

Abstract
Connections between deficient autophagy and insulin resistance have emerged, however, the mechanism through which reduced autophagy impairs insulin-signaling remains unknown. We examined mouse embryonic fibroblasts lacking Atg16l1 (ATG16L1 KO mouse embryonic fibroblasts (MEFs)), an essential autophagy gene, and observed deficient insulin and insulin-like growth factor-1 signaling. ATG16L1 KO MEFs displayed reduced protein content of insulin receptor substrate-1 (IRS1), pivotal to insulin signaling, whereas IRS1myc overexpression recovered downstream insulin signaling. Endogenous IRS1 protein content and insulin signaling were restored in ATG16L1 KO mouse embryonic fibroblasts (MEF) upon proteasome inhibition. Through proximity-dependent biotin identification (BioID) and co-immunoprecipitation, we found that Kelch-like proteins KLHL9 and KLHL13, which together form an E3 ubiquitin (Ub) ligase complex with cullin 3 (CUL3), are novel IRS1 interactors. Expression of Klhl9 and Klhl13 was elevated in ATG16L1 KO MEFs and siRNA-mediated knockdown of Klhl9, Klhl13, or Cul3 recovered IRS1 expression. Moreover, Klhl13 and Cul3 knockdown increased insulin signaling. Notably, adipose tissue of high-fat fed mice displayed lower Atg16l1 mRNA expression and IRS1 protein content, and adipose tissue KLHL13 and CUL3 expression positively correlated to body mass index in humans. We propose that ATG16L1 deficiency evokes insulin resistance through induction of Klhl9 and Klhl13, which, in complex with Cul3, promote proteasomal IRS1 degradation.
Keywords: Akt PKB, autophagy, cell signaling, insulin, insulin receptor substrate 1 (IRS-1), E3 ubiquitin ligase, autophagy-related protein 16L1 (ATG16L1), insulin signaling, Kelch-like gene family
Introduction
Obesity-associated type 2 diabetes (T2D),3 one of the most prevalent diseases in Western society, is defined by whole-body insulin resistance and insufficient compensatory insulin secretion (1). In mice and humans, obesity, insulin resistance, and T2D are often associated with defects in insulin receptor substrate 1 (IRS1) (2–7). IRS1 is the direct substrate of the insulin-sensing insulin receptor (IR) and insulin-like growth factor-1 receptor (IGF1R). Normally, insulin-stimulated IRS1 tyrosine phosphorylation promotes phosphoinositide 3-kinase (PI3K) binding and PI3K-induced conversion of PI(4,5)P2 to PI(3,4,5)P3 at the cell membrane. In turn, AKT binds PI(3,4,5)P3 via its pleckstrin homology (PH) domain, promoting AKT phosphorylation by mTOR complex 2 (mTORC2) at Ser-473 and by phosphoinositide-dependent kinase 1 (PDK1) at Thr-308 (8, 9). However, in insulin-resistant muscle, adipose and liver, IRS1 is altered through increased inhibitory serine phosphorylation and, notably, through diminished IRS1 protein content (2–7). One proposed mechanism for the obesity-linked reduction in IRS1 invokes inflammation and endoplasmic reticulum (ER) stress (10, 11). This leads to IRS1 serine-phosphorylation, and ubiquitination via a suppressor of cytokine signaling 1 (SOCS1)/SOCS3-containing E3 ubiquitin (Ub) ligase complex that targets IRS1 for proteasomal degradation (7, 12–14). However, it is unknown if these mechanisms account for IRS1 degradation under diverse conditions of insulin resistance, and moreover if it suffices to drive defects in downstream insulin signaling.
Recently, an association has emerged between insulin resistance and deficient macroautophagy (hereafter autophagy), a bulk degradation pathway. The expression of autophagy-related (ATG) proteins, which regulate the various steps of autophagy, is attenuated in liver of insulin-resistant mice (15, 16). Liver- or adipose tissue-specific Atg knockout mice display impaired tissue insulin action and whole-body insulin resistance (15, 17). Moreover, skeletal muscle-specific Atg7 knockout mice exhibit attenuated basal PI3K/AKT signaling (18). Conversely, hyper-activation of autophagy through a constitutively active beclin1 mutant, increases hepatic insulin signaling in mice (19). Lastly, insulin-dependent phosphorylation of IRS1 and AKT is reduced upon expression of the ATG5 dominant-negative mutant ATG5K130R in myoblasts (20). However, the mechanisms responsible for these observed defects in insulin signaling in the face of defective autophagy have yet to be described.
To fully elucidate the mechanisms responsible for deficient autophagy-induced insulin resistance, direct examination in cellular model systems presents logistical advantages to examine cause-effect relationships. Using Atg16l1 knockout mouse embryonic fibroblasts (ATG16L1 KO MEFs) as a cellular model of deficient autophagy (21), we analyzed the consequence on insulin signaling and possible underlying mechanisms. ATG16L1, in complex with ATG5 and ATG12, mediates lipidation of ATG8 homologs (including microtubule-associated light chain 3B, hereafter LC3) with phosphatidylethanolamine, an essential step in the autophagic process (22). Notably, inducible, adipocyte-specific Atg16l1 knockout mice display whole-body insulin resistance (17). We show that ATG16L1 deficiency causes insulin resistance in MEFs and propose a responsible mechanism implicating the KLHL9/KLHL13/CUL3 E3 Ub ligase complex in IRS1 degradation. These findings highlight a novel pathway leading to targeted degradation of IRS1 and causally to subsequent downstream defects in insulin signaling.
Results
ATG16L1-deficient MEFs display resistance to insulin and IGF1
ATG16L1 KO MEFs display ablated LC3 lipidation, determined by immunoblotting of LC3 (which distinguishes lipidated LC3-II from nonlipidated LC3-I) in the presence or absence of the autophagic flux inhibitor bafilomycin A1 (Fig. 1A). As well, LC3-GFP overexpression in ATG16L1 KO MEFs failed to induce fluorescent puncta accumulation that would normally correspond to autophagosome formation, which are, however, clearly observed in WT MEFs under these conditions (Fig. 1B) (23).
Figure 1.
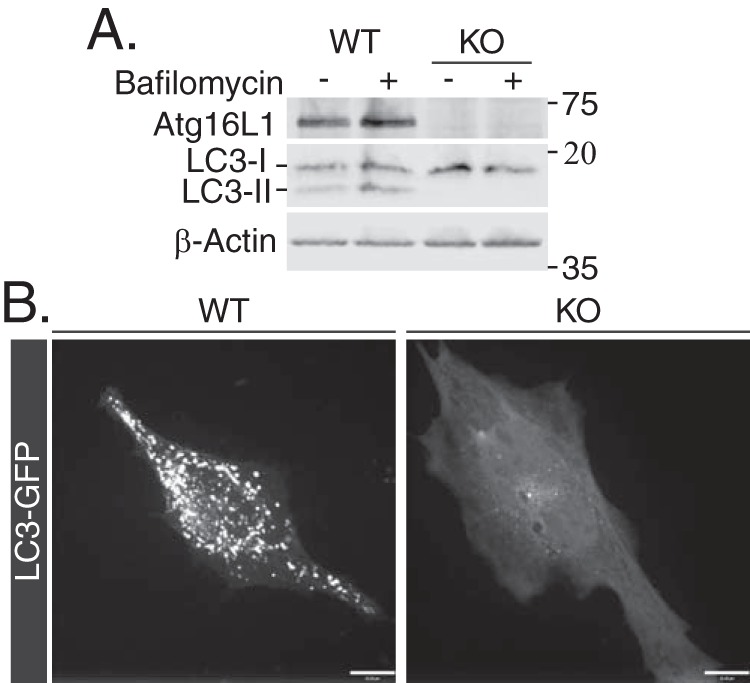
WT and ATG16L1 KO MEFs characterization. A, WT and ATG16L1 KO MEFs were treated with 500 nm bafilomycin for 4 h and immunoblotted for ATG16L1, LC3-I, and LC3-II to evaluate ATG16L1 KO and autophagic flux. B, MEFs were transfected with LC3-GFP (24 h) and live cell imaging was performed to evaluate autophagosome formation, seen as puncta. Scale bars: 10 μm.
AKT phosphorylation is commonly used as a measurement of signaling downstream of receptor tyrosine kinases. Insulin-stimulated AKT phosphorylation at Ser-473 and Thr-308 was significantly lower in ATG16L1 KO compared with WT MEFs (Fig. 2A). IGF1-induced AKT phosphorylation was also attenuated in ATG16L1 KO MEFs (Fig. 2B), suggesting that the signaling defect is likely at a step shared by insulin and IGF1 receptors' signaling. Importantly, re-expression of myc-ATG16L1 recovered insulin-stimulated AKT Ser-473 phosphorylation (Fig. S1).
Figure 2.

Insulin and IGF1 signaling is attenuated in ATG16L1 KO MEFs. Cell lysates were collected for immunoblotting of pAKT Ser-473 and Thr-308 residues from WT and ATG16L1 KO MEFs following treatment with (A) insulin (10 nm, 10 min, n = 4) or (B) IGF1 (100 ng/ml, 10 min, n = 5). Total AKT was also immunoblotted in each experiment and the pAKT/total AKT densities were calculated using Image Studio 5.2.5 software and compared with untreated (basal) cells. C, WT and ATG16L1 KO MEFs on coverslips were transfected with PH-Akt-GFP for 24 h. During experimentation, live cell imaging was performed for 4 min. 100 nm insulin was added to the media immediately following acquisition of the 0:00 time point (scale bars: 15 μm). Insets corresponding to the area within the box (scale bars: 3.75 μm), as well as the XZ plane cropped to display the same cell (scale bars: 7.5 μm), are shown. D, WT and ATG16L1 KO MEFs were treated with 100 nm insulin for 10 min, fixed, and stained with phalloidin-565 to examine insulin-stimulated cortical actin ruffling, observed as regions of enriched phalloidin labeling (scale bars: 30 μm). Insets of membrane actin organization are shown (scale bars: 5 μm). E, quantification of percentage of cells displaying actin remodeling (n = 3, with >20 cells examined per group in each independent experiment). For all measures, p value was calculated using a two-way ANOVA with Tukey's post hoc. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Downstream of AKT, we examined insulin-stimulated phosphorylation of GSK3β on Ser-9, which is enacted by AKT directly, and insulin-stimulated phosphorylation of P70S6K on Thr-389, which is enacted by mTORC1 downstream of AKT. Upon stimulation with insulin, ATG16L1 KO MEFs exhibited significantly diminished GSK3β and P70S6K phosphorylation compared with insulin-stimulated WT MEFs (Fig. S2, A and B).
ATG16L1 KO MEFs display defective insulin-dependent PI(3,4,5)P3 production, cortical actin remodeling, and ERK phosphorylation
We next sought to determine whether the diminished response of AKT to insulin stimulation occurs upstream of PI3K-dependent PI(3,4,5)P3 production. Conversion of PI(4,5)P2 to PI(3,4,5)P3 and subsequent AKT recruitment was examined using PH-AKT-GFP, a fluorescent construct containing the PI(3,4,5)P3-binding PH domain of AKT (24, 25). Insulin-induced relocalization of the PH-AKT-GFP probe to the cell periphery was observed in WT MEFs, but not in ATG16L1 KO MEFs (Fig. 2C). This suggests that PI(3,4,5)P3 is not generated by PI3K following insulin stimulation.
An additional outcome of insulin-dependent PI(3,4,5)P3 generation, independent of AKT, is the activation of the small GTPase Rac1 (26, 27) that leads to cortical actin remodeling (8, 9). Insulin-stimulated actin remodeling, forming regions of dorsally protruding ruffles, was attenuated in ATG16L1 KO MEFs compared with WT MEFs (Fig. 2, D and E). Moreover, insulin-stimulated ERK phosphorylation, which occurs downstream of the IR and IRS1, but independently of PI3K signaling, was also significantly attenuated in ATG16L1 KO MEFs (Fig. S2C). Together, these findings indicate that the defect in insulin signaling in ATG16L1 KO cells occurs upstream of PI3K and PI(4,5)P2 conversion to PI(3,4,5)P3.
ATG16L1 deficiency does not reduce insulin or IGF1 receptor expression
Insulin can bind to both the IR and IGF1R and therefore we considered if the receptor levels might be reduced in ATG16L1 KO MEFs. However, ATG16L1 KO MEFs displayed enhanced protein levels of IR-β, the transmembrane tyrosine kinase subunit of the IR, compared with WT MEFs (Fig. 3A), whereas IGF1R-β content was not significantly affected (Fig. 3B). Moreover, ATG16L1 KO had no effect on cell surface IR-β expression measured by Sulfo-NHS-SS-Biotin labeling of membrane proteins and immunoblotting (Fig. 3, C and D). Finally, radioactive 125I-insulin binding, which engages the α subunit of the IR, was unchanged in ATG16L1 KO compared with WT MEFs (Fig. 3E). Therefore, defects in insulin signaling at the level of AKT phosphorylation cannot be ascribed to changes in IR expression, membrane localization, or ligand binding.
Figure 3.
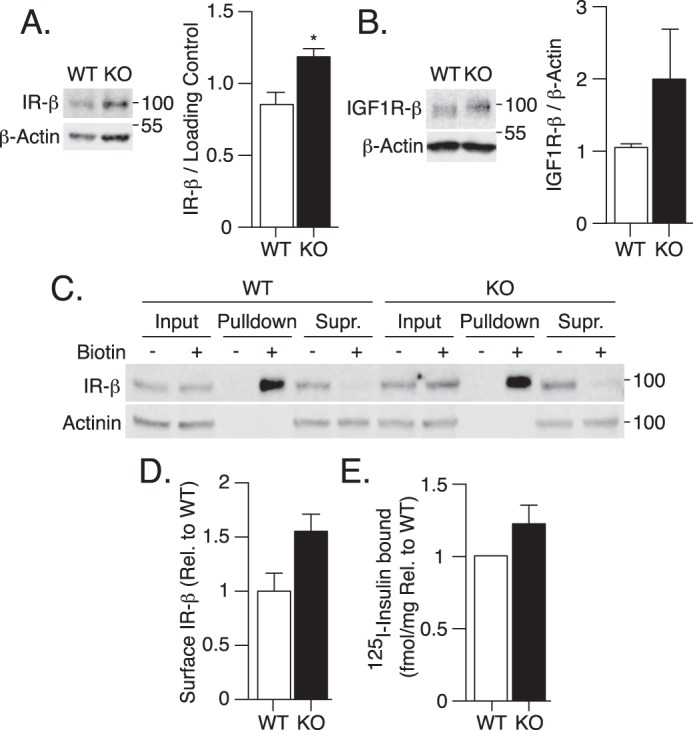
Attenuated insulin signaling in ATG16L1 KO MEFs is independent of changes at the level of the IR. A, IR-β, and B, IGF1R-β protein content was measured by immunoblotting WT and ATG16L1 KO MEF lysates (n = 3). C and D, cell surface expression of IR-β protein was measured by Sulfo-NHS-SS-Biotin labeling followed by streptavidin pulldown and immunoblotting of input lysates, pulldown and remaining supernatant following pulldown (Supr.) (n = 4). Immunoblots were quantified using Image Studio 5.2.5 software. E, WT and ATG16L1 KO MEFs were treated with 0.5 nm 125I-insulin on ice for 20 min to evaluate insulin binding. After washing away excess 125I-insulin, cells were lysed, radioactivity and protein content were measured, and the fmol/mg of bound 125I-insulin was calculated (n = 3). For IR-β expression, a one-tailed paired t test was performed, as others have shown the IR to be degraded by autophagy. For all other measures, p value was calculated using a two-tailed paired t test. *, p < 0.05.
ATG16L1 deficiency leads to loss of IRS1 protein
IRS1, immediately downstream of both the IR and IGF1R, regulates membrane recruitment of PI3K (8, 9). The protein content of IRS1 was significantly lower in ATG16L1 KO MEFs compared with WT MEFs (Fig. 4A), whereas Irs1 mRNA was not affected (Fig. 4B). This finding suggests that ATG16L1 deficiency induces decreased IRS1 protein levels, independent of changes in its gene expression. Importantly, when we reduced ATG16L1 expression in L6 myoblasts by siRNA targeting, we observed a significant correlation between ATG16L1 and IRS1 protein levels (Fig. S3). This finding shows that decreasing ATG16L1 induces a reduction in IRS1 levels also in a commonly studied, insulin-responsive cell type.
Figure 4.
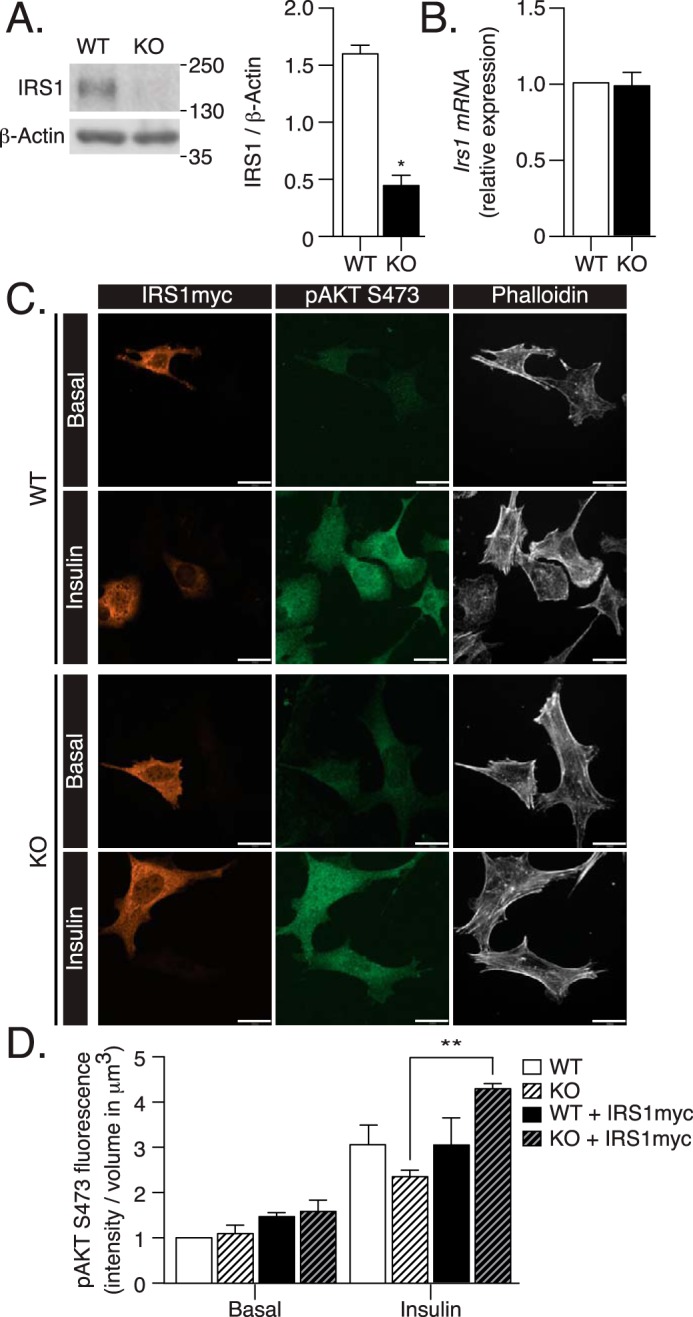
Defects in insulin signaling induced in ATG16L1 KO MEFs are mediated by decreased IRS1 expression. A, IRS1 protein content, and B, mRNA expression were analyzed by immunoblotting or qPCR in WT and ATG16L1 KO MEFs (n = 3). p value was calculated using a two-tailed paired t test. C, WT and ATG16L1 KO MEFs on coverslips were transfected with Irs1myc for 24 h, then treated with insulin (100 nm, 10 min), followed by fixation and immunostaining for myc, pAKT Ser-473, and with phalloidin 670. IRS1myc overexpressing cells were compared with non-overexpressing cells, identified by the presence of phalloidin 670 and lack of IRS1myc (n = 3). Scale bars: 25 μm. D, pAKT Ser-473 fluorescence was quantified as fluorescent intensity relative to cell volume using Velocity 6.3 software. Absolute values of insulin-stimulated pAKT Ser-473 intensity/μm2 (a.u.) for the representative images are as follows: WT, 35153.44; WT-IRS1myc, 40493.62; KO, 30533.00; and KO-IRS1myc. 56836.04. p value was calculated using a two-way ANOVA with Tukey's post hoc. *, p < 0.05; **, p < 0.01.
To elucidate the contribution of the diminished IRS1 protein content to downstream insulin resistance in ATG16L1 KO MEFs, we examined the potential to recover insulin action upon exogenous expression of myc-tagged IRS1. IRS1myc overexpression in ATG16L1 KO MEFs significantly increased insulin-stimulated AKT Ser-473 phosphorylation, compared with non-overexpressing ATG16L1 KO MEFs (Fig. 4C). The increase in this downstream response suggests that the drop in IRS1 contributes to ATG16L1 KO-induced defects in insulin signaling. As there is evidence for deficient autophagy inducing insulin resistance in vivo (15) and IRS1 defects are associated with diabetes (2–7), the mechanisms responsible for the loss of IRS1 in ATG16L1-deficient cells were further examined.
ATG16L1 deficiency leads to proteasomal degradation of IRS1
Unlike acute stimulation, prolonged exposure of cells to insulin, IGF1, or inflammatory stimuli can cause IRS1 degradation through the ubiquitin-proteasome system (14, 28–30). To examine the possible contribution of proteasomal degradation to decreased IRS1 in ATG16L1 KO MEFs, the cells were treated with the 20S proteasome inhibitor MG132. MG132 led to increased accumulation of ubiquitinated proteins in both WT and ATG16L1 KO MEFs, confirming attenuated proteasomal degradation (Fig. 5A). Importantly, MG132 treatment partially restored IRS1 expression in ATG16L1 KO MEFs (Fig. 5B). Moreover, immunoprecipitation of IRS1 following MG132 treatment revealed high levels of IRS1 ubiquitination in both WT and ATG16L1 KO MEFs (Fig. 5C). Insulin-stimulated phosphorylation of AKT on both Ser-473 and Thr-308 was significantly increased in ATG16L1 KO MEFs following MG132 treatment, compared with vehicle-treated, insulin-stimulated ATG16L1 KO MEFs (Fig. 5D). Therefore, ATG16L1 KO MEFs regained insulin signaling toward AKT upon partial inhibition of proteasomal degradation and consequent increases in IRS1 expression.
Figure 5.

Proteasome inhibition recovers IRS1 levels and insulin-stimulated AKT phosphorylation in ATG16L1 KO MEFs. WT and ATG16L1 KO MEFs were treated with 500 nm MG132, a 20S proteasome inhibitor, for 24 h. Cells were then lysed and immunoblotted for A, ubiquitin to evaluate proteasome inhibition, and B, IRS1 content (n = 6). C, IRS1 immunoprecipitation was performed with lysates from MEFs treated with or without MG132 for 24 h. Immunoblotting was performed for IRS1 and ubiquitin to examine IRS1 ubiquitination in WT and ATG16L1 KO MEFs (n = 4). D, MG132-treated MEFs were stimulated with insulin (10 nm, 10 min). Cell lysates were immunoblotted for total AKT, pAKT on Ser-473 and Thr-308 residues, and β-actin (n = 3). p value was calculated using a two-way ANOVA with Tukey's post hoc. *, p < 0.05; **, p < 0.01; ****, p < 0.0001.
IRS1 degradation and ensuing insulin resistance may involve several mediators, such as serine phosphorylation by c-Jun N-terminal kinase (JNK) or ubiquitination via CUL7, CBL-b, or SOCS1/SOCS3 (7, 12–14, 31–33). We found that Atg16l1 knockout-induced IRS1 degradation was independent of JNK activity or changes in Cul7, Cbl-b, or Socs1/Socs3 expression (Fig. S4, A–C). Specifically, JNK phosphorylation was increased in ATG16L1 KO MEFs compared with WT, however, inhibition of JNK activity with SP600125 (confirmed by immunoblotting for phosphorylated c-Jun) did not recover IRS1 protein content in ATG16L1 KO MEFs (Fig. S4, A and B). Interestingly, SP600125 treatment in WT MEFs did increase IRS1 protein content, suggesting that IRS1 protein turnover can be regulated by JNK in WT MEFs. Of the previously identified E3 Ub ligases that regulate IRS1 degradation, only Socs3 expression was up-regulated (Fig. S4C). However, siRNA-mediated knockdown of Socs3 did not significantly recover IRS1 expression in ATG16L1 KO MEFs (Fig. S4D). These findings suggest that previously established mechanisms of IRS1 degradation do not mediate IRS1 degradation in ATG16L1 KO MEFs.
ATG16L1 deficiency induces KLHL9 and KLHL13 expression, promoting degradation of IRS1
In search of the E3 Ub ligase complex responsible for IRS1 degradation we analyzed an ongoing BioID screen for interactors of E3 Ub ligases and their adaptors (34, 35). In particular, BioID analysis of the substrate-specific adaptor proteins Kelch-like 9 (KLHL9) and KLHL13 revealed their association with IRS1 (Tables 1 and 2; massIVE ID MSV000083653) and supported the previously established formation of a KLHL9, KLHL13, and Cullin 3 (CUL3) E3 Ub ligase complex (36). Co-immunoprecipitation experiments confirmed interactions of KLHL9, KLHL13, and CUL3 with IRS1 (Fig. 6A).
Table 1.
KLHL9 BioID
Expression of FLAGBirA*–KLHL9 or FLAGBirA* negative control from integrated cassettes in Flp-In 293 T-REx cells was induced with tetracycline in the presence of 50 μm biotin for 24 h. Biotinylated proteins were purified on streptavidin-Sepharose beads and recovered proteins were identified by LC-MS/MS. Total spectral counts for two independent experiments and two technical replicates are shown for the high-confidence interactors CUL3, KLHL13, and IRS1 (based on Bayesian False Discovery Rate (BFDR) <0.01), in comparison with 20 runs of FLAGBirA* negative control.
| Gene | Control peptide counts | Peptide counts | Sum | BFDR |
|---|---|---|---|---|
| Cul3 | 3 0 0 0 0 3 0 0 0 0 0 0 0 0 2 0 0 0 0 0 | 153 149 162 147 | 611 | 0 |
| Klhl13 | 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 | 110 110 110 110 | 440 | 0 |
| Irs1 | 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 | 12 11 16 11 | 50 | 0 |
Table 2.
KLHL13 BioID
As described in Table I, with FlagBirA*–KLHL13 as a bait.
| Gene | Control peptide counts | Peptide counts | Sum | BFDR |
|---|---|---|---|---|
| Cul3 | 3 0 0 0 0 3 0 0 0 0 0 0 0 0 2 0 0 0 0 0 | 219 225 245 239 | 611 | 0 |
| Klhl9 | 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 | 19 13 13 12 | 57 | 0 |
| Irs1 | 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 | 13 12 16 11 | 52 | 0 |
Figure 6.

KLHL9/KLHL13/CUL3 E3 Ub ligase complex components are novel IRS1 interactors up-regulated by ATG16L1 deficiency. A, co-immunoprecipitation of 3xHA-KLHL9, KLHL13-HA, and CUL3-HA with IRS1myc or the myc epitope alone in HEK293T cells. B, Klhl9, Klhl13, and Cul3 mRNA expression was examined in WT and ATG16L1 KO MEFs by qPCR (n = 3–4). p value was calculated using a two-tailed paired t test. *, p < 0.05; **, p < 0.01.
The regulation of E3 Ub ligase complexes is partially mediated by changes in the expression of substrate-specific adaptors (37). Consistent with the hypothesis that this regulatory mechanism would be up-regulated in ATG16L1 KO MEFs, Klhl9 and Klhl13 mRNA expression was higher in ATG16L1 KO MEFs compared with WT MEFs, although Cul3 mRNA was unaffected (Fig. 6B). Mechanistically, siRNA-mediated knockdown of Klhl9, Klhl13, or Cul3 significantly elevated the IRS1 protein content by ∼2-fold in ATG16L1 KO MEFs and by ∼1.5-fold in WT MEFs, compared with noncoding siRNA controls (Fig. 7, A–C). Increased IRS1 protein content was accompanied by augmented insulin-stimulated AKT phosphorylation at Ser-473 following Klhl13 or Cul3 siRNA knockdown in both WT and ATG16L1 KO MEFs (Fig. 7, E and F). However, Klhl9 siRNA knockdown-induced increases in AKT phosphorylation did not reach statistical significance (Fig. 7D). Together, these findings suggest that the KLHL9/KLHL13/CUL3 E3 Ub ligase complex contributes to IRS1 degradation, and that this function is exacerbated upon ATG16L1 depletion, translating into downstream insulin resistance.
Figure 7.

KLHL9/KLHL13/CUL3 E3 Ub ligase complex regulates IRS1 protein content and insulin action in ATG16L1 KO MEFs. A, Klhl9; B, Klhl13; and C, Cul3 siRNA-transfected WT and ATG16L1 KO MEFs (48 h) were examined for IRS1 expression (n = 3–4). p value was calculated using a two-tailed unpaired t test. D, Klhl9; E, Klhl13; and F, Cul3 siRNA-transfected WT and ATG16L1 KO MEFs were treated with insulin (10 nm, 10 min) and immunoblotted for pAKT on Ser-473 (n = 3–4). p value was calculated using a two-way ANOVA with Tukey's post hoc. siRNA knockdown efficiency: Klhl9 WT, 90.6 ± 2.3%; ATG16L1 KO, 84.7 ± 4.8%; Klhl13 WT, 96.7 ± 0.8%; ATG16L1 KO, 91.4 ± 2.5%; and Cul3 WT, 70.4 ± 2.0%; ATG16L1 KO, 66.9 ± 6.5%. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Obese human and high-fat fed mouse adipose tissue exhibits changes in Atg16l1 and E3 Ub ligase gene expression and IRS1 protein content
A hypothesis stemming from our findings in vitro is that insulin resistance in vivo may be associated with reduced levels of Atg16l1 and IRS1, and increased levels of Klhl9, Klhl13, or Cul3. To test this prediction we first examined mice fed a high-fat diet (HFD) that brings about insulin resistance (38). Notably, epididymal white adipose tissue of HFD-fed mice exhibited lower Atg16l1 mRNA expression compared with low-fat diet (LFD) controls (Fig. 8A). In parallel, IRS1 protein content was lower in HFD-fed relative to LFD adipose tissue (Fig. 8B). The change in IRS1 is in agreement with reductions previously observed in adipocytes of human diabetic individuals (2) and mice (39). These findings suggest that ATG16L1 deficiency induced by high-fat diet tracks with, and based on the cell cultures studies might contribute to, insulin resistance through the loss of IRS1 expression in vivo.
Figure 8.

Adipose tissue IRS1 protein content and Atg16l1, Klhl9, Klhl13, and Cul3 gene expression is altered upon high-fat feeding and in obesity. A, Atg16l1 mRNA expression was examined by qPCR in eWAT of LFD and HFD mice (n = 3/group). B, IRS1 protein content was analyzed by immunoblotting in epididymal white adipose tissue (eWAT) of LFD and HFD mice (n = 8/group). p value was calculated using a two-tailed unpaired Mann-Whitney t test. C, Klhl9, Klhl13, and Cul3 mRNA expression was examined by qPCR in eWAT of LFD and HFD mice (n = 3/group). p value was calculated using a two-tailed unpaired t test. *, p < 0.05. D, correlation analysis of BMI and ATG16L1, KLHL9, KLHL13, or CUL3 gene expression in publicly available datasets of human adipose tissue.
In adipose tissue of HFD mice, Klhl9 expression trended upwards without reaching statistical significance and Klhl13 and Cul3 remained unchanged compared with adipose tissue of LFD mice (Fig. 8C). On the other hand, analyzing published human transcriptomic datasets, a positive correlation emerged between adipose tissue KLHL13 mRNA and CUL3 mRNA with body mass index (BMI), an indicator of obesity (Fig. 8D). By this analysis, ATG16L1 and KLHL9 mRNA expression did not significantly correlate with BMI. Therefore, in human subjects, KLHL13 and CUL3 mRNA up-regulation in adipose tissue tracks with obesity.
Discussion
In this study, we sought to elucidate the mechanism responsible for attenuated insulin signaling to AKT described in autophagy-deficient murine and cellular models (15, 20). ATG16L1 KO MEFs displayed decreased insulin-stimulated PI(3,4,5)P3 generation that correlated with reductions in AKT phosphorylation and downstream GSK3β and P70S6K phosphorylation. Moreover, we observed attenuated insulin-stimulated ERK phosphorylation and loss of cortical actin remodeling, which are insulin responses independent of PI3K and AKT activity, respectively. Diminished actin dynamics contributes to insulin resistance additional to defects in AKT activity (8, 9, 26, 27) and might be an element in whole-body insulin resistance observed in models of deficient autophagy.
In principle, the various signaling defects observed in ATG16L1 KO MEFs could arise at a number of levels within the insulin signaling cascade. We considered it possible that the IR might be targeted following ATG16L1 depletion, by analogy to the effects of autophagy on other receptor tyrosine kinases and membrane proteins. This includes affecting c-MET phosphorylation and epidermal growth factor receptor stability (40), as well as expression of A disintegrin and metalloprotease 10 (Adam10) (41). However, reduced autophagy via deficient ATG16L1 did not lower IR or IGF1R content and in fact elevated the former. The higher IR-β protein levels of ATG16L1 KO MEFs are in line with previously reported autophagy-dependent regulation of the IR (42). This finding, together with the lack of changes in cell surface IR and insulin binding in ATG16L1 KO MEFs, excludes the possibility that defects at the level of the receptor play a causative role in the dampened AKT response to insulin observed in ATG16L1 KO cells.
Alternatively, we considered that ATG16L1 depletion may specifically target IRS1, a key nodule in insulin signaling (5, 8). Increased IRS1 inhibitory serine phosphorylation is a known manifestation of insulin resistance and T2D associated with decreased IRS1 tyrosine phosphorylation as well as diminished IRS1 protein content (2–7). However, others have shown that a nonphosphorylatable knock-in alanine mutation of a commonly affected IRS1 serine site (Ser-307) does not prevent high-fat diet-induced insulin resistance in mice and, in fact, induces markedly more severe insulin resistance (43). Therefore, inhibitory serine phosphorylation may not be sufficient to induce insulin resistance.
Here we show that ATG16L1 KO induces insulin resistance by diminishing IRS1 protein content, whereas overexpression of IRS1myc is sufficient to recover insulin signaling. Although Atg7 and Atg3 knockout and dominant-negative Atg5 are known to induce insulin resistance, to date the only connection between reduced autophagy and a defect in IRS1 is the observation that dominant-negative Atg5 decreases net insulin-stimulated IRS1 tyrosine phosphorylation in L6 myoblasts (20). However, IRS1 protein content was not investigated thereby leaving the underlying mechanism unresolved. As many studies continue to show that ATG proteins perform noncanonical roles (44, 45), it is imperative that future work examines IRS1 expression in other autophagy-deficient models. As such, our work indicates that ATG16L1 KO induces IRS1 loss, but does not discern whether this is a result of deficient autophagy or a noncanonical function of ATG16L1.
IRS1 protein content may be regulated by E3 Ub ligase-mediated ubiquitination, leading to its proteasomal degradation (5, 8, 12, 46, 47). Alternative mechanisms have also been proposed, such as proteasomal-independent degradation as observed under oxidative stress (46) and caspase 3-mediated IRS1 cleavage as observed under hypoxia (48). Hinting that the reduced IRS1 levels in ATG16L1 KO MEFs in the present study are proteasomal-dependent, inhibiting the 20S proteasome via MG132 resulted in increased IRS1 protein content. However, we found no involvement of the mechanisms previously described to signal or mediate proteasomal IRS1 degradation (involvement of JNK signaling and participation of E3 Ub ligases SOCS1 and SOCS3). Hence, we explored other potential E3 Ub ligase complexes that might regulate IRS1 protein levels, through mining of the BioID data of proteins associated with diverse E3 Ub ligase complexes. By this analysis the Kelch-like proteins KLHL9 and KLHL13, which form an E3 Ub ligase complex with CUL3, emerged as novel interactors of IRS1. Kelch-like proteins are a family of 42 proteins, several of which interact with the E3 Ub ligase CUL3 to provide substrate specificity for CUL3-mediated ubiquitination (49). To date, the only known target of the KLHL9/KLHL13/CUL3 complex is Aurora B, which is thereby ubiquitinated and destined for degradation as a requisite step in mitosis (36).
Consistent with the regulation of CUL3 activity through changes in the expression of its complex partner proteins (37), Klhl9 and Klhl13 expression was elevated in ATG16L1 KO MEFs concomitant with reduced IRS1 levels. Mechanistically, reducing the expression of each member of this E3 Ub ligase complex through siRNA-mediated knockdown increased IRS1 protein content. These findings support the notion that the interaction between KLHL9/KLHL13/CUL3 and IRS1 is of functional significance, promoting IRS1 degradation when expression of both KLHL proteins rises. Moreover, Klhl13 or Cul3 knockdown significantly recovered AKT phosphorylation in ATG16L1 KO MEFs. Therefore, we have identified a novel E3 Ub ligase complex that may contribute to insulin resistance through IRS1 degradation. A recent report indicates that CUL3, via KBTBD2 substrate recognition receptor binding, regulates p110-free p85 degradation, a component of PI3K that when unbound to p110 acts as a competitive inhibitor of PI3K for IRS1 binding, thus impairing insulin action (50). Taken together, these findings identify CUL3 as an important mediator in modulating insulin signaling, dependent on which substrate recognition receptor is present.
Given our findings of increased Klhl9 and Klhl13 mRNA in ATG16L1 KO MEFs, an interesting question arising is what regulates expression of these E3 Ub ligase components. To date, there are no studies examining the regulation of Klhl9 or Klhl13, and very few have searched for the proteins they target for degradation, with only Aurora B being identified (36). We speculate that, under conditions of deficient ATG16L1, the induction of Klhl9 and Klhl13 might be associated to induction of cellular stress responses such as ER stress, inflammation and/or oxidative stress. Nrf2, an antioxidant transcription factor, represents a promising potential regulator of these genes, as it is known to translocate to the nucleus and increase transcriptional activity in autophagy-deficient models, as well as under cellular stress (17, 51–53). Moreover, Nrf2 is associated with the development of insulin resistance (17, 54). Interestingly, KLHL13 is a predicted gene target of Nrf2 (55). However, over 1000 genes were identified as predicted targets, and this interaction has not been supported experimentally. Future work should examine the potential for the Nrf2 signaling pathway to mediate the induction of klhl9 and klhl13 mRNA expression and whether these E3 Ub ligase components mediate the insulin resistance associated with Nrf2 induction.
Our findings complement previous work on deficient autophagy-induced insulin resistance, in which autophagy depletion markedly decreased insulin-stimulated AKT phosphorylation in liver, adipose tissue, muscle, and L6 skeletal muscle cells (15, 17, 18, 20). Recent reports of increased insulin action in mice with hyperactive autophagy, induced by overexpression of constitutively active beclin 1, further support a role of autophagy in insulin signaling (19). As insulin signaling is known to inhibit autophagy through mTORC1 activation (56–58), these findings suggest a potential feedback mechanism whereby deficient autophagy inhibits insulin action at an upstream step. Although those studies were unable to elucidate a mechanistic link through which deficient autophagy induces insulin resistance, our results shed light on this connection.
The mechanistic findings presented here are of relevance to obesity and diabetes given the observations of attenuated autophagy in various tissues in these conditions (15, 16, 59–61). However, at present the association of deficient autophagy and diabetes remains unclear and likely dependent on the tissue examined and the model of insulin resistance (59, 62, 63). This highlights the complexity of establishing cause-effect mechanistic relationships in vivo. Therefore, we deemed it important to examine insulin action at the cellular level following autophagy depletion, in the interest of identifying autophagy-insulin signaling interactions directly and to separate the complications associated with tissue environments in vivo. Nevertheless, we found that adipose tissue from HFD-induced obese mice have lowered levels of IRS1 protein and Atg16l1 mRNA, and, in humans, increasing BMI was associated with higher adipose tissue KLHL13 and CUL3 mRNA expression.
Collectively, our findings show that ATG16L1 KO induces insulin resistance through a previously unrealized, targeted degradation of IRS1 mediated by the novel E3 Ub ligase complex KLHL9/KLHL13/CUL3. We hypothesize that this might contribute to the development of insulin resistance through reductions in IRS1 protein content that have been observed in adipocytes of human diabetic patients (2). As insulin resistance is associated with IRS1 dysfunction, the implication of deficient autophagy provoking IRS1 degradation highlights the potential for judicious targeting of autophagy and/or KLHL9/KLHL13/CUL3 to restore insulin action.
Experimental procedures
Materials
EZ-Link Sulfo-NHS-SS-Biotin (number 21331) and streptavidin-agarose beads (number 20349) were from Thermo Fisher Scientific. Human insulin analogue, Humulin R U-100, was from Eli Lilly. Human LR3 IGF1 was from GroPep (number AM001). [125I-TyrA14]insulin (125I-insulin) was from Perkin Elmer Life Sciences. Protease inhibitors (number P3840) were from Sigma. Odyssey blocking buffer (TBS; number 927-50000) and species-specific conjugated IRDye 800CW or IRDye 680LT secondary antibodies were from LI-COR. MG132 (number S2619) was from Selleckchem. Primary antibodies for immunoblotting and immunofluorescence experiments were: IRS1 (number 2382), IGF1R-β (number 3027), anti-rabbit β-actin (number 4970), pAKT S473 (number 4060), pAKT T308 (number 9275), pan-AKT (number 2920), pJNK T183/Y185 (number 9251), JNK (number 9252), pERK (number 9101), ERK (number 4696), pGSK3β (number 9323), GSK3β (number 9832), pP70S6K (number 9205) and P70S6K (number 9202) from Cell Signaling; anti-mouse β-actin (number A5441) from Sigma; LC3-B (number NB600-1384) from Novus Bio; IR-β (number SC-57342) and anti-myc 9E10 (number SC-40) from Santa Cruz; ATG16L1 (number M150-3) from MBL; FK2 antibody for mono- and poly-ubiquitinylated conjugates (number BML-PW8810) from Enzo Life Sciences; GAPDH (number MAB374) was from Millipore. Phalloidin-565, phalloidin-670, and all fluorescent secondary antibodies (Alexa Fluor conjugates) were from Invitrogen. GeneJuice transfection reagent (number 70967) was from Millipore. The following constructs were obtained as gifts: LC3-GFP (from Dr. T. Yoshimori, Osaka University, Japan (23)), PH-AKT-GFP (from Dr. S. Grinstein, The Hospital for Sick Children, Toronto (24, 25)), and IRS1myc (from Dr. S. I. Takahashi, University of Tokyo, Japan (64)). TaqMan qPCR gene expression primers for Irs1 (Mm01278327), Klhl9 (Mm01267656), Klhl13 (Mm00470674), Cul3 (Mm00516747), Socs1 (Mm00782550), Socs3 (Mm00545913), Cbl-b (Mm01343092), Cul7 (Mm00481653), and Abt1 (Mm00803824) were from Thermo Fisher Scientific, whereas custom qPCR gene expression primers for Hprt (forward, TCAGTCAACGGGGGACATAAA; reverse, GGGGCTGTACTGCTTAACCAG) and Atg16l1 (forward, CAGAGCAGCTACTAAGCGACT; reverse, AAAAGGGGAGATTCGGACAGA) were from Sigma. KLHL13 and CUL3, cloned into a pcDNA3.1 nHA-DEST vector, were obtained from the Human ORFeome via the SPARC BioCentre (Hospital for Sick Children). The KLHL9 gene sequence from BC039133 was cloned into a pcDNA3 3xHA plasmid. Myc-ATG16L1 was previously described (65).
Cell culture
ATG16L1 KO and corresponding wildtype (WT) MEF (21) and HEK293T cells were cultured in DMEM (Wisent) supplemented with 10% heat-inactivated FBS (Wisent) without antibiotics at 37 °C and 5% CO2. MEFs were serum starved for 3 h prior to treatment with insulin (10 nm for immunoblotting or 100 nm for immunofluorescence) or IGF1 (100 ng/ml; 13.3 nm) for 10 min. MG132 (500 nm) was administered for 24 h in regular media (DMEM with 10% FBS). For insulin stimulation in the presence of MG132, 3-h serum starvation was performed in the presence of MG132 following 21 h of treatment in regular media. Rat L6 muscle cells with stable expression of myc-tagged glucose transporter 4 (L6-GLUT4myc) were grown in α-minimal essential medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml of penicillin, 100 μg/ml of streptomycin, and 250 ng/ml of amphotericin B.
For siRNA-mediated knockdown, cells were seeded at 1.5 × 105 per well in 6-well plates 24 h before use. Cells were transfected using Lipofectamine RNAiMax (Invitrogen) as recommended by the manufacturer for 24 h, siRNA was removed, and cells were grown for an additional 24 h in regular media. The following siRNAs were obtained from Sigma: Atg16l1 (1: SASI_Rn02_00242286 and 2: (SASI_Mm01_00138429), Klhl13 (SASI_Mm01_00074699), Cul3 (SASI_Mm02_00323408), and Socs3 (SASI_Mm02_00312273). For control knockdown, MISSION® siRNA Universal Negative Control (Sigma, number SIC001) was used. qPCR was performed to evaluate knockdown efficiency, as noted in figure legends.
Animal model
For protein measurements, 8-week-old male C57BL/6 mice (Charles River) were randomly assigned to the following groups: a control LFD (10% kcal from fat; D12450Ji, Research Diets) or HFD (60% kcal from fat; D12492i, Research Diets) for 15 weeks, as previously described (38). Mice were caged in a temperature-controlled facility with a 12:12 h light:dark cycle. Mice were anesthetized with sodium pentobarbital and epididymal (abdominal) white adipose tissue was collected and flash frozen for immunoblotting analysis. Mice were then euthanized via cervical dislocation. Animal use protocols were approved by the University of Guelph Animal Care Committee and met Canadian Council on Animal Care (CCAC) guidelines.
For qPCR measurements, 8-week-old male C57BL/6 mice were fed LFD or HFD, as above, for 18 weeks. Mice were euthanized via cervical dislocation and epididymal (abdominal) white adipose tissue was collected and flash frozen for qPCR analysis. Animal use protocols were approved by The Hospital for Sick Children Animal Care Committee and met Canadian Council on Animal Care (CCAC) guidelines.
Immunoblotting
Cells were seeded at 1.5 × 105 per well in 6-well plates. Immunoblotting was performed as previously described (66). Briefly, cells rinsed with cold PBS and lysed on ice in RIPA buffer (50 mm Tris, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with 5 mm NaF, 1 mm EDTA, 1 mm PMSF, 5 mm Na3VO4 and protease inhibitors. Lysates were boiled for 5 min in 5× Laemmli sample buffer with β-mercaptoethanol. Equal amounts of protein were separated by SDS-PAGE, transferred to nitrocellulose membranes, blocked with LI-COR blocking buffer, and incubated overnight with primary antibodies in 1% BSA-TBST at 4 °C with gentle agitation. Membranes were then incubated with fluorescent secondary antibodies in TBST for 45 min protected from light at room temperature and developed using an Odyssey Fc Imager (LI-COR). Results were quantified using the Image Studio 4.0 software (LI-COR).
Biotin labeling of cell surface proteins
Biotin labeling of cell surface proteins was performed as previously described (66). MEFs in 6-well plates were washed with cold PBS, placed on ice for 10 min to block endocytosis, and then incubated at 4 °C with or without a solution of EZ-Link Sulfo-NHS-SS-Biotin (1 mg/ml in PBS) for 30 min with gentle rocking (3 wells/condition), followed by five PBS washes and a 45-min incubation on ice with 0.1 m glycine in PBS. Biotinylated cells were washed three times with cold PBS and lysed as described above. 15 μg of protein lysates were used as input and 150 μg were incubated overnight with 50 μl of 50% slurry streptavidin-agarose beads. After centrifugation, pelleted beads were washed 3 times with 1% Nonidet P-40 in PBS. Finally, biotinylated proteins (cell surface proteins) were eluted from the beads by adding 30 μl of 1× Laemmli buffer with β-mercaptoethanol and heating at 95 °C for 5 min. Immunoblotting for IR-β was performed using the input lysate, bead elution (pulldown), and the remaining supernatant following pulldown.
Immunoprecipitation for IRS1 ubiquitination
MEFs were seeded at 9 × 105 in 10-cm dishes, grown until 80–90% confluent, and then treated with DMSO or 500 nm MG132 for 24 h in regular media. After 24 h, cells were rinsed twice with PBS, lysed in nondenaturing buffer (20 mm Tris-HCl, pH 8, 137 mm NaCl, 1% Nonidet P-40, 10% glycerol) supplemented with 5 mm NaF, 1 mm PMSF, 5 mm Na3VO4 and protease inhibitors, diluted to 500 μg/300 μl and incubated with IRS1 antibody (Cell Signaling, number 2382) at 1:50 overnight with gentle rotation at 4 °C. 20 μl of 50:50 protein A bead slurry was added to tubes for 4 h. Samples were then centrifuged, supernatants were collected, and beads were washed five times with 1% Nonidet P-40–PBS. Beads were then diluted in 25 μl of 1× Laemmli buffer with β-mercaptoethanol and heated at 95 °C for 5 min. After centrifugation, immunoblotting was performed as described above.
Co-immunoprecipitation
HEK 293T cells were seeded in 10-cm tissue culture dishes. The following day, cells were co-transfected with Irs1myc or a control myc-tag plasmid and HA-tagged plasmids harboring the candidate target (6 μg of total DNA) for 24 h using GeneJuice (VWR) according to the manufacturer's instructions. Co-immunoprecipitation was performed according to the manufacturer's protocol for Myc-Trap_A (Chromotek). Briefly, cells were harvested with 1 ml of PBS, centrifuged at 500 × g for 3 min at 4 °C, washed three times in PBS, and resuspended in 200 μl of ice-cold lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100) supplemented with 1 mm PMSF, 5 mm NaF, 5 mm NaVO4, 10 μg/ml of aprotinin, 10 μg/ml of leupeptin, and 1 μm pepstatin A. Following mechanical lysis with a syringe, cells were centrifuged at 20,000 × g for 10 min at 4 °C and the supernatant was collected. A BCA assay was performed and 300 μg of protein was transferred to a new tube and incubated for 2 h with Myc-Trap_A (Chromotek) at 4 °C with end-over-end tumbling. 5× SDS-sample buffer was added to the remaining lysate, and boiled for 5 min to assess input. Following incubation, beads were centrifuged at 2,500 × g for 2 min at 4 °C, washed 3× in lysis buffer, eluted with 50 μl of 2× SDS-sample buffer, and boiled for 5 min prior to immunoblotting.
125I-Insulin–binding assay
MEFs were seeded at 2.5 × 104 cells per well in 24-well plates. Cells were serum starved for 3 h at 37 °C, placed on ice for 10 min, and then washed with cold PBS prior to incubation with 0.5 nm 125I-Insulin in HEPES-binding buffer for 20 min on ice. After incubation, cells were washed with cold PBS and lysed with 0.1 m NaOH. Radioactivity was measured in a Hidex 300 SL liquid scintillation counter (Hidex, Turku, Finland) that detects γ radiation. Background radiation count was determined using cells that were not incubated with 125I-Insulin, and the values were subtracted from all other samples. Protein was measured by BCA and insulin binding was expressed in fmol/mg, relative to WT.
qPCR
RNA was purified using TRIzol reagent and cDNA was synthesized by reverse transcription using the SuperScript VILO cDNA kit (Thermo Fisher Scientific). For cell culture and mouse tissue experiments, cDNAs were amplified using predesigned TaqMan probes according to the manufacturer's instructions. Abt1 (Taqman) was used as a housekeeping gene. For Hprt and Atg16l1, custom primers were designed, and qPCR was performed using Bio-Rad Ssofast Evagreen Supermix according to the manufacturer's protocols. Hprt was used as a housekeeping gene. Relative quantities of each mRNA were calculated by the comparative ΔΔCT method. Housekeeping gene expression did not change between groups.
Live cell imaging of PH-AKT-GFP
MEFs were plated at 1 × 105 cells per well in 6-well tissue culture plates with glass coverslips 48 h before imaging. At 24 h, cells were transfected with PH-Akt-GFP. On the day of experimentation, cells were serum starved for 3 h, coverslips were washed with PBS and transferred into microscope chambers with live cell imaging buffer (140 mm NaCl, 2.5 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 20 mm HEPES, 2 mm Na pyruvate, 20 mm glucose, pH 7.4). Quorum spinning disk confocal scan heads (Leica DMI 6000B inverted fluorescence microscope, Hamamatsu ImagEM x2 camera) equipped with a ×63 objective were used to acquire 1 image per minute for 5 min on Volocity 6 software. Immediately after capturing the first image (time 0.00), 100 nm insulin was added to the microscope chambers. Confocal images were imported into Adobe Photoshop and assembled in Adobe Illustrator for labeling.
Immunofluorescence of LC3-GFP, actin remodeling, IRS1myc, and Atg16l1myc
MEFs were seeded at 2 × 104 cells/well in 24-well plates with glass coverslips 48 h before fixation. For LC3-GFP, IRS1myc, and Atg16l1myc experiments, cells were transfected 24 h before fixation. For insulin treatment, cells were starved 3 h prior to incubation with 100 nm insulin for 10 min. Cells were fixed and immunostaining was conducted as previously described (67). Briefly, cells were permeabilized and blocked in PBS containing 0.2% saponin (Calbiochem) and 10% normal goat serum (SS-PBS) for 30 min. Subsequently cells were incubated for 1 h with primary antibodies in SS-PBS, washed three times with PBS, and incubated with secondary Alexa Fluor-conjugated antibodies for 1 h. Cells were washed three times with PBS and mounted in fluorescence mounting medium (Dako). Images were acquired using a Quorum spinning disk confocal scan head (Leica DMI 6000B inverted fluorescence microscope, Hamamatsu ORCA Flash 4 sCMOS and color camera) equipped with a ×63 objective. For IRS1myc expression experiments, fluorescent intensity of pAKT Ser-473 relative to region of interest volume was quantified using Volocity 6 software. Separately, cortical actin remodeling, observed as enriched regions of signal that extend dorsally above the cell, was quantified and the percent of cells displaying this phenotype was calculated. Confocal images were imported into Adobe Photoshop and assembled in Adobe Illustrator for labeling.
BioID sample preparation
BioID (34) was carried out essentially as described previously (35). In brief, full-length human KLHL9 (source DNA: BC039133) and KLHL13 (source DNA: BC064576) coding sequences were amplified by PCR and cloned into pcDNA5 FRT/TO FLAGBirA* expression vector. Using the Flp-In system (Invitrogen), Flp-In 293 T-REx cells (Thermo Fisher Scientific) stably expressing FLAGBirA* alone, FlagBirA*-KLHL9, or FLAGBirA*-KLHL13 were generated. After selection (DMEM + 10% FBS + 200 μg/ml of hygromycin B), two independent replicates of five 150-cm2 plates of subconfluent (60%) cells were incubated for 24 h in complete media supplemented with 1 μg/ml of tetracycline (Sigma) and 50 μm biotin (BioShop). Cells were collected and pelleted (2,000 rpm, 3 min), the pellet was washed twice with PBS, and dried pellets were snap frozen. The cell pellet was resuspended in 10 ml of lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 0.1% SDS, 1:500 protease inhibitor mixture (Sigma), 1:1,000 benzonase nuclease (Novagen) and incubated on an end-over-end rotator at 4 °C for 1 h, briefly sonicated to disrupt any visible aggregates, then centrifuged at 45,000 × g for 30 min at 4 °C. Supernatant was transferred to a fresh 15-ml conical tube. 30 μl of packed, pre-equilibrated streptavidin-Sepharose beads (GE Healthcare) were added and the mixture incubated for 3 h at 4 °C with end-over-end rotation. Beads were pelleted by centrifugation at 2,000 rpm for 2 min and transferred with 1 ml of lysis buffer to a fresh Eppendorf tube. Beads were washed once with 1 ml of lysis buffer and twice with 1 ml of 50 mm ammonium bicarbonate, pH 8.3. Beads were transferred in ammonium bicarbonate to a fresh centrifuge tube and washed two more times with 1 ml of ammonium bicarbonate buffer. Tryptic digestion was performed by incubating the beads with 1 μg of MS-grade TPCK-trypsin (Promega, Madison, WI) dissolved in 200 μl of 50 mm ammonium bicarbonate, pH 8.3, overnight at 37 °C. The following morning, 0.5 μg of MS-grade TPCK-trypsin was added, and beads were incubated 2 additional hours at 37 °C. Beads were pelleted by centrifugation at 2,000 × g for 2 min, and the supernatant was transferred to a fresh Eppendorf tube. Beads were washed twice with 150 μl of 50 mm ammonium bicarbonate, and these washes were pooled with the first eluate. The sample was lyophilized and resuspended in buffer A (0.1% formic acid). 1/5th of the sample was analyzed per MS run.
Mass spectrometry
HPLC was conducted using a 2-cm pre-column (Acclaim PepMap 50 mm × 100 μm inner diameter), and 50-cm analytical column (Acclaim PepMap, 500 mm × 75 μm diameter; C18; 2 um; 100 Å, Thermo Fisher Scientific, Waltham, MA), running a 120-min reversed-phase buffer gradient at 225 nl/min on a Proxeon EASY-nLC 1000 pump in-line with a Thermo Q-Exactive HF quadrupole-Orbitrap mass spectrometer. A parent ion scan was performed using a resolving power of 60,000, then up to 20 of the most intense peaks were selected for MS/MS (minimum ion count of 1,000 for activation) using higher energy collision-induced dissociation fragmentation. Dynamic exclusion was activated such that MS/MS of the same m/z (within a range of 10 ppm; exclusion list size = 500) detected twice within 5 s were excluded from analysis for 15 s. For protein identification, Thermo.RAW files were converted to the.mzXML format using Proteowizard (68), then searched using X!Tandem (69) and COMET (70) against the Human RefSeq Version 45 database (containing 36,113 entries). Data were analyzed using the trans-proteomic pipeline (71, 72) via the ProHits software suite (version 3.3) (73). Search parameters specified a parent ion mass tolerance of 10 ppm, and an MS/MS fragment ion tolerance of 0.4 Da, with up to 2 missed cleavages allowed for trypsin. Variable modifications of +16 at Met and Trp, +32 at Met and Trp, +42 at the N terminus, and +1 at the Asn and Gln were allowed. Proteins identified with an iProphet cut-off of 0.9 (corresponding to ≤1% FDR) and at least two unique peptides were analyzed with SAINT Express version 3.6. Twenty control runs (from cells expressing the FlagBirA* epitope tag) were collapsed to the two highest spectral counts for each prey and compared with the two technical of each of the two biological replicates of KLHL9 and KLHL13 BioID. High confidence interactors were defined as those with bayesian false discovery rate (BFDR) ≤0.01.
Human transcriptomic correlations to BMI
Data from 8 publicly available datasets of human obesity were downloaded from the GEO repository (GSE9624, GSE15773, GSE16415, GSE20950, GSE27949, GSE29231, and GSE29226). The analysis was performed using R 3.5.2. After quality control, datasets were annotated and merged according to official gene symbols. Raw data were then normalized using quantile normalization to allow comparisons across studies. BMI values were collected from the publications associated with each dataset. Adipose tissue expression data from ATG16L1, KLHL9, KLHL13, and CUL3 were then correlated with BMI using spearman correlations.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism version 7.0g. The mean ± S.E. is shown in figures and the statistical tests used to determine p values are described in detail in the corresponding figure legends. p < 0.05 was considered statistically significant. The degree of significance is denoted as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Author contributions
S. F.-C., J. H. B., and A. K. conceptualization; S. F.-C., N. J. P., J. H. B., and A. K. data curation; S. F.-C., J. R. J.-F., E. C., E. M. L., J. M. T., and N. J. P. formal analysis; S. F.-C., J. R. J.-F., E. C., E. M. L., L. K. T., J. M. T., N. J. P., J. H. B., and A. K. investigation; S. F.-C. visualization; S. F.-C., J. R. J.-F., E. C., E. M. L., L. K. T., J. M. T., R. J. X., N. J. P., B. R., and D. C. W. methodology; S. F.-C., J. H. B., and A. K. writing-original draft; S. F.-C. project administration; S. F.-C., J. R. J.-F., E. C., E. M. L., L. K. T., J. M. T., R. J. X., N. J. P., B. R., D. C. W., J. H. B., and A. K. writing-review and editing; R. J. X., B. R., D. C. W., J. H. B., and A. K. resources; B. R., D. C. W., J. H. B., and A. K. supervision; B. R., J. H. B., and A. K. funding acquisition.
Supplementary Material
Acknowledgments
We thank Dr. Philip J. Bilan for helpful advice throughout this study, Melanie Villanueva for help with construct cloning and sequencing, Parastoo Boroumand for providing tissues of mice fed high- and low-fat diets, and Zhi Liu for technical assistance.
This work was supported by Canadian Institutes of Health Research (CIHR) Grants FDN 154329 (to J. H. B.), FDN-143203 (to A. K.), and PJT 156093 (to B. R.), a John Evans Leadership Fund infrastructure grant from the Canadian Foundation for Innovation and the Ontario Innovation Trust (to J. H. B.), Natural Sciences and Engineering Research Council (NSERC) Alexander Graham Bell Canada Graduate Scholarship-Doctoral, a PricewaterhouseCoopers Student Bursary, and a Restracomp-Hospital for Sick Children scholarship (to S. F.-C.), a Natural Sciences and Engineering Research Council (NSERC) Post-graduate Scholarship award (to L. K. T.), a Tier II Canada Research Chair in Lipids, Metabolism and Health grant (to D. C. W.), a Tier I Canada Research Chair holder on Cell Biology of Insulin Action (to A. K.), a Canada Research Chair in Proteomics and Molecular Medicine (to B. R.), and the Pitblado Chair in Cell Biology at the Hospital for Sick Children (to J. H. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
The mass spectrometry data for KLHL9 and KLHL13 BioID has been deposited in the massIVE database and assigned the identifier MSV000083653.
- T2D
- type 2 diabetes
- IGF1
- insulin-like growth factor-1
- Ub
- ubiquitin
- ATG16L1
- autophagy-related 16L1
- KLHL
- Kelch-like
- IRS1
- insulin receptor substrate 1
- BioID
- proximity-dependent biotin identification
- IR
- insulin receptor
- CUL3
- Cullin 3
- LC3
- microtubule-associated light chain 3B
- PI3K
- phosphoinositide 3-kinase
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PI(3,4,5)P3
- phosphatidylinositol (3,4,5)-trisphosphate
- PH
- pleckstrin homology
- PDK1
- phosphoinositide-dependent kinase 1
- SOCS
- suppressor of cytokine signaling
- HFD
- high-fat diet
- LFD
- low-fat diet
- BMI
- body mass index
- PMSF
- phenylmethylsulfonyl fluoride
- TPCK
- l-1-tosylamido-2-phenylethyl chloromethyl ketone
- ER
- endoplasmic reticulum
- KO
- knock-out
- MEF
- mouse embryonic fibroblast
- FBS
- fetal bovine serum
- DMEM
- Dulbecco's modified Eagle's medium
- qPCR
- quantitative PCR
- ANOVA
- analysis of variance
- JNK
- c-Jun N-terminal kinase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GSK3β
- glycogen synthase kinase 3β.
References
- 1. Olokoba A. B., Obateru O. A., and Olokoba L. B. (2012) Type 2 diabetes mellitus: a review of current trends. Oman Med. J. 27, 269–273 10.5001/omj.2012.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carvalho E., Jansson P. A., Axelsen M., Eriksson J. W., Huang X., Groop L., Rondinone C., Sjöström L., and Smith U. (1999) Low cellular IRS 1 gene and protein expression predict insulin resistance and NIDDM. FASEB J. 13, 2173–2178 10.1096/fasebj.13.15.2173 [DOI] [PubMed] [Google Scholar]
- 3. Rondinone C. M., Wang L. M., Lonnroth P., Wesslau C., Pierce J. H., and Smith U. (1997) Insulin receptor substrate (IRS) 1 is reduced and IRS-2 is the main docking protein for phosphatidylinositol 3-kinase in adipocytes from subjects with non-insulin-dependent diabetes mellitus. Proc. Natl. Acad. Sci. U.S.A. 94, 4171–4175 10.1073/pnas.94.8.4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kerouz N. J., Hörsch D., Pons S., and Kahn C. R. (1997) Differential regulation of insulin receptor sibstrates-1 and -2 (IRS-1 and IRS-2) and phosphatidylinositol 3 kinase isoforms in liver and muscle of the obese diabetic (ob/ob) mouse. J. Clin. Invest. 100, 3164–3172 10.1172/JCI119872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thirone A. C., Huang C., and Klip A. (2006) Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol. Metab. 17, 72–78 10.1016/j.tem.2006.01.005 [DOI] [PubMed] [Google Scholar]
- 6. Björnholm M., Kawano Y., Lehtihet M., and Zierath J. R. (1997) Insulin receptor substrate-1 phosphorylation and phosphatidylinositol 3-kinase activity in skeletal muscle from NIDDM subjects after in vivo insulin stimulation. Diabetes 46, 524–527 10.2337/diab.46.3.524 [DOI] [PubMed] [Google Scholar]
- 7. Gual P., Le Marchand-Brustel Y., and Tanti J. F. (2005) Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie (Paris) 87, 99–109 10.1016/j.biochi.2004.10.019 [DOI] [PubMed] [Google Scholar]
- 8. Jaldin-Fincati J. R., Pavarotti M., Frendo-Cumbo S., Bilan P. J., and Klip A. (2017) Update on GLUT4 vesicle traffic: a cornerstone of insulin action. Trends Endocrinol. Metab. 28, 597–611 10.1016/j.tem.2017.05.002 [DOI] [PubMed] [Google Scholar]
- 9. Saltiel A. R., and Pessin J. E. (2002) Insulin signaling pathways in time and space. Trends Cell Biol. 12, 65–71 10.1016/S0962-8924(01)02207-3 [DOI] [PubMed] [Google Scholar]
- 10. Hotamisligil G. S. (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 10.1016/j.cell.2010.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boura-Halfon S., Shuster-Meiseles T., Beck A., Petrovich K., Gurevitch D., Ronen D., and Zick Y. (2010) A novel domain mediates insulin-induced proteasomal degradation of insulin receptor substrate 1 (IRS-1). Mol. Endocrinol. 24, 2179–2192 10.1210/me.2010-0072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hotamisligil G. S. (2005) Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and diabetes. Diabetes 54, S73–S78 10.2337/diabetes.54.suppl_2.S73 [DOI] [PubMed] [Google Scholar]
- 13. Aguirre V., Uchida T., Yenush L., Davis R., and White M. F. (2000) The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser 307*. J. Biol. Chem. 275, 9047–9054 10.1074/jbc.275.12.9047 [DOI] [PubMed] [Google Scholar]
- 14. Rui L., Yuan M., Frantz D., Shoelson S., and White M. F. (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 277, 42394–42398 10.1074/jbc.C200444200 [DOI] [PubMed] [Google Scholar]
- 15. Yang L., Li P., Fu S., Calay E. S., and Hotamisligil G. S. (2010) Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–478 10.1016/j.cmet.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu H. Y., Han J., Cao S. Y., Hong T., Zhuo D., Shi J., Liu Z., and Cao W. (2009) Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 284, 31484–31492 10.1074/jbc.M109.033936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cai J., Pires K. M., Ferhat M., Chaurasia B., Buffolo M. A., Smalling R., Sargsyan A., Atkinson D. L., Summers S. A., Graham T. E., and Boudina S. (2018) Autophagy ablation in adipocytes induces insulin resistance and reveals roles for lipid peroxide and Nrf2 signaling in adipose-liver crosstalk. Cell Rep. 25, 1708–1717.e5 10.1016/j.celrep.2018.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masiero E., Agatea L., Mammucari C., Blaauw B., Loro E., Komatsu M., Metzger D., Reggiani C., Schiaffino S., and Sandri M. (2009) Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–515 10.1016/j.cmet.2009.10.008 [DOI] [PubMed] [Google Scholar]
- 19. Yamamoto S., Kuramoto K., Wang N., Situ X., Priyadarshini M., Zhang W., Cordoba-Chacon J., Layden B. T., and He C. (2018) Autophagy differentially regulates insulin production and insulin sensitivity. Cell Rep. 23, 3286–3299 10.1016/j.celrep.2018.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Y., Palanivel R., Rai E., Park M., Gabor T. V., Scheid M. P., Xu A., and Sweeney G. (2015) Adiponectin stimulates autophagy and reduces oxidative stress to enhance insulin sensitivity during high-fat diet feeding in mice. Diabetes 64, 36–48 10.2337/db14-0267 [DOI] [PubMed] [Google Scholar]
- 21. Conway K. L., Kuballa P., Song J. H., Patel K. K., Castoreno A. B., Yilmaz O. H., Jijon H. B., Zhang M., Aldrich L. N., Villablanca E. J., Peloquin J. M., Goel G., Lee I. A., Mizoguchi E., Shi H. N., et al. (2013) Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 145, 1347–1357 10.1053/j.gastro.2013.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shibutani S. T., and Yoshimori T. (2014) A current perspective of autophagosome biogenesis. Cell Res. 24, 58–68 10.1038/cr.2013.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., and Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weernink P. A., Guo Y., Zhang C., Schmidt M., Von Eichel-Streiber C., and Jakobs K. H. (2000) Control of cellular phosphatidylinositol 4,5-bisphosphate levels by adhesion signals and Rho GTPases in NIH 3T3 fibroblasts. Eur. J. Biochem. 267, 5237–5246 10.1046/j.1432-1327.2000.01599.x [DOI] [PubMed] [Google Scholar]
- 25. Mallo G. V., Espina M., Smith A. C., Terebiznik M. R., Alemán A., Finlay B. B., Rameh L. E., Grinstein S., and Brumell J. H. (2008) SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J. Cell Biol. 182, 741–752 10.1083/jcb.200804131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sylow L., Kleinert M., Pehmøller C., Prats C., Chiu T. T., Klip A., Richter E. A., and Jensen T. E. (2014) Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell Signal. 26, 323–331 10.1016/j.cellsig.2013.11.007 [DOI] [PubMed] [Google Scholar]
- 27. Chiu T. T., Jensen T. E., Sylow L., Richter E. A., and Klip A. (2011) Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell Signal. 23, 1546–1554 10.1016/j.cellsig.2011.05.022 [DOI] [PubMed] [Google Scholar]
- 28. Lee A. V., Gooch J. L., Oesterreich S., Guler R. L., and Yee D. (2000) Insulin-like growth factor I-induced degradation of insulin receptor substrate 1 is mediated by the 26S proteasome and blocked by phosphatidylinositol 3′-kinase inhibition. Mol. Cell Biol. 20, 1489–1496 10.1128/MCB.20.5.1489-1496.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhande R., Mitchell J. J., Wu J., and Sun X. J. (2002) Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1. Mol. Cell. Biol. 22, 1016–1026 10.1128/MCB.22.4.1016-1026.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun X. J., Goldberg J. L., Qiao L. Y., and Mitchell J. J. (1999) Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes 48, 1359–1364 10.2337/diabetes.48.7.1359 [DOI] [PubMed] [Google Scholar]
- 31. Nakao R., Hirasaka K., Goto J., Ishidoh K., Yamada C., Ohno A., Okumura Y., Nonaka I., Yasutomo K., Baldwin K. M., Kominami E., Higashibata A., Nagano K., Tanaka K., Yasui N., et al. (2009) Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Mol. Cell Biol. 29, 4798–4811 10.1128/MCB.01347-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu X., Sarikas A., Dias-Santagata D. C., Dolios G., Lafontant P. J., Tsai S. C., Zhu W., Nakajima H., Nakajima H. O., Field L. J., Wang R., and Pan Z. Q. (2008) The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol. Cell 30, 403–414 10.1016/j.molcel.2008.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu X., Keshwani M., Meyer K., Sarikas A., Taylor S., and Pan Z. Q. (2012) Identification of the degradation determinants of insulin receptor substrate 1 for signaling Cullin-RING E3 ubiquitin ligase 7-mediated ubiquitination. J. Biol. Chem. 287, 40758–40766 10.1074/jbc.M112.405209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Coyaud E., Mis M., Laurent E. M., Dunham W. H., Couzens A. L., Robitaille M., Gingras A.-C., Angers S., and Raught B. (2015) BioID-based Identification of Skp Cullin F-box (SCF) β-TrCP1/2 E3 ligase substrates. Mol. Cell Proteomics 14, 1781–1795 10.1074/mcp.M114.045658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sumara I., Quadroni M., Frei C., Olma M. H., Sumara G., Ricci R., and Peter M. (2007) A Cul3-based E3 ligase removes aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev. Cell 12, 887–900 10.1016/j.devcel.2007.03.019 [DOI] [PubMed] [Google Scholar]
- 37. Petroski M. D., and Deshaies R. J. (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 10.1038/nrm1547 [DOI] [PubMed] [Google Scholar]
- 38. Frendo-Cumbo S., MacPherson R. E., and Wright D. C. (2016) Beneficial effects of combined resveratrol and metformin therapy in treating diet-induced insulin resistance. Physiol. Rep. 4, e12877 10.14814/phy2.12877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yin J., Gao Z., He Q., Zhou D., Guo Z., and Ye J. (2009) Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am. J. Physiol. Endocrinol. Metab. 296, E333–E342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fraser J., Cabodevilla A. G., Simpson J., and Gammoh N. (2017) Interplay of autophagy, receptor tyrosine kinase signalling and endocytic trafficking. Essays Biochem. 61, 597–607 10.1042/EBC20170091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maurer K., Reyes-Robles T., Alonzo F. 3rd, Durbin J., Torres V. J., and Cadwell K. (2015) Autophagy mediates tolerance to Staphylococcus aureus α-toxin. Cell Host Microbe. 17, 429–440 10.1016/j.chom.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou L., Zhang J., Fang Q., Liu M., Liu X., Jia W., Dong L. Q., and Liu F. (2009) Autophagy-mediated insulin receptor down-regulation contributes to endoplasmic reticulum stress-induced insulin resistance. Mol. Pharmacol. 76, 596–603 10.1124/mol.109.057067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Copps K. D., Hancer N. J., Opare-Ado L., Qiu W., Walsh C., and White M. F. (2010) Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 11, 84–92 10.1016/j.cmet.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Subramani S., and Malhotra V. (2013) Non-autophagic roles of autophagy-related proteins. EMBO Rep. 14, 143–151 10.1038/embor.2012.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Galluzzi L., and Green D. R. (2019) Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699 10.1016/j.cell.2019.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Potashnik R., Bloch-Damti A., Bashan N., and Rudich A. (2003) IRS1 degradation and increased serine phosphorylation cannot predict the degree of metabolic insulin resistance induced by oxidative stress. Diabetologia 46, 639–648 10.1007/s00125-003-1097-5 [DOI] [PubMed] [Google Scholar]
- 47. Pederson T. M., Kramer D. L., and Rondinone C. M. (2001) Serine/threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes 50, 24–31 10.2337/diabetes.50.1.24 [DOI] [PubMed] [Google Scholar]
- 48. Kang S. G., Brown A. L., and Chung J. H. (2007) Oxygen tension regulates the stability of insulin receptor substrate-1 (IRS-1) through caspase-mediated cleavage. J. Biol. Chem. 282, 6090–6097 10.1074/jbc.M610659200 [DOI] [PubMed] [Google Scholar]
- 49. Dhanoa B. S., Cogliati T., Satish A. G., Bruford E. A., and Friedman J. S. (2013) Update on the Kelch-like (KLHL) gene family. Hum. Genomics 7, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang Z., Turer E., Li X., Zhan X., Choi M., Tang M., Press A., Smith S. R., Divoux A., Moresco E. M., and Beutler B. (2016) Insulin resistance and diabetes caused by genetic or diet-induced KBTBD2 deficiency in mice. Proc. Natl. Acad. Sci. U.S.A. 113, E6418–E6426 10.1073/pnas.1614467113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Copple I. M., Lister A., Obeng A. D., Kitteringham N. R., Jenkins R. E., Layfield R., Foster B. J., Goldring C. E., and Park B. K. (2010) Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 285, 16782–16788 10.1074/jbc.M109.096545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y. S., Ueno I., Sakamoto A., Tong K. I., Kim M., Nishito Y., Iemura S., Natsume T., Ueno T., et al. (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223 10.1038/ncb2021 [DOI] [PubMed] [Google Scholar]
- 53. Bryan H. K., Olayanju A., Goldring C. E., and Park B. K. (2013) The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 85, 705–717 10.1016/j.bcp.2012.11.016 [DOI] [PubMed] [Google Scholar]
- 54. Xu J., Kulkarni S. R., Donepudi A. C., More V. R., and Slitt A. L. (2012) Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 61, 3208–3218 10.2337/db11-1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Papp D., Lenti K., Módos D., Fazekas D., Dúl Z., Türei D., Földvári-Nagy L., Nussinov R., Csermely P., and Korcsmáros T. (2012) The NRF2-related interactome and regulome contain multifunctional proteins and fine-tuned autoregulatory loops. FEBS Lett. 586, 1795–1802 10.1016/j.febslet.2012.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim J., Kundu M., Viollet B., and Guan K.-L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S. J., Di Lisi R., Sandri C., Zhao J., Goldberg A. L., Schiaffino S., and Sandri M. (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471 10.1016/j.cmet.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 58. Codogno P., and Meijer A. J. (2005) Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 12, 1509–1518 10.1038/sj.cdd.4401751 [DOI] [PubMed] [Google Scholar]
- 59. Zhang Y., Sowers J. R., and Ren J. (2018) Targeting autophagy in obesity: from pathophysiology to management. Nat. Rev. Endocrinol. 14, 356–376 10.1038/s41574-018-0009-1 [DOI] [PubMed] [Google Scholar]
- 60. Meng Q., and Cai D. (2011) Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IκB kinase β (IKKβ)/NF-κB pathway. J. Biol. Chem. 286, 32324–32332 10.1074/jbc.M111.254417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bollinger L. M., Powell J. J., Houmard J. A., Witczak C. A., and Brault J. J. (2015) Skeletal muscle myotubes in severe obesity exhibit altered ubiquitin-proteasome and autophagic/lysosomal proteolytic flux. Obesity 23, 1185–1193 10.1002/oby.21081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stienstra R., Haim Y., Riahi Y., Netea M., Rudich A., and Leibowitz G. (2014) Autophagy in adipose tissue and the beta cell: implications for obesity and diabetes. Diabetologia 57, 1505–1516 10.1007/s00125-014-3255-3 [DOI] [PubMed] [Google Scholar]
- 63. Maixner N., Bechor S., Vershinin Z., Pecht T., Goldstein N., Haim Y., and Rudich A. (2016) Transcriptional dysregulation of adipose tissue autophagy in obesity. Physiology 31, 270–282 10.1152/physiol.00048.2015 [DOI] [PubMed] [Google Scholar]
- 64. Ozoe A., Sone M., Fukushima T., Kataoka N., Chida K., Asano T., Hakuno F., and Takahashi S.-I. (2014) Insulin receptor substrate-1 associates with small nucleolar RNA which contributes to ribosome biogenesis. Front. Endocrinol. (Lausanne) 5, 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kuballa P., Huett A., Rioux J. D., Daly M. J., and Xavier R. J. (2008) Impaired autophagy of an intracellular pathogen induced by a Crohn's disease associated ATG16L1 variant. PLoS ONE 3, e3391 10.1371/journal.pone.0003391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jaldin-Fincati J. R., Pereira R. V. S., Bilan P. J., and Klip A. (2018) Insulin uptake and action in microvascular endothelial cells of lymphatic and blood origin. Am. J. Physiol. Endocrinol. Metab. 315, E204–E217 10.1152/ajpendo.00008.2018 [DOI] [PubMed] [Google Scholar]
- 67. Czuczman M. A., Fattouh R., van Rijn J. M., Canadien V., Osborne S., Muise A. M., Kuchroo V. K., Higgins D. E., and Brumell J. H. (2014) Listeria monocytogenes exploits efferocytosis to promote cell-to-cell spread. Nature 509, 230–234 10.1038/nature13168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kessner D., Chambers M., Burke R., Agus D., and Mallick P. (2008) ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 10.1093/bioinformatics/btn323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Craig R., and Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 10.1093/bioinformatics/bth092 [DOI] [PubMed] [Google Scholar]
- 70. Eng J. K., Jahan T. A., and Hoopmann M. R. (2013) Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 10.1002/pmic.201200439 [DOI] [PubMed] [Google Scholar]
- 71. Pedrioli P. G. (2010) Trans-proteomic pipeline: a pipeline for proteomic analysis. Methods Mol. Biol. 604, 273–283 10.1007/978-1-60761-444-9_15 [DOI] [PubMed] [Google Scholar]
- 72. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., and Aebersold R. (2010) A guided tour of the trans-proteomic pipeline. Proteomics 10, 1150–1159 10.1002/pmic.200900375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu G., Zhang J., Larsen B., Stark C., Breitkreutz A., Lin Z. Y., Breitkreutz B. J., Ding Y., Colwill K., Pasculescu A., Pawson T., Wrana J. L., Nesvizhskii A. I., Raught B., Tyers M., and Gingras A. C. (2010) ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat. Biotechnol. 28, 1015–1017 10.1038/nbt1010-1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.