

SUMMARY
There is considerable debate about the functionality of long non-coding RNAs (lncRNAs). Lack of sequence conservation has been used to argue against functional relevance. We investigate antisense lncRNAs called COOLAIR at the A. thaliana FLC locus and have experimentally determined their secondary structure. The major COOLAIR variants are highly structured, organized by exon. The distally polyadenylated transcript has a complex multi-domain structure, altered by a single non-coding SNP defining a functionally distinct A. thaliana FLC haplotype. The A. thaliana COOLAIR secondary structure was used to predict COOLAIR exons in evolutionarily divergent Brassicaceae species. These predictions were validated through chemical probing and cloning. Despite relatively low nucleotide sequence identity, the structures, including multi-helix junctions, show remarkable evolutionary conservation. In a number of places the structure is conserved through covariation of non-contiguous DNA sequence. This structural conservation supports a functional role for COOLAIR transcripts themselves, rather than, or in addition to antisense transcription.
Keywords: RNA structure, antisense transcripts, COOLAIR, FLC, chemical probing, A. thaliana, evolution, shotgun secondary structure (3S)
INTRODUCTION
Long non-coding RNAs (lncRNAs) have recently emerged as potentially important players in the epigenetic regulation of development and disease in many organisms. These RNAs are typically 1–10 kb in length, polyadenylated, capped and alternatively spliced (Guttman and Rinn, 2012; Ulitsky and Bartel, 2013). They can be cis- or trans-acting and have been associated with gene regulation in mechanisms including chromatin scaffolding, Polycomb complex (PRC2) recruitment to chromatin, mRNA decay, and decoys for proteins and miRNAs. Specific functional studies have shown lncRNAs to be essential for Xist regulation, paraspeckle formation, lineage commitment, stem cell development, cancer associated effects, co-activation of hormone response, and brain development (Klattenhoff et al., 2013; Novikova et al., 2012; Sauvageau et al., 2013).
While the functional importance of lncRNAs such as Xist is well accepted, more general roles are still controversial, especially in the light of low primary sequence conservation through evolution (Graur et al., 2013; Nitsche et al., 2015). Interestingly, conserved RNA secondary structure can occur despite weak conservation of primary sequence. For example, riboswitches (regulatory RNAs with exquisite control over metabolism in bacteria) typically have nucleic acid sequence identities of only 50–65%, but secondary and tertiary structures conserved across many species (Mandal and Breaker, 2004; Nawrocki et al., 2015; Roth and Breaker, 2009). Likewise, the U2 and U4 spliceosomal RNAs, 5S ribosomal RNA, and group I introns have low sequence identities but highly conserved structures (Nawrocki et al., 2015). In such cases of low sequence identity sequence-based search algorithms (e.g. BLAST) are not generally productive; however, a strategy that aligns syntenic sequences according to chemically-probed structures has proved successful for identifying riboswitch RNAs (Cheah et al., 2007; Weinberg et al., 2009). While few in vivo chemical probing studies on riboswitches have been performed, almost every in vitro chemical probing derived riboswitch structure has been validated with high resolution crystallographic structures (Roth and Breaker, 2009). Nature often evolves structural RNAs with significant changes in helical length or addition/subtraction of entire helices, presenting a formidable challenge for computation. We therefore adopt an integrative approach, proven to be accurate for riboswitches and ribosomes, of time-consuming iteration between chemical probing, secondary structure refinement, sequence alignment refinement and functional studies.
Over the past few years, researchers have been laying the groundwork for lncRNA structure-function studies. Genome-wide studies suggest lncRNAs are more structured than mRNAs, but less structured than ribosomal RNAs (Ding et al., 2014; Quinn et al., 2016; Wan et al., 2012). Studies of MALAT1 and related RNAs show the 3’-end forms a triple helix protecting it from RNase degradation (Brown et al., 2014). Other pioneering studies have examined stem loop related structures (Quinn et al., 2016) and lncRNA-protein interactions (Davidovich et al., 2013). Very few, however, have attempted to determine the secondary structure of complete, intact single lncRNA systems. Those that have revealed hierarchically structured RNAs with sub-domains containing modular RNA secondary structure motifs (Ilik et al., 2013; Novikova et al., 2012; Somarowthu et al., 2015).
We chose to investigate Arabidopsis thaliana antisense lncRNAs, named COOLAIR, which are important in the regulation of a major plant developmental gene FLOWERING LOCUS C (FLC). These initiate just downstream of the protein-coding sense transcript poly(A) site and are alternatively spliced and polyadenylated; either at a proximal site to give ~ 400 nucleotide (nt) class I transcripts or at a distal site within the FLC promoter region to give ~ 750 nt class II transcripts (Figure 1A). These transcripts act in a feedback mechanism linking COOLAIR processing to FLC gene body histone demethylation, reduced FLC transcription, and earlier flowering (Liu et al., 2010). COOLAIR is upregulated during prolonged cold, contributing to a Polycomb-mediated epigenetic switch between opposing chromatin states (Csorba et al., 2014). Whilst the COOLAIR promoter region is evolutionarily conserved, sequence conservation is low in regions corresponding to FLC 5’ and 3’ untranslated and intronic sequences (Castaings et al., 2014; Li et al., 2015b). Whether it is COOLAIR transcription or the COOLAIR transcripts themselves, or both, which are functionally important is not yet known.
Figure 1: Shotgun secondary structure (3S) determination via SHAPE probing of the distal A. thaliana COOLAIR lncRNA.
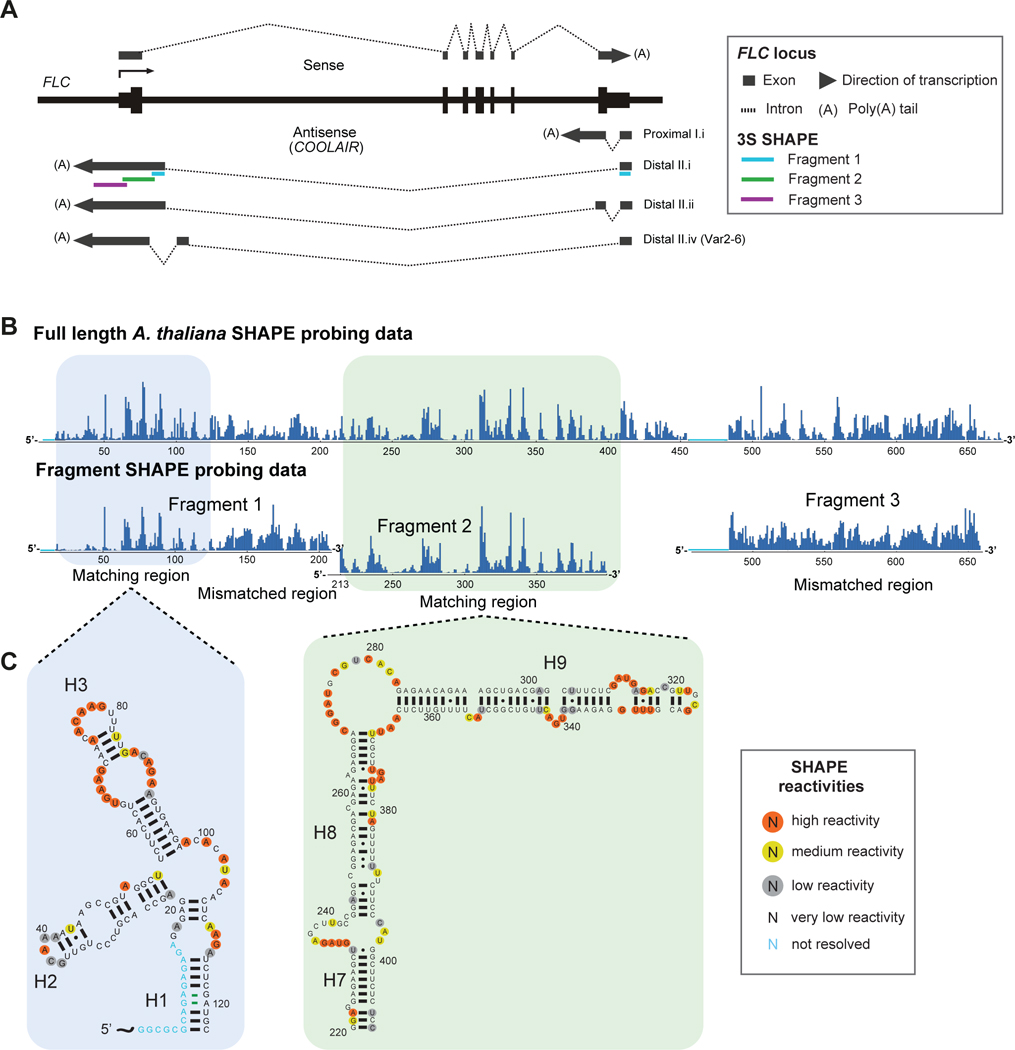
(A) Schematic representation of FLC and COOLAIR transcripts at the FLC locus, with 3S fragment positions mapped. (B) SHAPE reactivities for the full length A. thaliana distal COOLAIR (class II.i) transcript are compared with shorter fragments 1–3 for 3S determination. (C) Modular secondary structure corresponding to reactivity data of the boxed regions in (B).
Here, we apply the riboswitch strategy supplemented with the shotgun secondary structure determination method (3S) to determine the secondary structure of the COOLAIR transcripts (Novikova et al., 2013; Weinberg et al., 2007). We find the distal COOLAIR transcript is highly structured in A. thaliana with numerous secondary structure motifs, an intricate multi-way junction and two unusual asymmetric 5’ internal loops (right-hand turn motifs). Part of this structure is altered by a single non-coding SNP that has been shown to confer functional cis-regulatory variation to a naturally occurring FLC haplotype. The secondary structure was used to predict COOLAIR exonic sequences in a range of evolutionary distinct Brassicaceae species, including Arabidopsis lyrata, Capsella rubella and Brassica rapa, which were then validated in vivo.
RESULTS
Shotgun secondary structure (3S) chemical probing of A. thaliana COOLAIR transcripts
COOLAIR transcripts were probed in vitro using selective 2’-OH acylation analysed by primer extension (SHAPE) (Merino et al., 2005). In addition, to isolate modularly folded regions within COOLAIR, fragments of the full-length distal transcript were probed using 3S (Novikova et al., 2013). We divided the distal class II.i isoform into 3 segments of ~200–250 nts (positions 1–235, 211–433, and 403–658, respectively). In the first fragment, the SHAPE reactivity profile of the 5’ region had significant overlap with the SHAPE reactivity profile of the full RNA (positions ~1–125), suggesting this region possesses an autonomous, modular fold in the context of the full COOLAIR with a well-defined 3-way junction (Figure 1). While the relative ratio in reactivity differed slightly the positions of base paired nucleotides remained the same. The reactivity profile of the 3’ half of fragment 1 (~125–235) differed significantly suggesting that this region forms interactions outside of fragment 1 positions. The reactivity profile of the vast majority of fragment 2 agreed with the full COOLAIR profile suggesting a modular fold with two well-defined helices joined by a large internal loop. Combining 3S fold information from fragments 1 and 2 with SHAPE probing data from the full-length transcript allowed us to produce the secondary structure for the distal COOLAIR II.i transcript (Figure S1), further confirmed by CMCT probing data (Figure 2A).
Figure 2: Secondary structure of the distal and proximal A. thaliana COOLAIR lncRNAs.

(A) Secondary structure of the distal class II.i lncRNA from the A. thaliana Col accession, based on SHAPE and CMCT probing experiments. Normalized SHAPE reactivity represented as colored circles and normalized CMCT reactivity as colored diamonds. A short segment of sequence was replaced with B. rapa sequence to improve reactivity data reads, and to confirm the predicted fold in Figure S1. For the rarity of the structural motifs see Figure S1 and Table S3. (B) Secondary structure of the proximal class I.i lncRNA from the A. thaliana Col accession, based on SHAPE probing experiments. The potential pseudo-knot may be conserved across species (see Figure S4). (C) Secondary structure of the distal class II.iv lncRNA from the A. thaliana Var2–6 accession, based on SHAPE probing experiments.
The distal COOLAIR transcript has a complex structural architecture organized into three distinct domains
The distal COOLAIR lncRNA structure is arranged into 12 helices, seven stem loops, a three-way junction, a five-way junction and two rare right-hand turn motifs (r-turns) (Figure 2A). Nucleotides that exhibit high SHAPE reactivities are mainly located in the terminal loops, internal loops and junction regions, e.g. the terminal loops of helix 3 (H3) and H12, the internal loop separating H7 and H8, and the multi-way junction connecting H5, H6, H7, H10 and H11. Many of these single-stranded regions are purine-rich. This is consistent with the secondary structures of ribosomal RNAs, riboswitch RNAs, SRA-1, and Braveheart, which each show a similar propensity for purine-rich single-stranded locations. Nucleotides restrained by base pairing interactions generally show a much lower tendency toward modification. There are a few select instances where nucleotides involved in base pairing, located close to the single-stranded regions or bulges, can also be reactive towards the SHAPE reagent, such as in H8 and H9. This was observed from SHAPE probing of the 16S rRNA, the secondary structure of which is well known (Noller and Woese, 1981). Minor instances of SHAPE-reactive nucleotides positioned in the central part of helices have also been previously observed in rRNA (Deigan et al., 2009).
COOLAIR appears to be organized into three major domains: the 5’ domain in exon 1 characterised by a 3-way junction, the 3’ major domain (3’ M or ‘central domain’) in exon 2 containing the long helix H4, r-turn and 5-way junction, and the 3’ minor domain (3’ m or ‘stalk’) also in exon 2 and containing the two long helices H8 and H9 connected by the second r-turn. Interestingly, the majority of distal COOLAIR structural features do not correspond to FLC protein exonic regions apart from the stalk domain, which is formed from sequences within exon 1 of the sense transcript. The extensive distal H4 corresponds to sense intronic regions. The sequence underlying the first exon (H1–3) of both the proximal and distal transcripts also corresponds with non-coding sequence.
The right hand turn motif (r-turn)
The secondary structure motifs of COOLAIR are found in many instances of ribosomal RNAs and RNase P RNAs (Table S3), with the exception of the two ‘right-hand turn’ (r-turn) motifs. These are internal loop structures consisting of a large single-stranded region (19 and 13 nts) on the 5’ side and a very short single-stranded region (2 and 3 nts) on the 3’ side, corresponding to type 1 and type 2 r-turns, respectively. Internally, this motif contains two adjacent pairs consisting of either non-canonical GA or canonical Watson-Crick or GU base pairs. We have followed the definition of ‘motif’ used by Moore and co-workers (Klein, et al., 2001). Since the r-turn is well-defined and recurrent, it may play a role in function either directly through binding protein or ligand or indirectly, through positioning helices or engaging in tertiary contacts (Yesselman and Das, 2015). Two recent crystallographic studies revealed similar motifs (Figure S1) in the U6 small nuclear ribonucleoprotein and pistol ribozyme (Montemayor et al., 2014; Ren et al., 2016). For U6, the r-turn is a receptor for a protein forming an extensive interface with multiple RRM regions of the Prp24 protein. In pistol, the r-turn is a receptor for a pseudo-knot interaction. Interestingly, the r-turn occurs in two other lncRNA systems, the steroid receptor RNA activator (SRA-1) and Braveheart AGIL (Novikova et al., 2012; Ren et al., 2016).
The proximal isoform shares the distal 5’-domain structure
We also performed SHAPE probing on the proximally polyadenylated COOLAIR I.i transcript (Figure 2B). This transcript was substantially disordered (high reactivity) with three localized regions of secondary structure. The secondary structure of the 5’-domain for the proximal transcript was identical to that of the distal transcript (H1–3 in both) as they share a common first exon. The 3’-domain of the proximal transcript consists of three helical structures (H4-H6), each capped by a stem loop, with H4 underlying exon 7 of the protein-coding sense transcript. H4 contains three internal loops and an 8-member stem loop. The potential for a pseudo-knot interaction, consistent with the probing data, exists between a stretch of sequence (GGUGGCU) spanning the exon 1/exon 2 splice junction and the first internal loop (AGUCACC) of H4.
Functionally important natural cis polymorphism influences COOLAIR secondary structure
In order to investigate the functional significance of the COOLAIR secondary structure we took advantage of natural variation at FLC. In the experiments above, we probed COOLAIR RNA from the widely used Columbia (Col) accession. Other functionally distinct FLC haplotypes exist in A. thaliana accessions from different parts of the world. Haplotype 11, characterised in the Var2–6 accession from Northern Sweden, contains a SNP that changes the splicing pattern of COOLAIR causing a shift to a downstream distal splice acceptor site and inclusion of an internal exon (Li et al., 2015a). This distal isoform (class II.iv; Figure 1A) co-transcriptionally increases transcription of the FLC nascent transcript thus delaying flowering. We compared the secondary structure of the functionally distinct Var2–6 distal transcript with the Col transcript using SHAPE analysis (Figure 2C vs. 2A). The structure was nearly identical, including the 5’-domain, the first r-turn and multi-way junction, and the stalk. However, there were several significant differences. In the Var2–6 isoform, the 3’ end of the additional exon forms one half of H4 – thus a shorter H4 is maintained in a similar position but composed of entirely different sequence. This supports the need for H4 to be maintained, as without it the first r-turn would not form. In addition to its shorter length (17 vs. 37 bp), H4 contains a large highly reactive internal loop (14 bases) making it likely less stable. The 5’ end of the additional exon is apparently unstructured with many highly reactive bases creating a longer distance between H1 and H4. The 3’ end is more structured, with H12 bifurcated and an additional H13. While four additional polymorphisms found within the Var2–6 haplotype group have been highlighted in Figure 2C, the most significant alteration to the structure is caused by the splice site shift. The U-A SNP in H3 disrupts base pairing and promotes one large terminal loop, in contrast to the internal and smaller terminal loop in Col. Although structurally interesting, this SNP is found in a large number of accessions and is not responsible for the Var2–6 phenotype. The changed functionality of the Var2–6 COOLAIR transcript is therefore most likely to be due to the structural changes associated with H4.
Use of secondary structure to identify COOLAIR in other plant species
Although distal and/or proximal isoforms of COOLAIR have been identified in A. lyrata, A. alpina and B. rapa, low sequence conservation complicated the identification of all isoforms (Castaings et al., 2014; Li et al., 2015b). We derived COOLAIR secondary structures for five Brassicaceae species, A. lyrata, A. alpina, C. rubella, E. salsugineum and B. rapa, representing ca. 13–43 million years divergence from A. thaliana (Beilstein et al., 2010; Koch and Kiefer, 2005). Following the strategy of Weinberg et al., 2007, we scanned syntenic regions for stretches of sequence identity, and then improved the alignment with the chemically-determined A. thaliana structure, using covariant base pairs to help validate helices.
COOLAIR helices H8-H9 are antisense to a highly conserved coding region of FLC (containing the MADS box motif) and were therefore used to align homologous sequences across the five species. Next, stretches of sequence flanking H8-H9 were shifted to improve alignment with helices in A. thaliana. This was repeated and iterated outwards towards the 5’ and 3’ ends. The resulting secondary structures show a high degree of similarity with A. thaliana, maintaining the majority of structural elements (Figure 3). We find the 5’-domain, the two r-turns, the stalk and the terminal region of H4 to be conserved across all six species. These each contain covariant base pair flips in the helices and greater variation in the single stranded regions, supporting the conservation of secondary structure.
Figure 3: Predicted COOLAIR distal lncRNA secondary structures for five Brassicaceae species.
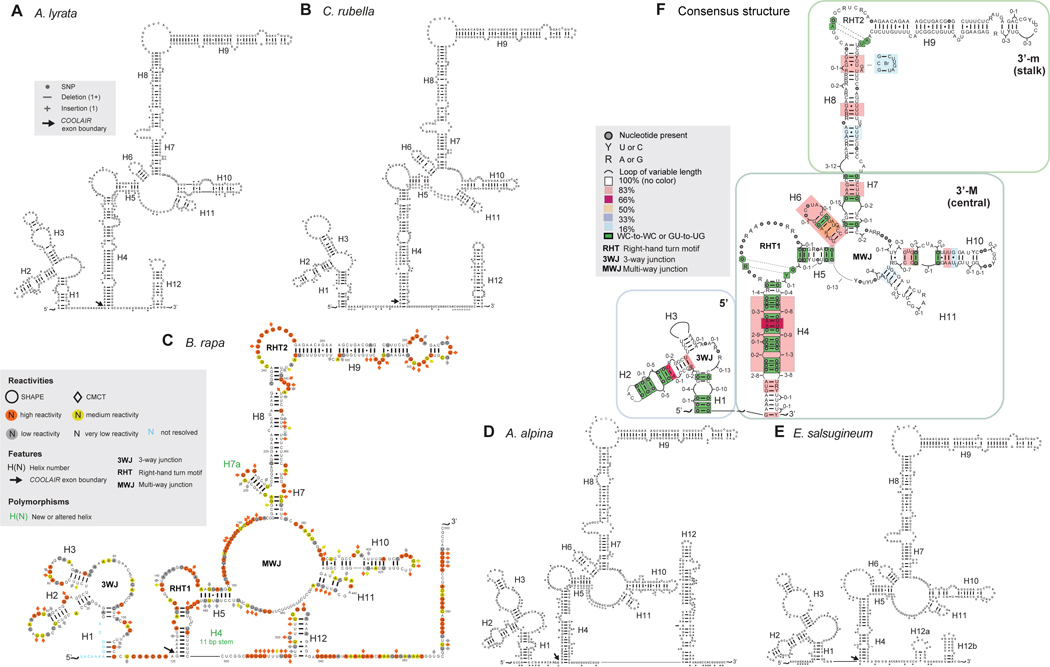
Predicted secondary structure of the distal class II.i transcript of COOLAIR for (A) A. lyrata (FLC1), (B) C. rubella, (C) B. rapa (FLC3), (D) A. Alpina, and (E) E. salsugineum. (C) is annotated with SHAPE and CMCT data, and a variant structure for a different accession is given in Figure S3. Polymorphisms are mapped from pairwise alignment with the A. thaliana COOLAIR distal class II.i transcript, and sequence divergence represented in more detail in Figure S2. (F) Consensus diagram combining structural information from the five species plus A. thaliana shows conservation of secondary structure, where coloured boxes represent the percentage of conservation across species, i.e. pink box = 83% = 5/6 species conserve that structural element. Dots in a looping region signify that the length of the loop is conserved, but the sequence varies. Dots paired to dots in helices signify that a base pair is always present but the sequence varies (i.e., a covariant base pair).
Looking at the consensus structure in Figure 3F, five species contain a 5-way junction, whilst one species (B. rapa, later SHAPE-probed to produce Figure 3C) contains a 4-way junction in the central domain due to lack of H6. H7, H8 and H10 are conserved across 6 species but exhibit some length variation. H11 exists in all six species. Covariant base pairs were found in all helices apart from H3, H8, H9 and H11. The terminal four base pairs of H3 are conserved across all six species. While this helix does not exhibit covariant base pairing from species to species, the length of the helix and loop varies, supporting its existence. A similar situation occurs for H11. As H9 overlaps with a coding region in the sense transcript, it exhibits minimal sequence variation, and therefore has almost no opportunity for base pair covariance. While H11 does not exhibit covariant base pairing, A. lyrata does contain an insertion of 3 base pairs in the helix.
Conservation of key structural features, despite low sequence similarity in non-protein-coding regions (Figure S2), strongly supports a functional role. In effect, nature has maintained these structural features even from sequence of largely, or (in the case of Var2–6 helix H4) completely different composition. Whilst a role for the distal COOLAIR transcript in the cold-induced epigenetic silencing of FLC is perhaps less likely because the two perennial species (A. alpina and A. lyrata) do not exhibit distinct structural features, the Var2–6 data are supportive of a role in setting initial levels of FLC expression in the warm.
Validation of COOLAIR spliced transcripts in evolutionarily diverse species
Primers designed from the predicted secondary structures in Figure 3 confirmed the in vivo presence of the proximal and distal COOLAIR isoforms in A. lyrata, C. rubella and B. rapa (Figure 4A). Three major splice variants were identified and classified according to their similarity to A. thaliana: the proximal class I.i, and the distal II.i and II.ii transcripts (Figure 1A). Splice sites are largely conserved, with the exception of the proximal 3’ acceptor splice site in B. rapa, and the distal class II.ii terminal exon 3’ acceptor site in C. rubella (Figure 4B).
Figure 4: Experimental validation of COOLAIR transcripts in A. lyrata, C. rubella and B. rapa.

(A) RT-PCR experiments probing for the proximal (left) and distal (right) forms of COOLAIR from non-vernalized A. lyrata and C. rubella, and two weeks vernalized B. rapa leaf tissue. Initial RT-PCR (green line) was followed by two rounds of nested PCR (purple and blue lines) to amplify a specific band, where the + column is the cDNA sample and the -RT column is the DNA contamination control. Different splice variants have been labeled, according to A. thaliana classes in Figure 1A. (B) Sequencing the RT-PCR products revealed the major COOLAIR splicing isoforms, with grey boxes in the schematic representing exon positions and triangles representing primer positions. Sequences were aligned with A. thaliana to compare splice sites, highlighted in blue.
Whereas the same proximal isoform is conserved in all four species, two different distal isoforms were identified. Differential distal splicing in A. thaliana accessions (Var2–6 vs. Col) resulted in changes in FLC expression, and so may be equally important across species. Comparison of the loci is complicated by ancient polyploidization and tandem duplication events creating multiple copies of FLC in B. rapa and A. lyrata. We analysed one of four B. rapa copies (FLC3) and one of two A. lyrata copies (FLC1). As loci diverge independently over time, it may be that each expresses unique splicing isoforms. Indeed, a distal COOLAIR isoform with an alternate 3’ acceptor site was recently identified at the FLC2 locus in B. rapa (Li et al., 2015b).
Detection of COOLAIR isoforms with similar architecture to A. thaliana supported the predicted conservation of secondary structure. To further validate this we performed SHAPE and CMCT analysis on the more diverged B. rapa distal COOLAIR (Figure 3C). This class II.i transcript is spliced in the same way as the A. thaliana Col isoform, but contains multiple polymorphisms. We know from Var2–6 that a single SNP can significantly alter secondary structure, but covariance analysis predicted the B. rapa structure would be maintained. We found the 5’-domain, including the 3-way junction, and stalk were conserved, with covariant base pair flips in the helices and greater variation in the single stranded regions. Strong sequence conservation of the protein-coding exon (H8 and H9) retains the second r-turn. Similar to the A. thaliana Var2–6 isoform and the A. alpina and E. salsugineum transcripts, H4 was significantly shorter, partially due to an 11 bp deletion disrupting its 5’ side. The 17 bp stem of H4 in Var2–6 could be responsible for its altered behaviour and late flowering phenotype; the B. rapa distal COOLAIR, with its even shorter stem, may therefore behave more similarly to Var2–6 than Col. B. rapa genotypes exhibit a wide range of morphological and flowering phenotypes; it is interesting to speculate that this could, in part, be a consequence of sequence polymorphism between COOLAIR transcripts. Indeed, we have identified a SNP within H4 between two B. rapa FLC3 alleles that correlates with differences in FLC sense expression and flowering time (Figure S3). Maintenance of even a short H4 preserves the first r-turn, connecting H4 and H5. The multi-way junction is present but contains one less helix (H6) relative to the other species. In addition, H1 and H7 are less stable than for other species, and some helices have shifted or changed in length. H3 has a large terminal loop and no internal loop, reminiscent of the Var2–6 structure. Potential base pairing between the loop of the multi-way junction (which contains 13 nts with low SHAPE reactivities) and the nucleotides forming the 3’ side of H12 could impact tertiary folding.
We have confirmed that covariation of physically separated regions of the primary COOLAIR sequence has maintained COOLAIR secondary structures over evolutionary time. The conserved H8/H9 structure, flanked by a robust and complicated secondary structure unit that shows covariance (first r-turn plus multi-way junction) suggests an important functional role, reinforced by the finding that this region associates with FLC chromatin in chromatin isolation by RNA purification (ChIRP) experiments (Csorba et al., 2014).
DISCUSSION
COOLAIR is a set of antisense RNAs expressed from the A. thaliana FLC locus, different components of which have been shown to regulate expression of FLC. In order to further investigate COOLAIR function we determined the secondary structure of the COOLAIR transcripts using chemical probing experiments. The transcripts were found to be highly modular and organized by exon, suggesting a mix-and-match strategy for lncRNA structure that was also observed in the SRA and HOTAIR lncRNA structures, conserved throughout mammals (Novikova et al., 2012; Somarowthu et al., 2015). The first exon of both the proximal and distal transcripts of COOLAIR is shared whilst their distinct second exons display a conserved core with variations in certain structural elements.
Overall, we find intricate (e.g., multiway junctions as opposed to single stem loops) secondary structures to be conserved in spite of low sequence conservation. Similar phenomena occur in domain IV of the steroid receptor RNA activator, SRA-1, across vertebrates (Sanbonmatsu, 2016). Although commonplace in other RNA systems, this may have implications for lncRNAs, a large number of which have been dismissed as non-conserved. Interestingly, the Bartel lab identified lncRNAs of over 2 kb (megamind and cyrano) that were functionally conserved from zebrafish to human, despite only a 26 nt conserved stretch of sequence (Ulitsky et al., 2011). Likewise, human and mouse local repeats within the mammalian FIRRE lncRNA have only 68% nucleic acid sequence identity and yet share protein-binding functions (Hacisuleyman et al., 2016), while orthologs of the D. melanogaster roX system have low sequence homology but conserved structure and function (Quinn et al., 2016). We have shown that the 3S method finds conserved secondary structures when faced with lncRNAs containing short patches of conserved sequence surrounded by regions with much lower sequence conservation. In light of the large number of such low sequence identity syntenic lncRNAs recently identified, this approach might be useful for other systems (Ulitsky et al., 2015).
COOLAIR exons largely correspond to non-coding sequences from the sense strand, and are relatively poorly conserved by sequence in evolutionarily distant plant relatives. We characterized COOLAIR from a range of species within the Brassicaceae, using the Weinberg et al., 2007 strategy of experimentally probing an RNA of one species to determine its secondary structure and then using this to find COOLAIR in other species We then validated the RNAs through cloning and chemical structure probing of the most evolutionarily distant species analysed. In vivo chemical probing will be an essential tool to complement methods used in the present study (Ding et al., 2014). However, in vivo it is difficult to assign protected bases to RNA helices as protein binding can give similar protection. While in vivo chemical probing will help to further validate these in vitro structures, we emphasize that many in vitro determined structures have been proven in vivo and in crystallographic studies (Noller and Woese, 1981; Roth and Breaker, 2009). The in vitro secondary structure is also a critical step for cryo-EM and crystal structures. Recently, modular domains of the in vitro determined secondary structure of human SRA-1 were validated via binding studies (Arieti et al., 2014; Huet et al., 2014). Additionally, the in vitro secondary structure of the well-characterized mammalian HOTAIR lncRNA was determined to gain insight into how it functions on a molecular level (Somarowthu et al., 2015).
The conservation of COOLAIR structural features, from A. thaliana to B. rapa, suggests they may be involved in FLC regulation. The proximal transcripts are functional in the autonomous pathway mechanism that results in restraint of FLC expression. In addition, an R-loop formed over the COOLAIR promoter represses COOLAIR and FLC expression (Sun et al., 2013). The H1-H3 helices, combined with proximal H4-H6 could be involved in these mechanisms. From our functional (Li et al., 2015a) and structural analysis the distal H4 appears to be an important component of the regulation of FLC transcription in the warm. Its length and stability are significantly altered by the SNP responsible for the Var2–6 late-flowering phenotype. This helix is also shorter in A. alpina, E. salsugineum and B. rapa, the more distant species in our study. We propose that the changed functionality of the Var2–6 COOLAIR transcript therefore results from the structural changes associated with H4. Identification of the COOLAIR interacting protein complex(es) will help us to determine whether this is correct. H4, plus other structures in the distal COOLAIR transcript, including the multi-way junction, may also play a role during vernalization, the process where prolonged cold epigenetically silences FLC. Distal COOLAIR associates with the FLC locus near the nucleation region where chromatin modifications switch from an active H3K36me3 state to an inactive H3K27me3 state (Csorba et al., 2014). By analogy, it is interesting to note that one of the only large RNA crystal structures solved to date (the ribosome) possesses a highly conserved core along with separate variable structures that allow for adaptation. Further COOLAIR studies, including motif deletion and compensatory mutations, will aid in interrogating structure-function relationships including roles in temperature perception. Identifying COOLAIR in more species will allow consensus secondary structure refinement.
In summary, the central domain and stalk of COOLAIR have withstood evolutionary selection, while the variation in H4 length, linked to trait variation, has varied potentially allowing for adaptation to a changing environment. By solving the in vitro secondary structure of COOLAIR, we move a step closer to understanding its role in establishing expression levels of the floral repressor FLC. Clarifying the role of COOLAIR in monitoring long-term exposure to fluctuating temperatures experienced by plants during winter, and how this function has evolved during adaptation, will provide an important paradigm for lncRNA studies.
EXPERIMENTAL PROCEDURES
RNA synthesis, chemical probing and capillary electrophoresis analysis
RNA was synthesized using the Standard RNA IVT kit (CELLSCRIPT, USA) for run-off transcription. For SHAPE probing, folded RNA was probed using 1M7. Parallel RNA samples were treated with DMSO as a blank. For CMCT, 1-cyclohexyl-(2-morpholinoethyl) carbodiimide metho-p-toluene sulfonate (Sigma-Aldrich) was added to 50 mM. Both were reacted for 5 min. at 22 °C and precipitated. The modified sites of RNA were analysed by reverse transcription using site-specific 5’-fluorophore-labeled primers and SuperScript III reverse transcriptase (Life Technologies, USA). The samples, supplemented with the dideoxy terminate sequencing products of Cy3-labeled primer extension, were denatured and loaded on an ABI PRISM 3100-Avant genetic analyzer. Capillary electrophoresis traces will be deposited online in the repository of RNA structure probing (RNA Mapping Database, rmdb.stanford.edu) (Cordero, et al., 2012).
Shotgun secondary structure determination (3S) analysis
In combination with full-length lncRNA analysis, three overlapping fragments covering the COOLAIR distal RNA were probed as in Novikova et al., 2013. Modular regions were determined by comparison to the full-length RNA; non-modular regions were searched for long-range interactions.
Conservation and covariance of secondary structure across species
A. lyrata MN47, C. rubella Monte Gargano, and E. salsugineum Pall. FLC sequences were obtained from Phytozome, A. alpina FJ543377.1 from GenBank, and B. rapa R018 from in-house sequencing. Multiple sequence alignments for a conserved ~250 nt region of the sense coding region of FLC were used as an initial alignment, and improved manually using the 3S secondary structure of A. thaliana COOLAIR, according to Weinberg et al. (2007) and Griffiths-Jones (2005). For the consensus structure (Figure 3F), a conservative approach was used. Only Watson-Crick (WC) base pairs and GU wobble base pairs are reported as pairs. They are not defined as base pairs in the consensus structure if any mutation in any of the six species causes a pair to break (i.e., no bar between bases). Covariant base pairs were reported where at least one base pair flip occurs (WC to WC, or GU to UG; GU-AU and GC-GU transitions are not counted). Only one covariant pair included a GU to UG flip; all others were WC to WC.
RT-PCR analysis of COOLAIR in three species
Total RNA was extracted from non-vernalized A. lyrata MN47 and C. rubella Cr22.5, and vernalized (for two weeks at 4 °C) B. rapa R018 leaf tissue as in Box et al. (2011), DNA removed with TURBO DNA-free™ kit (Ambion), and RNA reverse transcribed with SuperScript® III (Invitrogen) and gene-specific primers. cDNA was amplified by touchdown PCR using GoTaq® DNA Polymerase (Promega), followed by two nested PCRs (Table S1 and S2). RT-PCR products were gel-purified, cloned and sequenced.
Supplementary Material
ACKNOWLEDGEMENTS
KS acknowledges funding from LANL LDRD. CD acknowledges funding from the European Research Council (ERC) under the European Union’s Seventh Framework Programme, the UK Biotechnology and Biological Sciences Research Council (BBSRC) Institute Strategic Program grant BB/C517633/1 (CD and JAI) and BBSRC iCASE Studentship BB/K012371/1 with Bayer CropScience to EH. We would also like to thank Julia Questa, Rebecca Bloomer, and Yiliang Ding for useful contributions and editing of the manuscript and Nicola Stacey for providing C. rubella leaf tissue. We thank Peijin Li, Katie Sanbonmatsu, Bette Korber, Zasha Weinberg, and Ron Breaker for useful discussions on the Var2–6 Arabidopsis accession, covariance and phylogenetics.
REFERENCES
- Arieti F, Gabus C, Tambalo M, Huet T, Round A, and Thore S (2014). The crystal structure of the Split End protein SHARP adds a new layer of complexity to proteins containing RNA recognition motifs. Nucleic Acids Res 42, 6742–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilstein MA, Nagalingum NS, Clements MD, Manchester SR, and Mathews S (2010). Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc Natl Acad Sci USA 107, 18724–18728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Box MS, Coustham V, Dean C, and Mylne JS (2011). Protocol: A simple phenol-based method for 96-well extraction of high quality RNA from Arabidopsis. Plant methods 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Bulkley D, Wang J, Valenstein ML, Yario TA, Steitz TA, and Steitz JA (2014). Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 21, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaings L, Bergonzi S, Albani MC, Kemi U, Savolainen O, and Coupland G (2014). Evolutionary conservation of cold-induced antisense RNAs of FLOWERING LOCUS C in Arabidopsis thaliana perennial relatives. Nat Commun 5, 4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah MT, Wachter A, Sudarsan N, and Breaker RR (2007). Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature 447, 497–500. [DOI] [PubMed] [Google Scholar]
- Cordero P, Lucks JB, and Das R (2012). An RNA Mapping DataBase for curating RNA structure mapping experiments. Bioinformatics 28, 3006–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csorba T, Questa JI, Sun Q, and Dean C (2014). Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc Natl Acad Sci USA 111, 16160–16165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovich C, Zheng L, Goodrich KJ, and Cech TR (2013). Promiscuous RNA binding by Polycomb repressive complex 2. Nat. Struct. Mol. Biol. 20, 1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deigan KE, Li TW, Mathews DH, and Weeks KM (2009). Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci USA 106, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, and Assmann SM (2014). In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700. [DOI] [PubMed] [Google Scholar]
- Graur D, Zheng Y, Price N, Azevedo RB, Zufall RA, and Elhaik E (2013). On the immortality of television sets: “function” in the human genome according to the evolution-free gospel of ENCODE. Genome biology and evolution 5, 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S (2005). RALEE--RNA ALignment editor in Emacs. Bioinformatics 21, 257–259. [DOI] [PubMed] [Google Scholar]
- Guttman M, and Rinn JL (2012). Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacisuleyman E, Shukla CJ, Weiner CL, and Rinn JL (2016). Function and evolution of local repeats in the Firre locus. Nat Commun 7, 11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet T, Miannay FA, Patton JR, and Thore S (2014). Steroid receptor RNA activator (SRA) modification by the human pseudouridine synthase 1 (hPus1p): RNA binding, activity, and atomic model. PLoS One 9, e94610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S, Wan Y, Spitale RC, Luscombe N, Backofen R, et al. (2013). Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol Cell 51, 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, et al. (2013). Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152, 570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DJ, Schmeing TM, Moore PB, and Steitz TA (2001). The kink-turn: a new RNA secondary structure motif. Embo J. 20, 4214–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, and Kiefer M (2005). Genome evolution among cruciferous plants: a lecture from the comparison of the genetic maps of three diploid species - Capsella rubella, Arabidopsis lyrata subsp. petraea, and A. thaliana. Am. J. Bot. 92, 761–767. [DOI] [PubMed] [Google Scholar]
- Li P, Tao Z, and Dean C (2015a). Phenotypic evolution through variation in splicing of the noncoding RNA COOLAIR. Genes Dev. 29, 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang S, Bai J, and He Y (2015b). Tuning growth cycles of Brassica crops via natural antisense transcripts of BrFLC. Plant Biotechnol. J. 14, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Marquardt S, Lister C, Swiezewski S, and Dean C (2010). Targeted 3’ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 327, 94–97. [DOI] [PubMed] [Google Scholar]
- Mandal M, and Breaker RR (2004). Gene regulation by riboswitches. Nat. Rev. Mol. Cell Biol. 5, 451–463. [DOI] [PubMed] [Google Scholar]
- Merino EJ, Wilkinson KA, Coughlan JL, and Weeks KM (2005). RNA structure analysis at single nucleotide resolution by selective 2’-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 127, 4223–4231. [DOI] [PubMed] [Google Scholar]
- Montemayor EJ, Curran EC, Liao HH, Andrews KL, Treba CN, Butcher SE, and Brow DA (2014). Core structure of the U6 small nuclear ribonucleoprotein at 1.7-Å resolution. Nat. Struct. Mol. Biol. 21, 544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, Floden EW, Gardner PP, Jones TA, Tate J, et al. (2015). Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche A, Rose D, Fasold M, Reiche K, and Stadler PF (2015). Comparison of splice sites reveals that long noncoding RNAs are evolutionarily well conserved. RNA 21, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noller HF, and Woese CR (1981). Secondary structure of 16S ribosomal RNA. Science 212, 403–411. [DOI] [PubMed] [Google Scholar]
- Novikova IV, Hennelly SP, and Sanbonmatsu KY (2012). Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res. 40, 5034–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikova IV, Hennelly SP, and Sanbonmatsu KY (2013). 3S: Shotgun Secondary Structure determination for long non-coding RNAs. Methods 63, 170–177. [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Zhang QC, Georgiev P, Ilik IA, Akhtar A, and Chang HY (2016). Rapid evolutionary turnover underlies conserved lncRNA-genome interactions. Genes Dev 30, 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren A, Vusurovic N, Gebetsberger J, Gao P, Juen M, Kreutz C, Micura R, and Patel DJ (2016). Pistol ribozyme adopts a pseudoknot fold facilitating site-specific in-line cleavage. Nat. Chem. Biol. doi: 10.1038/nchembio.2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, and Breaker RR (2009). The structural and functional diversity of metabolite-binding riboswitches. Annu. Rev. Biochem. 78, 305–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanbonmatsu KY (2016). Towards structural classification of long non-coding RNAs. Biochim Biophys Acta 1859, 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, et al. (2013). Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2, e01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somarowthu S, Legiewicz M, Chillon I, Marcia M, Liu F, and Pyle AM (2015). HOTAIR forms an intricate and modular secondary structure. Mol. Cell 58, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Csorba T, Skourti-Stathaki K, Proudfoot NJ, and Dean C (2013). R-loop stabilization represses antisense transcription at the Arabidopsis FLC locus. Science 340, 619–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, and Bartel DP (2013). lincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, and Bartel DP (2011). Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147, 1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, and Bartel DP (2015). Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Reports 11, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Qu K, Ouyang Z, Kertesz M, Li J, Tibshirani R, Makino DL, Nutter RC, Segal E, and Chang HY (2012). Genome-wide Measurement of RNA Folding Energies. Molecular cell 48, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, Gore J, Wang JX, Lee ER, Block KF, Sudarsan N, et al. (2007). Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 35, 4809–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Perreault J, Meyer MM, and Breaker RR (2009). Exceptional structured noncoding RNAs revealed by bacterial metagenome analysis. Nature 462, 656–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesselman JD, and Das R (2015). Modeling Small Non-canonical RNA Motifs with the Rosetta FARFAR Server. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.