol synthesis present in A549 lung cancer cells. Finally, erythrose reductase activity in A549 cells was not detected when excess NADH was added to the cell lysate, which contrasts the observation that the recombinant human ADH1 and SORD proteins were capable of using NADH as a cofactor in vitro. This could be due to differences in binding affinity of NADH and NADPH for ADH1 and/or SORD. Although outside the scope of the current study, this is worth further characterization in other cell types, especially noncancer cell types.
In summary, we identified two human enzymes, ADH1 and SORD, that catalyze the NADPH-dependent reduction of erythrose to erythritol in humans. These studies have not only identified previously unknown metabolic activities for these enzymes, but have further characterized this novel human metabolic pathway. Based on the in vitro kinetic properties obtained for human SORD and the ADH1 variants, catalysis of erythritol formation by SORD is more efficient than that of ADH1. In addition, SORD meaningfully contributes to intracellular erythritol production in A549 lung cancer cells. Further studies are needed to determine the contribution of allelic variants of ADH1 to intracellular erythritol formation in ADH1-expressing tissues, such as liver. Importantly, these findings also suggest that the relationships among ADH1 variants and/or SORD expression, glucose utilization, and ultimately energy balance may contribute, in part, to individual differences in response to diet.
Experimental procedures
Cell culture and preparation of A549 cell extract
Human A549 lung cancer cells were cultured in Dulbecco's modified Eagle's medium (DMEM D5796) supplemented with 10% fetal bovine serum and grown in a humidified incubator at 37 °C, 5% CO2. To prepare sufficient amounts of cell extract, 106 A549 lung cancer cells were seeded on a 10-cm cell culture dish until confluence reached 90 to 100%. Culture medium was removed and cells were washed with PBS once prior to cell lysis. 800 μl of M-PER protein extraction reagent (Thermo) completed with protease inhibitor mixture (Roche) was applied and cells were detached from the plate by scraping on ice. The liquid was collected in a 1.5-ml reaction tube and centrifuged for 5 min at 15,000 × g and 4 °C. The supernatant was directly used in the enzyme assay.
Enzyme activity assay using A549 cell extract
The enzymatic assay was performed in cuvettes at 37 °C with a total volume of 1 ml. The assay mixture was composed of 15 mm sodium pyrophosphate buffer, pH 8.5, and 150 μl of A549 cell extract. The cofactors NADH and NADPH (final concentration: 400 μm) and substrates erythrose (final concentration: 3 mm) and erythrose-4-phosphate (final concentration: 400 μm) were tested, and enzyme activity was determined spectrophotometrically by monitoring A340 upon oxidation of NADH/NADPH.
Time-resolved metabolite extraction
To confirm the formation of erythritol during the enzymatic assay, we performed a metabolite extraction at several time points (0, 20, 60, and 120 min) during the enzyme assay. To do so, 10 μl of assay solution was added to 90 μl of ice-cold extraction fluid composed of methanol and water in a ratio of 9:1. The mixture was vortexed for 5 min at 1,400 rpm and 4 °C and subsequently centrifuged for 5 min at 16,000 × g and 4 °C. 70 μl of the supernatant were transferred in a glass vial with inlet and dried in a refrigerated vacuum centrifuge at −4 °C overnight.
Knockdown of ADH and SORD in A549 cells using siRNA
Nontargeting siRNA as well as siRNAs for ADH1B (L-008112-00-0005), ADH1C (L-006521-00-0005), and SORD (L-008323-00-0005) were purchased from Dharmacon and sequences are given in Table S1. A549 cells were reversely transfected using RNAiMAX (Thermo Fisher Scientific) according to manufacturer's instructions. Briefly, per well (12-well plate), 1 μl of 20 μm siRNA and 2.5 μl of RNAiMAX were diluted in 200 μl of OptiMEM (Thermo Fisher Scientific), mixed gently, and incubated for 20 min at room temperature. The solution was applied to the well 5 min prior to cell seeding (100,000 cells in 800 μl of Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum). Transfected cells were incubated for 48 h until metabolite extraction or cell counting.
Stable isotope labeling of A549 cells with [U-13C]glucose
17 h post-transfection, the nonlabeled standard culture medium was removed and replaced by the tracer medium containing 25 mm [U-13C]glucose. The cells were incubated with the tracer for an additional 30 h.
Extraction of intracellular metabolites
Intracellular metabolites were extracted as described by Sapcariu et al. (29). Briefly, medium was removed and cells were washed with 0.9% NaCl. 200 μl of ice-cold methanol and ddH2O containing 2 μg/ml of d6-pentanedioic acid as internal standard was added to the well and cells were scraped on a cold plate. The liquid was transferred to a 2-ml reaction tube containing 200 μl of chloroform. The mixture was then vortexed for 20 min at 4 °C and 1400 rpm and subsequently centrifuged for 5 min at 4 °C and 17,000 × g. 200 μl of the upper, polar phase were transferred to a glass vial and dried in a refrigerated vacuum centrifuge overnight at 4 °C.
RNA extraction and cDNA production
RNA was extracted from the interphase obtained during “Extraction of intracellular metabolites” using the NucleoSpin RNA kit (Macherey and Nagel); it was then reversely transcribed to cDNA using qScript cDNA SuperMix (Quantabio) according to the manufacturer's instructions.
Knockdown verification by qPCR
Individual 20-μl qPCR consisted of 10 μl of 2× iTaqTM Universal Probes Supermix (Bio-Rad), 4 μl of RNase-free H2O, 4 μl of diluted cDNA, and 1 μl each of dye-labeled ×20 TaqManTM Gene Expression Assay primer mix (forward and reverse; ADH1B (Hs00605175_m1), ADH1C (Hs02383872_s1), SORD (Hs00162091_m1), S18 (Hs01375212_g1)), where the housekeeping gene ribosomal protein S18 (RPS18) was labeled with VIC fluorescent dye, the target genes were labeled with FAM fluorescent dye to enable multiplexing. qPCR was carried out on a QuantStudio 5 (Thermo Fisher), using a comparative CT program provided by the manufacturer. All experiments were performed in three biological replicates and included three controls. RPS18 was amplified simultaneously for normalization and ΔCT was calculated for the relative quantification of the targeted genes. Data were analyzed using a Welch's t test and Bonferroni correction (n = 5) to adjust for multiple comparison.
Derivatization and GC-MS measurement of erythritol
Prior to GC-MS measurement, the dried extracts were derivatized using a Gerstel MPS as described previously (30). Briefly, dried polar metabolites were dissolved in 15 μl of 2% methoxyamine hydrochloride in pyridine and incubated for 90 min at 40 °C under continuous shaking. Thereafter, 15 μl of N-methyl-N-TMS-fluoroacetamide was added and incubated for 30 min at 40 °C under continuous shaking. For erythritol, the 4TMS derivative was detected (M: C16H42O4Si4, m/z 410), however, the fragment M-90 (C13H32O3Si3, m/z 320) was used to calculate the MID. The respective structure and resulting fragment are provided in Fig. S5.
GC method 1
1 μl of sample was injected into an SSL injector at 270 °C in split mode (1:10). GC was performed using an Agilent 7890A GC equipped with a 30m DB-35MS capillary column (0.25 mm internal diameter, 0.25 μm film thickness). We used helium as the carrier gas at a flow rate of 1.0 ml/min. The GC-MS oven temperature was held at 80 °C. After 6 min at 80 °C, the temperature was increased to 300 °C at a rate of 6 °C/min. When 300 °C was reached, the temperature was further increased to 325 °C at a rate of 10 °C/min and held for 4 min. This temperature profile results in a total run time of 59.167 min.
GC method 2
1 μl of sample was injected into an SSL injector at 270 °C in splitless mode. GC-MS analysis was performed using an Agilent 7890A GC equipped with a 30m DB-35MS + 5m Duraguard capillary column (0.25 mm inner diameter, 0.25 μm film thickness). Helium was used as the carrier gas at a flow rate of 1.0 ml/min. The GC oven temperature was held at 90 °C for 1 min, subsequently increased to 270 °C at 9 °C/min, and then further increased to 320 °C at 25 °C/min and held for 7 min resulting in a total run time of 30 min per sample.
Mass spectrometry
Tuning
According to the supplier's instruction, an automated tuning routine is applied every 150 injections.
SCAN mode
The GC was connected to an Agilent 5975C inert XL MSD. The transfer line temperature was set to 280 °C and the MSD was operating under electron ionization at 70 eV. The MS source was held at 230 °C and the quadrupole at 150 °C. Full scan mass spectra were acquired from m/z 70 to 700 at a scan rate of 5.2 scans/s and a solvent delay of 5 min. GC-MS chromatograms were analyzed using the MetaboliteDetector software package (31).
Protein purification from rabbit liver
Preparation of rabbit liver extract
Four frozen rabbit livers were thawed at room temperature, and homogenized in 50 mm sodium phosphate buffer, pH 6.8, containing β-mercaptoethanol (0.1%, v/v) and PEG3350 (10%, w/v) in a Waring blender. The homogenate was found to have NADPH- and NADH-dependent erythrose dehydrogenase activity. The suspension was clarified by centrifugation at 9000 rpm at 4 °C for 30 min. The pellet did not contain erythritol synthesis activity and was discarded. PEG3350 was added to the supernatant to a concentration of 50% (w/v). The resulting suspension was stirred at room temperature for 30 min and clarified by centrifugation at 9000 rpm and 4 °C for 30 min. The pellet, but not the supernatant, was found to have erythritol synthesis activity and was dissolved in 20 mm sodium phosphate buffer, pH 6.8.
The dissolved pellet was applied to a CM Sephadex C50 column (bed size: height, 17 cm; radius, 1.5 cm) equilibrated with 10 mm sodium phosphate buffer, pH 6.8. The column was washed with 20 mm sodium phosphate buffer, pH 6.8, until the absorbance at 280 nm was less than 0.1. Proteins that bound were eluted with 200 mm potassium phosphate buffer, pH 7.5. The activity that bound to the CM column was labeled as “protein A,” and the activity present in the flow-through was labeled as “protein B.”
Purification of protein A
Ammonium sulfate fractionation
The solutions containing proteins A and B were precipitated using ammonium sulfate (30%, w/v) and the suspension was clarified by centrifugation (9000 rpm, 4 °C, 30 min). Both proteins A and B were present in the protein pellet. The pellets were dissolved in 50 mm sodium phosphate buffer, pH 6.8, and dialyzed overnight against 20 mm potassium phosphate buffer, pH 7.2.
Heat treatment
The dialyzed fraction of protein A was heat treated at 60 °C and denatured protein was removed by centrifugation at 12,000 rpm and 4 °C for 20 min. The ammonium sulfate concentration of the supernatant was adjusted to 50% (w/v) and the suspension was clarified by centrifugation at 12,000 rpm and 4 °C for 20 min. The pellet was dissolved in 10 mm potassium phosphate buffer, pH 7.2, and dialyzed overnight against 10 mm potassium phosphate buffer, pH 7.2.
Size exclusion chromatography
The dialyzed fraction of protein A was loaded onto a Sephadex G 100–120 column (particle size: 40–200 μm, bed size: h, 43 cm; r, 1 cm) equilibrated with 10 mm potassium phosphate buffer, pH 7.2. Fractions were collected until the absorbance at 280 nm was less than 0.1. Active fractions were pooled.
Hydroxyapaptite chromatography
The active fraction containing protein A from the size exclusion column was diluted 1:2 with 10 mm potassium phosphate buffer, pH 6.8, and loaded on a hydroxyapatite column (BioGel, Bio-Rad; bed size: h, 10 cm; r, 1 cm). A 10–200 mm phosphate gradient was used for the elution of the binding proteins, and fractions were collected until the absorbance at 280 nm was less than 0.1. The active fractions were combined and concentrated in a size exclusion filter tube with a cut off of 5 kDa (Spin-X UF, 5K MWCO PES, 431487 Corning). The concentrated fraction was used for further characterization.
Purification of protein B
DEAE Sephadex chromatography
The dialyzed fraction containing protein B was applied to a DEAE Sephadex column (bed size: h, 9 cm; r, 2.75 cm) equilibrated with 10 mm potassium phosphate buffer, pH 6.8, and active fractions of the DEAE flow-through were combined. The ammonium sulfate concentration was adjusted to 50% (w/v) and the suspension was clarified by centrifugation at 12,000 rpm and 4 °C for 30 min. The pellet was dissolved in 10 mm potassium phosphate buffer at pH 6.8 and the PEG3350 concentration was adjusted to 40% (w/v). The precipitated protein was harvested by centrifugation at 12,000 rpm and 4 °C for 30 min and dissolved in 10 mm potassium phosphate buffer, pH 6.8.
Size exclusion chromatography
The active fraction containing protein B from the DEAE Sephadex column was applied to a Sephadex G 100–120 column (particle size: 40–200 μm, bed size: h, 43 cm; r, 1 cm) equilibrated with 10 mm potassium phosphate buffer, pH 7.2. Fractions were collected until the absorbance at 280 nm was less than 0.1 and active fractions were pooled.
Hydroxyapatite chromatography
The active fraction containing protein B from the size exclusion column was diluted 1:2 with 10 mm potassium phosphate buffer, pH 6.8, and applied to a hydroxyapatite column (BioGel, Bio-Rad; bed size: h, 5 cm; r, 1.25 cm). The active protein B was present in the flow-through. Fractions were collected until the absorbance at 280 nm was less than 0.1 and all active fractions were combined.
Phenyl-Sepharose chromatography
The salt content of the flow-through fraction containing the active protein B from the hydroxyapatite column was increased by adjusting to a NaCl concentration of 2 m. The high salt fraction was applied to a phenyl-Sepharose column (bed size: h, 10 cm; r, 1 cm) equilibrated with 2 m NaCl in 10 mm potassium phosphate buffer, pH 6.8. Protein B bound to the phenyl-Sepharose column and was eluted with 10 mm potassium phosphate buffer, pH 6.8. Fractions were collected until the absorbance at 280 nm was less than 0.1 and the active fractions were pooled and concentrated using a size exclusion concentrator with a cutoff of 5 kDa (Spin-X UF, 5K MWCO PES, Corning). The concentrated fraction was used for further characterization.
Activity assay
Throughout purification, erythrose reduction activity was determined sprectrophotometrically by monitoring A340 upon oxidation of NADPH at room temperature to identify fractions with the activity of interest. During purification, the assay mixture contained 900 μl of 50 mm sodium phosphate buffer, pH 6.8, 200 μm NADPH (Sigma), 50 μl of enzyme solution, and 11.6 mm erythrose (Sigma). The reaction was initiated by addition of the substrate. Kinetic constants were also determined for the proteins that were subject to SDS-PAGE and subsequent proteomic identification. In this assay, the reaction buffer consisted of 50 mm sodium phosphate buffer, pH 6.8, 200 μm NADPH or NADH, and at least 5 concentrations of erythrose ranging from 0.5 to 100 mm. Reactions were initiated with addition of purified protein; protein concentration was determined as previously described (32). Final concentrations of protein were 49 μm fraction A (ADH1) for NADPH, 25 μm fraction A (ADH1) for NADH, 0.7 μm fraction B (SORD) for NADPH, and 2.6 μm fraction B (SORD) for NADH. Kinetic constants were determined for the NAD-dependent conversion of ethanol to acetaldehyde for fraction A (ADH) by monitoring production of NADH at 340 nm. Kinetic constants were also determined for the NAD-dependent conversion of sorbitol to fructose for fraction B (SORD) by monitoring production of NADH at 340 nm. Reactions were initiated with addition of purified protein (final concentration 1.3 μm for fraction A (ADH) and 0.13 μm for fraction B (SORD)). Km and Vmax values determined from fitting the data to Y = Vmax × X/(Km + X), where Y represents reaction velocity and X indicates substrate concentration using GraphPad Prism software (Michaelis-Menten kinetics), and kcat values were determined by dividing Vmax by the concentration of purified protein in the assay, which was determined as previously described (32).
Identification of proteins isolated from rabbit liver
In-gel trypsin digestion of SDS gel bands
The protein bands from an SDS-PAGE gel (3 slices: A1, B1, and B2) were cut into ∼1 mm cubes and subjected to in-gel digestion followed by extraction of the tryptic peptide as reported previously (33). The excised gel pieces were washed consecutively in 200 μl of distilled water, 100 mm ammonium bicarbonate (Ambic)/acetonitrile (1:1), and acetonitrile (ACN). The gel pieces were reduced with 70 μl of 10 mm DTT in 100 mm Ambic for 1 h at 56 °C, alkylated with 100 μl of 55 mm iodoacetamide in 100 mm Ambic at room temperature in the dark for 60 min. After the wash steps as described above, the gel slices were dried and rehydrated with 50 μl of trypsin in 50 mm Ambic, 10% ACN (20 ng/μl) at 37 °C for 16 h. The digested peptides were extracted twice with 70 μl of 50% acetonitrile, 5% formic acid (FA), and once with 70 μl of 90% acetonitrile, 5% FA. Extracts from each sample were combined and lyophilized.
Protein identification by nano-LC/MS/MS analysis
The in-gel tryptic digests were reconstituted in 20 μl of 0.5% FA for nano-LC-ESI-MS/MS analysis, which was carried out using an Orbitrap FusionTM TribridTM (Thermo Fisher Scientific, San Jose, CA) mass spectrometer equipped with a nanospray Flex Ion Source, and coupled with a Dionex UltiMate 3000RSLC nano system (Thermo, Sunnyvale, CA) (34, 35). The gel-extracted peptide samples (19 μl) were injected onto a PepMap C-18 RP Nano-Trap column (5 μm, 100 μm inner diameter × 20 mm, Dionex) with nanoViper Fittings at 20 μl/min flow rate for on-line desalting and then separated on a PepMap C-18 RP nano column (2 μm, 75 μm × 25 cm) at 35 °C, and eluted in a 120-min gradient of 5 to 38% ACN in 0.1% FA at 300 nl/min, followed by an 8-min ramping to 90% ACN, 0.1% FA, and a 9-min hold at 90% ACN, 0.1% FA. The column was re-equilibrated with 0.1% FA for 25 min prior to the next run. The Orbitrap Fusion is operated in positive ion mode with spray voltage set at 1.6 kV and source temperature at 275 °C. External calibration for FT, IT, and quadrupole mass analyzers was performed. In data-dependent acquisition (DDA) analysis, the instrument was operated using FT mass analyzer in MS scan to select precursor ions followed by 3-s “Top Speed” data-dependent CID ion trap MS/MS scans at 1.6 m/z quadrupole isolation for precursor peptides with multiple charged ions above a threshold ion count of 10,000 and normalized collision energy of 30%. MS survey scans were done at a resolving power of 120,000 (full width at half-maximum at m/z 200), for the mass range of m/z 375–1575. Dynamic exclusion parameters were set at 50 s of exclusion duration with ±10 ppm exclusion mass width. All data were acquired under Xcalibur 3.0 operation software (Thermo Fisher Scientific).
Data analysis
The DDA raw files for CID MS/MS only were subjected to database searches using Proteome Discoverer (PD) 2.2 software (Thermo Fisher Scientific, Bremen, Germany) with the Sequest HT algorithm. The database search was conducted against an NCBI database for Oryctolagus cuniculus that contained 38,600 entries (downloaded on 4/4/2017 from NCBInr) with two missed trypsin-cleavage sites allowed. The peptide precursor tolerance was set to 10 ppm and fragment ion tolerance was set to 0.6 Da. Variable modification of methionine oxidation, N-terminal acetylation, and deamidation of asparagines/glutamine and fixed modification of cysteine carbamidomethylation, were set for the database search. Only high confidence peptides defined by Sequest HT with a 1% FDR by Percolator were considered for the peptide identification. The final protein IDs contained protein groups that were filtered with at least 2 peptides per protein. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (36) partner repository with the dataset identifier PXD015178.
Cloning, expression, and purification of recombinant human ADH1 and SORD proteins
ADH1B1 (clone ID 4828235), ADH1C2 (clone ID 30408761), and SORD (clone ID 2989137) cDNA constructs were purchased from GE Dharmacon. ADH1B1 was cloned into the pet28a(+) vector using EcoRI and XhoI restriction sites (forward primer: 5′-aagaattcatgagcacagcaggaaaagtaatcaaatg-3′; reverse primer: 5′-aactcgagtcaaaacgtcaggacggtacggata-3′). ADH1C2 was cloned into the pet28a(+) vector using EcoRI and XhoI restriction sites (forward primer: 5′-aagaattcatgagcacagcaggaaaagtaatcaaatg-3′; reverse primer: 5′-aactcgagtcaaaacgtcaggacggtacggata-3′). SORD was cloned into the pet28a(+) vector using SacI and XhoI restriction sites (forward primer: 5′-aagagctcatggcggcggcggccaa-3′; reverse primer: 5′-aactcgagtcagggattctggtcactggggtc-3′). PCR was performed using GoTaq (Promega) according to the manufacturer's instructions. Pet28a-ADH1B2 was generated by mutating the Arg-48 codon of pet28a-ADH1B1 to His-48 using the QuikChange II kit (Stratagene) per the manufacturer's instructions (forward primer: 5′-ctgtaggaatctgtcacacagatgaccacgtg-3′; reverse primer: 5′-cacgtggtcatctgtgtgacagattcctacag-3′). Pet28a-SORD-R208H was also generated using the QuikChange II kit with primers 5′-gatctgtctgctacccatttgtccaaagccaagg-3′ and 5′-ccttggctttggacaaatgggtagcagacagatc-3′. All constructs were sequence verified and then transfected into BL21(DE3) cells (Invitrogen) for recombinant protein expression. His-ADH1B1, His-ADH1B2, His-ADH1C2, and His-SORD (WT and R208H) were expressed in BL21(DE3) cells grown to mid-late log phase in Luria-Bertani broth containing 50 μg/ml of kanamycin. Expression was induced using 750 μm isopropyl β-d-thiogalactopyranoside after which bacteria were grown overnight at 16 °C. Purification was carried out as previously described for His-MTHFS (37). The erythrose reduction activity was determined spectrophotometrically by monitoring A340 upon oxidation of NADPH at room temperature. The assay buffer consisted of 50 mm sodium phosphate buffer, pH 6.8, 200 μm NADPH (or NADH), and at least 5 different concentrations of erythrose (measured in duplicate) ranging from 17.5 to 1120 mm. The reaction was initiated by addition of 100 nm recombinant protein. For ADH1B2, ADH1C2, and SORD (WT and R208H) Km and Vmax values and their associated standard deviations were determined from fitting the data to Y = Vmax × X/(Km + X), where Y represents reaction velocity and X indicates substrate concentration using GraphPad Prism software. For ADH1B1, the Km, Vmax, and Ki values from each independent experiment was determined by fitting the data to Y = Vmax × X/(Km + X × (1 + X/Ki)), where Y represents reaction velocity and X indicates substrate concentration using GraphPad Prism software. kcat values were determined by dividing Vmax by the enzyme concentration (determined as previously described (32)). Activity at each substrate concentration was measured in duplicate. Data are reported as average and standard deviation of two independent determinations.
Model construction
A model of ADH1B1 with bound Zn2+, NAD+, and erythrose was constructed from PDB entry 1U3U, which contains bound Zn2+, NAD+, and an N-benzylformamide inhibitor, by replacing the inhibitor with d-erythrose. Erythrose was placed manually, using Coot (38), in position to accept a hydrogen from NADH, as expected according to the reaction mechanism proposed by Agarwal et al. (39). A second model, with NAD+ replaced by NADPH, was generated by matching NADPH from the HIC-Up database (40) with NAD+ and manually adjusting it to optimize fit in the binding site. Similar models were constructed for ADH1B2 and ADH1C2, starting with PDB entries 1HDY and 1U3W, respectively.
For SORD, structures available in the PDB include 1PL6, with Zn2+ and NAD+ bound, and 1PL8, with Zn2+, NAD+, and an inhibitor bound. From these, plus the structure of sorbitol from PDB entry 5A06, a model was first constructed for SORD, Zn2+-NAD+-sorbitol. In this case, the sorbitol was positioned so that a primary hydroxyl was adjacent to the Zn2+, whereas the adjacent secondary hydroxyl was in position for hydride transfer to NAD+. This is different from the ADH case, where the same primary hydroxyl in the canonical substrate ethanol interacts with both Zn2+ and NAD+. From the sorbitol-containing version, models were constructed, replacing sorbitol with erythrose, and NAD+ with NADPH, using the same procedure as for ADH.
For each model, hydrogen atoms were generated in riding positions using REFMAC (41) and the structure was relaxed to minimize energy using Rosetta (42). An attempt to place l-erythrose in the active site of ADH1B1 showed that steric hindrance prevented positioning the molecule correctly for reaction with NADH.
Author contributions
L. S., K. H., and M. S. F. conceptualization; L. S., D. M. E. S., S. R. O., A. H., K. H., and M. S. F. formal analysis; L. S. funding acquisition; L. S., D. M. E. S., S. R. O., A. H., K. H., and M. S. F. investigation; L. S., D. M. E. S., S. R. O., K. H., and M. S. F. methodology; L. S., D. M. E. S., and M. S. F. writing-original draft; K. H. writing-review and editing; M. S. F. supervision; M. S. F. project administration.
Supplementary Material
Acknowledgments
We thank the Proteomics Facility of Cornell University for providing the MS data used for protein identification and National Institutes of Health SIG Grant 1S10 OD017992-01 support for the Orbitrap Fusion mass spectrometer. We thank Rodrigo Gutierrez and Hannah Stein for technical assistance. We also thank Annegrät Daujeumont for help with the A549 cell extracts, enzymatic assays, and metabolite extraction.
This work was supported by a Joachim Herz Stiftung “Add-On Fellowship for Interdisciplinary Science” (to L. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5 and Table S1.
The Proteomics data are available via ProteomeXchange with identifier PXD015178.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- T2DM
- type 2 diabetes mellitus
- ACN
- acetonitrile
- ADH1
- alcohol dehydrogenase 1
- DDA
- data-dependent acquisition
- E4P
- erythrose-4-phosphate
- G6P
- glucose 6-phosphate
- MID
- mass isotopomer distribution
- PPP
- pentose phosphate pathway
- RPS18
- ribosomal protein S18
- SORD
- sorbitol dehydrogenase
- FA
- formic acid
- qPCR
- quantitative PCR
- PDB
- Protein Data Bank.
References
- 1. Rzechonek D. A., Dobrowolski A., Rymowicz W., and Mirończuk A. M. (2018) Recent advances in biological production of erythritol. Crit. Rev. Biotechnol 38, 620–633 10.1080/07388551.2017.1380598 [DOI] [PubMed] [Google Scholar]
- 2. Munro I., Bernt W., Borzelleca J., Flamm G., Lynch B. S., Kennepohl E., Bär E. A., Modderman J., and Bernt W. O. (1998) Erythritol: an interpretive summary of biochemical, metabolic, toxicological and clinical data. Food Chem. Toxicol. 36, 1139–1174 10.1016/S0278-6915(98)00091-X [DOI] [PubMed] [Google Scholar]
- 3. Hootman K. C., Trezzi J.-P., Kraemer L., Kraemer L., Burnwell L. S., Dong X., Guertin K. A., Jaeger C., Stober P. J., Hiller K., and Cassano P. A. (2017) Erythritol is a pentose-phosphate pathway metabolite and associated with adiposity gain in young adults. Proc. Natl. Acad. Sci. U.S.A. 114, E4233–E4240 10.1073/pnas.1620079114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rebholz C. M., Yu B., Zheng Z., Chang P., Tin A., Köttgen A., Wagenknecht L. E., Coresch J., Boerwinkle E., and Selvin E. (2018) Serum metabolomic profile of incident diabetes. Diabetologia 61, 1046–1054 10.1007/s00125-018-4573-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hiele M., Ghoos Y., Rutgeerts P., and Vantrappen G. (1993) Metabolism of erythritol in humans: Comparison with glucose and lactitol. Br. J. Nutr. 69, 169 10.1079/BJN19930019 [DOI] [PubMed] [Google Scholar]
- 6. Tylki-Szymanska A., Wamelink M. M., Stradomska T. J., Salomons G. S., Taybert J., Dabrowska-Leonik N., and Rurarz M. (2014) Clinical and molecular characteristics of two transaldolase-deficient patients. Eur. J. Pediatr. 173, 1679–1682 10.1007/s00431-014-2261-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ishikawa M., Miyashita M., Kawashima Y., Nakamura T, Saitou N., and Modderman J. (1996) Effects of oral administration of erythritol on patients with diabetes. Regul. Toxicol. Pharmacol. 24, S303–S308 10.1006/rtph.1996.0112 [DOI] [PubMed] [Google Scholar]
- 8. Wen H., Tang B., Stewart A. J., Tao Y., Shao Y., Cui Y., Yue H., Pei J., Liu Z., Mei L., Yu R., and Jiang L. (2018) Erythritol attenuates postprandial blood glucose by inhibiting α-glucosidase. J. Agric Food Chem. 66, 1401–1407 10.1021/acs.jafc.7b05033 [DOI] [PubMed] [Google Scholar]
- 9. Flint N., Hamburg N. M., Holbrook M., Dorsey P. G., LeLeiko R. M., Berger A., de Cock P., Bosscher D., and Vita J. A. (2014) Effects of erythritol on endothelial function in patients with type 2 diabetes mellitus: a pilot study. Acta Diabetol. 51, 513–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Donnell S., Baudier K., and Marenda D. R. (2016) Non-nutritive polyol sweeteners differ in insecticidal activity when ingested by adult Drosophila melanogaster (Diptera: Drosophilidae). J. Insect Sci. 16, pii47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baudier K. M., Kaschock-Marenda S. D., Patel N., Diangelus K. L., O'Donnell S., and Marenda D. R. (2014) Erythritol, a non-nutritive sugar alcohol sweetener and the main component of truvia®, is a palatable ingested insecticide. PLoS ONE 9, e98949 10.1371/journal.pone.0098949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moon H.-J., Jeya M., Kim I.-W., and Lee J.-K. (2010) Biotechnological production of erythritol and its applications. Appl. Microbiol. Biotechnol. 86, 1017–1025. 10.1007/s00253-010-2496-4 [DOI] [PubMed] [Google Scholar]
- 13. Lee D.-H., Lee Y.-J., Ryu Y.-W., and Seo J.-H. (2010) Molecular cloning and biochemical characterization of a novel erythrose reductase from Candida magnoliae JH110. Microb. Cell Fact. 9, 43 10.1186/1475-2859-9-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Janek T., Dobrowolski A., Biegalska A., and Mirończuk A. M. (2017) Characterization of erythrose reductase from Yarrowia lipolytica and its influence on erythritol synthesis. Microb. Cell Fact. 16, 118 10.1186/s12934-017-0733-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jovanović B., Mach R. L., and Mach-Aigner A. R. (2013) Characterization of erythrose reductases from filamentous fungi. AMB Express 3, 43 10.1186/2191-0855-3-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mato J. M., Martínez-Chantar M. L., and Lu S. C. (2008) Methionine metabolism and liver disease. Annu. Rev. Nutr. 28, 273–293 10.1146/annurev.nutr.28.061807.155438 [DOI] [PubMed] [Google Scholar]
- 17. Polimanti R., and Gelernter J. (2018) ADH1B: from alcoholism, natural selection, and cancer to the human phenome. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 177, 113–125 10.1002/ajmg.b.32523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oates P. J. (2002) Polyol pathway and diabetic peripheral neuropathy. Int. Rev. Neurobiol. 50, 325–392 10.1016/S0074-7742(02)50082-9 [DOI] [PubMed] [Google Scholar]
- 19. Crabb D. W., Matsumoto M., Chang D., and You M. (2004) Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 63, 49–63 10.1079/PNS2003327 [DOI] [PubMed] [Google Scholar]
- 20. Circu M. L., Maloney R. E., and Aw T. Y. (2011) Disruption of pyridine nucleotide redox status during oxidative challenge at normal and low-glucose states: implications for cellular adenosine triphosphate, mitochondrial respiratory activity, and reducing capacity in colon epithelial cells. Antioxid. Redox Signal. 14, 2151–2162 10.1089/ars.2010.3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. El-Kabbani O., Darmanin C., and Chung R. (2004) Sorbitol dehydrogenase: structure, function and ligand design. Curr. Med. Chem. 11, 465–476 10.2174/0929867043455927 [DOI] [PubMed] [Google Scholar]
- 22. Carugo O., and Argos P. (1997) NADP-dependent enzymes: I, conserved stereochemistry of cofactor binding. Proteins 28, 10–28 [DOI] [PubMed] [Google Scholar]
- 23. Höög J. O., and Östberg L. J. (2011) Mammalian alcohol dehydrogenases: a comparative investigation at gene and protein levels. Chem. Biol. Interact. 191, 2–7 10.1016/j.cbi.2011.01.028 [DOI] [PubMed] [Google Scholar]
- 24. Ramchandani V. A., Bosron W. F., and Li T. K. (2001) Research advances in ethanol metabolism. Pathol. Biol. 49, 676–682 10.1016/S0369-8114(01)00232-2 [DOI] [PubMed] [Google Scholar]
- 25. Gould R. M., and Plapp B V. (1990) Substitution of arginine for histidine-47 in the coenzyme binding site of yeast alcohol dehydrogenase I. Biochemistry 29, 5463–5468 10.1021/bi00475a009 [DOI] [PubMed] [Google Scholar]
- 26. Pauly T. A., Ekstrom J. L., Beebe D. A., Chrunyk B., Cunningham D., Griffor M., Kamath A., Lee S. E., Madura R., Mcguire D., Subashi T., Wasilko D., Watts P., et al. (2003) X-ray crystallographic and kinetic studies of human sorbitol dehydrogenase. Structure 11, 1071–1085 10.1016/S0969-2126(03)00167-9 [DOI] [PubMed] [Google Scholar]
- 27. Chung S. S., Ho E. C., Lam K. S., Chung S. K. (2003) Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 14, S233–S236 10.1097/01.ASN.0000077408.15865.06 [DOI] [PubMed] [Google Scholar]
- 28. Winnier D. A., Fourcaudot M., Norton L., Abdul-Ghani M. A., Hu S. L., Farook V. S., Coletta D. K., Kumar S., Puppala S., Chittoor G., Dyer T. D., Arya R., Carless M., Lehman D. M., et al. (2015) Transcriptomic identification of adh1b as a novel candidate gene for obesity and insulin resistance in human adipose tissue in Mexican Americans from the Veterans Administration Genetic Epidemiology Study (VAGES). PLoS ONE 10, e0119941 10.1371/journal.pone.0119941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sapcariu S. C., Kanashova T., Weindl D., Ghelfi J., Dittmar G., and Hiller K. (2014) Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX 1, 74–80 10.1016/j.mex.2014.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krämer L., Jäger C., Trezzi J.-P., Jacobs D. M., and Hiller K. (2018) Quantification of stable isotope traces close to natural enrichment in human plasma metabolites using gas chromatography-mass spectrometry. Metabolites 8, pii E15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hiller K., Hangebrauk J., Jäger C., Spura J., Schreiber K., and Schomburg D. (2009) Metabolite detector: comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Anal. Chem. 81, 3429–3439 10.1021/ac802689c [DOI] [PubMed] [Google Scholar]
- 32. Bensadoun A., and Weinstein D. (1976) Assay of proteins in the presence of interfering materials. Anal. Biochem. 70, 241–250 10.1016/S0003-2697(76)80064-4 [DOI] [PubMed] [Google Scholar]
- 33. Yang Y., Thannhauser T. W., Li L., and Zhang S. (2007) Development of an integrated approach for evaluation of 2-D gel image analysis: impact of multiple proteins in single spots on comparative proteomics in conventional 2-D gel/MALDI workflow. Electrophoresis 28, 2080–2094 10.1002/elps.200600524 [DOI] [PubMed] [Google Scholar]
- 34. Yang Y., Anderson E., and Zhang S. (2018) Evaluation of six sample preparation procedures for qualitative and quantitative proteomics analysis of milk fat globule membrane. Electrophoresis 39, 2332–2339 10.1002/elps.201800042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thomas C. J., Cleland T. P., Zhang S., Gundberg C. M., and Vashishth D. (2017) Identification and characterization of glycation adducts on osteocalcin. Anal. Biochem. 525, 46–53 10.1016/j.ab.2017.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anguera M. C., Liu X., and Stover P. J. (2004) Cloning, expression, and purification of 5,10-methenyltetrahydrofolate synthetase from Mus musculus. Protein Expr. Purif. 35, 276–283 10.1016/j.pep.2004.02.010 [DOI] [PubMed] [Google Scholar]
- 38. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agarwal P. K., Simon Webb P., and Hammes-Schiffer S. (2000) Computational studies of the mechanism for proton and hydride transfer in liver alcohol dehydrogenase. J. Am. Chem. Soc. 122, 4803–4812 10.1021/JA994456W [DOI] [PubMed] [Google Scholar]
- 40. Kleywegt G. J. (2007) Crystallographic refinement of ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 63, 94–100 10.1107/S0907444906022657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Murshudov G. N., Vagin A. A., Dodson E. J., IUCr. (1997) Refinement of macromolecular Structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 42. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman D., Renfrew P. D., Smith C. A., Sherrler W., Davis I. W., Cooper S., et al. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 10.1016/B978-0-12-381270-4.00019-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjostedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics: tissue-based map of the human proteome. Science 347, 1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.